Metabolic Activation of the Tumorigenic Pyrrolizidine Alkaloid, Retrorsine, Leading to DNA Adduct Formation In Vivo

Abstract
:Introduction
Materials and Methods
Materials
Animals and Treatment
In vitro Metabolism of Retrorsine
Preparation of DHP-Modified Calf thymus DNA
Metabolism of Retrorsine in the Presence of Calf Thymus DNA
Liver DNA Isolation
Results
In Vitro Metabolism by Extrahepatic Tissue Microsomes
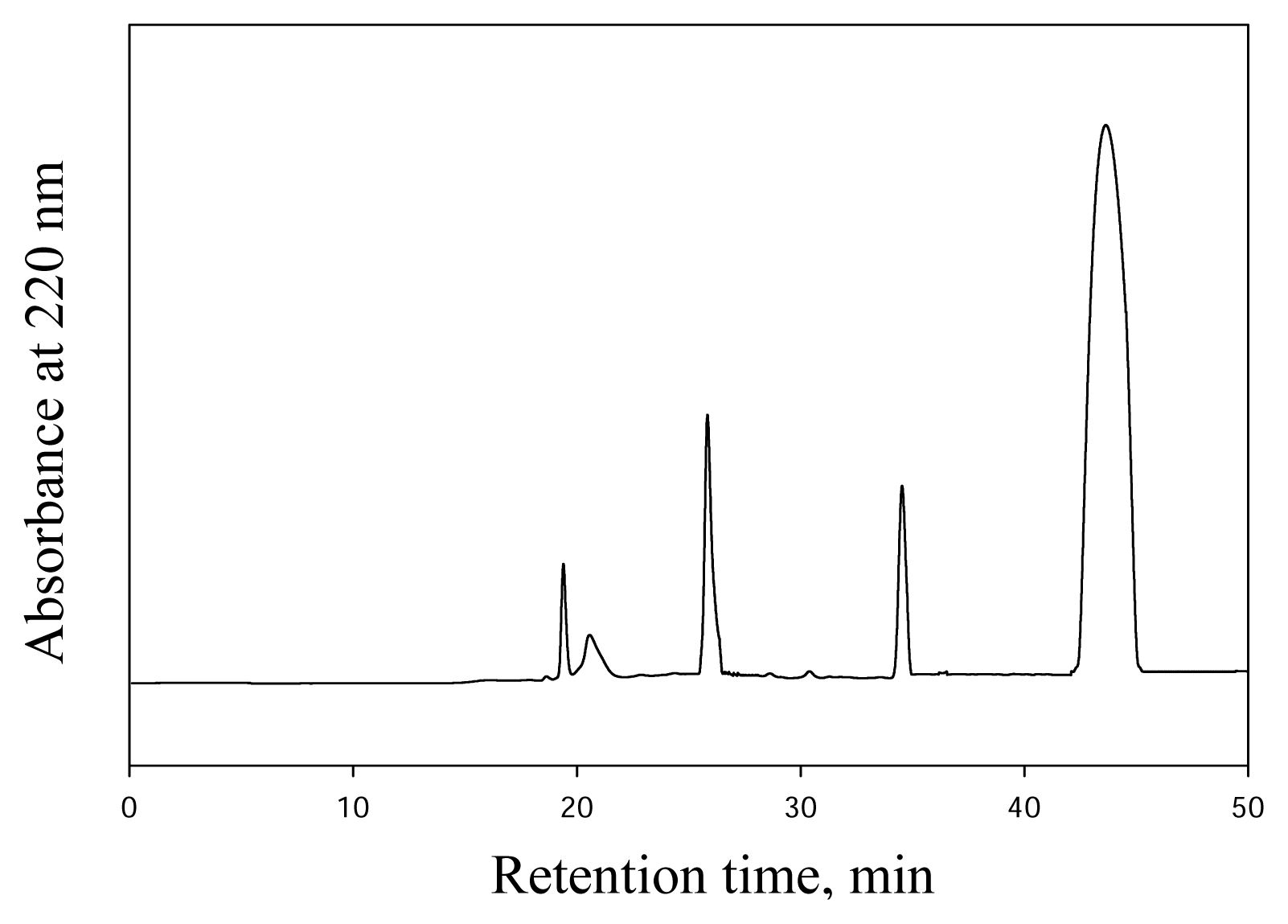
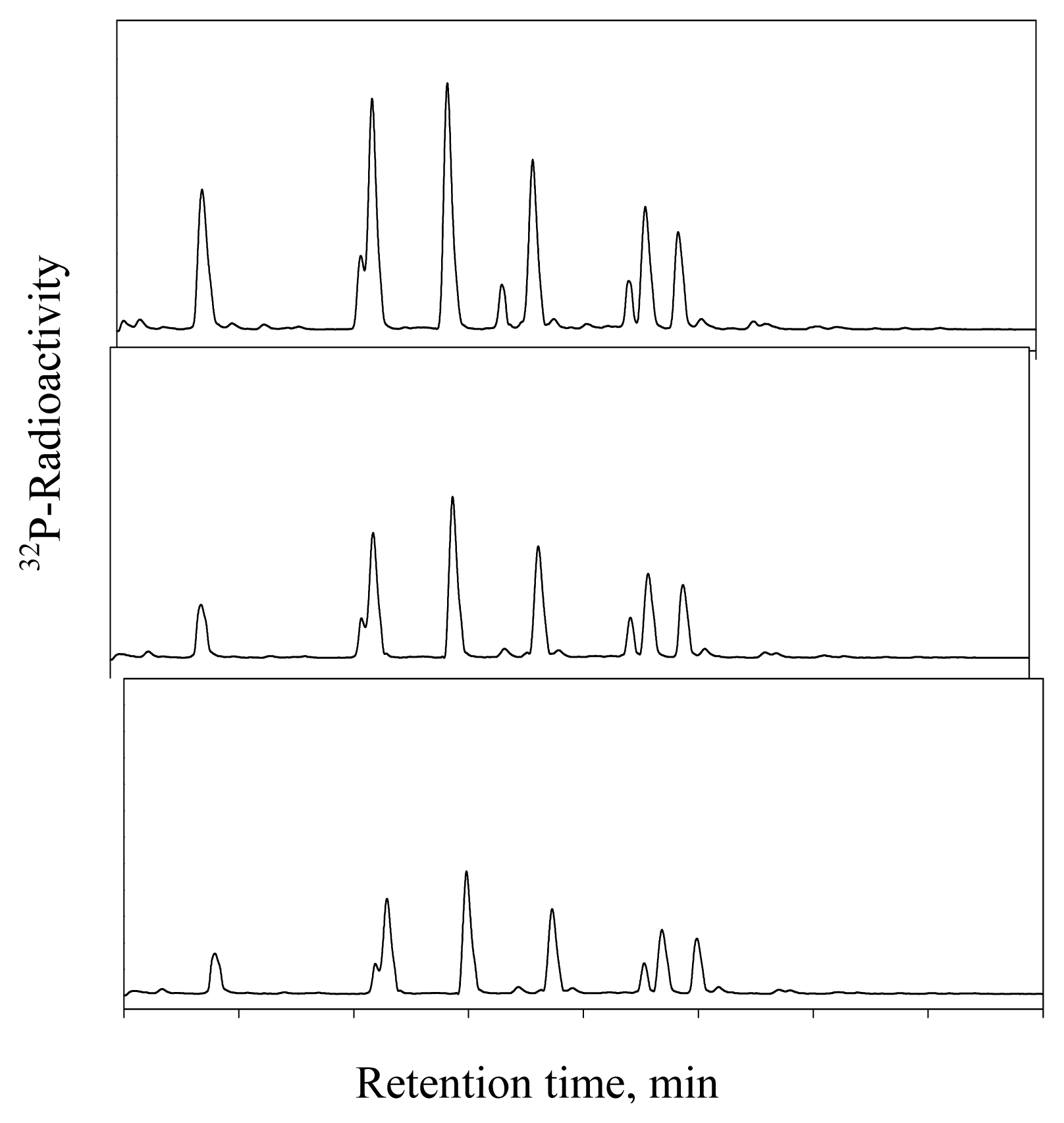
DHP-Derived DNA Adduct Formation
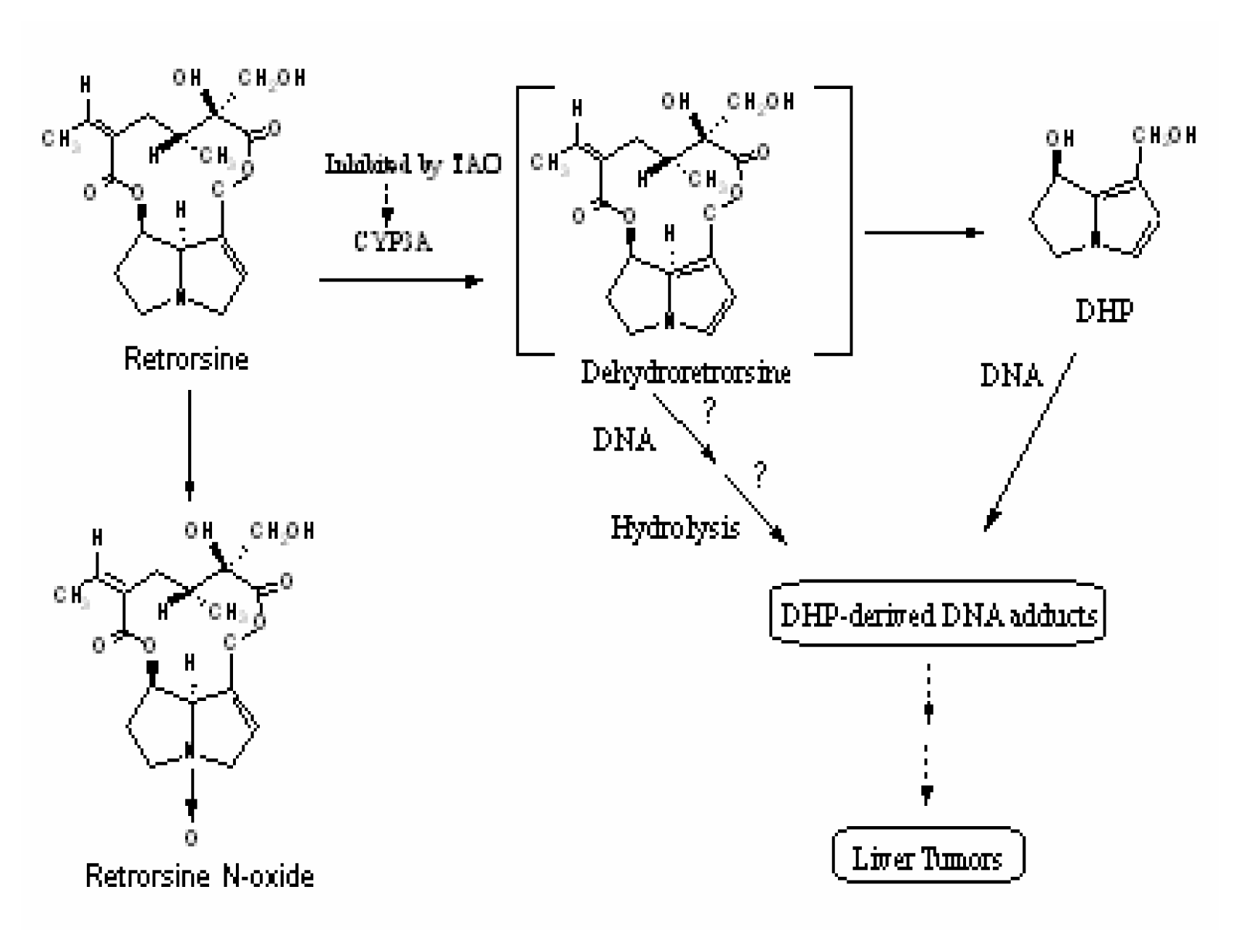
Discussion




{kind=link}
{kind=link}
{kind=link}
| Control-microsomes | Dexamethasone-microsomes | |||
|---|---|---|---|---|
| Substrate | DHP (nmol/mg/min) | N-oxide (nmol/mg/min) | DHP (nmol/mg/min) | N-oxide (nmol/mg/min) |
| Retrorsine | 4.8±0.11,2 | 17.6±0.51,3 | 8.3±0.21,2 | 25.1±0.91,3 |
| Retrorsine + TAO | 1.1±0.041 | 12.4±0.61 | 2.7±0.71 | 17.7±0.91 |
| Microsomes | DHP (pmol/mg/min) | Retrorsine N-oxide (pmol/mg/min) |
|---|---|---|
| Lung | 510 ± 40 | 727 ± 26 |
| Kidney | 79 ± 3 | 627 ± 6 |
| Spleen | 128 ± 6 | 702 ± 39 |
Acknowledgement
References
- Bull, L. B.; Culvenor, C. C.; Dick, A. J. The pyrrolizidine alkaloids, their chemistry, pathogenicity and other biological properties; North-Holland: Amsterdam, 1968. [Google Scholar]
- International Agency for Research in Cancer (IARC), Pyrrolizidine alkaloids. In IARC Monograph on the evaluation of carcinogenic risk of chemicals to man - Some naturally occurring substance; International Agency for Research in Cancer: Lyon, France, 1976.
- Mattocks, A. R. Chemistry and Toxicology of Pyrrolizidine Alkaloids; London, New York: Academic Press, 1986. [Google Scholar]
- Woo, Y. T.; Lai, D. Y.; Arcos, J. C.; Argus, M. F. Chemical Induction of Cancer; Academic Press Inc: San Diego, 1988. [Google Scholar]
- International Programme on Chemical Safety (IPCS), Pyrrolizidine Alkaloids Health and Safety Guide. In Health and Safety Criteria Guide 26; WHO: Geneva, 1989.
- Roeder, E. Medicinal Plans in Europe Containing Pyrrolizidine Alkaloids. Pharmazie 1995, 50(2), 83–98. [Google Scholar]
- Stegelmeier, B. L.; Edgar, J. A.; Colegate, S. M.; Gardner, D. R.; Schoch, T. K.; Coulombe, R. A., Jr.; Molyneux, R. J. Pyrrolizidine Alkaloid Plants, Metabolism and Toxicity. J. Nat. Toxins 1999, 8(1), 95–116. [Google Scholar]
- Coulombe, R. A., Jr.; Drew, G. L.; Stermitz, F. R. Pyrrolizidine Alkaloids Crosslink DNA with Actin. Toxicol. Appl. Pharmacol 1999, 154, 198–202. [Google Scholar]
- Steenkamp, V.; Stewart, M. J.; Zuckerman, M. Clinical and Analytical Aspects of Pyrrolizidine Poisoning Caused by South African Traditional Medicine. Ther. Drug Monit 2000, 22(3), 302–306. [Google Scholar]
- Fu, P. P.; Chou, M. W.; Xia, Q.; Yang, Y. C.; Yan, J.; Doerge, D. R.; Chan, P. C. Genotoxic Pyrrolizidine Alkaloids and Pyrrolizidine Alkaloid N-oxides-Mechanism Leading to DNA Adduct Formation and Tumorgenicity. Environ. Carcinogen Ecotoxicol. Rev 2001, 19, 353–385. [Google Scholar]
- Smith, L. W.; Culvenor, C. C. Plant Sources of Hepatotoxic Pyrrolizidine Alkaloids. J. Nat. Prod 1981, 44(2), 129–152. [Google Scholar]
- Chan, P. C. NTP Technical Report on Toxicology and Carcinogenesis Studies of Riddelliine in F344/N Rats and B6C3F1 Mice. NTP TR 2001, 508. [Google Scholar]
- Yang, Y. C.; Yan, J.; Doerge, D. R.; Chan, P. C.; Fu, P. P.; Chou, M. W. Metabolic Activation of the Tumorgenic Pyrrolizidine Alkaloid, Riddelliine, Leading to DNA adduct Formation in Vivo. Chem. Res. Toxicol 2001, 14(1), 101–109. [Google Scholar]
- Yang, Y. C.; Yan, J.; Churchwell, M.; Beger, R.; Chan, P.; Doerge, D. R.; Fu, P. P.; Chou, M. W. Development of 32P-Postlabeling/ HPLC method for Detection of Dehydroretronecine-Derived DNA Adduct in Vitro and in Vivo. Chem. Res. Toxicol 2001, 14(1), 91–100. [Google Scholar]
- Chou, M. W.; Yan, J.; Williams, L.; Xia, Q.; Churchwell, M.; Doerge, D. R.; Fu, P. P. Identification of DNA Adduct Derived from Riddelliine, a Carcinogenic Pyrrolizidine Alkaloid. Chem. Res. Toxicol 2003, 16(9), 1130–1137. [Google Scholar]
- Chou, M. W.; Yan, J.; Nichols, J.; Xia, Q.; Beland, F. A.; Chan, P. C.; Fu, P. P. Correlation of DNA Adduct Formation and Riddelliine-induced Liver Tumorigenesis in F344 Rats and B6C3F1 Mice. Cancer Lett 2003, 193(2), 119–125. [Google Scholar]
- Chu, P. S.; Lame, M. W.; Segall, H. J. In Vivo Metabolism of Retrorsine and Retrorsine-N-oxide. Arch Toxicol 1993, 67(1), 39–43. [Google Scholar]
- Mattocks, A. R.; White, I. N. Toxic Effects and Pyrrolic Metabolites in the Liver of Young Rats Given the Pyrrolizidine Alkaloid Retrorsine. Chem-Biol. Interactions 1973, 6(5), 297–306. [Google Scholar]
- Chou, M. W.; Wang, Y. P.; Yan, J.; Yang, Y. C.; Beger, R.; Williams, L. D.; Doerge, D. R.; Fu, P. P. Riddelliine N-oxide is a Photochemical and Mammalian Metabolite with Genotoxic Activity That is Comparable to the Parent Pyrrolizidine Alkaloid Riddelliine. Toxicol. Lett 2003, 145(3), 239–247. [Google Scholar]
- Hincks, J. R.; Kim, H. Y.; Segall, H. J.; Molyneux, R. J.; Stermitz, F. R.; Coulombe, R. A., Jr. DNA Cross-linking in Mammalian Cells by Pyrrolizidine Alkaloids, Structure-Activity Relationships. Toxicol Appl Pharmacol 1991, 111(1), 90–98. [Google Scholar]
- Kedzierskiand, B.; Buhler, D. R. The Formation of 6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizine, a Metabolite of Pyrrolizidine Alkaloids. Anal Biochem 1986, 152(1), 59–65. [Google Scholar]
- Chan, M. Y.; Zhao, X. L.; Ogle, C. W. A Comparative Study on the Hepatic Toxicity and Metabolism of Crotalaria Assamica and Eupatorium Species. Am J Chin Med 1989, 17(3–4), 165–170. [Google Scholar]
- Couet, C. E.; Hopley, J.; Hanley, A. B. Metabolic Activation of Pyrrolizdine Alkaloids by Human, Rat and Avocado Microsomes. Toxicon 1996, 34(9), 1058–1061. [Google Scholar]
- Schoental, R.; Head, M. A.; Peacock, P. R. Senecio Alkaloids: a Primary Liver Tumours in Rats as a Results of Treatment with (1) A Mixture of Alkaloids from S. Jakabae Lin; (2) Retrorsine; (3) Isatidine. Br. J. Cancer 1954, 8(3), 458–465. [Google Scholar]
- Reid, M. J.; Lame, M. W.; Morin, D.; Wilson, D. W.; Segall, H. J. Involvement of Cytochrome P450 3A in the Metabolism and Covalent Binding of 14C-Monocrotaline in Rats Liver Microsomes. J. Biochem Mol Toxicol 1998, 12(3), 157–166. [Google Scholar]
- Chung, W. G.; Buhler, D. R. The Effect of Spironolacton Treatment on the Cytochome P450-mediated Metabolism of the Pyrrolizidine Alkaloid Senecionine by Hepatic Microsomes from Rats and Guinea Pigs. Toxicol. Appl. Pharmacol 1994, 127(2), 314–319. [Google Scholar]
- Xia, Q.; Chou, M. W.; Kadlubar, F. F.; Chan, P. C.; Fu, P. P. Human Liver Microsomal Metabolism and DNA Adduct Formation of Tumorigenic Pyrrolizidine Alkaloid, Riddelliine. Chem Res Toxicol 2003, 16(1), 66–73. [Google Scholar]
© 2005 MDPI. All rights reserved.
Share and Cite
Wang, Y.-P.; Fu, P.P.; Chou, M.W. Metabolic Activation of the Tumorigenic Pyrrolizidine Alkaloid, Retrorsine, Leading to DNA Adduct Formation In Vivo. Int. J. Environ. Res. Public Health 2005, 2, 74-79. https://doi.org/10.3390/ijerph2005010074
Wang Y-P, Fu PP, Chou MW. Metabolic Activation of the Tumorigenic Pyrrolizidine Alkaloid, Retrorsine, Leading to DNA Adduct Formation In Vivo. International Journal of Environmental Research and Public Health. 2005; 2(1):74-79. https://doi.org/10.3390/ijerph2005010074
Chicago/Turabian StyleWang, Yu-Ping, Peter P. Fu, and Ming W. Chou. 2005. "Metabolic Activation of the Tumorigenic Pyrrolizidine Alkaloid, Retrorsine, Leading to DNA Adduct Formation In Vivo" International Journal of Environmental Research and Public Health 2, no. 1: 74-79. https://doi.org/10.3390/ijerph2005010074