Protein-Energy Wasting and Mortality in Chronic Kidney Disease

{kind=link}
{kind=link}
Abstract
:1. Protein Energy Wasting (PEW) in CKD: Clinical Implications
2. Biomarkers/Proxies for Malnutrition and Mortality in CKD
2.1. Hypoalbuminemia
2.2. Oxidative Stress
2.3. Microinflammation
3. Effect of Body Composition on the Risk of Mortality of CKD Patients
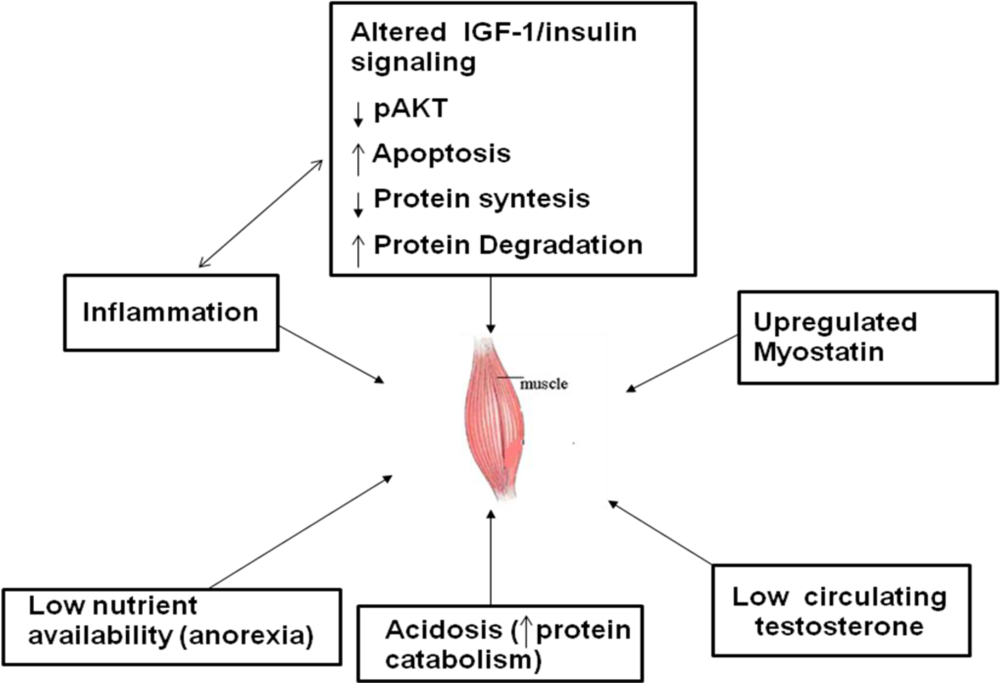
4. Mechanisms of PEW
4.1. Altered Amino Acid and Protein Handling by the Diseased Kidney
4.2. The Kidney and Methionine Transmethylation
4.3. The Kidney and Nitric Oxide (NO) Synthesis
4.4. Resistance to GH/IGF-1
4.5. Low Testosterone
4.6. Insulin Resistance and Altered Insulin Signaling
4.7. Anorexia
5. Ageing and Protein Metabolism
6. Infections
7. A New Perspective: Accelerated Loss of Myonuclei and Defective Regenerative Potential in the Skeletal Muscle of CKD Patients
Acknowledgments
References
- Kalantar-Zadeh, K; Abbott, KC; Kronenberg, F; Anker, SD; Horwich, TB; Fonarow, GC. Epidemiology of dialysis patients and heart failure patients. Semin. Nephrol 2006, 26, 118–133. [Google Scholar]
- Culleton, BF; Larson, MG; Wilson, PW; Evans, JC; Parfrey, PS; Levy, D. Cardiovascular disease and mortality in a community-based cohort with mild renal insufficiency. Kidney Int 1999, 56, 2214–2219. [Google Scholar]
- Al-Aly, Z; Zeringue, A; Fu, J; Rauchman, MI; McDonald, JR; El-Achkar, TM; Balasubramanian, S; Nurutdinova, D; Xian, H; Stroupe, K; et al. Rate of Kidney function decline associates with mortality. J. Am. Soc. Nephrol 2010, 21, 1961–1969. [Google Scholar]
- Himmelfarb, J; Ikizler, TA. Hemodialysis. N. Engl. J. Med 2010, 363, 1833–1845. [Google Scholar]
- Foque, D; Kalantar-Zadeh, K; Kopple, JD; Canau, N; Chauveau, P; Cuppari, L; Franch, H; Guarnieri, G; Ikizler, TA; Kaysen, G; et al. A proposed nomenclature and diagnostic criteria for protein-energy wasting in acute and chronic kidney disease. Kidney Int 2008, 73, 391–398. [Google Scholar]
- Locatelli, F; Manzoni, C; Del Vecchio, L; Di Filippo, S. Changes in the clinical condition of haemodialisys patients. J. Nephrol 1999, 12, S82–S91. [Google Scholar]
- Kalantar-Zadeh, K; Block, G; Humphreys, MH; Kopple, JD. Reverse epidemiology of cardiovascular risk factors in maintenance dialysis patients. Kidney Int 2003, 63, 793–808. [Google Scholar]
- Kalantar-Zadeh, K; Abbott, KC; Salahudeen, AK; Kilpatrick, RD; Horwich, TB. Survival advantages of obesity in dialysis patients. Am. J. Clin. Nutr 2005, 81, 543–554. [Google Scholar]
- Kovesdy, CP; Kalantar-Zadeh, K. Why is protein-energy wasting associated with mortality in chronic kidney disease? Semin. Nephrol 2009, 29, 3–14. [Google Scholar]
- Stenvinkel, P. Inflammation in end-stage renal failure: could it be treated. Nephrol Dial Transpl 2002, 17, S33–S38. [Google Scholar]
- Chmielewski, M; C̲arrero, JJ; Qureshi, AR; Axelsson, J; Heimburger, O; Berglund, L; Barany, P; Rutkowski, B; Lindholm, B; Stenvinkel, P. Temporal discrepancies in the association between the apoB/apoA-I ratio and mortality in incident dialysis patients. J. Int. Med 2009, 265, 708–716. [Google Scholar]
- Chmielewski, M; Verduijn, M; Drechsler, C; Lindholm, B; Stenvinkel, P; Rutkowski, B; Boeschoten, EW; Krediet, R; Dekker, FW. Low Cholesterol in dialysis patients-causal factor for mortality or an effect of confounding. Nephrol Dial Transpl 2011. [Google Scholar] [CrossRef]
- Liu, Y; Coresh, J; Eustace, JA; Longenecker, JC; Jaar, B; Fink, NE; Tracy, RP; Powe, NR; Klag, MJ. Association between cholesterol level and mortality in dialysis patients: Role of inflammation and malnutrition. JAMA 2004, 291, 451–459. [Google Scholar]
- Contrerars, G; Hu, B; Astor, BC; Greene, T; Erlinger, T; Kusek, JW; Lipkowitz, M; Lewis, JA; Randall, OS; Hebert, L; et al. Malnutrition-Inflammation modifies the relationship of cholesterol with cardiovascular disease. J. Am. Soc. Nephrol 2010, 21, 2131–2142. [Google Scholar]
- Topinkovà, E. Aging, disability and frailty. Ann. Nutr. Metab 2008, 52, S6–S11. [Google Scholar]
- Kalantar-Zadeh, K; Block, G; Horwich, T; Fonarow, GC. Reverse epidemiology of conventional cardiovascular risk factors in patients with chronic heart failure. J. Am. Coll. Cardiol 2004, 21, 1439–1444. [Google Scholar]
- Kalantar-Zadeh, K; Kilpatrick, RD; Kuwae, N. Revisiting mortality predictability of serum albumin in the dialysis population: time dependency, longitudinal changes and population-attributable fraction. Nephrol. Dial. Transpl 2005, 20, 1880–1888. [Google Scholar]
- Kaysen, GA; Eiserich, JP. The role of oxidative stress-altered lipoprotein structure and function and microinflammation on cardiovascular risk in patients with minor renal dysfunction. J. Am. Soc. Nephrol 2004, 15, 538–548. [Google Scholar]
- Lowrie, EG; Lew, NL. Death risk in hemodialysis patients: the predictive value of commonly measured variables and an evaluation of death rate differences between facilities. Am. J. Kidney Dis 1990, 15, 458–482. [Google Scholar]
- Reaich, D; Price, SR; England, BK; Mitch, WE. Mechanisms causing muscle loss in chronic renal failure. Am. J. Kidney Dis 1995, 26, 242–247. [Google Scholar]
- Khaw, KT; Dowsett, M; Folkerd, E; Bingham, S; Wareham, N; Luben, R; Welch, A; Day, N. Endogenous testosterone and mortality due to all causes, cardiovascular disease, and cancer in men: European prospective investigation into cancer in Norfolk (EPIC-Norfolk) Prospective Population Study. Circulation 2007, 116, 2694–2701. [Google Scholar]
- Kaisar, OM; Johnson, DW; Prins, JB; Isbel, N. The role of novel biomarkers of cardiovascular disease in chronic kidney disease: focus on adiponectin and leptin. Curr. Cardiol. Rev 2008, 4, 287–292. [Google Scholar]
- Axelsson, J; Witasp, A; Carrero, JJ; Qureshi, AR; Suliman, ME; Heimbürger, O; Bárány, P; Lindholm, B; Alvestrand, A; Schalling, M. Circulating levels of visfatin/pre-B-cell colony-enhancing factor 1 in rekation to genotype, GFR, body composition, and survival in patients with CKD. Am. J. Kidney Dis 2007, 49, 237–244. [Google Scholar]
- Zoccali, C; Mallamaci, F; Tripepi, G; Cutrupi, S; Pizzini, P. Low triiodothyronine and survival in end-stage renal disease. Kidney Int 2006, 70, 523–528. [Google Scholar]
- Carrero, JJ; Qureshi, AR; Axelsson, J; Yilmaz, MI; Rehnmark, S; Witt, MR; Barany, P; Heimburger, O; Suliman, ME; et al. Clinical and biochemical implications of low thyroid hormone levels (total and free forms) in euthyroid patients with chronic kidney disease. J. Int. Med 2007, 262, 690–701. [Google Scholar]
- Ikizler, TA; Wingard, RL; Harvell, J; Shyr, Y; Hakim, R. Association of morbidity with markers of nutrition and inflammation in chronic hemodialysis patients: A prospective study. Kidney Int 1999, 55, 1945–1951. [Google Scholar]
- Foley, RN; Pafrey, PS; Harnett, JD; Kent, GM; Murray, DC. Hypoalbuminemia, cardiac morbidity, and mortality in end-stage renal disease. J. Am. Soc. Nephrol 1996, 7, 728–736. [Google Scholar]
- Chertow, GM; Goldstein-Fuchs, DJ; Lazarus, JM; Kaysen, GA. Prealbumin, mortality, and cause-specific hospitalization in hemodialysis patients. Kidney Int 2005, 68, 2794–2800. [Google Scholar]
- Keys, A; Brozek, J; Henschel, A; Mickelsen, O; Taylor, H. The Biology of Human Starvation, Minneapolis; University of Minnesota Press: Minneapolis, MN, USA, 1950. [Google Scholar]
- Kopple, JD; Levey, AS; Greene, T; Chumlea, WC; Gassman, JJ; Hollinger, DL; Maroni, BJ; Merrill, D; Scherch, LK; Shulman, G; et al. Effect of dietary protein restriction on nutritional status in the modification of diet in renal diseases study. Kidney Int 1997, 52, 778–791. [Google Scholar]
- Aparicio, M; Chauveau, P; De Precigout, V; Bouchet, JL; Lasseur, C; Combe, C. Nutrition and outcome on renal replacement therapy of patients with chronic renal failure treated by a supplemented very low protein diet. J. Am. Soc. Nephrol 2000, 11, 708–716. [Google Scholar]
- Gil, HW; Yang, JO; Lee, EY; Lee, EM; Choi, JS; Hong, SY. The effect of dialysis membrane flux on amino acid loss in haemodialysis patients. J. Korean Med. Sci 2007, 22, 598–603. [Google Scholar]
- Kario, K; Matsuo, T; Kobyasahi, H. Heparin cofactor II deficiency in elderly: Comparison with antithrombin II. Thromb. Res 1992, 66, 489–498. [Google Scholar]
- Joles, JA; Willekes-Koolschij, N; Koomas, HA. Hypoalbuminemia causes high blood pressure by increasing red cell lysophosphatidylcholine. Kidney Int 1997, 52, 761–770. [Google Scholar]
- Emerson, TE. Unique features of albumin: A brief review. Crit. Care Med 1989, 17, 690–694. [Google Scholar]
- Bergstrom, J. Nutrition and mortality in hemodialysis. J. Am. Soc. Nephrol 1995, 6, 1329–1341. [Google Scholar]
- Nguyen-Khoa, T; Massay, ZA; De Bandt, JP; Kebede, M; Salama, L; Lambrey, G; Witko-Sarsat, V; Drüeke, TB; Lacour, B; Thévenin, M. Oxidative stress and haemodialysis: Role of inflammation and duration of dialysis treatment. Nephrol. Dial. Transpl 2001, 16, 335–340. [Google Scholar]
- Zoccali, C; Mallamaci, F; Asahia, K; Benedetto, FA; Tripepi, G; Tripepi, R; Nicocia, G; Buemi, M; Miyata, T. Pentosidine, carotid atherosclerosis and alterations in left ventricular geometry in hemodialysis patients. J. Nephrol 2001, 14, 293–298. [Google Scholar]
- Koyama, H; Nishizawa, Y. AGEs/RAGE in CKD: Irreversible metabolic memory road toward CVD? Eur. J. Clin. Invest 2010, 40, 623–635. [Google Scholar]
- Nakashima, A; Carrero, JJ; Qureshi, AR; Miyamoto, T; Anderstam, B; Barany, P; Heimburger, O; Stenvinkel, P; Lindholm, B. Effect of circulating soluble receptor for Advanced glycation End products (sRAGE) and the proinflammatory RADE Ligand (EN-RAGE, S100A12) on mortality in hemodialysis patients. Clin. J. Am. Soc. Nephrol 2010, 5, 2213–2219. [Google Scholar]
- Zhou, LL; Hou, FF; Wang, GB; Yang, F; Xie, D; Wang, YP; Tian, JW. Accumulation of advanced oxidation protein products induces podocyte apoptosis and deletion through NADPH-dependent mechanisms. Kidney Int 2009, 76, 1125–1127. [Google Scholar]
- Witko-Sarsat, V; Gausson, V; Nguyen, AT; Touam, M; Drüeke, T; Santangelo, F; Descamps-Latscha, B. AOPP-induced activation of human neutrophil and monocyte oxidative metabolism: A potential target for N-acetylcysteine treatment in dialysis patients. Kidney Int 2003, 64, 82–91. [Google Scholar]
- Descamps-Latscha, B; Witko-Sarsat, V; Nguyen-Khoa, T; Nguyen, AT; Gausson, V; Mothu, N; London, GM; Jungers, P. Advanced oxidation protein products as risk factors for cardiovascular events in nondiabetic predialysis patients. Am. J. Kidney Dis 2005, 45, 39–47. [Google Scholar]
- Valli, A; Mohamed, ES; Meert, S; Vanholder, R; Lindholm, B; Stenvinkel, P; Watanabe, M; Barany, P; Alvestrand, A; Anderstam, B. Overestimation of advanced oxidation protein products in uremic plasma due to presence of triglycerides and other endogenous factors. Clin. Chim. Acta 2007, 379, 87–94. [Google Scholar]
- Panichi, V; Migliori, M; De Pietro, S; Taccola, D; Bianchi, AM; Giovannini, L; Norpoth, M; Metelli, MR; Cristofani, R; Bertelli, AA; et al. C-reactive protein and interleukin-6 levels are related to renal function in predialytic chronic renal failure. Nephron 2002, 91, 594–600. [Google Scholar]
- Stenvinkel, P; Haimburger, O; Paultre, F; Diczfalusy, U; Wang, T; Berglund, L; Jogestrand, T. Strong association between malnutrition, inflammation, and atherosclerosis in chronic renal failure. Kidney Int 1999, 55, 1899–1911. [Google Scholar]
- Yeun, JY; Levine, RA; Mantadilok, V; Kaysen, GA. C-reactive protein predicts all-cause and cardiovascular mortality in haemodialysis patients. Am. J. Kidney Dis 2000, 35, 469–476. [Google Scholar]
- Eustace, JA; Astor, B; Muntner, PM; Ikizler, TA; Coresh, J. Prevalence of acidosis and inflammation and their association with low serum albumin in chronic kidney disease. Kidney Int 2004, 65, 1031–1040. [Google Scholar]
- Schiffl, H; Lang, SM; Stratakis, D; Fischer, R. Effects of ultrapure dialysis fluid on nutritional status and inflammatory parameters. Nephrol. Dial. Transpl 2001, 16, 1863–1869. [Google Scholar]
- Cuneo, RC; Salomon, F; Wiles, CM; Hesp, R; Sönksen, PH. Growth hormone treatment in growth hormone–deficient adults. I. Effects on muscle and strength. J. Appl. Physiol 1991, 70, 688–694. [Google Scholar]
- Bistrian, BR; Schwartz, J; Istifan, NW. Cytokines, muscle proteolysis and the catabolic response to infection and inflammation. Proc. Soc. Exp. Boil. Med 1992, 200, 220–223. [Google Scholar]
- Zwaka, PT; Hombach, V; Torzewski, J. C-reactive protein-mediated low-density lipoprotein uptake by macrophages. Circulation 2001, 103, 1194–1197. [Google Scholar]
- Boeheme, M; Kaehene, F; Kuehne, A; Bernhardt, W; Shroder, M; Pommer, W; Fiscer, C; Becker, H; Muller, C; Schindler, R. Pentraxin 3 is elevated in haemodialysis patients and is associated with cardiovascular disease. Nephrol. Dial. Transpl 2007, 22, 2224–2229. [Google Scholar]
- Tong, M; Carrero, JJ; Qureshi, AR; Anderstam, B; Heimburger, O; Barany, P; Axelsson, J; Alvestrand, A; Stenvinkel, P; Lindholm, B; et al. Plasma Pentraxin 3 in chronic kidney disease patients: Associations with renal function, protein-energy wasting, cardiovascular disease and mortality. Clin. J. Am. Soc. Nephrol 2007, 2, 889–897. [Google Scholar]
- Yilmaz, MI; Sonmez, A; Ortiz, A; Saglam, M; kilic, S; Eyileten, T; Caglar, K; Oguz, Y; Vural, A; Cakar, M; et al. Soluble TWEAK and PTX3 in nondialysis CKD patients: impact on endothelial Dysfunction and cardiovascular outcomes. Clin. J. Am. Soc. Nephrol 2011, 6, 785–792. [Google Scholar]
- Alberti, L; Gilardini, L; Zulian, A; Micheletto, G; Peri, G; Doni, A; Mantovani, A; Invitti, C. Expression of long pentraxin PTX3 in human adipose tissue and its relation with cardiovascular risk factors. Atherosclerosis 2009, 202, 455–460. [Google Scholar]
- Raj, DS; Carrero, JJ; Shah, VO; Qureshi, AR; Bárány, P; Heimbürger, O; Lindholm, B; Ferguson, J; Moseley, PL; Stenvinkel, P. Soluble CD14 levels, interleukin 6, and mortality among prevalent hemodialysis patiient. Am. J. Kidney Dis 2009, 54, 1072–1080. [Google Scholar]
- Shoji, T; Shinohara, K; Hatsuda, S; Kimoto, E; Fukumoto, S; Emoto, M; Tahara, H; Koyama, H; Ishimura, E; Miki, T; et al. Altered relationship between body fat and plasma adiponectin in end-stage renal disease. Metabolism 2005, 54, 330–334. [Google Scholar]
- Honda, H; Qureshi, AR; Axelsson, J; Heimburger, O; Suliman, ME; Barany, P; Stenvinkel, P; Lindholm, B. Obese sarcopenia in patients with end-stage renal disease is associated with inflammation and increased mortality. Am. J. Clin. Nutr 2007, 86, 633–638. [Google Scholar]
- Kakiya, R; Shoji, T; Tsujimoto, Y; Tatsumi, N; Hatsuda, S; Shinohara, K; Kimoto, E; Tahara, H; Koyama, H; Emoto, M; et al. Body fat mass and lean mass as predictors of survival in hemodialysis patients. Kidney Int 2006, 70, 549–556. [Google Scholar]
- Kalantar-Zadeh, K; Kuwae, N; Wu, DY; Shantouf, RS; Fouque, D; Anker, SD; Block, G; Kopple, JD. Associations of body fat and its changes over time with quality of life and prospective mortality in hemodialysis patients. Am. J. Clin. Nutr 2006, 83, 202–210. [Google Scholar]
- Leinig, C; Pecoits-Filho, R; Nascimento, MM; Goncalves, S; Riella, MC; Martins, C. Association between body mass index and body fat in chronic kidney disease stages 3 to 5, hemodialysis, and peritoneal dialysis patients. J. Ren. Nutr 2008, 18, 424–429. [Google Scholar]
- Carrero, JJ; Cordeiro, AC; Lindholm, B; Stenvinkel, P. the emerging pleiotrophic role of adipokines in the uremic phenotype. Curr. Opin. Nephrol. Hypertens 2010, 19, 37–42. [Google Scholar]
- Zoccali, C; Postorino, M; Marino, C; Pizzini, P; Cutrupi, S; Tripepi, G. CREDIT working group abdominal obesity and all-cause mortality in end-stage renal disease. J. Am. Coll. Cardiol 2009, 53, 1265–1272. [Google Scholar]
- Knudson, JD; Payne, GA; Borbouse, L; Tune, JD. Leptin and mechanisms of endothelial dysfunction and cardiovascular disease. Curr. Hypertens. Rep 2008, 10, 434–439. [Google Scholar]
- Liu, SW; Qiao, SB; Yuan, JS; Liu, DQ. Association of plasma visfatin levels with inflammation, atherosclerosis and acute coronary syndrome(ACS) in humans. Clin. Endocrinol 2009, 71, 202–207. [Google Scholar]
- Schneiderman, J; Simon, AJ; Schroeter, MR; Flugelman, MY; Konstantinides, S; Schaefer, K. Leptin receptor is elevated in carotid plaques from neurologically symptomatic patients and positively correlated with augmented machrophage density. J. Vasc. Surg 2008, 48, 1146–1155. [Google Scholar]
- Dahl, TB; Yndestand, A; Skjelland, M; Oie, E; Dahl, A; Michelsen, A; Damas, JK; Tunheim, SH; Ueland, T; Smith, C; et al. Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis. Possible role in inflammation and plaque destabilization. Circulation 2007, 115, 972–980. [Google Scholar]
- Ramkumar, N; Pappas, LM; Beddhu, S. Effect of body size and body composition on survival in peritoneal dialysis patients. Perit. Dial. Int 2005, 25, 461–469. [Google Scholar]
- Huang, CX; Tighiouart, H; Beddhu, S; Cheung, AK; Dwyer, JT; Eknoyan, G; Beck, GJ; Levey, AS; Sarnak, MJ. Both low muscle mass and low fat are associated with higher all-cause mortality in hemodialysis patients. Kidney Int 2010, 77, 624–629. [Google Scholar]
- Noori, N; Kovesdy, CP; Dukkipati, R; Kim, Y; Duong, U; Bross, R; Oreopoulos, A; Luna, A; Benner, D; Kopple, JD; et al. Survival predictability of lean and fat mass in men and women undergoing maintenance hemodialysis. Am. J. Clin. Nut 2010, 92, 1060–1070. [Google Scholar]
- Tessari, P; Deferrari, G; Robaudo, C; Vettore, M; Pastorin, N; De Biasi, L; Garibotto, G. Phenylalanine hydroxylation across the kidney in humans. Kidney Int 1999, 56, 2168–2172. [Google Scholar]
- Kopple, JD. Phenylalanine and tyrosine metabolism in chronic kidney failure. J. Nutr 2007, 137, 1586S–1590S. [Google Scholar]
- Garibotto, G; Sofia, A; Saffioti, S; Russo, R; Deferrari, G; Rossi, D; Verzola, D; Gandolfo, MT; Sala, MR. Interorgan exchange of aminothiols in humans. Am. J. Physiol. Endocrinol. Metab 2003, 284, E757–E763. [Google Scholar]
- Garibotto, G; Valli, A; Anderstam, B; Eriksson, M; Suliman, M; Balbi, M; Rollando, D; Vigo, E; Lindholm, B. The kidney is a major site of S-adenosylhomocysteine disposal in humans. Kidney Int 2009, 76, 293–296. [Google Scholar]
- Garibotto, G; Sofia, A; Saffioti, S; Bonanni, A; Mannucci, I; Verzola, D. Amino acid and protein metabolism in the human kidney and in patients with chronic kidney disease. Clin. Nutr 2010, 29, 424–433. [Google Scholar]
- van de Poll, MC; Soesters, PB; Deutz, NE; Fearon, KC; Dejong, CH. Renal metabolism of amino acids: Its role in inter-organ amino acid exchange. Am. J. Clin. Nutr 2004, 79, 185–197. [Google Scholar]
- Lee, ME; Wang, H. Homocysteine and hypomethylation: a novel link to vascular disease. Trends Cardiovasc. Med 1999, 9, 49–54. [Google Scholar]
- Cantoni, GL. Experimental and clinical roles of S-Adenosylmethionine. In Biological Methylation and Drug Design; Borchardt, RT, Creveling, CR, Ueland, PM, Eds.; Humana Press: Clifton, NJ, USA, 1986; pp. 227–238. [Google Scholar]
- Liu, C; Wang, Q; Guo, H; Xia, M; Yuan, Q; Hu, Y. Plasma S-adenosylhomocysteine is a better biomarker of atherosclerosis than homocysteine in apolipoprotein E-deficient mice fed high dietary methionine. J. Nutr 2008, 138, 311–315. [Google Scholar]
- Valli, A; Carrero, JJ; Qureshi, AR; Garibotto, G; Barany, P; Axelsson, J; Lindholm, B; Stenvinkel, P; Anderstam, B; Suliman, ME. Elevated serum levels of S-adenosylhomocysteine, but not homocysteine, are associated with cardiovascular disease in stage 5 chronic kidney disease patients. Clin. Chim. Acta 2008, 395, 106–110. [Google Scholar]
- Ingrosso, D; Cimmino, A; Perna, AF; Masella, L; De Santo, NG; De Bonis, ML; Vacca, M; D’Esposito, M; D’Urso, M; Galletti, P; et al. Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet 2003, 17, 1693–1694. [Google Scholar]
- Baylis, C. Arginine, arginine analogous and nitric oxid production in chronic kidney disease. Nat. Clin. Pract. Nephrol 2006, 2, 209–220. [Google Scholar]
- Matsuoka, H. Endothelial dysfunction associated with oxidative stress in human. Diabetes Res. Clin. Pract 2001, 54, S65–S72. [Google Scholar]
- Kielstein, JT; Zoccali, C. Asymmetric dimethylarginine: A novel marker of risk and potential target for therapy in chronic kidney disease. Curr. Opin. Nephrol. Hyperten 2008, 17, 609–615. [Google Scholar]
- Vallance, P; Leone, A; Calver, A; Collier, J; Moncada, S. Accumulation of an endogenous inhibitor on nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar]
- Mihout, F; Shweke, N; Bigé, N; Jouanneau, C; Dussaule, JC; Ronco, P; Chatziantoniou, C; Boffa, JJ. Asymmetric dimethylarginine (ADMA) induces chronic kidney disease through a mechanism involving collagen and TGF-ß1 synthesis. J. Pathol 2011, 223, 37–45. [Google Scholar]
- Cross, JM; Donald, A; Vallance, P; Deanfield, JE; Woolfson, RG; MacAllister, RJ. Hemodialysis improves endothelial function in humans. Nephrol. Dial. Transpl 2001, 16, 1823–1829. [Google Scholar]
- Kaskel, F. Chronic renal disease: A growing problem. Kidney Int 2003, 64, 1141–1151. [Google Scholar]
- Qing, DP; Ding, HU; Vagdama, J. Elevated myocardial cytosolic calcium impairs insulin-like growth factor-1 stimlated protein synthesis in chronic renal failure. J. Am. Soc. Nephrol 1999, 10, 84–91. [Google Scholar]
- Blum, WF; Ranke, MB; Kietzmann, K; Tönshoff, B; Mehls, O. growth hormone resistance and inhibition of somatomedin activity by excess of insulin-like growth factor binding protein in uremia. Pediatr. Nephrol 1991, 5, 539–544. [Google Scholar]
- Sun, DF; Zheng, Z; Tummala, P; Oh, J; Schaefer, F; Rabkin, R. Chronic uremia attenuates growth hormone-induced signal transduction in skeletal muscle. J. Am. Soc. Nephrol 2004, 15, 2630–2646. [Google Scholar]
- Chen, Y; Biada, J; Sood, S; Rabkin, R. Uremia attenuates growth hormone-stimulated insulin-like growth factor-1 expression, a process worsened by inflammation. Kidney Int 2010, 78, 89–95. [Google Scholar]
- Cooney, RN; Shumate, M. The inhibitory effects of interleukin-1 on growth hormone action during catabolic illness. Vitam. Horm 2006, 74, 317–340. [Google Scholar]
- Brungger, M; Hulter, HN; Krapf, R. Effect of chronic metabolic acidosis on the growth hormone/IGF-1 endocrine axis: new cause of growth hormone insensitivity in humans. Kidney Int 1997, 51, 216–221. [Google Scholar]
- Garibotto, G; Russo, R; Sofia, A; Ferone, D; Fiorini, F; Cappelli, V; Tarroni, A; Gandolfo, MT; Vigo, E; Valli, A; et al. Effects of uremia and inflammation on growth hormone resistance in patients with chronic kidney disease. Kidney Int 2008, 74, 937–945. [Google Scholar]
- Bhasin, S; Taylor, WE; Singh, R; Artaza, J; Sinha-Hikim, I; Jasuja, R; Choi, H; Gonzalez-Cadavid, NF. The mechanisms of androgen effects on body composition: mesenchymal pluripotent cell as the target of androgen action. J. Gerontol. Med. Sci 2003, 58, 1103–1110. [Google Scholar]
- Wolfe, R; Ferrando, A; Sheffield-Moore, M; Urban, R. Testosterone and muscle protein metabolism. Mayo Clin. Proc 2000, 75, S55–S59. [Google Scholar]
- Harman, SM; Metter, EJ; Tobin, JD; Pearson, J; Blackman, MR. Baltimore longitudinal study of aging. Longitudinal effects of aging on serum total and free testosterone levels in healthy men. J. Clin. Endocrinol. Metab 2001, 86, 724–731. [Google Scholar]
- Morley, JE; Melmed, S. Gonadal dysfunction in systemic disorders. Metabolism 1979, 28, 1051–1073. [Google Scholar]
- Roubenoff, R; Parise, H; Payette, HA; Abad, LW; D’Agostino, R; Jacques, PF; Wilson, PW; Dinarello, CA; Harris, TB. Cytokines, insulin-like growth factor 1, sarcopenia, and mortality in very old communitydwelling men and women: the Framingham Heart Study. Am. J. Med 2003, 115, 429–435. [Google Scholar]
- Haring, R; Nauck, M; Volzke, H; Endlich, K; Lendeckel, U; Friedrich, N; Dorr, M; Rettig, R; Kroemer, HK; Wallaschofski, H. Low serum testosterore is associated with increased mortality in men with stage 3 or greater nephropathy. Am. J. Nephrol 2011, 33, 209–217. [Google Scholar]
- Khaw, KT; Dowsett, M; Folkerd, E; Bingham, S; Wareham, N; Luben, R; Welch, A; Day, N. Endogenous testosterone and mortality due to all causes, cardiovascular disease, and cancer in men: European prospective investigation into cancer in Norfolk (EPIC-Norfolk) prospective population study. Circulation 2007, 116, 2694–2701. [Google Scholar]
- Carrero, JJ; Qureshi, AR; Parini, P; Arver, S; Lindholm, B; Bárány, P; Heimbürger, O; Stenvinkel, P. Low serum testosterone increases mortality risk among male dialysis patients. J. Am. Soc. Nephrol 2009, 20, 613–620. [Google Scholar]
- Gungor, O; Kircelli, F; Carrero, JJ; Asci, G; Toz, H; Tatar, E; Hur, E; Sever, MS; Arinsoy, T; Ok, E. Endogenous testosterone and mortality in male hemodialysis patients: Is this the resul of aging? Clin. J. Am. Soc. Nephrol 2010, 5, 2018–2023. [Google Scholar]
- Alvestrand, A; DeFronzo, RA; Smith, D; Wahren, J. Influence of Hyperinsulinemia on intracellular amino acids levels and amino acid exchange across splanchnic and leg tissues in uremia. Clin. Sci 1988, 74, 155–162. [Google Scholar]
- Bailey, JL; Zheng, B; Hu, Z; Price, SR; Mitch, WE. Chronic kidney disease causes defects in signalling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: Implications for muscle atrophy. J. Am. Soc. Nephrol 2006, 17, 1388–1394. [Google Scholar]
- Pupim, LB; Flakoll, PJ; Majchrzak, KM; Aftab Guy, DL; Stenvinkel, P; Ikizler, TA. Increased muscle protein breakdown in chronic hemodialysis patients with type 2 diabetes mellitus. Kidney Int 2005, 68, 1857–1865. [Google Scholar]
- Rajan, V; Mitch, WE. Ubiquitin, proteasomes and proteolytic mechanisms activated by kidney disease. Biochim. Biophys. Acta 2008, 1782, 795–799. [Google Scholar]
- Reaich, D; Channo, SM; Scrimgeour, CM; Daley, SE; Wilkinson, R; Goodship, TH. Correction of acidosis in humans with CRF decreases protein degradation and amino acid oxidation. Am. J. Physiol 1993, 265, E230–E235. [Google Scholar]
- Burrowes, JD; Larive, B; Chertow, GM. Self-reported appetite, hospitalization and death in haemodialysis patients: findings from the haemodialysis (HEMO) study. Nephrol. Dial. Transpl 2005, 20, 2765–2774. [Google Scholar]
- Cheung, W; Yu, PX; Little, BM. Role of leptin and melanocortin signaling in uremia-associated cachexia. J. Clin. Invest 2005, 115, 1659–1665. [Google Scholar]
- Kalantar Zadeh, K; Block, G; Mc Allister, CJ; Humphreys, MH; Kopple, JD. Appetite and inflammation, nutrition, anemia, and clinical outcome in hemodialysis patients. Am. J. Clin. Nutr 2004, 80, 299–307. [Google Scholar]
- Carrero, JJ; Qureshi, AR; Axelsson, J; Avesani, CM; Suliman, ME; Kato, S; Bárány, P; Snaedal-Jonsdottir, S; Alvestrand, A; Heimbürger, O; et al. Comparison of nutritional and inflammatory markers in dialysis patients with reduced appetite. Am. J. Clin. Nutr 2007, 85, 695–701. [Google Scholar]
- Gayle, DA; Desai, M; Casillas, E; Belooseski, R; Ross, MG. Gender specific orexigenic and anorexigenic mechanisms in rats. Life Sci 2006, 79, 1531–1536. [Google Scholar]
- Latos, DL. Chronic dialysis in patients over age 65. J. Am. Soc. Nephrol 1996, 7, 637–646. [Google Scholar]
- Lindeman, RD. Overview: Renal physiology and pathophysiology of aging. Am. J. Kidney Dis 1990, 16, 275–282. [Google Scholar]
- Timiras, PS. Aging of the Skeleton, Joints and Muscles. In Physiological Basis of Aging and Geriatrics, 2nd ed; Timiras, PD, Ed.; CRC Press: Boca Raton, FL, USA, 1994; pp. 259–272. [Google Scholar]
- Rothstein, M. Altered proteins, errors and aging. In Protein Metabolism in Aging; Segal, HL, Rothstein, M, Bergamini, P, Eds.; Wiley-Liss Inc: New York, NY, USA, 1990; pp. 3–14. [Google Scholar]
- Gersovitz, M; Bier, D; Matthews, D; Udall, J; Munro, HN; Young, VR. Dynamic aspects of whole body glycine metabolism. Influence of protein intake in young adult and elderly males. Metabolism 1980, 29, 1087–1094. [Google Scholar]
- Robert, JJ; Bier, D; Schoeller, D; Wolfe, R; Matthews, DE; Munro, HN; Young, VR. Effect of intravenous glucose on whole body leucine dynamics, studied with 1-13C-leucine, in healthy, young and elderly adults. J. Gerontol 1984, 39, 673–681. [Google Scholar]
- Nair, KS. Age-related changes in muscle. Mayo. Clin. Proc 2000, 75S, S14–S18. [Google Scholar]
- Boirie, Y; Gachon, P; Cordat, N; Ritz, P; Beaufrère, B. Differential insulin sensitivities of glucose, amino acid, and albumin metabolism in elderly men and women. J. Clin. Endocrinol. Metab 2001, 86, 638–644. [Google Scholar]
- Oreopoulos, A; Kalantar-Zadeh, K; Sharma, AM; Fonarow, GC. The obesity paradox in the elderly: Potential mechanisms and clinical implications. Clin. Geriatr. Med 2009, 25, 643–659. [Google Scholar]
- Miller, SL; Wolfe, RR. The danger of weight loss in the elderly. J. Nutr. Health Aging 2008, 12, 487–489. [Google Scholar]
- Vanholder, R; Ringoir, S. Infectious morbidity and defects of phagocytic function in end-stage renal disease: A review. J. Am. Soc. Nephrol 1993, 3, 1541–1554. [Google Scholar]
- Dalrymple, LS; Go, AS. Epidemiology of acute infections among patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol 2008, 3, 1487–1493. [Google Scholar]
- Allon, M; Depner, TA; Radeva, M; Bailey, J; Beddhu, S; Butterly, D; Coyne, DW; Gassman, JJ; Kaufman, AM; Kaysen, GA; et al. Impact of dialysis dose and membrane on infection-related hospitalization and death: Results of the HEMO Study. J. Am. Soc. Nephrol 2003, 14, 1863–1870. [Google Scholar]
- Chinen, J; Shearer, WT. Secondary immunodeficiencies, including HIV infection. J. Allergy Clin. Immunol 2008, 121, S388–392. [Google Scholar]
- Hulsewe, KW; van Acker, BA; Von Meyenfeldt, MF; Soeters, PB. Nutritional depletion and dietary manipulation: Effects on the immune response. World J. Surg 1999, 23, 536–544. [Google Scholar]
- Souba, WW. Nutritional support. N. Engl. J. Med 1997, 336, 41–48. [Google Scholar]
- Alexander, JW. Immunoenhancement via enteral nutrition. Arch. Surg 1993, 128, 1242–1245. [Google Scholar]
- Kimmel, PL; Phillips, TM; Lew, SQ; Langman, CB. Zinc modulates mononuclear cellular calcitriol metabolism in peritoneal dialysis patients. Kidney Int 1996, 49, 1407–1412. [Google Scholar]
- Erten, Y; Kayatas, M; Sezer, S; Kayataş, M; Haberal, A; Güz, G; Erten, Y; Hizel, N; Boyacioğlu, S; Gülmüş, S; et al. Zinc deficiency: Prevalence and causes in hemodialysis patients and effect on cellular immune response. Transpl. Proc 1998, 30, 850–851. [Google Scholar]
- Casciato, DA; McAdam, LP; Kopple, JD; Bluestone, R; Goldberg, LS; Clements, PJ; Knutson, DW. Immunologic abnormalities in hemodialysis patients: Improvement after pyridoxine therapy. Nephron 1984, 38, 9–16. [Google Scholar]
- Dobbelstein, H; Korner, WF; Mempel, W; Grosse-Wilde, H; Edel, HH. Vitamin B6 deficiency in uremia and its implications for the depression of immune responses. Kidney Int 1974, 5, 233–239. [Google Scholar]
- Minnaganti, VR; Cunha, BA. Infections associated with uremia and dialysis. Infect. Dis. Clin. North Am 2001, 15, 385–406. [Google Scholar]
- Cohen, G; Haag-Weber, M; Horl, WH. Immune dysfunction in uremia. Kidney Int. Supp 1997, 62, S79–S82. [Google Scholar]
- Pesanti, EL. Immunologic defects and vaccination in patients with chronic renal failure. Infect. Dis. Clin. North Am 2001, 15, 813–832. [Google Scholar]
- Eleftheriadis, T; Antoniadi, G; Liakopoulos, V; Kartsios, C; Stefanidis, I. Disturbances of acquired immunity in hemodialysis patients. Semin. Dial 2007, 20, 440–451. [Google Scholar]
- Lim, WH; Kireta, S; Leedham, E; Russ, GR; Coates, PT. Uremia impairs monocyte and monocyte-derived dendritic cell function in hemodialysis patients. Kidney Int 2007, 72, 1138–1148. [Google Scholar]
- Fantuzzi, G; Faggioni, R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. J. Leukoc. Biol 2000, 68, 437–446. [Google Scholar]
- Matarese, G; La Cava, A; Sanna, V; Lord, GM; Lechler, RI; Fontana, S; Zappacosta, S. Balancing susceptibility to infection and autoimmunity: A role for leptin? Trends Immunol 2002, 23, 182–187. [Google Scholar]
- Madej, T; Boguski, MS; Bryant, SH. Threading analysis suggests that the obese gene product may be a helical cytokine. FEBS Lett 1995, 373, 13–18. [Google Scholar]
- Baumann, H; Morella, KK; White, DW; Dembski, M; Bailon, PS; Kim, H; Lai, CF; Tartaglia, LA. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc. Natl. Acad. Sci. USA 1996, 93, 8374–8378. [Google Scholar]
- Lord, GM; Matarese, G; Howard, JK; Baker, RJ; Bloom, SR; Lechler, RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998, 394, 897–901. [Google Scholar]
- Loffreda, S; Yang, SQ; Lin, HZ; Karp, CL; Brengman, ML; Wang, DJ; Klein, AS; Bulkley, GB; Bao, C; Noble, PW; Lane, MD; Diehl, AM. Leptin regulates proinflammatory immune responses. FASEB J 1998, 12, 57–65. [Google Scholar]
- Gabay, C; Dreyer, MG; Pellegrinelli, N; Chicheportiche, R; Meier, CA. Leptin directly induces the secretion of interleukin 1 receptor antagonist in human monocytes. Clin. Endocrinol. Metab 2001, 86, 783–791. [Google Scholar]
- Santos-Alvarez, J; Goberna, R; Sanchez-Margalet, V. Human leptin stimulates proliferation and activation of human circulating monocytes. Cell Immunol 1999, 194, 6–11. [Google Scholar]
- Sanchez-Margalet, V; Martin-Romero, C; Santos-Alvarez, J; Goberna, R; Najib, S; Gonzalez-Yanes, C. Role of leptin as an immunomodulator of blood mononuclear cells: Mechanism of action. Clin. Exp. Immunol 2003, 133, 11–19. [Google Scholar]
- Matarese, G; La Cava, A. The intricate interface between immune system and metabolism. Trends Immunol 2004, 25, 193–200. [Google Scholar]
- Rajala, MW; Scherer, PE. The adipocyte. At the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 2003, 144, 3765–3773. [Google Scholar]
- Diez, JJ; Iglesias, P; Fernandez-Reyes, MJ; Aguilera, A; Bajo, MA; Alvarez-Fidalgo, P; Codoceo, R; Selgas, R. Serum concentrations of leptin, adiponectin and resistin, and their relationship with cardiovascular disease in patients with end-stage renal disease. Clin. Endocrinol 2005, 62, 242–249. [Google Scholar]
- McTernan, PG; Fisher, FM; Valsamakis, G; Chetty, R; Harte, A; McTernan, CL; Clark, PM; Smith, SA; Barnett, AH; Kumar, S. Resistin and type 2 diabetes: regulation of resistin expression by insulin and rosiglitazone and the effects of recombinant resistin on lipid and glucose metabolism in human differentiated adipocytes. J. Clin. Endocrinol. Metab 2003, 88, 6098–6106. [Google Scholar]
- Cohen, G; Ilic, D; Raupachova, J; Hörl, WH. Resistin inhibits essential functions of polymorphonuclear leukocytes. J. Immunol 2008, 181, 3761–3768. [Google Scholar]
- Lim, VS; Kopple, JD. Protein metabolism in patients with chronic renal failure: Role of uremia and dialysis. Kidney Int 2000, 58, 1–10. [Google Scholar]
- Mitch, WE. Malnutrition: A frequent misdiagnosis for hemodialysis patients. J. Clin. Invest 2002, 110, 437–439. [Google Scholar]
- Stevinkel, P; Heimburger, O; Lindholm, B. Wasting, but not malnutrition, predicts cardiovascular mortality in end-stage renal disease. Nephrol. Dial. Transpl 2004, 19, 2181–2183. [Google Scholar]
- Mitch, WE; Goldberg, AL. Mechanisms of muscle wasting: The role of ubiquitin-proteasome system. N. Engl. J. Med 1996, 335, 1897–1905. [Google Scholar]
- Lecker, SH; Goldberg, AL; Mitch, WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol 2006, 17, 1807–1819. [Google Scholar]
- Dupont-Versteegden, EE. Apoptosis in skeletal muscle and its relevance to atrophy. World J. Gastroenterol 2006, 12, 7463–7466. [Google Scholar]
- Gundersen, K; Bruusgaard, JC. Nuclear domains during muscle atrophy: Nuclei lost or paradigm lost? J. Physiol 2008, 586, 2675–2681. [Google Scholar]
- Frost, RA; Lang, CH. Protein kinase B/Akt: A nexus of growth factor and cytokine signaling in determining muscle mass. J. Appl. Physiol 2007, 103, 378–387. [Google Scholar]
- Yamaguchi, H; Wang, HG. The protein kinase PKB/Akt regulates cell servival and apoptosis by inhibiting Bax conformational change. Oncogene 2001, 20, 7779–7786. [Google Scholar]
- Lee, SW; Dai, G; Hu, Z; Wang, X; Du, J; Mitch, WE. Regulation of muscle protein degradation: Coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidynositol 3 kinase. J. Am. Soc. Nephrol 2004, 15, 1537–1545. [Google Scholar]
- Zhang, L; Wang, XH; Wang, H; Du, J; Mitch, WE. Satellite cell dysfunction and impaired IGF-I signaling cause CKD-induced muscle atrophy. J. Am. Soc. Nephrol 2010, 21, 419–427. [Google Scholar]
- Zhang, L; Ran, L; Garcia, GE; Wang, XH; Han, S; Du, J; Mitch, WE. Chemokine CXCL16 regulates neutrophil and macrophage infiltration into injured muscle promoting muscle regeneration. Am. J. Pathol 2009, 175, 2518–2527. [Google Scholar]
- Verzola, D; Procopio, V; Sofia, A; Villaggio, B; Tarroni, A; Bonanni, A; Mannucci, I; De Cian, F; Gianetta, E; Saffioti, S; Garibotto, G. Apoptosis and myostatin mRNA are upregulated in skeletal muscle of patients with chronic kidney disease. Kidney Int 2011, 79, 773–782. [Google Scholar]
- Garibotto, G; Russo, R; Sofia, A; Sala, MR; Robaudo, C; Moscatelli, P; Deferrari, G; Tizianello, A. Skeletal muscle protein synthesis and degradation in patients with chronic renal failure. Kidney Int 1994, 45, 1432–1439. [Google Scholar]
- Hara, Y; May, RC; Kelly, RA; Mitch, WE. Acidosis, not azotemia, stimulates branched-chain, amino acid catabolism in uremic rats. Kidney Int 1987, 32, 808–814. [Google Scholar]
- Reaich, D; Channon, SM; Scrimgeour, CM; Daley, SE; Wilkinson, R; Goodship, TH. Correction of acidosis in humans with CRF decreases protein degradation and amino acid oxidation. Am. J. Physiol 1993, 265, E230–E235. [Google Scholar]
- Young, GA; Swanepoel, CR; Croft, MR; Hobson, SM; Parsons, FM. Anthropometry and plasma valine, amino acids, and proteins in the nutritional assessment of hemodialysis patients. Kidney Int 1982, 21, 492–499. [Google Scholar]
- Kornasion, R; Riederer, I; Butler-Browne, G; Mouly, V; Uni, Z; Halevy, O. Beta-hydroxy-beta-methylbutyrate (HMB) stimulates myogenic cell proliferation, differentiation and survival via the MAPK/ERK and PI3K/Akt pathways. Biochim. Biophys. Acta 2009, 1793, 755–763. [Google Scholar]


© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bonanni, A.; Mannucci, I.; Verzola, D.; Sofia, A.; Saffioti, S.; Gianetta, E.; Garibotto, G. Protein-Energy Wasting and Mortality in Chronic Kidney Disease. Int. J. Environ. Res. Public Health 2011, 8, 1631-1654. https://doi.org/10.3390/ijerph8051631
Bonanni A, Mannucci I, Verzola D, Sofia A, Saffioti S, Gianetta E, Garibotto G. Protein-Energy Wasting and Mortality in Chronic Kidney Disease. International Journal of Environmental Research and Public Health. 2011; 8(5):1631-1654. https://doi.org/10.3390/ijerph8051631
Chicago/Turabian StyleBonanni, Alice, Irene Mannucci, Daniela Verzola, Antonella Sofia, Stefano Saffioti, Ezio Gianetta, and Giacomo Garibotto. 2011. "Protein-Energy Wasting and Mortality in Chronic Kidney Disease" International Journal of Environmental Research and Public Health 8, no. 5: 1631-1654. https://doi.org/10.3390/ijerph8051631