Arsenic Trioxide Modulates DNA Synthesis and Apoptosis in Lung Carcinoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
Cell Lines and Chemicals
Cell Culture
[3H]Thymidine Incorporation Assay
Caspase-3 FITC Assay
Cell Cycle Analysis
P38 MAP Kinase Assay
Statistical Analysis
3. Results
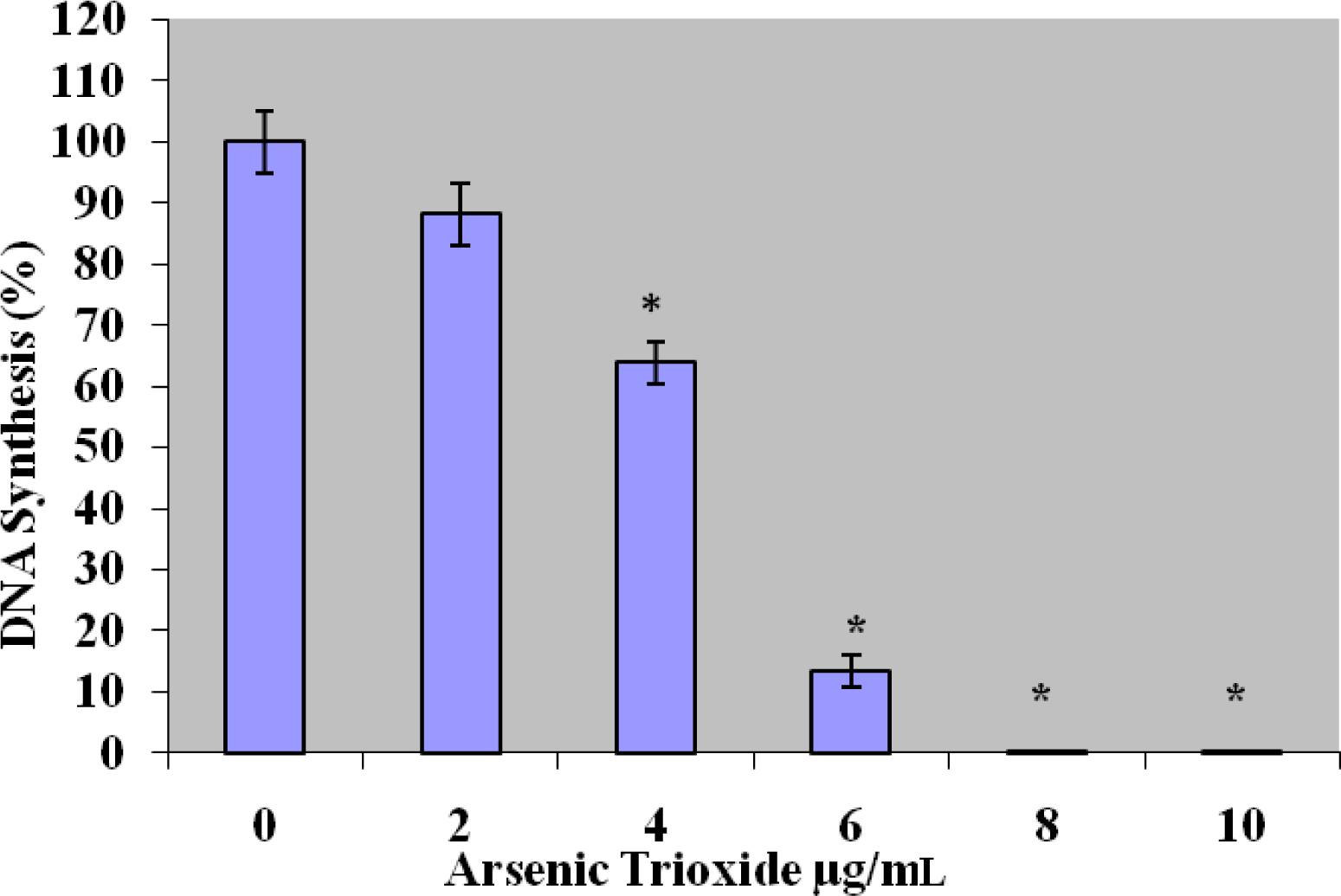
[3H]Thymidine Incorporation Assay
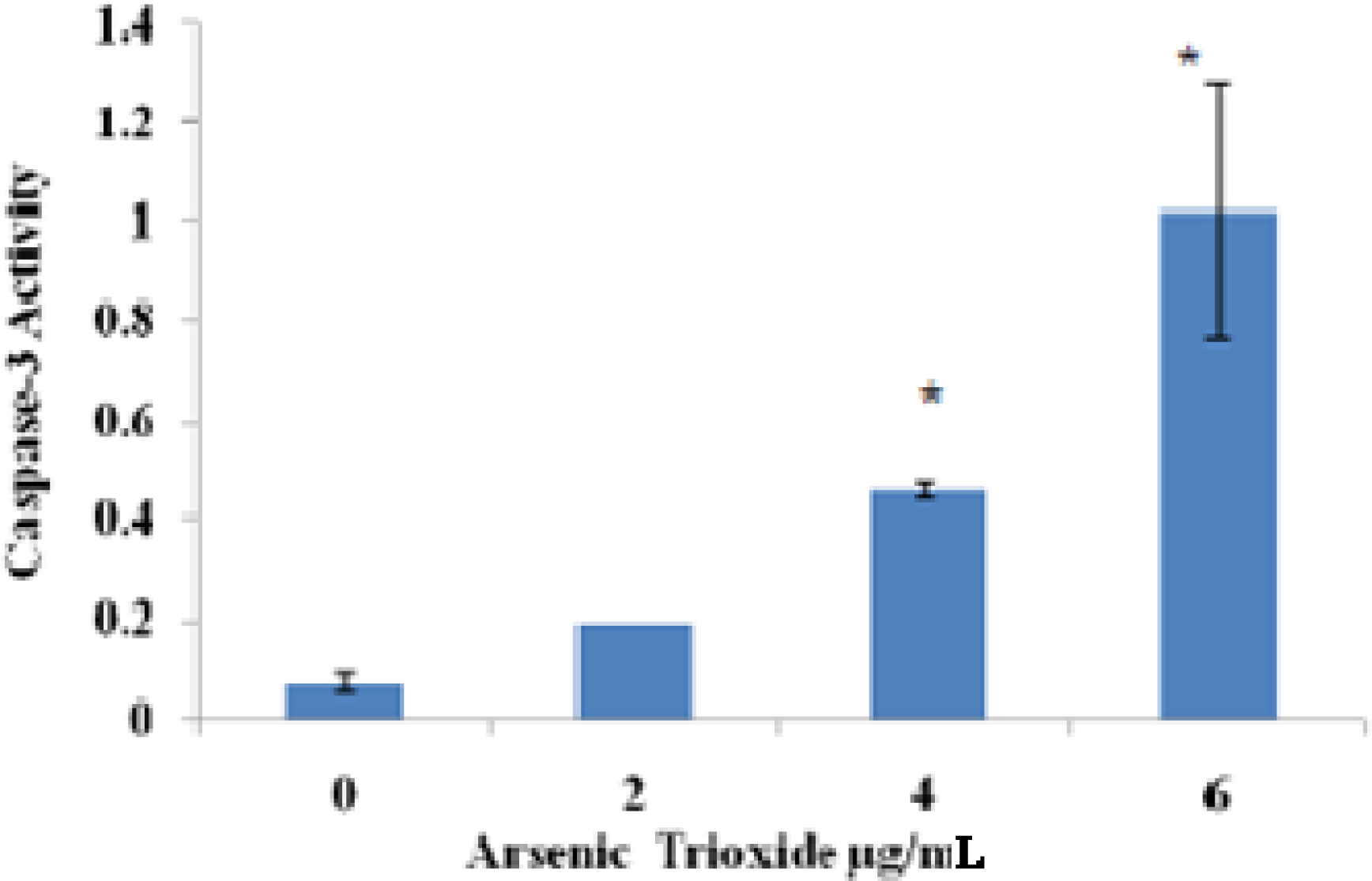
Caspase 3-FITC Analysis
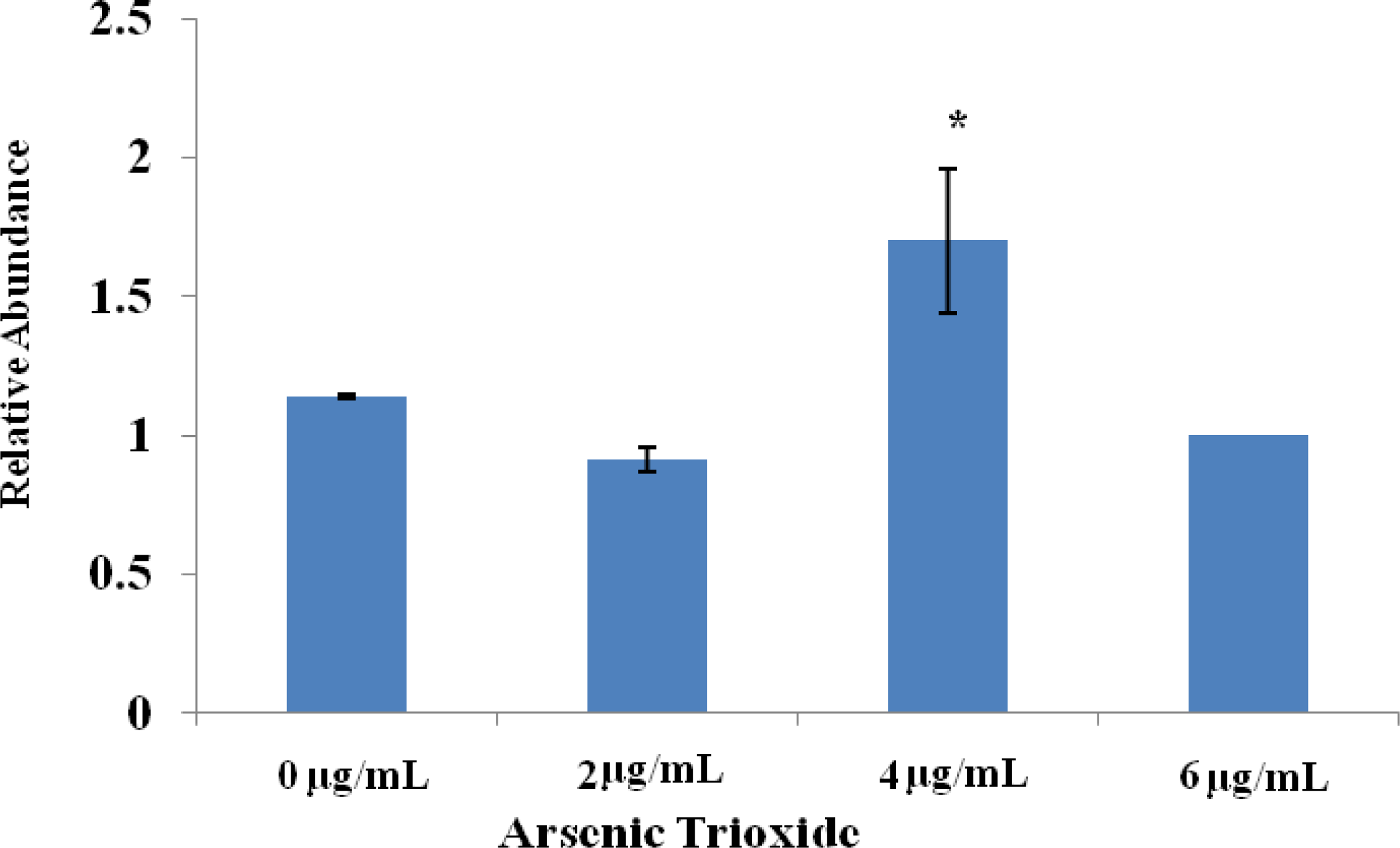
Effect of Arsenic Trioxide on p38 MAPK Activity
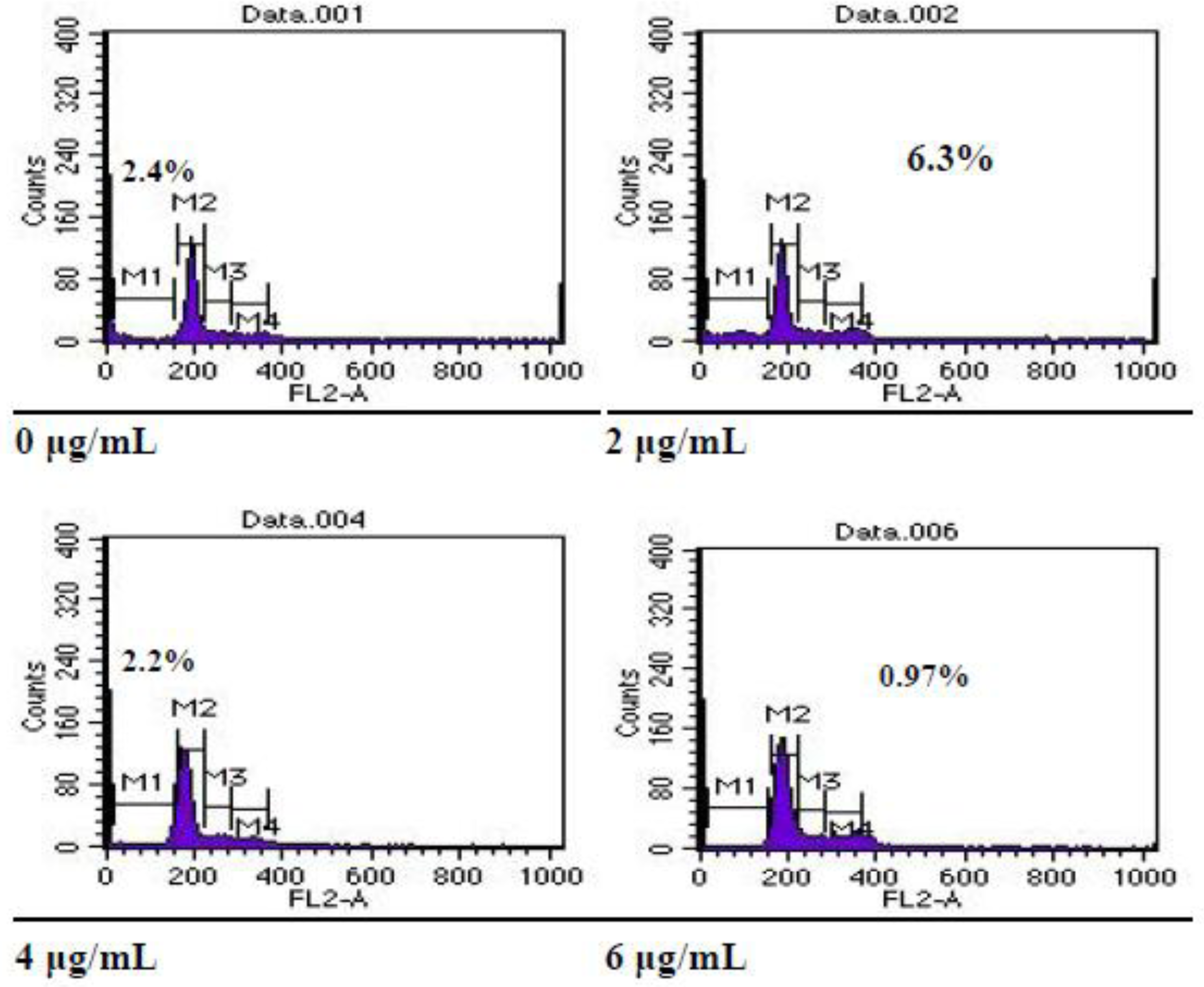
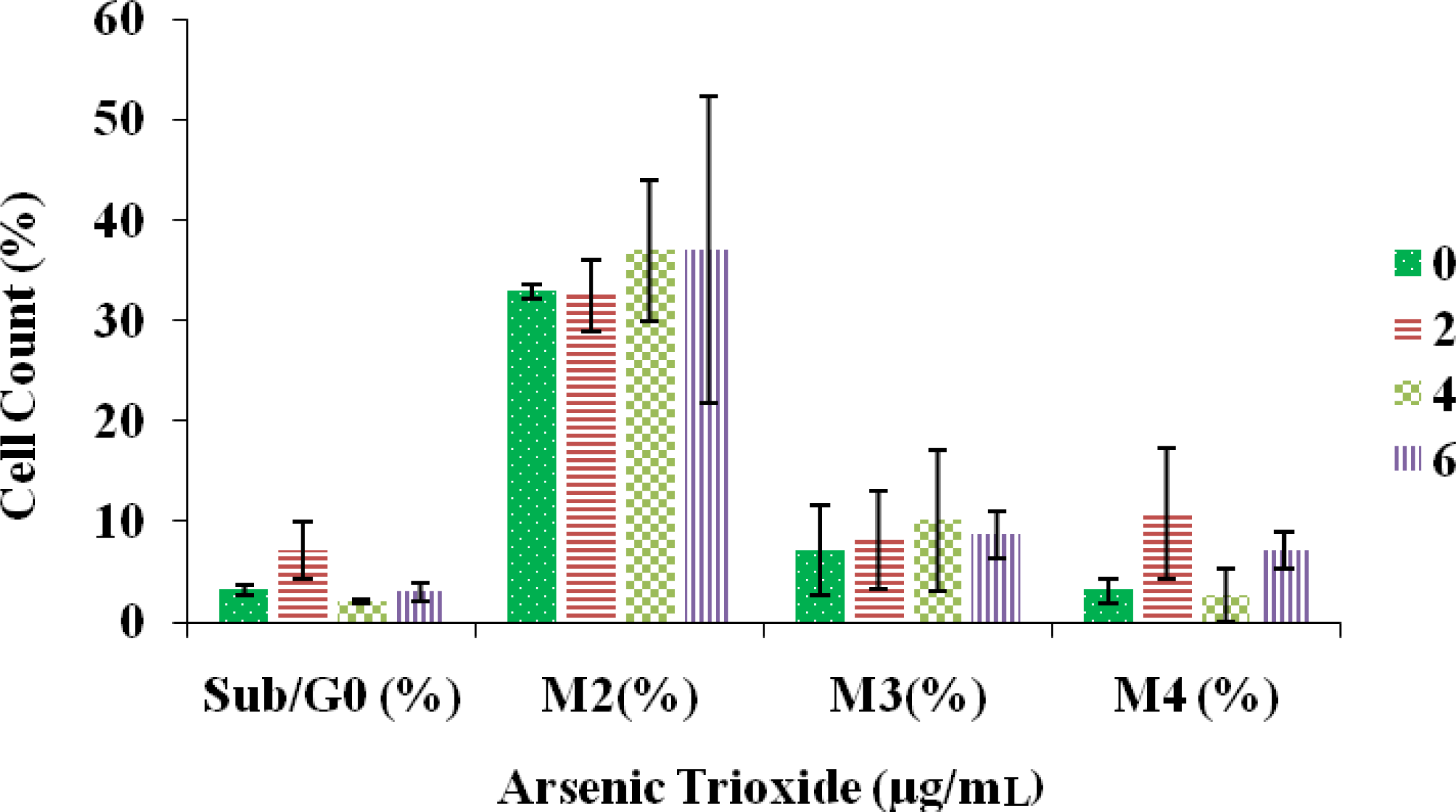
Effect of Arsenic Trioxide on Cell Cycle Distribution
4. Discussion
5. Conclusions
Acknowledgments
References
- Agency for Toxic Substances and Disease Registry (ATDR). Toxicological Profile for Arsenic (update); U.S. Public Health Service, U.S. DHHS: Atlanta, GA, USA, 2005. [Google Scholar]
- Jimi, S; Uchiyama, M; Takaki, A; Suzumiya, J; Hara, S. Mechanisms of cell death induced by cadmium and arsenic. Ann. N.Y. Acad. Sci 2004, 1101, 325–331. [Google Scholar]
- Lau, AT; He, QY; Chui, JF. A proteome analysis of the arsenite response in culture lung cells; evidence for in vitro oxidative stress-induced apoptosis. Biochem. J 2004, 382, 641–650. [Google Scholar]
- Rojewski, MT; Korper, S; Thiel, E; Schrezenmeir, H. Arsenic trioxide-induced apoptosis is independent of CD95 in lymphocytic cell line. Oncol. Rep 2004, 11, 509–513. [Google Scholar]
- Miller, WH; Schipper, HM; Lee, JS; Waxman, S. Mechanisms of action of arsenic trioxide. Cancer Res 2002, 62, 3893–3903. [Google Scholar]
- Zhang, TC; Cao, EH; Li, JF; Ma, W; Qin, JF. Induction of apoptosis and inhibition of human gastric cancer MGC-803 cell growth by arsenic trioxide. Eur. J. Cancer 1999, 35, 1258–1263. [Google Scholar]
- Florea, AM; Yamoah, EN; Dopp, E. Intracellular calcium disturbances induced by arsenic and its methylated derivatives in relation to genomic damage and apoptosis induction. Environ. Health Perspect 2005, 113, 659–664. [Google Scholar]
- Yedjou, CG; Tchounwou, PB. In vitro cytotoxic and genotoxic effects of arsenic trioxide on human leukemia (HL-60) cells using the MTT and alkaline single cell gel electrophoresis. Mol. Cell. Biochem 2007, 1, 123–30. [Google Scholar]
- Akao, Y; Yamada, H; Nakagawa, Y. Arsenic induced apoptosis in malignant cells in vitro. Leuk. Lymphoma 2005, 17, 1333–1337. [Google Scholar]
- Cai, X; Yu, Y; Huang, Y; Zhang, I; Jia, PM; Zhao, Q; Chen, Z; Tong, JH; Dai, W; Chen, GQ. Arsenic trioxide-induced mitotic arrest and apoptosis in acute promyleocytic leukemia cells. Leukemia 2003, 17, 1333–1337. [Google Scholar]
- Iwana, K; Nakajo, S; Aiuchi, T; Nakaya, K. Apoptosis induced by arsenic trioxide in leukemia U937 cells are dependent on activation of p38, inactivation of ERK and the Ca2+-dependent of superoxide. Int. J. Cancer 2001, 92, 518–526. [Google Scholar]
- Shen, ZY; Shen, J; Cai, WJ; Hong, C; Zheng, MH. The alteration of mitochondria is an early event of arsenic trioxide induced apoptosis in esophageal carcinoma cells. Int. J. Mol. Med 2000, 5, 155–158. [Google Scholar]
- Lowe, SW; Lin, AW. Apoptosis I cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar]
- Gewie, A. Introduction to apoptosis. Apo Review. 2003, pp. 4–26.
- Stellar, H. Mechanisms and cell suicide. Science 1995, 267, 1445–1449. [Google Scholar]
- Hockenberry, DE. Defining apoptosis. Am. J. Pathology 1995, 146, 16–19. [Google Scholar]
- Verma, A; Mohindru, M; Deb, DK; Sassano, A; Kambhampati, S; Ravandi, F; Minucci, S; Kalvakolanu, DV; Platanias, LC. Activation of Rac1 and the p38 mitogen-activated protein kinase pathway in response to arsenic trioxide. J. Biol. Chem 2002, 22, 44988–44995. [Google Scholar]
- Ndebele, K; Tchounwou, PB; McMurray, PW. Effects of Xenoestrogens on T lymphocytes: Modulation of bcl-2, p53, and apoptosis. Int. J. Mol. Med 2003, 4, 45–61. [Google Scholar]
- Jenkins, JK; Suwannaroj, S; Elbourne, KB; Ndebele, K; McMurray, RW. 17-β-estradiol alters Jurkat lymphocyte cell cycling and induces apoptosis through suppression of bcl-2 and cyclin A. Internat. J. Immunopharmacol 2001, 11, 1897–1911. [Google Scholar]
- McMurray, RW; Suwannaroj, S; Ndebele, K; Jenkins, JK. Differential effects of sex steroids on T and B lymphocytes: modulation of cell cycling, apoptosis, and bcl-2. Pathobiol 2001, 69, 44–58. [Google Scholar]
- Harnagea-Theophilus, E; Miller, MR. Acetaminophen alters estrogenic response in vitro: stimulation of DNA synthesis in estrogen-responsive human breast cancer cells. Toxicol. Sci 1998, 46, 38–44. [Google Scholar]
- Tchounwoun, PB; Yedjou, CG; Dorsey, WC. Arsenic trioxide induced transcriptional activation of stress genes and expression related proteins in human liver carcinoma cells (HepG2). Cell. Mol. Biol 2003, 47, 1071–1079. [Google Scholar]
- Chow, SKY; Chan, JYW; Fung, KP. Inhibition of cell proliferation and the action mechanisms of arsenic trioxide As2O3 on human breast cancer cells. J. Cell. Biochem 2004, 93, 173–187. [Google Scholar]
- Lau, AT; Li, M; Xie, R; He, QY; Chiu, J. Opposed arsenite-induced signaling pathways promote cell proliferation and apoptosis I cultured lung cells. Carcinogenesis 2004, 25, 21–28. [Google Scholar]
- Li, X; Ding, X; Adrian, TE. Arsenic trioxide causes redistribution of cell cycle, caspase activation, and GADD expression in human colonic, breast and pancreatic cancer cells. Cancer Invest 2004, 22, 389–400. [Google Scholar]
- Park, WH; Cho, YH; Won Jung, C; Park, JO; Kim, K; Im, YH; Lee, MH; Kang, WK; Park, K. Arsenic trioxide inhibits the growth of A498 renal cell carcinoma cells via cell cycle arrest or apoptosis. Biochem. Biophys. Res. Commun 2003, 300, 230–235. [Google Scholar]
- Ya Ming, W; Ying-Xian, O; Hai, B; Lu, JH; Rong-Liang, Z. Down-regulation of four arsenic antagonists on apoptosis and telomerase activity induced by arsenic trioxide in three myelocytic leukemia cell lines. Acta. Pharmacol. Sin 2001, 8, 725–730. [Google Scholar]
- Walker, AM; Stevens, JJ; Tchounwou, PB. Arsenic trioxide mediated apoptosis in breast (MCF-7) and lung (A549) carcinoma cells. Metal Ions Biol. Med 2008, 10, 135–139. [Google Scholar]
- Zheng, J; Deng, YP; Lin, C; Fu, M; Xiao, PG; Wu, M. Arsenic trioxide induces apoptosis of HPV 16 DNA—immortalized human cervical epithelial cells and selectively inhibits viral gene expression. Int. J. Cancer 1999, 82, 286–297. [Google Scholar]
- Cohen, GM. Caspases; the executioners of apoptosis. Biochem. J 1997, 326, 1–16. [Google Scholar]
- Han, YH; Kim, SZ; Kim, SH; Park, WH. Arsenic trioxide inhibits the growth of As4.1 juxtaglomerular cells via cell cycle and caspase-independent apoptosis. Am. J. Physiol. Renal. Physiol 2007, 293, 511–520. [Google Scholar]
- Namgung, U; Xia, Z. Arsenic induces apoptosis in rat cerebellar neurons via activation of JNK3 and p38 MAP kinases. Toxicol. Appl. Pharmacol 2001, 174, 130–138. [Google Scholar]
- Han, YH; Kim, SZ; Kim, SH; Park, WH. Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a G2 arrest of the cell cycle and apoptosis accompanied with the depletion of GSH. Cancer Letters 2008, 270, 40–55. [Google Scholar]
- Liu, Q; Hilsenbeck, S; Gazitt, Y. Arsenic trioxide induces apoptosis in myeloma cells; p53 dependent G1 or G2/M cell cycle arrest, activation of caspase 8 or caspase 3 and synergy with APO2/TRAIL. Blood 2003, 101, 4078–4087. [Google Scholar]






© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Walker, A.M.; Stevens, J.J.; Ndebele, K.; Tchounwou, P.B. Arsenic Trioxide Modulates DNA Synthesis and Apoptosis in Lung Carcinoma Cells. Int. J. Environ. Res. Public Health 2010, 7, 1996-2007. https://doi.org/10.3390/ijerph7051996
Walker AM, Stevens JJ, Ndebele K, Tchounwou PB. Arsenic Trioxide Modulates DNA Synthesis and Apoptosis in Lung Carcinoma Cells. International Journal of Environmental Research and Public Health. 2010; 7(5):1996-2007. https://doi.org/10.3390/ijerph7051996
Chicago/Turabian StyleWalker, Alice M., Jacqueline J. Stevens, Kenneth Ndebele, and Paul B. Tchounwou. 2010. "Arsenic Trioxide Modulates DNA Synthesis and Apoptosis in Lung Carcinoma Cells" International Journal of Environmental Research and Public Health 7, no. 5: 1996-2007. https://doi.org/10.3390/ijerph7051996