Alcoholic Pancreatitis: Pathogenesis, Incidence and Treatment with Special Reference to the Associated Pain

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
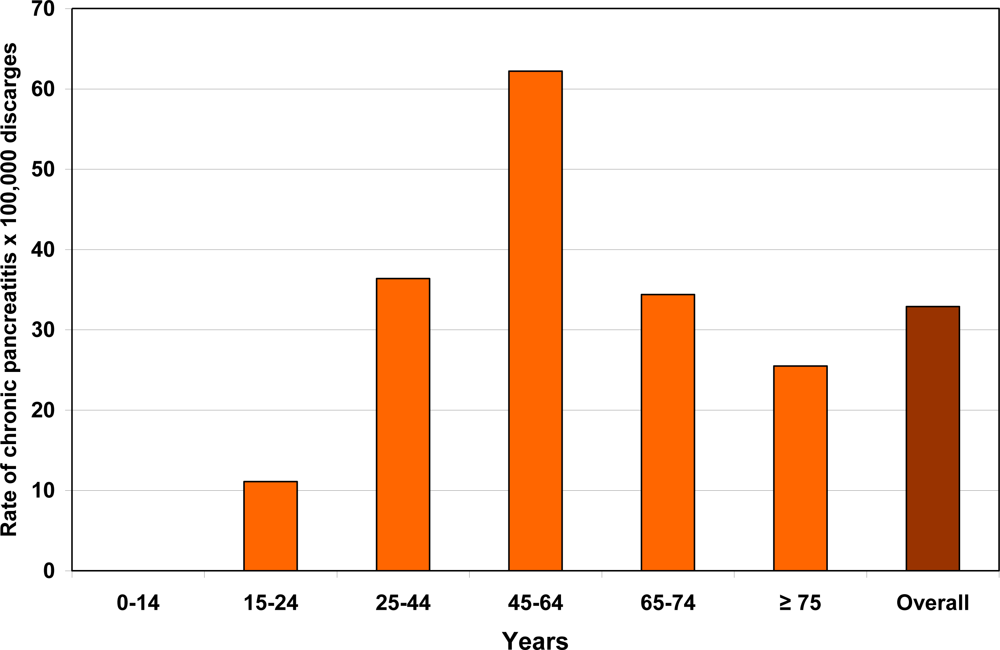
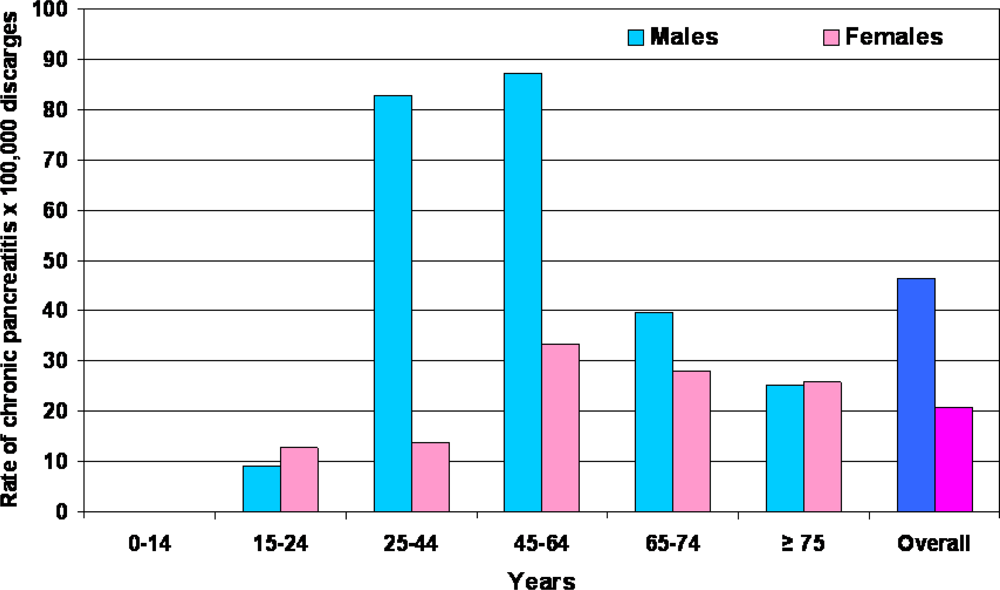
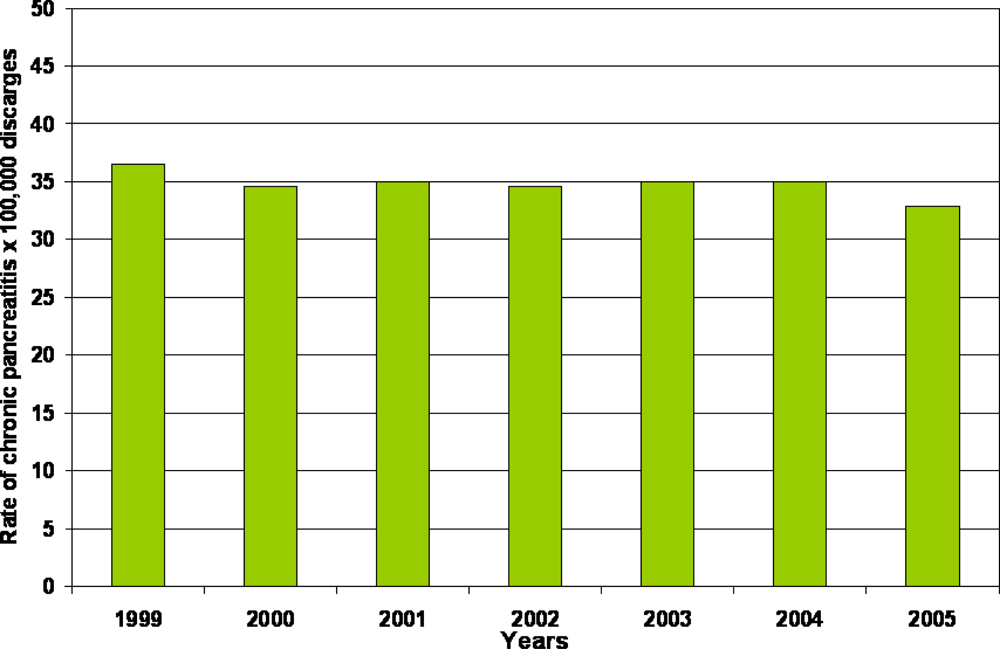
2. Epidemiology
3. The Importance of Alcohol as an Etiological Factor
4. Changing Lifestyle Modifies the Alcoholic Etiology of Chronic Pancreatitis
5. Genetic Factors
6. Modification of the Etiology
7. Pathogenesis of Alcoholic Pancreatitis
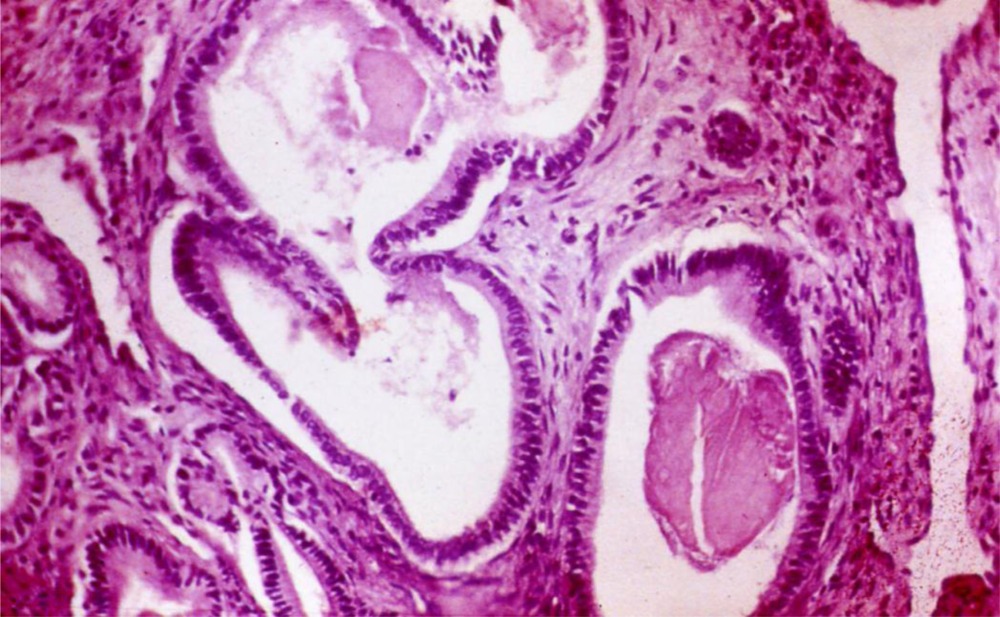
8. Histology of Alcoholic Pancreatitis
9. Alcohol and Pancreatic Inflammation
10. Alcohol and Pancreatic Pain
11. Other Causes of Pain in Chronic Pancreatitis
12. Role of Alcoholic Chronic Pancreatitis in Pancreatic Cancer
13. A New Perspective for the Treatment of Intractable Pain in Chronic Alcoholic Pancreatitis Patients
13.1. Control of the Central Nervous System
13.2. Antoxidants
13.3. Surgical or Endoscopic Approach?
14. Conclusions
References
- Sakorafas, GH; Tsiotou, AG; Peros, G. Mechanisms and natural history of pain in chronic pancreatitis: a surgical perspective. J. Clin. Gastroenterol 2007, 41, 689–699. [Google Scholar]
- Pezzilli, R; Morselli Labate, AM; Ceciliato, R; Frulloni, L; Cavestro, GM; Comparato, G; Ferri, B; Corinaldesi, R; Gullo, L. Quality of life in patients with chronic pancreatitis. Dig. Liver Dis 2005, 37, 181–189. [Google Scholar]
- Frulloni, L; Gabbrielli, A; Pezzilli, R; Zerbi, A; Cavestro, GM; Marotta, F; Falconi, M; Gaia, E; Uomo, G; Maringhini, A; Mutignani, M; Maisonneuve, P; di Carlo, V; Cavallini, G. Chronic pancreatitis: report from a multicenter Italian survey (PanCroInfAISP) on 893 patients. Dig. Liver Dis 2009, 41, 311–317. [Google Scholar]
- Fantini, L; Zanini, N; Fiscaletti, M; Calculli, L; Casadei, R; Campana, D; Pezzilli, R. Autoimmune pancreatitis: the classification puzzle. Adv. Med. Sci 2007, 52, 71–75. [Google Scholar]
- Lowenfels, AB; Maisonneuve, P. Epidemiology and risk factors for pancreatic cancer. Best Pract. Res. Clin. Gastroenterol 2006, 20, 197–209. [Google Scholar]
- Lankisch, PG; Banks, PA. Pancreatitis, 1st Ed ed; Springer-Verlag: Berlin, Germany, 1998. [Google Scholar]
- .
- O’Sullivan, JN; Nobrega, FT; Morlock, CG; Brown, AL, Jr; Bartholomew, LG. Acute and chronic pancreatitis in Rochester, Minnesota, 1940 to 1969. Gastroenterology 1972, 62, 373–379. [Google Scholar]
- James, O; Agnew, JE; Bouchier, IA. Chronic pancreatitis in England: a changing picture. Br. Med. J 1974, 2, 34–38. [Google Scholar]
- Gullo, L; Costa, PL; Labò, G. Chronic pancreatitis in Italy. Aetiological, clinical and histological observations based on 253 cases. Rendic. Gastroenterol 1977, 9, 97–104. [Google Scholar]
- Durbec, JP; Sarles, H. Multicenter survey of the etiology of pancreatic diseases. Relationship between the relative risk of developing chronic pancreatitis and alcohol, protein and lipid consumption. Digestion 1978, 18, 337–350. [Google Scholar]
- Sarles, H; Cros, RC; Bidart, JM. A multicenter inquiry into the etiology of pancreatic diseases. Digestion 1979, 19, 110–125. [Google Scholar]
- Pedersen, TN; Andersen, NB; Pedersen, G; Worning, H. Chronic pancreatitis in Copenhagen. A retrospective study of 64 consecutive patients. Scand. J. Gastroenterol 1982, 17, 925–931. [Google Scholar]
- Ammann, RW; Akovbiantz, A; Largiader, F; Schueler, G. Course and outcome of chronic pancreatitis. Longitudinal study of a mixed medical-surgical series of 245 patients. Gastroenterology 1984, 86, 820–828. [Google Scholar]
- Dzieniszewski, J; Jarosz, M; Ciok, J. Chronic pancreatitis in Warsaw. Mater Med. Pol 1990, 22, 202–204. [Google Scholar]
- Johnson, CD; Hosking, S. National statistics for diet, alcohol consumption, and chronic pancreatitis in England and Wales, 1960–88. Gut 1991, 32, 1401–1405. [Google Scholar]
- Jaakkola, M; Nordback, I. Pancreatitis in Finland between 1970 and 1989. Gut 1993, 34, 1255–1260. [Google Scholar]
- Cavallini, G; Frulloni, L; Pederzoli, P; Talamini, G; Bovo, P; Bassi, C; Di Francesco, V; Vaona, B; Falconi, M; Sartori, N; Angelini, G; Brunori, MP; Filippini, M. Long-term follow-up of patients with chronic pancreatitis in Italy. Scand. J. Gastroenterol 1998, 33, 880–889. [Google Scholar]
- Díte, P; Starý, K; Novotný, I; Precechtelová, M; Dolina, J; Lata, J; Zboril, V. Incidence of chronic pancreatitis in the Czech Republic. Eur. J. Gastroenterol. Hepatol 2001, 13, 749–750. [Google Scholar]
- Dani, R; Mott, CB; Guarita, DR; Nogueira, CE. Epidemiology and etiology of chronic pancreatitis in Brazil: a tale of two cities. Pancreas 1990, 5, 474–478. [Google Scholar]
- Smith, DI; Burvill, PW. Relationship between male pancreatitis morbidity and alcohol consumption in Western Australia, 1971–84. Br. J. Addict 1990, 85, 655–658. [Google Scholar]
- Marks, IN; Girdwood, AH; Bank, S; Louw, JH. The prognosis of alcohol-induced calcific pancreatitis. S. Afr. Med. J 1980, 57, 640–643. [Google Scholar]
- Otsuki, M. Chronic pancreatitis in Japan: epidemiology, prognosis, diagnostic criteria, and future problems. J. Gastroenterol 2003, 38, 315–326. [Google Scholar]
- Lin, Y; Tamakoshi, A; Hayakawa, T; Ogawa, M; Ohno, Y. Associations of alcohol drinking and nutrient intake with chronic pancreatitis: findings from a case-control study in Japan. Am. J. Gastroenterol 2001, 96, 2622–2627. [Google Scholar]
- Das, SK; Balakrishnan, V; Vasudevan, DM. Alcohol: its health and social impact in India. Natl. Med. J. India 2006, 19, 94–99. [Google Scholar]
- WHO Global Status Report on Alcohol 2004; World Health Organization Department of Mental Health and Substance Abuse: Geneva, Switzerland, 2004.
- Gullo, L; Barbara, L; Labò, G. Effect of cessation of alcohol use on the course of pancreatic dysfunction in alcoholic pancreatitis. Gastroenterology 1988, 95, 1063–1068. [Google Scholar]
- Le Bodic, L; Bignon, JD; Raguenes, O; Mercier, B; Georgelin, T; Schnee, M; Soulard, F; Gagne, K; Bonneville, F; Muller, JY; Bachner, L; Férec, C. The hereditary pancreatitis gene maps to long arm of chromosome 7. Hum. Mol. Genet 1996, 5, 549–554. [Google Scholar]
- Whitcomb, DC; Preston, RA; Aston, CE; Sossenheimer, MJ; Barua, PS; Zhang, Y; Wong-Chong, A; White, GJ; Wood, PG; Gates, LK, Jr; Ulrich, C; Martin, SP; Post, JC; Ehrlich, GD. A gene for hereditary pancreatitis maps to chromosome 7q35. Gastroenterology 1996, 110, 1975–1980. [Google Scholar]
- Whitcomb, DC; Gorry, MC; Preston, RA; Furey, W; Sossenheimer, MJ; Ulrich, CD; Martin, SP; Gates, LK, Jr; Amann, ST; Toskes, PP; Liddle, R; McGrath, K; Uomo, G; Post, JC; Ehrlich, GD. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet 1996, 14, 141–145. [Google Scholar]
- Witt, H; Luck, W; Hennies, HC; Classen, M; Kage, A; Lass, U; Landt, O; Becker, M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat. Genet 2000, 25, 213–216. [Google Scholar]
- Keim, V. Genetic risk factors in pancreatic diseases—significance for general practice. Med. Klin. (Munich) 2002, 97, 278–284. [Google Scholar]
- Pezzilli, R; Morselli-Labate, AM; Mantovani, V; Romboli, E; Selva, P; Migliori, M; Corinaldesi, R; Gullo, L. Mutations of the CFTR gene in pancreatic disease. Pancreas 2003, 27, 332–346. [Google Scholar]
- Verlaan, M; Te Morsche, RH; Roelofs, HM; Laheij, RJ; Jansen, JB; Peters, WH; Drenth, JP. Genetic polymorphisms in alcohol-metabolizing enzymes and chronic pancreatitis. Alcohol Alcohol 2004, 39, 20–24. [Google Scholar]
- Ryu, JK; Lee, JK; Kim, YT; Lee, DK; Seo, DW; Lee, KT; Seo, DW; Lee, KT; Kim, HG; Kim, JS; Lee, HS; Kim, TN; Rho, MH; Moon, JH; Lee, J; Choi, HS; Lee, WJ; Yoo, BM; Yoon, YB. Clinical features of chronic pancreatitis in Korea: a multicenter nationwide study. Digestion 2005, 72, 207–211. [Google Scholar]
- Garg, PK; Tandon, RK. Survey on chronic pancreatitis in the Asia-Pacific region. J. Gastroenterol. Hepatol 2004, 19, 998–1004. [Google Scholar]
- Pezzilli, R; Morselli-Labate, AM; Fantini, L; Campana, D; Corinaldesi, R. Assessment of the quality of life in chronic pancreatitis using Sf-12 and EORTC QLQ-C30 questionnaires. Dig. Liver Dis 2007, 39, 1077–1086. [Google Scholar]
- Montalto, G; Carroccio, A; Soresi, M; Ficano, L; Notarbartolo, A. Chronic pancreatitis in Sicily. Preliminary reports. Ital. J. Gastroenterol 1990, 22, 33–35. [Google Scholar]
- Balakrishnan, V; Unnikrishnan, AG; Thomas, V; Choudhuri, G; Veeraraju, P; Singh, SP; Garg, P; Pai, CG; Devi, RN; Bhasin, D; Jayanthi, V; Premalatha, N; Chacko, A; Kar, P; Rai, RR; Rajan, R; Subhalal, N; Mehta, R; Mishra, SP; Dwivedi, M; Vinayakumar, KR; Jain, AK; Biswas, K; Mathai, S; Varghese, J; Ramesh, H; Alexander, T; Philip, J; Raj, VV; Vinodkumar, A; Mukevar, S; Sawant, P; Nair, P; Kumar, H; Sudhindran, S; Dhar, P; Sudheer, OV; Sundaram, KR; Tantri, BV; Singh, D; Nath, TR. Chronic pancreatitis. A prospective nationwide study of 1,086 Subjects from India. JOP. J. Pancreas (Online) 2008, 9, 593–600. [Google Scholar]
- Talukdar, R; Saikia, N; Singal, DK; Tandon, R. Chronic pancreatitis: evolving paradigms. Pancreatology 2006, 6, 440–449. [Google Scholar]
- Morselli-Labate, AM; Fantini, L; Pezzilli, R. Hydrogen sulfide, nitric oxide and a molecular mass 66 u substance in the exhaled breath of chronic pancreatitis patients. Pancreatology 2007, 7, 497–504. [Google Scholar]
- Watari, N; Hotta, Y; Mabuchi, Y. Morphological studies on a vitamin A-storing cell and its complex with macrophage observed in mouse pancreatic tissues following excess vitamin A administration. Okajimas Folia Anat. Jpn 1982, 58, 837–858. [Google Scholar]
- Apte, MV; Haber, PS; Applegate, TL; Norton, ID; McCaughan, GW; Korsten, MA; Pirola, RC; Wilson, JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation and culture. Gut 1998, 43, 128–133. [Google Scholar]
- Bachem, MG; Schneider, E; Gross, H; Weidenbach, H; Schmid, RM; Menke, A; Siech, M; Beger, H; Grünert, A; Adler, G. Identification, culture and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432. [Google Scholar]
- Apte, MV; Phillips, PA; Fahmy, RG; Darby, SJ; Rodgers, SC; McCaughan, GW; Korsten, MA; Pirola, RC; Naidoo, D; Wilson, JS. Does alcohol directly stimulate pancreatic fibrogenesis? Studies with rat pancreatic stellate cells. Gastroenterology 2000, 118, 780–794. [Google Scholar]
- Saotome, T; Inoue, H; Fujiyama, Y; Bamba, T. Morphological and immunocytochemical identification of periacinar fibroblast-like cells derived from human pancreatic acini. Pancreas 1997, 14, 373–382. [Google Scholar]
- Schneider, E; Schmid-Kotsas, A; Zhao, J; Weidenbach, H; Schmid, RM; Menke, A; Adler, G; Waltenberger, J; Grünert, A; Bachem, MG. Identification of mediators stimulating proliferation and matrix synthesis of rat pancreatic stellate cells. Am. J. Physiol. Cell. Physiol 2001, 281, C532–543. [Google Scholar]
- Apte, MV; Haber, PS; Darby, SJ; Rodgers, SC; McCaughan, GW; Korsten, MA; Pirola, RC; Wilson, JS. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut 1999, 44, 534–41. [Google Scholar]
- Phillips, PA; Wu, MJ; Kumar, RK; Doherty, E; McCarroll, JA; Park, S; Pirola, RC; Wilson, JS; Apte, MV. Cell migration: a novel aspect of pancreatic stellate cell biology. Gut 2003, 52, 677–682. [Google Scholar]
- Phillips, PA; McCarroll, JA; Park, S; Wu, MJ; Pirola, R; Korsten, M; Wilson, JS; Apte, MV. Rat pancreatic stellate cells secrete matrix metalloproteinases: implications for extracellular matrix turnover. Gut 2003, 52, 275–82. [Google Scholar]
- Shimizu, K; Kobayashi, M; Tahara, J; Shiratori, K. Cytokines and peroxisome proliferator-activated receptor gamma ligand regulate phagocytosis by pancreatic stellate cells. Gastroenterology 2005, 128, 2105–2118. [Google Scholar]
- Lopez-Ilasaca, M. Signaling from G-protein-coupled receptors to mitogen-activated protein (MAP)-kinase cascades. Biochem. Pharmacol 1998, 56, 269–277. [Google Scholar]
- Masamune, A; Kikuta, K; Satoh, M; Satoh, A; Shimosegawa, T. Alcohol activates activator protein-1 and mitogen-activated protein kinases in rat pancreatic stellate cells. J. Pharmacol. Exp. Ther 2002, 302, 36–42. [Google Scholar]
- McCarroll, JA; Phillips, PA; Park, S; Doherty, E; Pirola, RC; Wilson, JS; Apte, MV. Pancreatic stellate cell activation by ethanol and acetaldehyde: is it mediated by the mitogen-activated protein kinase signaling pathway? Pancreas 2003, 27, 150–160. [Google Scholar]
- McCarroll, JA; Phillips, PA; Kumar, RK; Park, S; Pirola, RC; Wilson, JS; Apte, MV. Pancreatic stellate cell migration: role of the phosphatidylinositol 3-kinase(PI3-kinase) pathway. Biochem. Pharmacol 2004, 67, 1215–1225. [Google Scholar]
- Jaster, R; Sparmann, G; Emmrich, J; Liebe, S. Extracellular signal regulated kinases are key mediators of mitogenic signals in rat pancreatic stellate cells. Gut 2002, 51, 579–584. [Google Scholar]
- Lewis, TS; Shapiro, PS; Ahn, NG. Signal transduction through MAP kinase cascades. Adv. Cancer Res 1998, 74, 49–139. [Google Scholar]
- Chang, L; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar]
- Rebollo, A; Martinez, AC. Ras proteins: recent advances and new functions. Blood 1999, 94, 2971–2980. [Google Scholar]
- Casey, PJ; Solski, PA; Der, CJ; Buss, JE. p21ras is modified by a farnesyl isoprenoid. Proc. Natl. Acad. Sci. USA 1989, 86, 8323–8327. [Google Scholar]
- Goldstein, JL; Brown, MS. Regulation of mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar]
- Ohnishi, H; Miyata, T; Yasuda, H; Satoh, Y; Hanatsuka, K; Kita, H; Ohashi, A; Tamada, K; Makita, N; Iiri, T; Ueda, N; Mashima, H; Sugano, K. Distinct roles of Smad2-, Smad3-, and ERK-dependent pathways in transforming growth factor-beta1 regulation of pancreatic stellate cellular functions. J. Biol. Chem 2004, 279, 8873–8878. [Google Scholar]
- Masamune, A; Kikuta, K; Satoh, M; Sakai, Y; Satoh, A; Shimosegawa, T. Ligands of peroxisome proliferator-activated receptor-gamma block activation of pancreatic stellate cells. J. Biol. Chem 2002, 277, 141–147. [Google Scholar]
- Masamune, A; Kikuta, K; Satoh, M; Suzuki, N; Shimosegawa, T. Protease-activated receptor-2-mediated proliferation and collagen production of rat pancreatic stellate cells. J. Pharmacol. Exp. Ther 2005, 312, 651–658. [Google Scholar]
- Suda, K; Fukumura, Y; Takase, M; Kashiwagi, S; Izumi, M; Kumasaka, T; Suzuki, F. Activated perilobular, not periacinar, pancreatic stellate cells contribute to fibrogenesis in chronic alcoholic pancreatitis. Pathol. Int 2007, 57, 21–25. [Google Scholar]
- Kuroda, J; Suda, K; Hosokawa, Y. Periacinar collagenization in patients with chronic alcoholism. Pathol. Int 1998, 48, 857–868. [Google Scholar]
- Martin, E; Bedossa, P. Diffuse fibrosis of the pancreas: a peculiar pattern of pancreatitis in alcoholic cirrhosis. Gastroenterol. Clin. Biol 1989, 13, 579–584. [Google Scholar]
- Suda, K; Takase, M; Takei, K; Nakamura, T; Akai, J; Nakamura, T. Histopathologic study of coexistent pathologic states in pancreatic fibrosis in patients with chronic alcohol abuse: two distinct pathologic fibrosis entities with different mechanisms. Pancreas 1996, 12, 69–72. [Google Scholar]
- Machado, MCC; Monteiro, JE; Bacchella, T; de Barros Mott, C; Duarte, I; Bettarello, A. Acute pancreatic necrosis in chronic alcoholic pancreatitis. Dig. Dis. Sci 1984, 29, 709–713. [Google Scholar]
- Migliori, M; Manca, M; Santini, D; Pezzilli, R; Gullo, L. Does acute alcoholic pancreatitis precede the chronic form or is the opposite true? A histological study. J. Clin. Gastroenterol 2004, 38, 272–275. [Google Scholar]
- Skinazi, F; Levy, P; Bernades, P. Les pancréatites aigues alcooliques révèlent-elles toujours une pancréatite chronique? Gastroenterol. Clin. Biol 1995, 19, 266–269. [Google Scholar]
- Hanck, C; Singer, MV. Does acute alcoholic pancreatitis exist without preexisting chronic pancreatitis? Scand. J. Gastroenterol 1997, 32, 625–626. [Google Scholar]
- Comfort, M; Gambill, E; Baggenstoss, A. Chronic relapsing pancreatitis: a study of 29 cases without associated disease of the biliary or gastro-intestinal tract. Gastroenterology 1946, 6, 239–285. [Google Scholar]
- Ammann, RW; Heitz, PU; Kloppel, G. Course of alcoholic chronic pancreatitis: a prospective clinicomorphological long-term study. Gastroenterology 1996, 111, 224–231. [Google Scholar]
- Oruc, N; Whitcomb, DC. Theories, mechanisms, and models of alcoholic chronic pancreatitis. Gastroenterol. Clin. N. Am 2004, 33, 733–750. [Google Scholar]
- Pandol, SJ; Raraty, M. Pathobiology of alcoholic pancreatitis. Pancreatology 2007, 7, 105–114. [Google Scholar]
- Singh, M. Alcoholic pancreatitis in rats fed ethanol in a nutritionally adequate liquid diet. Int. J. Pancreatol 1987, 2, 311–324. [Google Scholar]
- Gukovsky, I; Lugea, A; Shahsahebi, M; Cheng, JH; Hong, PP; Jung, YJ; Deng, QG; French, BA; Lungo, W; French, SW; Tsukamoto, H; Pandol, SJ. A rat model reproducing key pathological responses of alcoholic chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol 2008, 294, G68–79. [Google Scholar]
- Michalski, CW; Selvaggi, F; Bartel, M; Mitkus, T; Gorbachevski, A; Giese, T; Sebastiano, PD; Giese, NA; Friess, H. Altered anti-inflammatory response of mononuclear cells to neuropeptide PACAP is associated with deregulation of NF-kappaB in chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol 2008, 294, G50–57. [Google Scholar]
- Bockman, DE; Buchler, M; Malfertheiner, P; Beger, HG. Analysis of nerves in chronic pancreatitis. Gastroenterology 1988, 115, 1459–1469. [Google Scholar]
- Xu, GY; Winston, JH; Shenoy, M; Yin, H; Pendyala, S; Pasricha, PJ. Transient receptor potential vanilloid 1 mediates hyperalgesia and is up-regulated in rats with chronic pancreatitis. Gastroenterology 2007, 133, 1282–1292. [Google Scholar]
- Wick, EC; Hoge, SG; Grahn, SW; Kim, E; Divino, LA; Grady, EF; Bunnett, NW; Kirkwood, KS. Transient receptor potential vanilloid 1, calcitonin gene-related peptide, and substance P mediate nociception in acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol 2006, 290, G959–569. [Google Scholar]
- Vera-Portocarrero, L; Westlund, KN. Role of neurogenic inflammation in pancreatitis and pancreatic pain. Neurosignals 2005, 14, 158–165. [Google Scholar]
- Dimcevski, G; Sami, SA; Funch-Jensen, P; Le Pera, D; Valeriani, M; Arendt-Nielsen, L; Drewes, AM. Pain in chronic pancreatitis: the role of reorganisation in the central nervous system. Gastroenterology 2007, 132, 1546–1556. [Google Scholar]
- Drewes, AM; Krarup, AL; Detlefsen, S; Malmstrøm, ML; Dimcevski, G; Funch-Jensen, P. Pain in chronic pancreatitis: the role of neuropathic pain mechanisms. Gut 2008, 57, 616–627. [Google Scholar]
- Drewes, AM; Gratkowski, M; Sami, SA; Dimcevski, G; Funch-Jensen, P; Arendt-Nielsen, L. Is the pain in chronic pancreatitis of neuropathic origin? Support from EEG studies during experimental pain. World J. Gastroenterol 2008, 14, 4020–4027. [Google Scholar]
- Vijungco, JD; Prinz, RA. Management of biliary and duodenal complications of chronic pancreatitis. World J. Surg 2003, 27, 1258–1270. [Google Scholar]
- Lowenfels, AB; Maisonneuve, P; Lankisch, PG. Chronic pancreatitis and other risk factors for pancreatic cancer. Gastroenterol. Clin. North Am 1999, 28, 673–685. [Google Scholar]
- Lowenfels, AB; Maisonneuve, P; Cavallini, G; Ammann, RW; Lankisch, PG; Andersen, JR; Dimagno, EP; Andrén-Sandberg, A; Domellöf, L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N. Engl. J. Med 1993, 328, 1433–1437. [Google Scholar]
- Lieb, JG, II; Shuster, JJ; Theriaque, D; Curington, C; Cintrón, M; Toskes, PP. A pilot study of Octreotide LAR® vs. Octreotide tid for pain and quality of life in chronic pancreatitis. JOP. J. Pancreas (Online) 2009, 10, 518–522. [Google Scholar]
- Kongkam, P; Wagner, DL; Sherman, S; Fogel, EL; Whittaker, SC; Watkins, JL; McHenry, L; Lehman, GA. Intrathecal narcotic infusion pumps for intractable pain of chronic pancreatitis: a pilot series. Am. J. Gastroenterol 2009, 104, 1249–1255. [Google Scholar]
- Dahm, P; Nitescu, P; Appelgren, L; Curelaru, I. Efficacy and technical complications of long-term continuous intraspinal infusions of opioid and/or bupivacaine in refractory nonmalignant pain: a comparison between the epidural and the intrathecal approach with externalized or implanted catheters and in. Clin. J. Pain 1998, 14, 4–16. [Google Scholar]
- Pezzilli, R; Fantini, L. Antioxidants and pain control in patients with chronic pancreatitis: a never-ending story. Turk. J. Med. Sci 2007, 37, 3–6. [Google Scholar]
- Bhardwaj, P; Garg, PK; Maulik, SK; Saraya, A; Tandon, RK; Acharya, SK. A randomized controlled trial of antioxidant supplementation for pain relief in patients with chronic pancreatitis. Gastroenterology 2009, 136, 149–159. [Google Scholar]
- Dite, P; Ruzicka, M; Zboril, V; Novotny, I. A prospective, randomized trial comparing endoscopic and surgical therapy for chronic pancreatitis. Endoscopy 2003, 35, 553–558. [Google Scholar]
- Costamagna, G; Bulajic, M; Tringali, A; Pandolfi, M; Gabbrielli, A; Spada, C; Petruzziello, L; Familiari, P; Mutignani, M. Multiple stenting of refractory pancreatic duct strictures in severe chronic pancreatitis: long-term results. Endoscopy 2006, 38, 254–259. [Google Scholar]
- Weber, A; Schneider, J; Neu, B; Meining, A; Born, P; Schmid, RM; Prinz, C. Endoscopic stent therapy for patients with chronic pancreatitis: results from a prospective follow-up study. Pancreas 2007, 34, 287–294. [Google Scholar]
- Cahen, DL; Gouma, DJ; Nio, Y; Rauws, EA; Boermeester, MA; Busch, OR; Stoker, J; Laméris, JS; Dijkgraaf, MG; Huibregtse, K; Bruno, MJ. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N. Engl. J. Med 2007, 356, 676–684. [Google Scholar]




© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pezzilli, R.; Morselli-Labate, A.M. Alcoholic Pancreatitis: Pathogenesis, Incidence and Treatment with Special Reference to the Associated Pain. Int. J. Environ. Res. Public Health 2009, 6, 2763-2782. https://doi.org/10.3390/ijerph6112763
Pezzilli R, Morselli-Labate AM. Alcoholic Pancreatitis: Pathogenesis, Incidence and Treatment with Special Reference to the Associated Pain. International Journal of Environmental Research and Public Health. 2009; 6(11):2763-2782. https://doi.org/10.3390/ijerph6112763
Chicago/Turabian StylePezzilli, Raffaele, and Antonio M. Morselli-Labate. 2009. "Alcoholic Pancreatitis: Pathogenesis, Incidence and Treatment with Special Reference to the Associated Pain" International Journal of Environmental Research and Public Health 6, no. 11: 2763-2782. https://doi.org/10.3390/ijerph6112763