Application of Oligonucleotide Microarrays for Bacterial Source Tracking of Environmental Enterococcus sp. Isolates

Abstract
:Introduction
Materials and Methods
Bacterial Isolates
PCR Amplification and Labelling
Microarray Oligonucleotide Probes and Fabrication
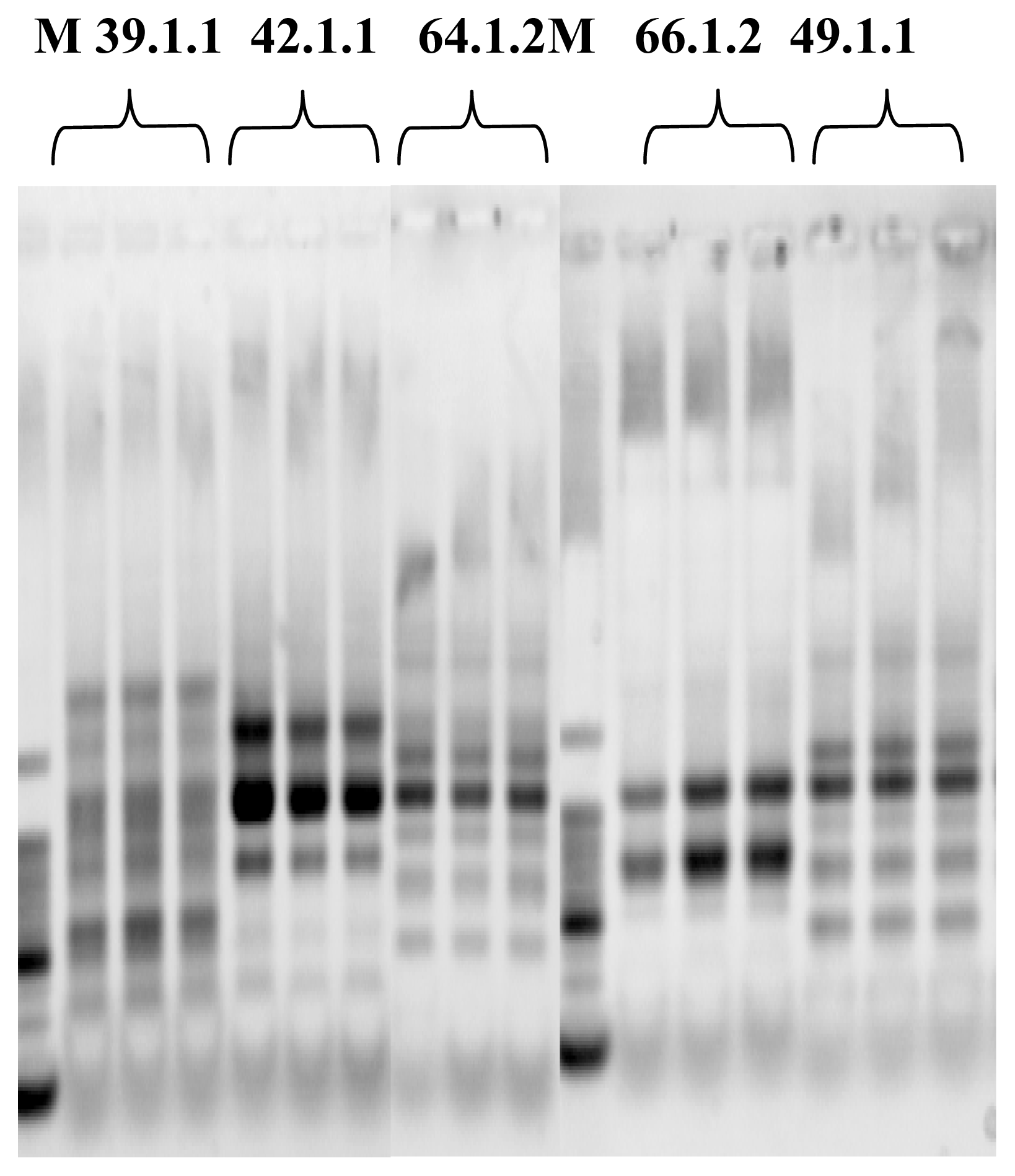
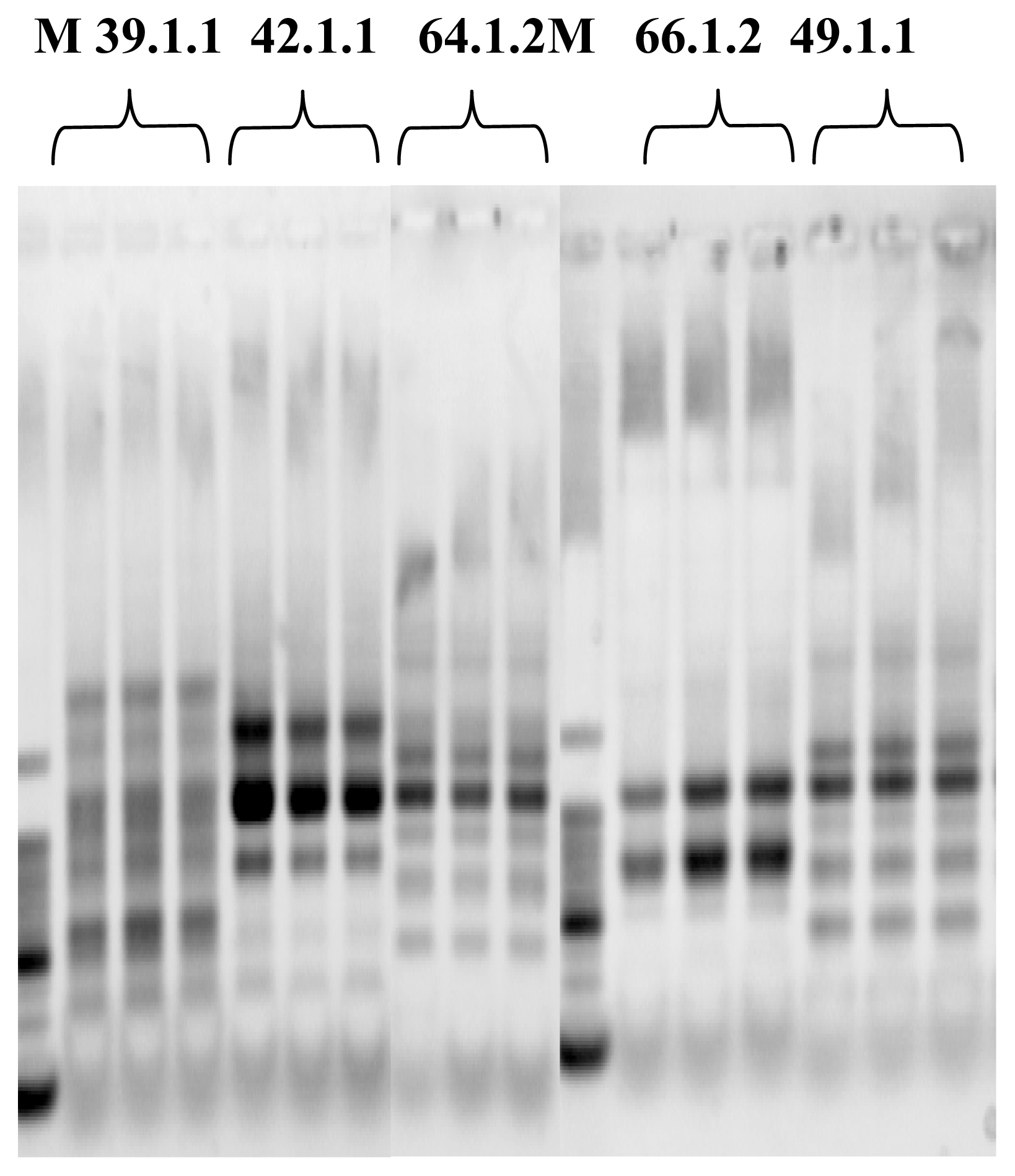
Microarray Hybridization
Image Analysis and Statistics
Results
Oligonucleotide Microarray Bacterial Source Tracking
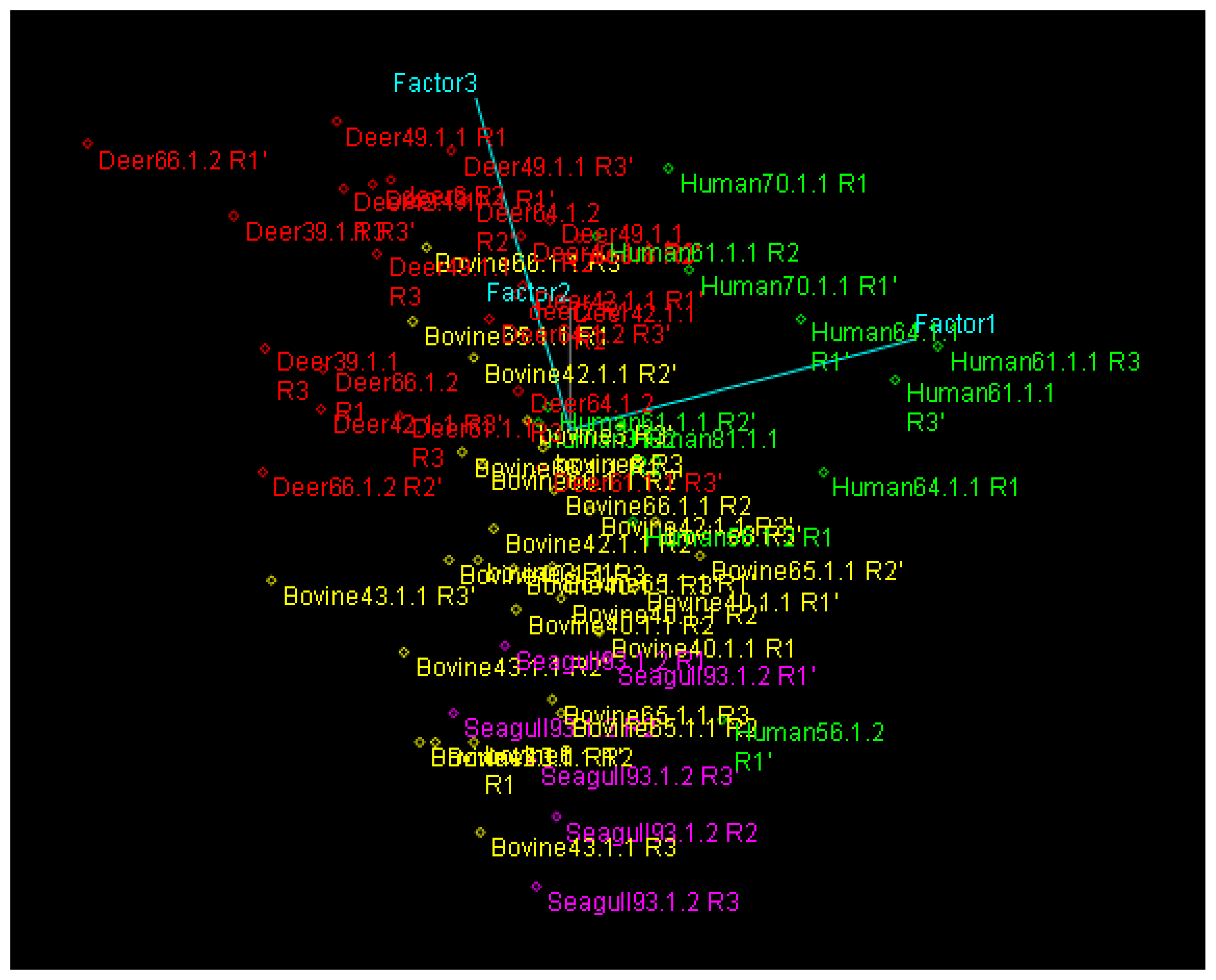
PCA and HCA Analysis
Discussion




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
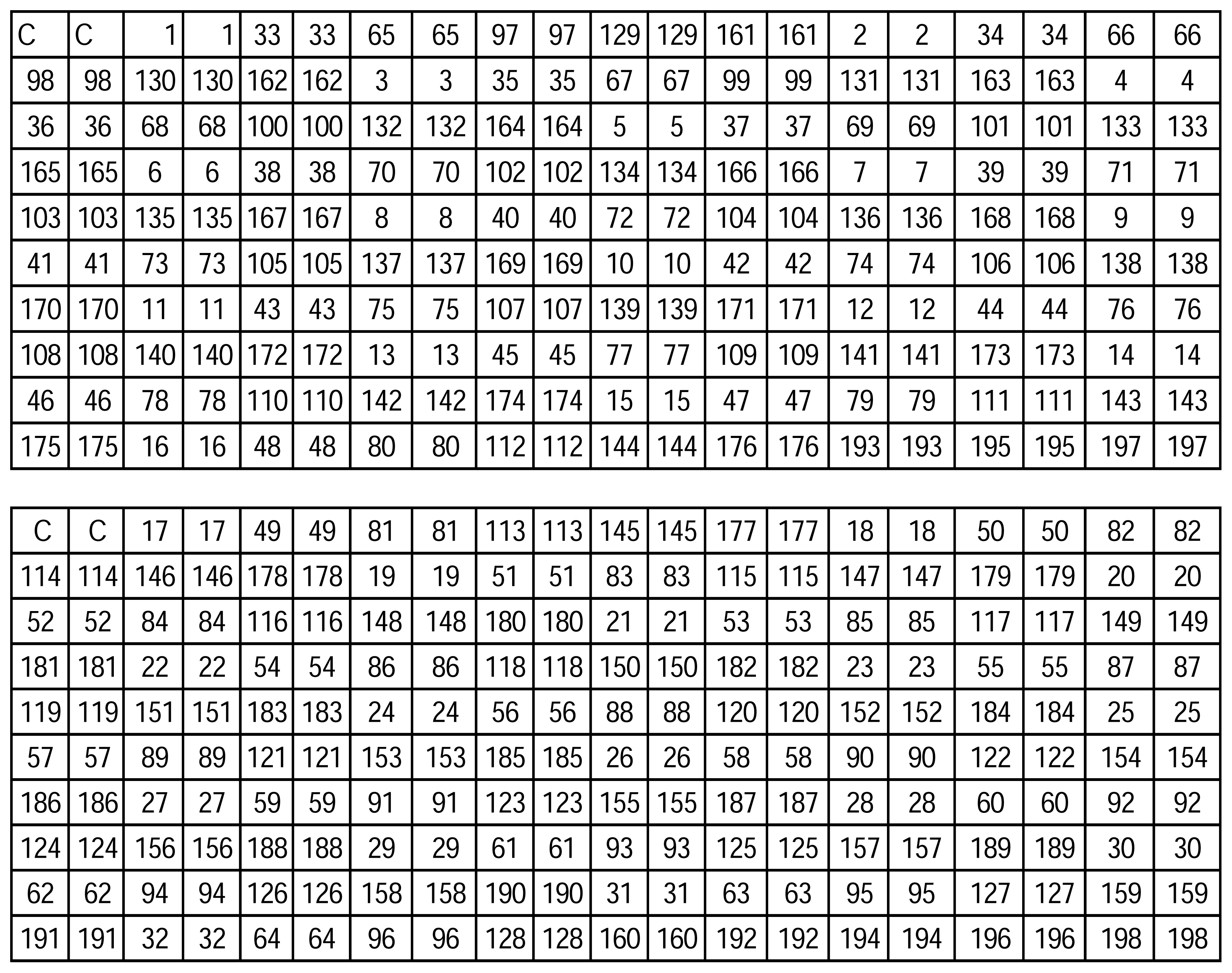
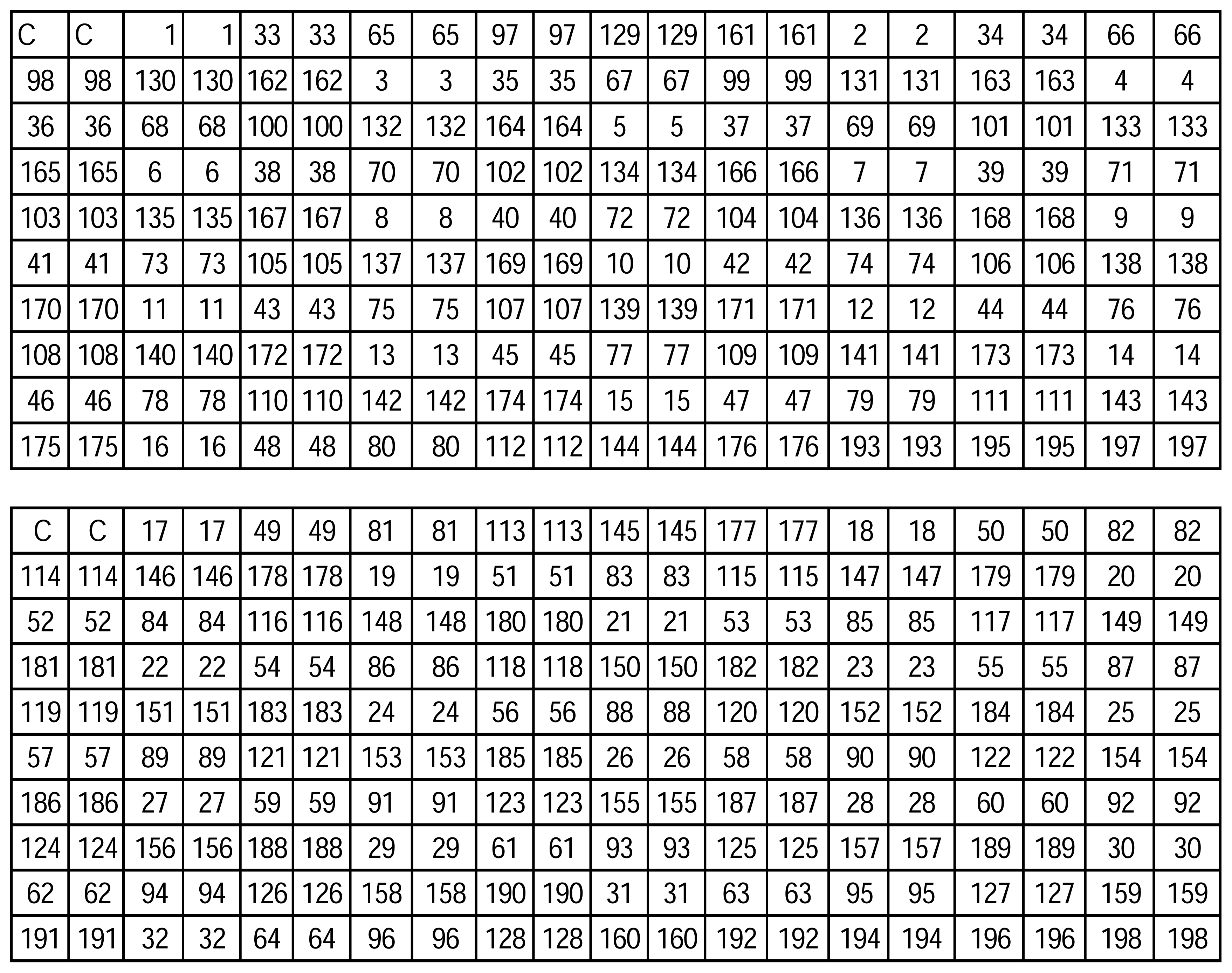
| ID No. | Sequences (5′-3′) |
|---|---|
| 1 | AAATACCCG |
| 2 | CAAATACCC |
| 3 | AATTGCCCT |
| 4 | GGGCCATTT |
| 5 | GACGAGCTT |
| 6 | AGCAGATAG |
| 7 | CTTTCCAGG |
| 8 | ATGACAGAC |
| 9 | TGAGAGGCT |
| 10 | GGTAGTGCT |
| 11 | CATTGTCCG |
| 12 | ATCTCTTGC |
| 13 | CTACCAAGG |
| 14 | AACACTACC |
| 15 | CCATAATCC |
| 16 | GAACTGGCA |
| 17 | CAAATCTGG |
| 18 | GCGATGTTG |
| 19 | AGAGAAGCC |
| 20 | TCAGCGCAT |
| 21 | GCAACCAAA |
| 22 | CTTGATTCC |
| 23 | TACCCACTG |
| 24 | TTACACCGC |
| 25 | CTGCGATCA |
| 26 | GAGCTGTCA |
| 27 | TGGGCGTTT |
| 28 | GGGCGTTTA |
| 29 | CATCTGTCG |
| 30 | AAGTAGCCC |
| 31 | AATATGCGG |
| 32 | GTACGGAGT |
| 33 | TCTGCTATG |
| 34 | CAAATGTCC |
| 35 | AAATCTCGC |
| 36 | AATTTCGGC |
| 37 | ACTCTCCCT |
| 38 | CCAAGTTCT |
| 39 | GAAAGAGCA |
| 40 | CCCTTTCCA |
| 41 | ACCTATGCG |
| 42 | TTGGGTTCG |
| 43 | ATACCGATG |
| 44 | TGCTTCACA |
| 45 | ACGCTACGA |
| 46 | TACTGTCGG |
| 47 | GCTGCTACA |
| 48 | TCCAACTAG |
| 49 | CCGCAAAGT |
| 50 | GATTAGCGC |
| 51 | GGATAGCGA |
| 52 | TATTGGTCG |
| 53 | AAGCAGCAG |
| 54 | CAGACACGA |
| 55 | AAAGTGCCC |
| 56 | CAATCGTTC |
| 57 | AATCCGTAG |
| 58 | CAAGAGGGT |
| 59 | AATGGAACC |
| 60 | CATATCCTC |
| 61 | AACTTGCCG |
| 62 | CATCTTGAC |
| 63 | AAGACAGTG |
| 64 | CACTACGCA |
| 65 | AAGGGATGA |
| 66 | CACGAATCC |
| 67 | ATATCACGG |
| 68 | CAGATGACC |
| 69 | ATAGTCCAG |
| 70 | CAGCAGATG |
| 71 | ATTCACACC |
| 72 | CAGGTGTGT |
| 73 | ATTGGTGGG |
| 74 | CTATACGCA |
| 75 | ATCAGGGAA |
| 76 | CTACACGCA |
| 77 | ATCGAGCCT |
| 78 | CTTATAGGG |
| 79 | ATGTCAAGG |
| 80 | CTTCCATAC |
| 81 | ATGCCGGTT |
| 82 | CTTGGAACC |
| 83 | ATGGACACC |
| 84 | CTCATAGGT |
| 85 | ATGGGTACG |
| 86 | CTCTGTTCC |
| 87 | ACATGACAG |
| 88 | CTCCTTTGC |
| 89 | ACACACCAT |
| 90 | CTCGATCAC |
| 91 | ACAGTCTCA |
| 92 | CTGAGTACA |
| 93 | ACTAAGCGC |
| 94 | CTGTAGACC |
| 95 | ACTTAGCCA |
| 96 | CTGCTACAC |
| 97 | ACTTCGTCG |
| 98 | CTGCTGTGT |
| 99 | ACTCTCTCT |
| 100 | CTGGCTTCT |
| 101 | TGGCTACGT |
| 102 | GGGCTGAAT |
| 103 | TGGCTCGAA |
| 104 | GGCCCATAT |
| 105 | TGCGTACAT |
| 106 | GGCTCAAGA |
| 107 | TGCCCAAGA |
| 108 | GGTTCTGTA |
| 109 | TGCAGAACG |
| 110 | GGTTTGTGT |
| 111 | TGCAAGTTC |
| 112 | GGTAGTTTC |
| 113 | TGTCTATCG |
| 114 | GGACCTAAC |
| 115 | TGAGGATAG |
| 116 | GGAAATCTG |
| 117 | TGATGAGAC |
| 118 | GCGATATTC |
| 119 | GCCAATGTT |
| 120 | TCGCCCTTA |
| 121 | TCGTTATGG |
| 122 | GCTTCCGTT |
| 123 | TCCGAGACT |
| 124 | GCTTGTGAT |
| 125 | TCCGTCAAG |
| 126 | GCTACCTTC |
| 127 | TCCTTGGTT |
| 128 | GCACTCTAA |
| 129 | TCCATCGTG |
| 130 | GCATGTAGG |
| 131 | TCTCGTACC |
| 132 | GTGGGCATT |
| 133 | GCAAAGCCT |
| 134 | TCTTCCTAC |
| 135 | GTGCTGGAT |
| 136 | TCTACCCAC |
| 137 | GTGTAGAAC |
| 138 | TCAGCATAG |
| 139 | GTGAGGTTC |
| 140 | TCAACCTTC |
| 141 | GTCACGTTA |
| 142 | TTGAGCTGA |
| 143 | GTTTCGTGT |
| 144 | TTCGCACTC |
| 145 | GTTAGGGTG |
| 146 | TTCTAGCGC |
| 147 | GTATCGCTA |
| 148 | TTAGCGTGC |
| 149 | GTAACTGTC |
| 150 | TTACCTGGC |
| 151 | GAGGAGATA |
| 152 | TAGCGAGTG |
| 153 | GAGTTTCAG |
| 154 | TAGTCGTCT |
| 155 | GAGAGAAAC |
| 156 | TAGCATAGG |
| 157 | GACTCTACG |
| 158 | TACGGTTCT |
| 159 | GACAGTTCA |
| 160 | TACCCAGTT |
| 161 | GATGATACC |
| 163 | GAAGGAAAG |
| 164 | TAAGCCGCA |
| 165 | GAACTAAGC |
| 166 | AGGCTGTTC |
| 167 | CGGTCAGAT |
| 168 | AGGTAGGAA |
| 169 | CGCCTATGT |
| 170 | AGCCGTACA |
| 171 | CGTGTTCTC |
| 172 | AGCTATGCG |
| 173 | CGTGGTTAT |
| 174 | AGCAAGTGT |
| 175 | CGTCTAACC |
| 176 | AGTCTCAAG |
| 177 | CGAAGTTTG |
| 179 | CGACTGGAA |
| 180 | AGTTACCCT |
| 181 | CGAAACAGG |
| 182 | AGAGTTCGA |
| 183 | CCGTGGAAA |
| 184 | CACAACTCT |
| 185 | AACGAAACG |
| 186 | AACACGCTT |
| 187 | CATGAGGTT |
| 188 | CATAGCGAA |
| 189 | CAAACGAGG |
| 190 | AAACACGTC |
| 191 | CTGCTACGA |
| 192 | ACTAGCGGT |
| 193 | ACAACACTC |
| 194 | CTATGTCGG |
| 195 | CTATCAACC |
| 196 | AACGATACC |
| 197 | CAAACGGGA |
| 198 | CTGTCACTG |
| probe I.D.# | Factor1 | Factor2 | Factor3 | Factor4 | Factor5 |
|---|---|---|---|---|---|
| 10 | −0.0693 | −0.0124 | −0.1724 | 0.3365 | 0.1906 |
| 101 | −0.2427 | −0.0110 | 0.1314 | −0.0107 | −0.0573 |
| 103 | −0.0893 | −0.0051 | 0.1625 | 0.0274 | 0.0169 |
| 109 | 0.0946 | −0.1203 | 0.0935 | −0.2996 | 0.2562 |
| 110 | 0.1979 | 0.1221 | 0.0276 | 0.1051 | −0.0005 |
| 116 | 0.1914 | 0.0207 | 0.1926 | 0.0159 | 0.1193 |
| 120 | −0.0518 | 0.0044 | 0.2388 | 0.1803 | 0.1916 |
| 129 | −0.0098 | −0.1774 | −0.2056 | 0.1891 | 0.0237 |
| 135 | 0.2337 | −0.0282 | −0.0125 | 0.1349 | 0.1671 |
| 139 | −0.0159 | 0.1796 | −0.0037 | −0.0411 | 0.2386 |
| 143 | −0.0029 | −0.1737 | −0.0814 | −0.1052 | −0.2598 |
| 148 | −0.1357 | 0.0204 | 0.2510 | 0.1294 | 0.0086 |
| 151 | 0.0471 | −0.0152 | −0.0363 | 0.3958 | 0.1300 |
| 152 | −0.0195 | −0.2508 | 0.1577 | 0.0328 | −0.1484 |
| 156 | −0.1935 | −0.0510 | 0.1517 | 0.0772 | −0.0967 |
| 16 | 0.2880 | −0.0279 | 0.0481 | 0.0090 | 0.0807 |
| 163 | −0.1135 | −0.1457 | 0.1707 | 0.2306 | −0.0090 |
| 164 | −0.0643 | 0.1756 | 0.1276 | −0.2373 | 0.1324 |
| 173 | −0.0276 | −0.0829 | −0.1689 | −0.1325 | 0.2091 |
| 179 | 0.1752 | 0.1325 | 0.2503 | 0.0267 | 0.0454 |
| 183 | −0.1061 | −0.0186 | 0.1716 | 0.3182 | 0.1273 |
| 188 | 0.0372 | −0.1136 | 0.2749 | −0.0896 | −0.1253 |
| 197 | 0.1127 | 0.1055 | 0.1382 | 0.0709 | −0.2228 |
| 2 | 0.3161 | −0.1013 | −0.0028 | 0.0365 | −0.0345 |
| 23 | 0.0120 | 0.0386 | 0.1942 | −0.2457 | 0.0996 |
| 24 | 0.0043 | −0.1837 | 0.1502 | −0.0565 | −0.1489 |
| 27 | 0.0415 | 0.2624 | 0.0418 | 0.1381 | −0.0770 |
| 3 | −0.1492 | −0.1004 | 0.1142 | 0.0727 | −0.1953 |
| 31 | −0.1750 | −0.0222 | 0.1739 | −0.0174 | −0.0754 |
| 39 | 0.0871 | −0.2652 | 0.0902 | 0.1966 | 0.0277 |
| 42 | −0.0778 | −0.0590 | 0.1760 | −0.0517 | 0.2568 |
| 43 | 0.0425 | −0.2962 | −0.0494 | 0.1284 | 0.0829 |
| 51 | 0.2070 | −0.0761 | 0.0827 | 0.1584 | −0.0281 |
| 52 | 0.1503 | 0.2894 | −0.0144 | 0.0820 | −0.1347 |
| 54 | −0.1305 | 0.1739 | 0.0558 | 0.0927 | 0.2616 |
| 61 | −0.0134 | 0.1594 | 0.3046 | −0.0007 | 0.0549 |
| 63 | 0.2623 | −0.1179 | 0.0312 | 0.0517 | 0.1014 |
| 65 | 0.2654 | 0.1653 | 0.0571 | −0.0874 | −0.0411 |
| 67 | 0.1681 | −0.2348 | −0.0132 | −0.1448 | 0.1331 |
| 7 | 0.2416 | −0.2058 | 0.0358 | −0.0196 | −0.0125 |
| 72 | −0.0466 | 0.1237 | −0.3268 | 0.0042 | 0.1066 |
| 74 | 0.1058 | 0.2964 | 0.0585 | 0.1051 | −0.1957 |
| 76 | −0.0709 | −0.1606 | 0.0171 | −0.1116 | −0.1814 |
| 85 | 0.2414 | 0.0085 | 0.1413 | −0.0517 | −0.0178 |
| 97 | −0.1241 | −0.0890 | 0.1126 | −0.1321 | 0.3621 |
Acknowledgements
References
- Natural Resource Defence Council (NRDC). Testing the Waters 2001; Natural Resources Defense Council: New York, NY, 2002. http://www.nrdc.org/water/oceans/ttw/titinx.asp.
- USEPA, Protocol for Developing Pathogen TMDLs; U. S. Environmental Protection Agency: Washington DC, EPA 841-R-00-00; 2001b.
- For list of impairment by state visit http://www.epa.gov/OWOW/tmdl/
- Mostaghimi, S.; Brannan, K. M.; Dillaha, T. A. Fecal Colifrom TMDL Development: Case Study and Ramifications. Water Resources Update 2002, 122(March 2002), 27–33. [Google Scholar]
- Term first used by Hagedorn and Wiggins at http://www.bsi.vt.edu/biol_4684/BST/BST.html
- Scott, T. M.; Rose, J. B.; Jenkins, T. M.; Farrah, S. R.; Lukasik, J. Microbial Source Tracking: Current Methodology and Future Directions. Appl. Environ. Microbiol 2002, 68, 5796–5803. [Google Scholar]
- Simpson, J. M.; Santo Domingo, J. W.; Reasoner, D. J. Microbial Source Tracking: State of the Science. Environ. Sci. Technol 36, 5279–5288.
- Harwood, V. J.; Whitlock, J.; Withington, V. Classification of antibiotic resistance patterns of indicator bacteria by discriminant analysis: use in predicting the source of fecal contamination in subtropical waters. Appl. Environ. Microbiol 2000, 66, 3698–3704. [Google Scholar]
- Wiggins, B. A. Discriminant analysis of antibiotic resistance patterns in fecal streptococci, a method to differentiate human and animal sources of fecal pollution in natural waters. Appl. Environ. Microbiol 1996, 62, 3997–4002. [Google Scholar]
- Wiggins, B. A.; Andrews, R. W.; Conway, R. A.; Corr, C. L.; Dobratz, E. J.; Dougherty, D. P.; Eppard, J. R.; Knupp, S. R.; Limjoco, M. C.; Mettenburg, J. M.; Rinehardt, J. M.; Sonsino, J.; Torrijos, R. L.; Zimmerman, M. E. Use of antibiotic resistance analysis to identify nonpoint sources of fecal pollution. Appl. Environ. Microbiol 1999, 65, 3483–3486. [Google Scholar]
- Hagedorn, C. S.; Robinson, S. L.; Filtz, J. R.; Grubbs, S. M.; Angier, T. A.; Reneau, R. B. Using antibiotic resistance patterns in the fecal streptococci to determine sources of fecal pollution in a rural Virginia watershed. Appl. Environ. Microbiol 1999, 65, 5522–5531. [Google Scholar]
- Hardwood, V. J. Lessons learned and questions unanswered from 5 years of Bacterial Source Tracking; Marriott Hotel and Conference Center: Irvine, CA, February 5 2002. [Google Scholar]
- Samadpour, M. Microbial Source Tracking: Principles and Practice. U. S. EPA Workshop on Microbial Source Tracking, Marriott Hotel and Conference Center, Irvine, CA, February 5; 2002. [Google Scholar]
- Carson, C. A.; Shear, B. L.; Ellersieck, M. R.; Asfaw, A. Identification of fecal Escherichia coli from humans and animals by ribotyping. Appl. Environ. Microbiol 2001, 67, 1503–1507. [Google Scholar]
- Hartel, P. G.; Summer, J. D.; Hill, J. L.; Collins, J. V.; Entry, J. A.; Segars, W. I. Geographic variability of Escherichia coli ribotypes from animals in Idaho and Georgia. J. Environ. Qual 2002, 31, 1273–1278. [Google Scholar]
- Parveen, S.; Portier, K. M.; Robinson, K.; Edmiston, L.; Tamplin, M. L. Discriminant analysis of ribotype profiles of Escherichia coli for differentiating human and nonhuman sources of fecal pollution. Appl. Environ. Microbiol 1999, 65, 3142–3147. [Google Scholar]
- Bernhard, A. E.; Field, K. G. A PCR assay to discriminate human and ruminant feces on the basis of host differences in Bacteroides-Prevotella genes encoding 16S rRNA. Appl. Environ. Microbiol 2000, 66, 4571–4574. [Google Scholar]
- Bernhard, A. E.; Field, K. G. Identification of nonpoint sources of fecal pollution in coastal waters by using host-specific 16S ribosomal DNA genetic markers from fecal anaerobes. Appl. Environ. Microbiol 2000, 66, 1587–1594. [Google Scholar]
- Dombek, P. E.; Johnson, L. K.; Zimmerley, S. T.; Sadowsky, M. J. Use of repetitive DNA sequences and the PCR to differentiate Escherichia coli isolates from human and animal sources. Appl. Environ. Microbiol 2000, 66, 2572–2577. [Google Scholar]
- Buchan, A.; Alber, M.; Hodson, R. E. Strain-specific differentiation of environmental Escherichia coli isolates via denaturing gradient gel electrophoresis (DGGE) analysis of the 16S–23S intergenic spacer region. FEMS. Microbiol. Ecol 2001, 35, 313–321. [Google Scholar]
- Farnleitner, A. H.; Kreuzinger, N.; Kavka, G. G.; Grillenberger, S.; Rath, J.; Mach, R. L. Simultaneous detection and differentiation of Escherichia coli populations from environmental freshwaters by means of sequence variations in a fragment of the beta-D-glucuronidase gene. Appl. Environ. Microbiol 2000, 66, 1340–1346. [Google Scholar]
- Kariuki, S.; Gilks, C.; Kimari, J.; Obanda, A.; Muyodi, J.; Waiyaki, P.; Hart, C. A. Genotype analysis of Escherichia coli strains isolated from children and chickens living in close contact. Appl. Environ. Microbiol 1999, 65, 472–476. [Google Scholar]
- Dicuonzo, G.; Gherardi, G.; Lorino, G.; Angeletti, S.; Battistoni, F.; Bertuccini, L.; Creti, R.; Di Rosa, R.; Venditti, M.; Baldassarri, L. Antibiotic resistance and genotypic characterization by PFGE of clinical and environmental isolates of enterococci. FEMS. Microbiol. Lett 2001, 201, 205–211. [Google Scholar]
- Parveen, S.; Hodge, N. C.; Stall, R. E.; Farrah, S. R.; Tamplin, M. L. Phenotypic and genotypic characterization of human and nonhuman. Escherichia coli. Water Res 2001, 35, 379–386. [Google Scholar]
- Guan, S.; Xu, R.; Chen, S.; Odumeru, J.; Gyles, C. Development of a procedure for discriminating among Escherichia coli isolates from animal and human sources. Appl. Environ. Microbiol 2002, 68, 2690–2698. [Google Scholar]
- Sharkey, F. H.; Banat, I.; Marchant, R. Detection and Quantification of gene expression in environmental bacteriology. Appl. Environ. Microbiol 2004, 70, 3795–3806. [Google Scholar]
- Willse, A.; Straub, T. M.; Wunschel, S. C.; Small, J. A.; Call, D. R.; Daly, D. S.; Handler, D. P. Quantitative oligonucleotide microarray fingerprinting of Salmonella enterica isolates. Nucleic Acids Res 2004, 32, 1848–1856. [Google Scholar]
- Kingsley, M. T.; Straub, T. M.; Call, D. R.; Daly, D. S.; Wunschel, S. C.; Chandler, D. P. Fingerprinting closely related xanthomonas pathovars with random nonamer oligonucleotide microarrays. Appl. Environ. Microbiol 2002, 68, 6361–6370. [Google Scholar]
- Hassan, W. M.; Wang, S. Y.; Ellender, R. D. Methods to increase the fidelity of rep-PCR fingerprint-based bacterial source tracking efforts. Appl. Environ. Microbiol.
- Sambrook, J.; Fritsch, E. F.; Maniatis, T. Molecularcloning: a laboratory manual, 2nd ed; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, N.Y.
- Gilbert, J.; Hasseman, J.; Cline, R. Microbial genomic DNA aminoallyl labelling for microarrays. The Institute for Genomic Research Standard Operating Procedure #M009, ver 0.3. http://pfgrc.tigr.org/protocols.shtml.
- Butte, A. J.; Kohane, I. S. Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. Pac. Symp. Biocomput 2000, 418–429. [Google Scholar]
- Stoeckel, D. M.; Mathes, M. V.; Hyer, K. E.; Hagedorm, C.; Kator, H.; Lukasik, J.; O’Brien, T. L.; Fenger, T. W.; Samadpour, M.; Stickler, K. M.; Wiggens, B. A. Comparison of seven protocols to identify fecal contamination sources using. Escherichia coli. Environ. Sci. Technol 2004, 38, 6109–6117. [Google Scholar]
© 2005 MDPI. All rights reserved.
Share and Cite
Indest, K.J.; Betts, K.; Furey, J.S. Application of Oligonucleotide Microarrays for Bacterial Source Tracking of Environmental Enterococcus sp. Isolates. Int. J. Environ. Res. Public Health 2005, 2, 175-185. https://doi.org/10.3390/ijerph2005010175
Indest KJ, Betts K, Furey JS. Application of Oligonucleotide Microarrays for Bacterial Source Tracking of Environmental Enterococcus sp. Isolates. International Journal of Environmental Research and Public Health. 2005; 2(1):175-185. https://doi.org/10.3390/ijerph2005010175
Chicago/Turabian StyleIndest, Karl J., Kelley Betts, and John S. Furey. 2005. "Application of Oligonucleotide Microarrays for Bacterial Source Tracking of Environmental Enterococcus sp. Isolates" International Journal of Environmental Research and Public Health 2, no. 1: 175-185. https://doi.org/10.3390/ijerph2005010175