2.1. Amorphous a-Mn(BH4)2-Type Hydride after Mechanochemical Synthesis by Ball Milling (BM)
The SEM micrographs of the morphology of the as-received ultrafine filamentary Ni and graphene additives are shown in
Figure 1a,b, respectively. The agglomerated and entangled filamentary nature of the Ni additive can be clearly seen in
Figure 1a. However, plate-like graphene powder particles are rather well dispersed (
Figure 1b). The SEM micrographs for the as-received NaBH
4 and MnCl
2 primary reactants were reported by Varin et al. [
11] and are not shown here. Suffice it to say that both primary reactants are very coarse, with a globular morphology [
11].
Changing the angular positions of one or two strong NdFeB magnets and altering the number of hard steel balls in a milling vial can control the milling energy in the magneto ball mill Uni-Ball-Mill 5 [
3,
7,
8,
9,
10,
13]. As recently reported by Parviz et al. [
13], the quantity of milling energy, Q
TR(R) per unit mass·hour (kJ/gh), which was injected into and stored in a milled powder for each particular milling mode with a fixed ball-to-powder mass ratio, R, in the magneto ball mill Uni-Ball-Mill 5, can be calculated via a semi-empirical method. In the present work, we solely used the milling mode IMP68-4B-R132 for 0.5 and 1 h with the constant total milling energy input, Q
TR = 36.4 and 72.8 kJ/g, respectively (see Table 5 [
13]).
Figure 1c,d show the evolution during ball milling of the powder morphology for the additive-free (2NaBH
4 + MnCl
2), ((2NaBH
4 + MnCl
2) + 5 wt. % graphene) and ((2NaBH
4 + MnCl
2) + 5 wt. % Ni) samples, respectively. A refinement of the initial powder mixture with some pronounced agglomerations can be seen, in comparison with the large-size of the as-received NaBH
4 and MnCl
2 particulates, already shown by Varin et al. [
11]. However, it appears from
Figure 1c that the entangled filamentary Ni morphology is preserved during ball milling to an extent, and does not disperse into individual, smaller particles of Ni as expected.
Figure 1d suggests that the graphene additive is quite thoroughly and intimately milled with the reactants.
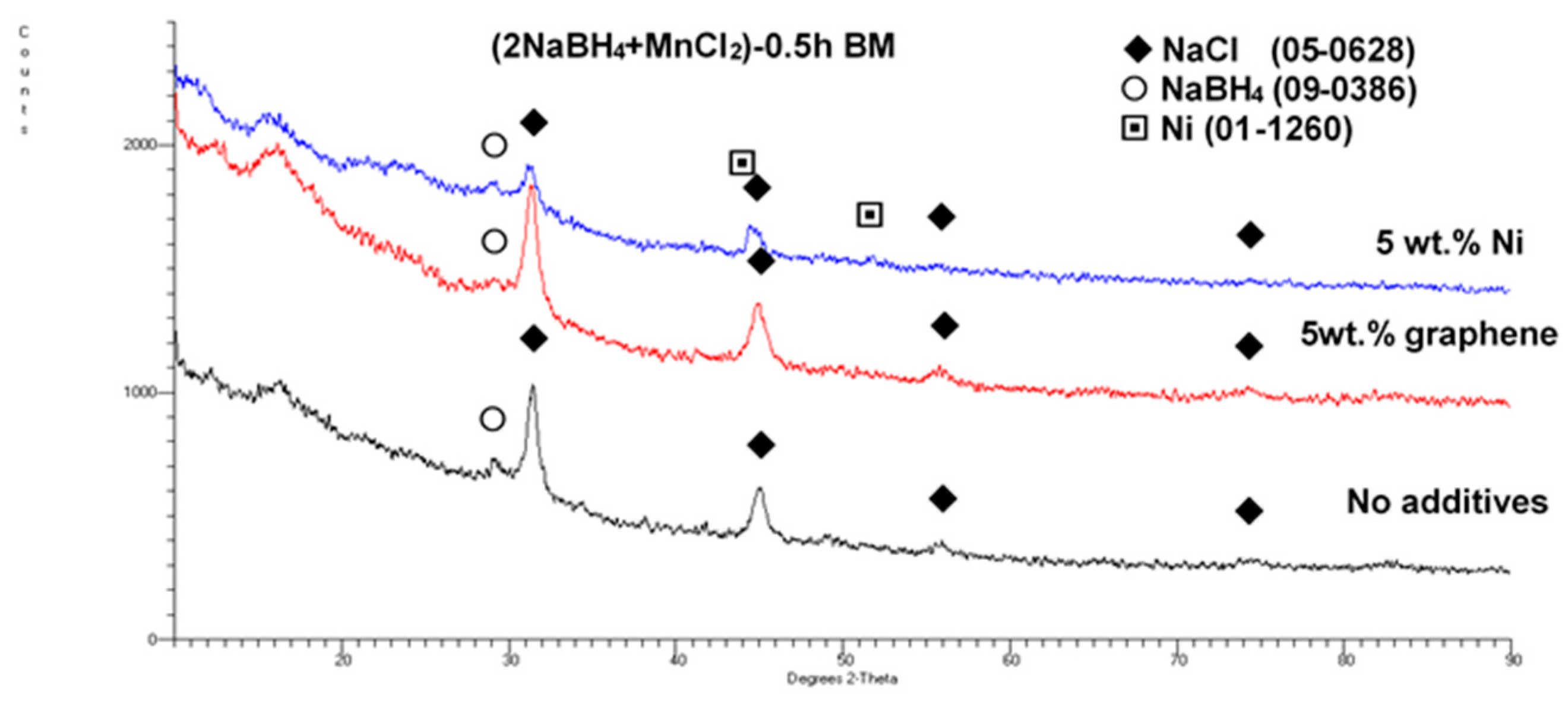
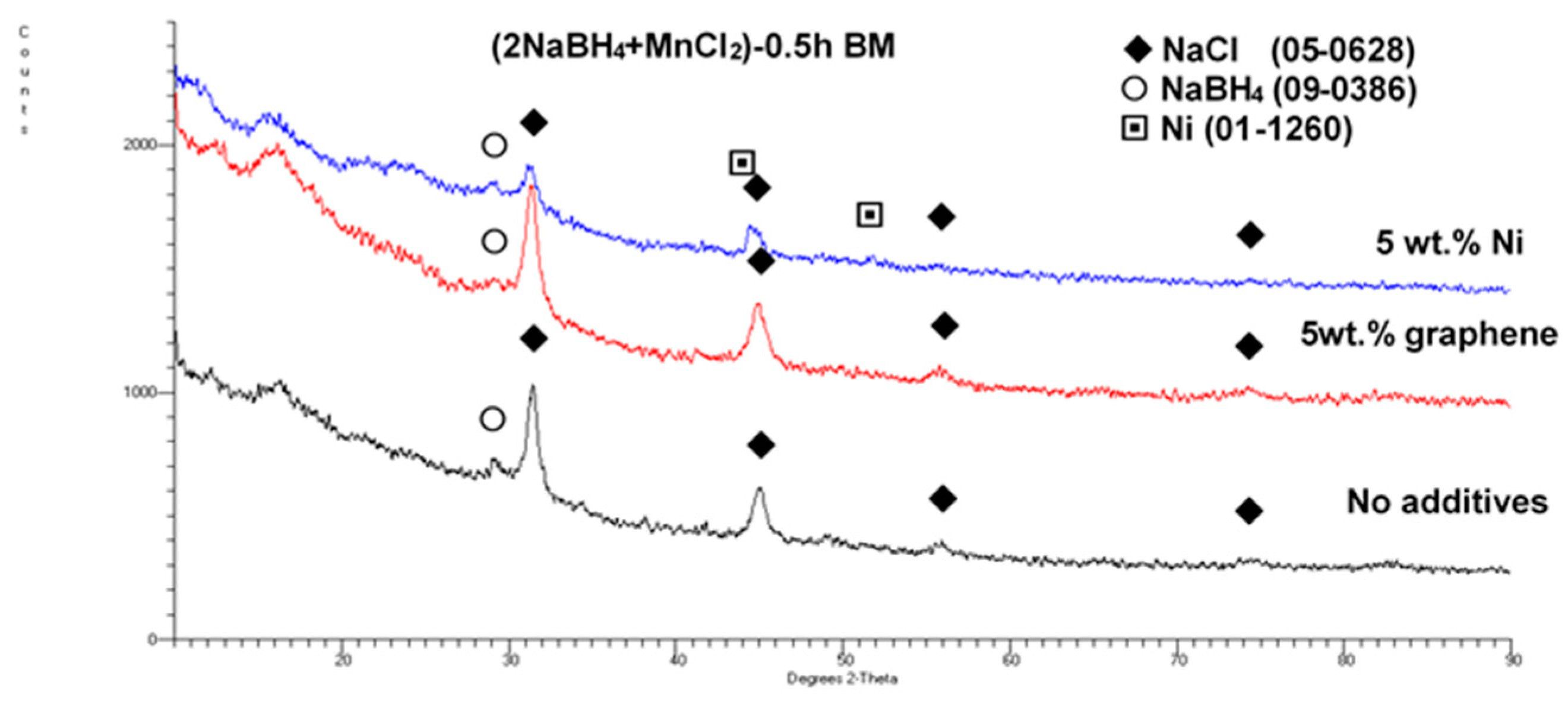
Figure 2 shows the XRD patterns of the (2NaBH
4 + MnCl
2) mixture ball milled (BM) together with 5 wt. % of the graphene and filamentary Ni additives, as compared to an XRD pattern for an additive-free sample. Only the very strong NaCl-based diffraction peaks and a weak diffraction peak of retained NaBH
4 can be clearly seen. These are accompanied by the discernible Ni peaks in this particular sample. Apparently, a mechanochemical reaction occurred between the NaBH
4 and MnCl
2 reactants during ball milling, which produced a NaCl-type salt accompanied by an
amorphous a-Mn(BH
4)
2-type hydride, as reported by Varin et al. [
11]. This issue will be discussed in more detail later.
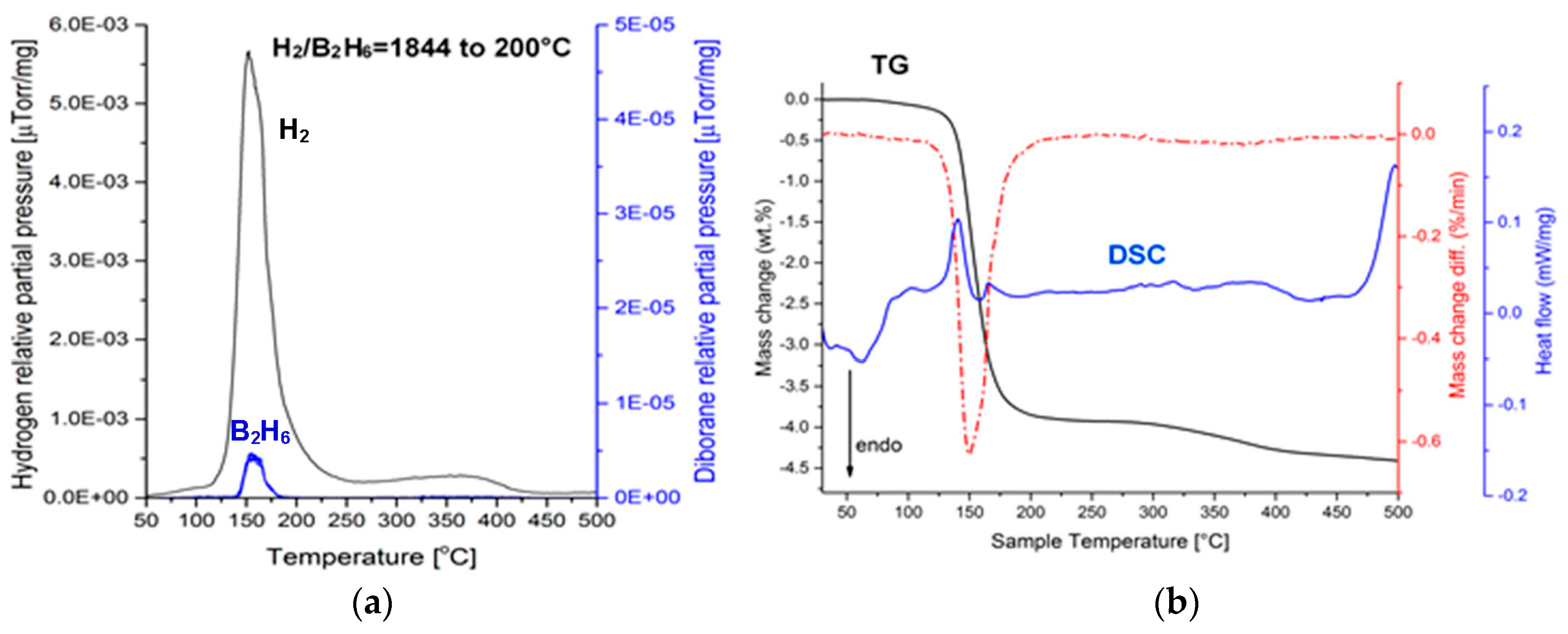
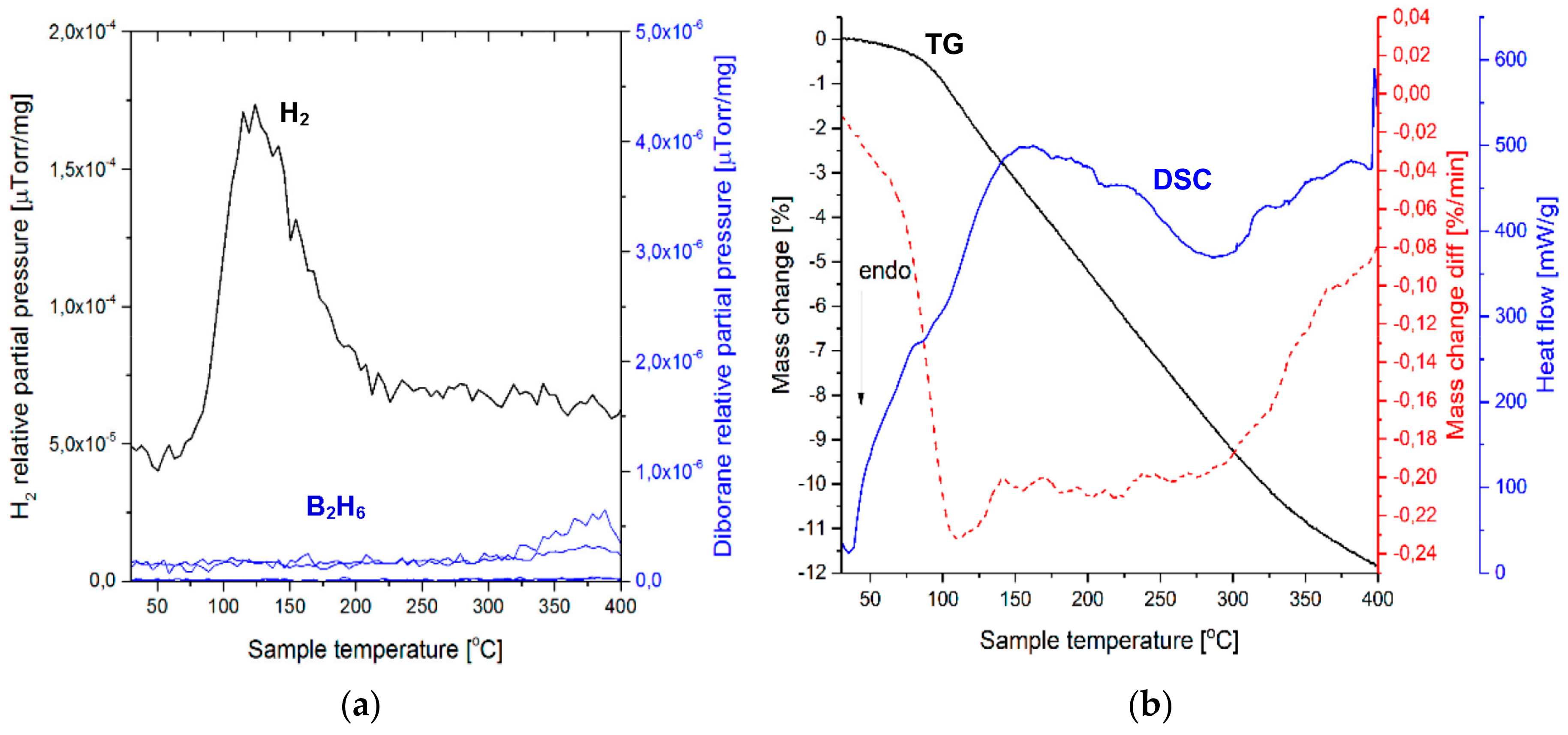
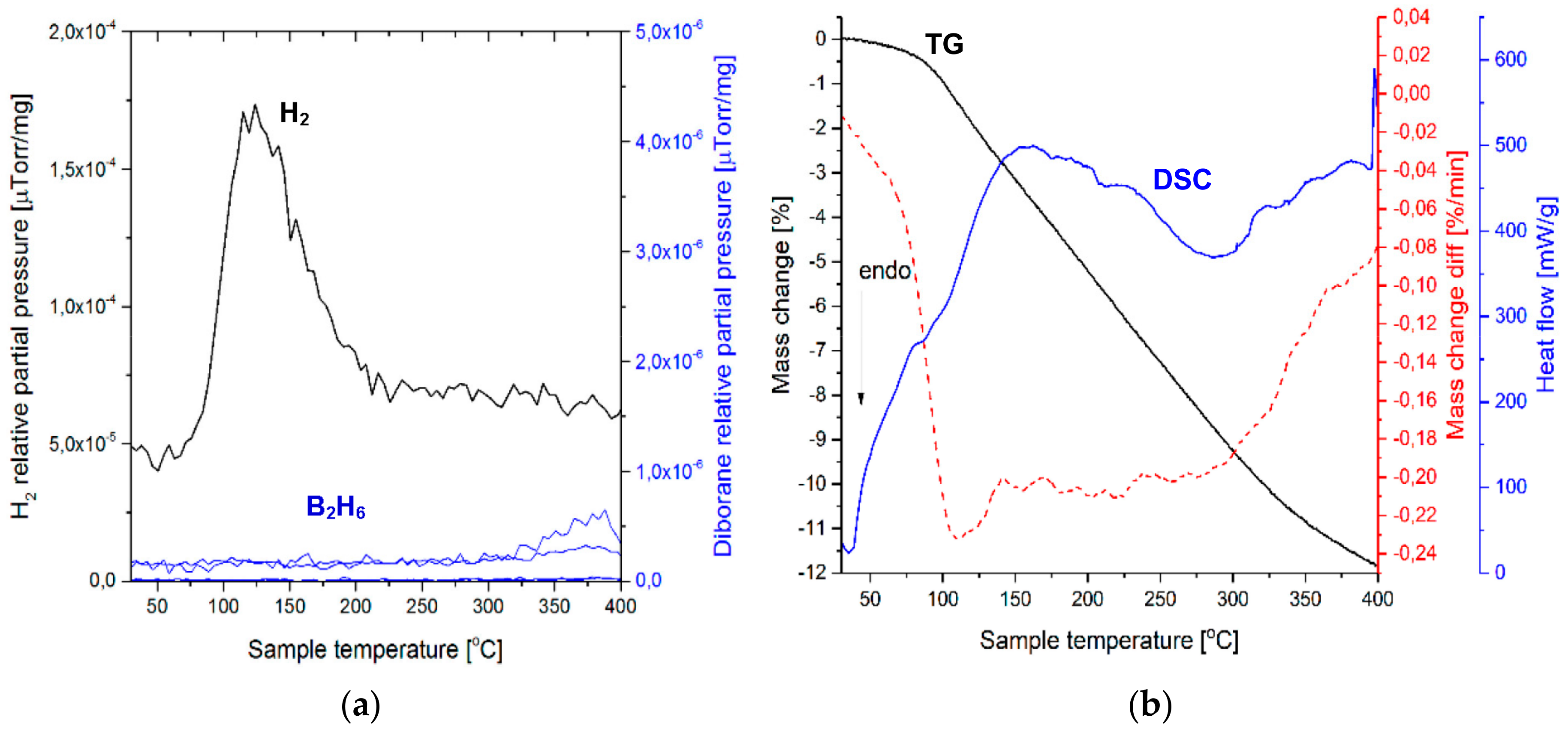
Figure 3a shows the mass spectrometry (MS) gas desorption spectra for a ((2NaBH
4 + MnCl
2) + 5 wt. % graphene) sample synthesized for 0.5 h (Q
TR = 36.4 kJ/g). It is seen that the principal peak corresponds to H
2 within the range from ~100 to 200 °C (a maximum at ~160 °C), which is accompanied by a very small peak of diborane (B
2H
6) that commences at ~135 °C and terminates at ~180 °C, with a peak maximum at ~160 °C. The intensity ratio of the MS H
2/B
2H
6 signal up to 200 °C (the threshold temperature for the end of the B
2H
6 release) equals 1844, which can be compared to the ratio of 948 for the additive-free 0.5 h BM (2NaBH
4 + MnCl
2) sample that was investigated by Varin et al. [
11]. This shows that the quantity of B
2H
6 released up to ~200 °C, from the sample with graphene, is smaller than that released from an additive-free sample. The curves in
Figure 3b show thermogravimetric (TG) and DSC results for a sample with a 5 wt. % graphene additive, which was extracted from the 0.5 h BM sample. A black curve in
Figure 3 shows that a rapid TG mass loss, which attains 3.84 wt. % up to 200 °C, is observed within a narrow temperature range of 150–175 °C. Nevertheless, a slow TG mass loss is still observable beyond 200 °C, which is most likely related to a gradual decomposition of retained NaBH
4 (
Figure 2), albeit without the release of B
2H
6, which already terminated at ~200 °C. At ~160 °C, one can see a small endothermic peak on a DSC curve (blue). A range of temperatures within which the mass loss occurs is indicated very well by a first derivative of the TG line (dTG/dT where T-temperature) (broken red line in
Figure 3b). It clearly shows that the occurrence of the endothermic peak at ~160 °C is linked to the TG mass loss in
Figure 3b. For the powder with 5 wt. % graphene that was ball milled for 0.5 h, we investigated a second, random sample that could unequivocally confirm the correctness of the MS and TGA/DSC results in
Figure 3b. We found that the TGA/DSC curves for the second sample (a plot not included here) were nearly identical to
Figure 3, and exhibited a mass loss of 4.09 wt. % up to 200 °C, for which a DSC curve also showed an endothermic event. In addition, this second sample exhibited a very similar MS pattern (an MS plot not included here) to that in
Figure 3a, having the MS peak intensity ratio H
2/B
2H
6 = 1256, again, much higher than that reported for the additive-free material [
11].
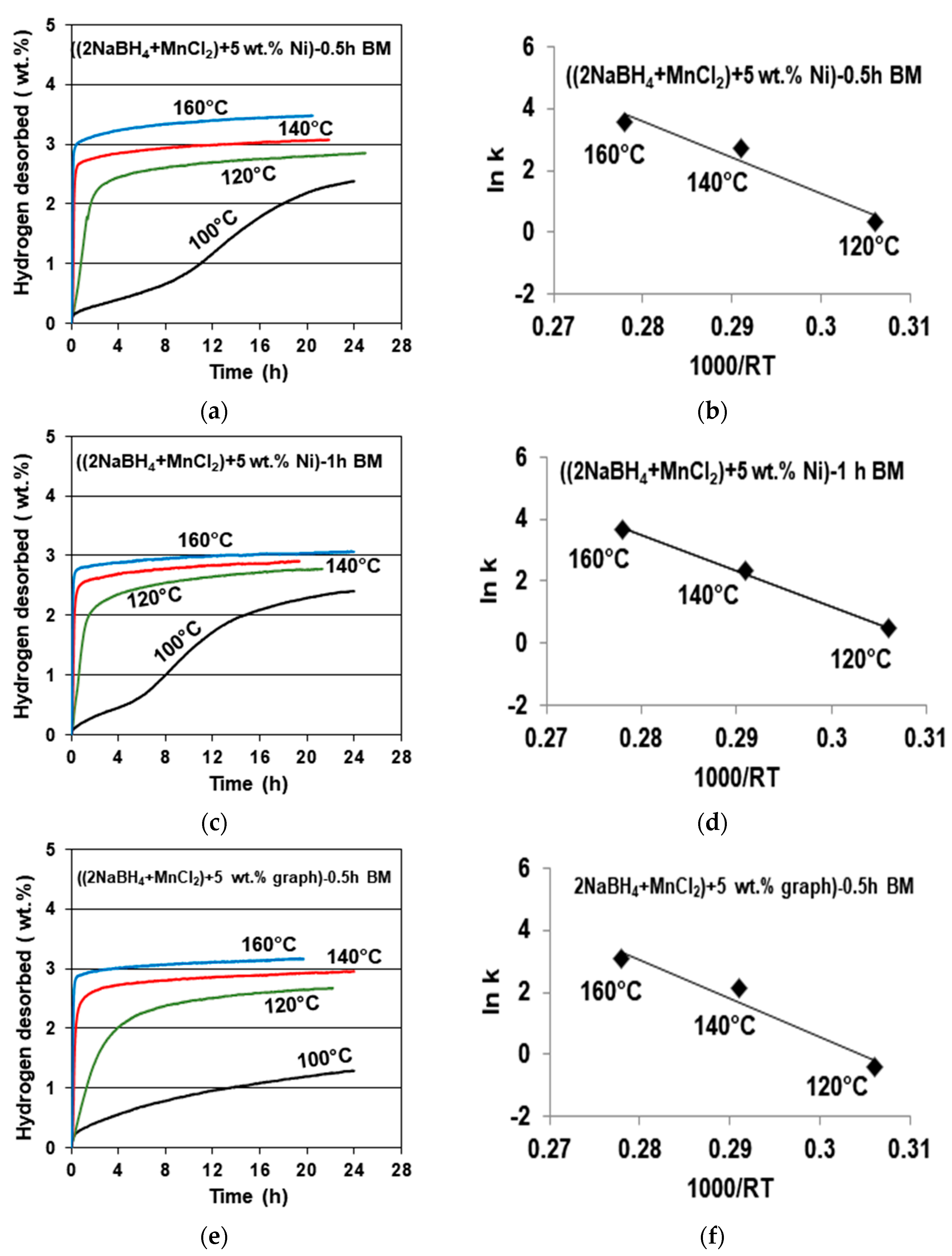
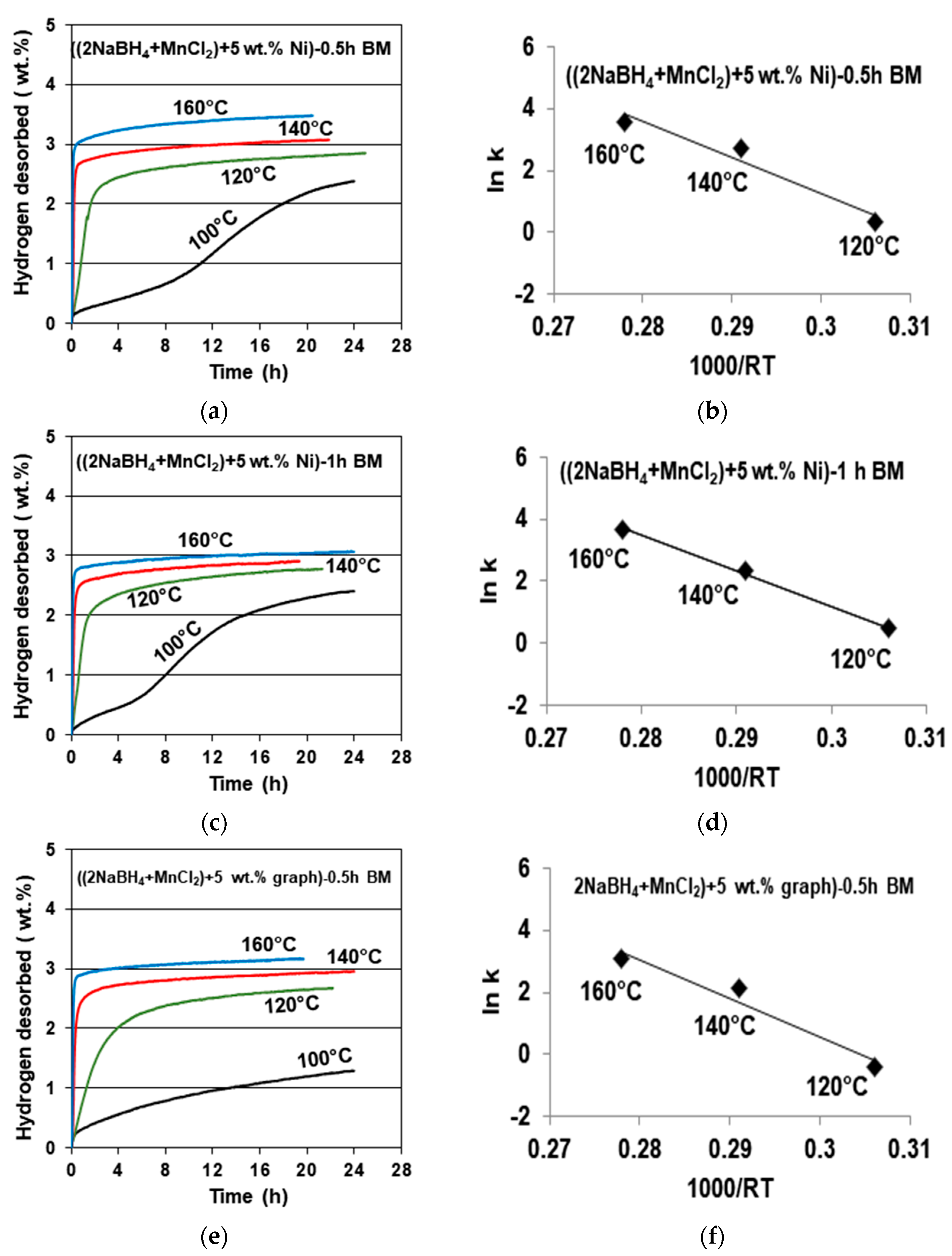
Figure 4 shows examples of gas desorption curves for the samples containing a 5 wt. % Ni additive ball milled for 0.5 (
Figure 4a) and 1 h (
Figure 4c), together with accompanying Arrhenius plots for the estimate of the apparent activation energy (AAE) of desorption (
Figure 4b,d). A desorption curve for a sample with a 5 wt. % graphene additive ball milled for 0.5 h (
Figure 4e) with an accompanying Arrhenius plot for the AAE (
Figure 4f) is also shown. It must be pointed out that, as stated earlier, the released gas contained a miniscule quantity of B
2H
6. It was so small that, for simplicity, the
y axis for the gas desorption curves in
Figure 4 is designated as desorbing solely H
2. In all three cases, gas desorption at 100 °C is very sluggish, and that particular curve was not taken into consideration for the AAE calculations. It is also seen that for the 5 wt. % Ni sample, the duration of ball milling for either 0.5 or 1 h has no measurable effect on both the quantity of desorbed H
2 (3–3.5 wt. %) and the values of the AAE for desorption, which are equal to 116.68 for a 0.5 h BM sample, and 115.19 kJ/mol for a 1 h BM sample (with an error of ±15.1 kJ/mol).
The desorption curves for the sample with a 5 wt. % graphene additive (
Figure 4e) shows the same H
2 desorption capacity of about 3 wt. %, and a slightly higher AAE of about 125.93 kJ/mol (
Figure 4f), with an estimated error of ±13.4 kJ/mol. For comparison, the AAE values for the additive-free samples ball milled for 0.5 and 1 h were reported as 118.21 and 123.09 kJ/mol, respectively [
11]. Apparently, the additives of filamentary Ni and graphene do not measurably affect the AAE of desorption.
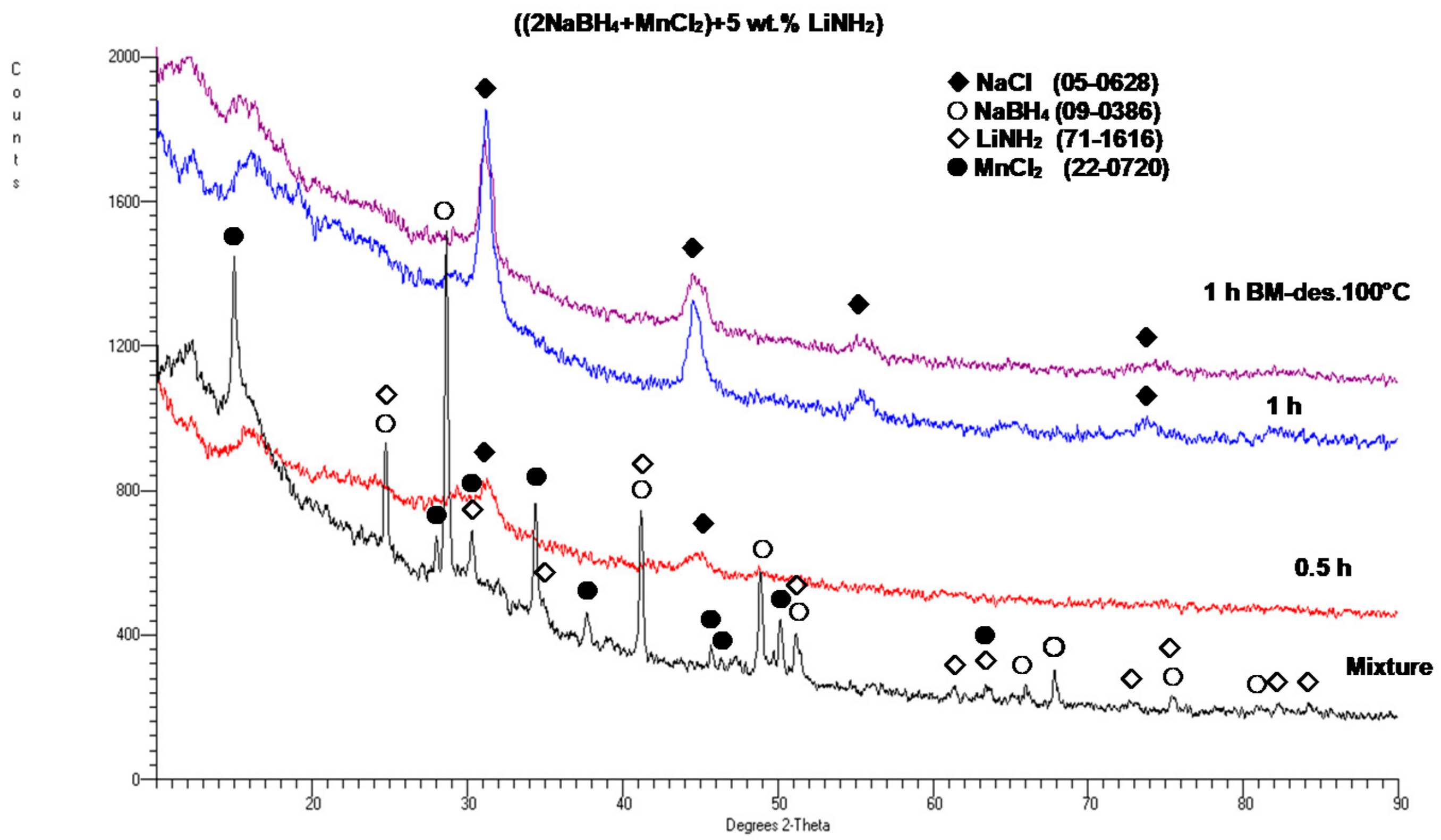
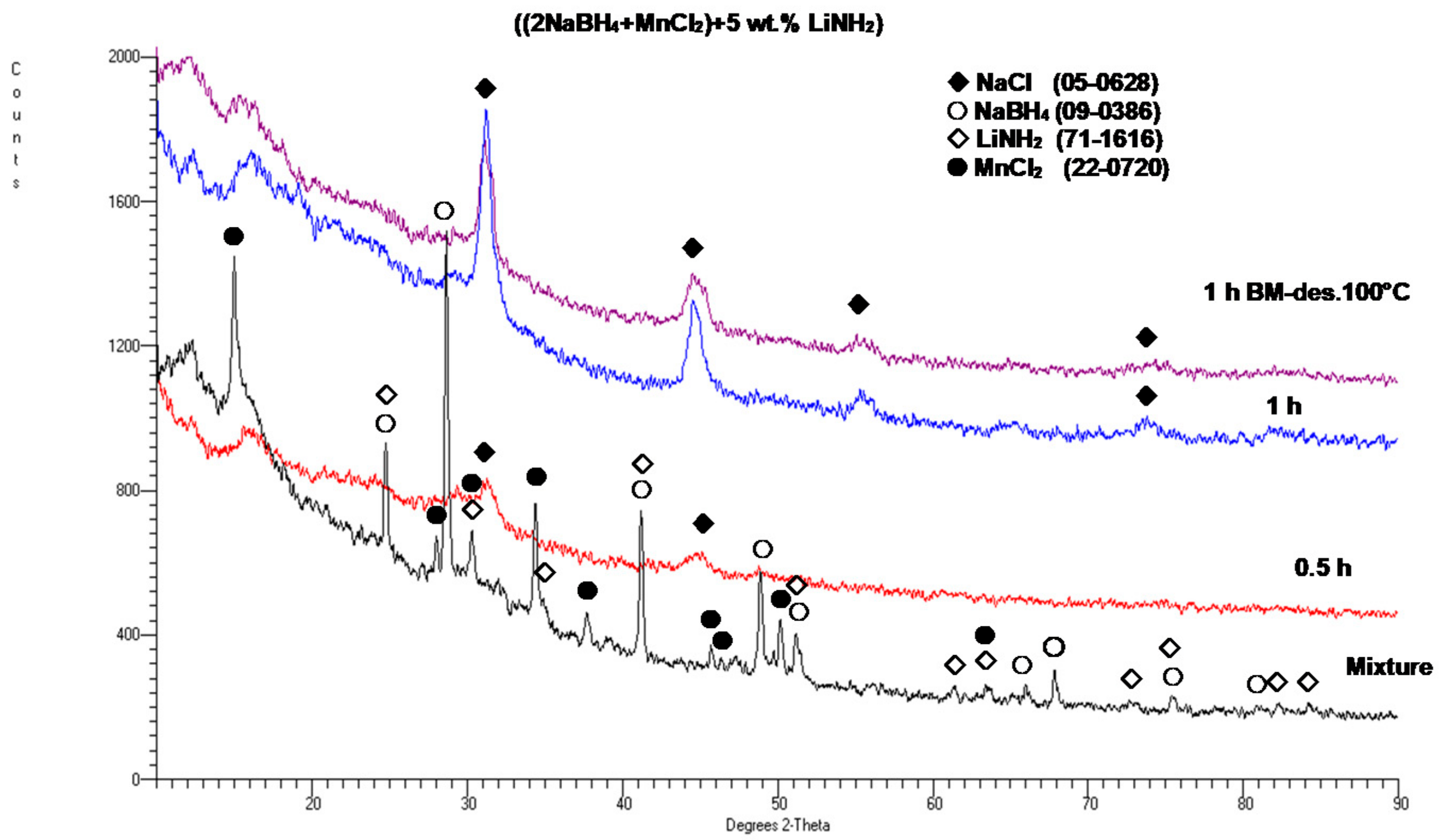
Figure 5 shows the XRD patterns for a sample with a 5 wt. % LiNH
2 additive after 0.5 and 1 h BM, as compared to the XRD patterns for the initial reactants NaBH
4 and MnCl
2. For comparison, an XRD pattern for the 1 h synthesized sample after desorption at 100 °C for 36 h is also shown, and no change in the XRD pattern is observed. However, as will be shown later, the synthesized sample released some quantity of H
2 gas. It is observed that after barely 0.5 h of BM, the diffraction peaks of the reactants disappear, and the new diffraction peaks of the NaCl-type salt appear. This clearly confirms that a mechanochemical reaction must have occurred during ball milling between the NaBH
4 and MnCl
2 reactants, which produced a NaCl-type salt accompanied by an
amorphous a-Mn(BH
4)
2-type hydride in the same way as reported by Varin et al. [
11]. This behavior is apparently common to the (2NaBH
4 + MnCl
2) additive-free samples [
11] and those with the 5 wt. % graphene and filamentary Ni additives (
Figure 2). In addition, the LiNH
2 diffraction peaks, which are visible in a mixture of reactants and additives, disappeared after BM, which indicates that the LiNH
2 additive was likely amorphized during BM.
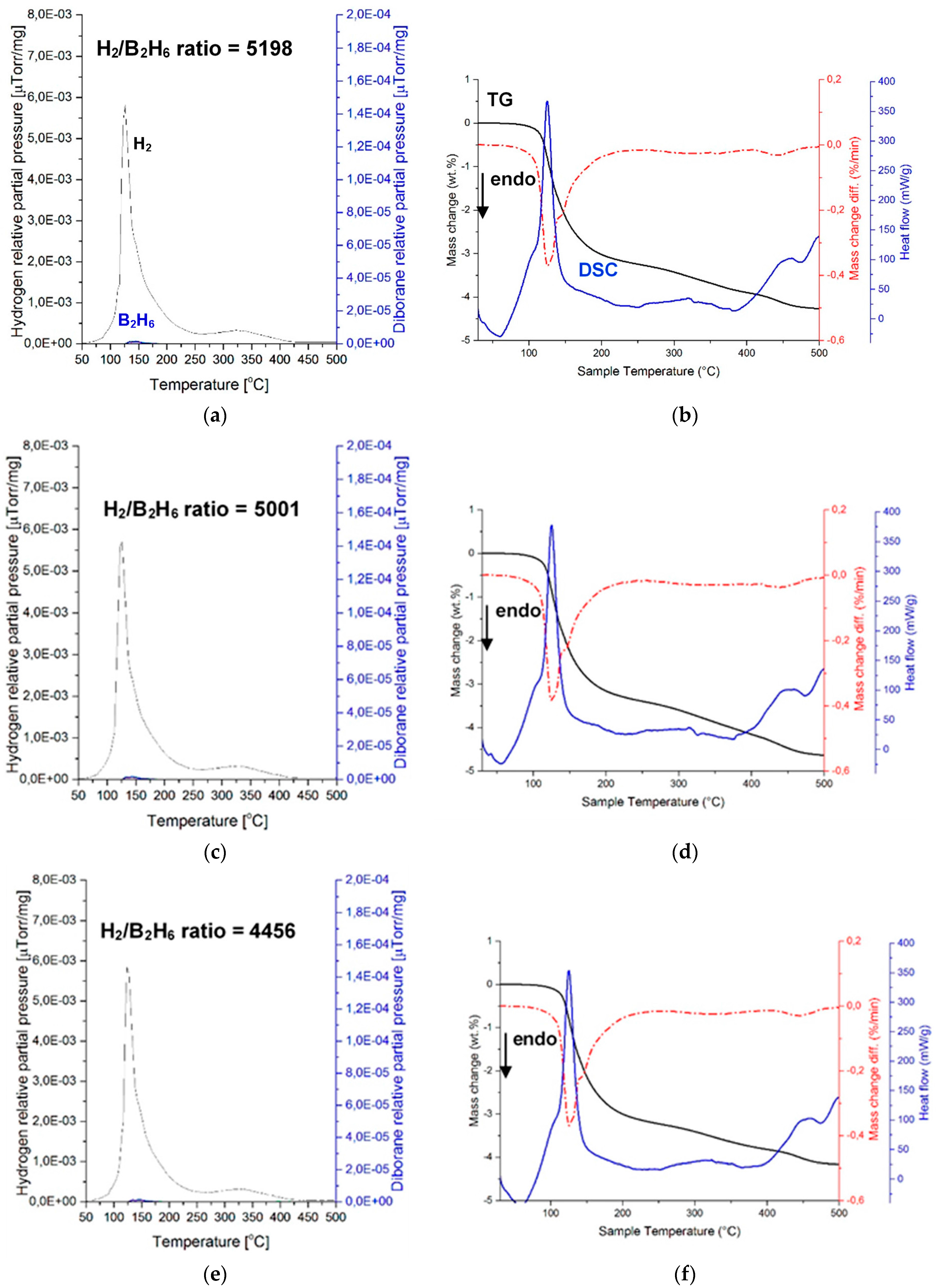
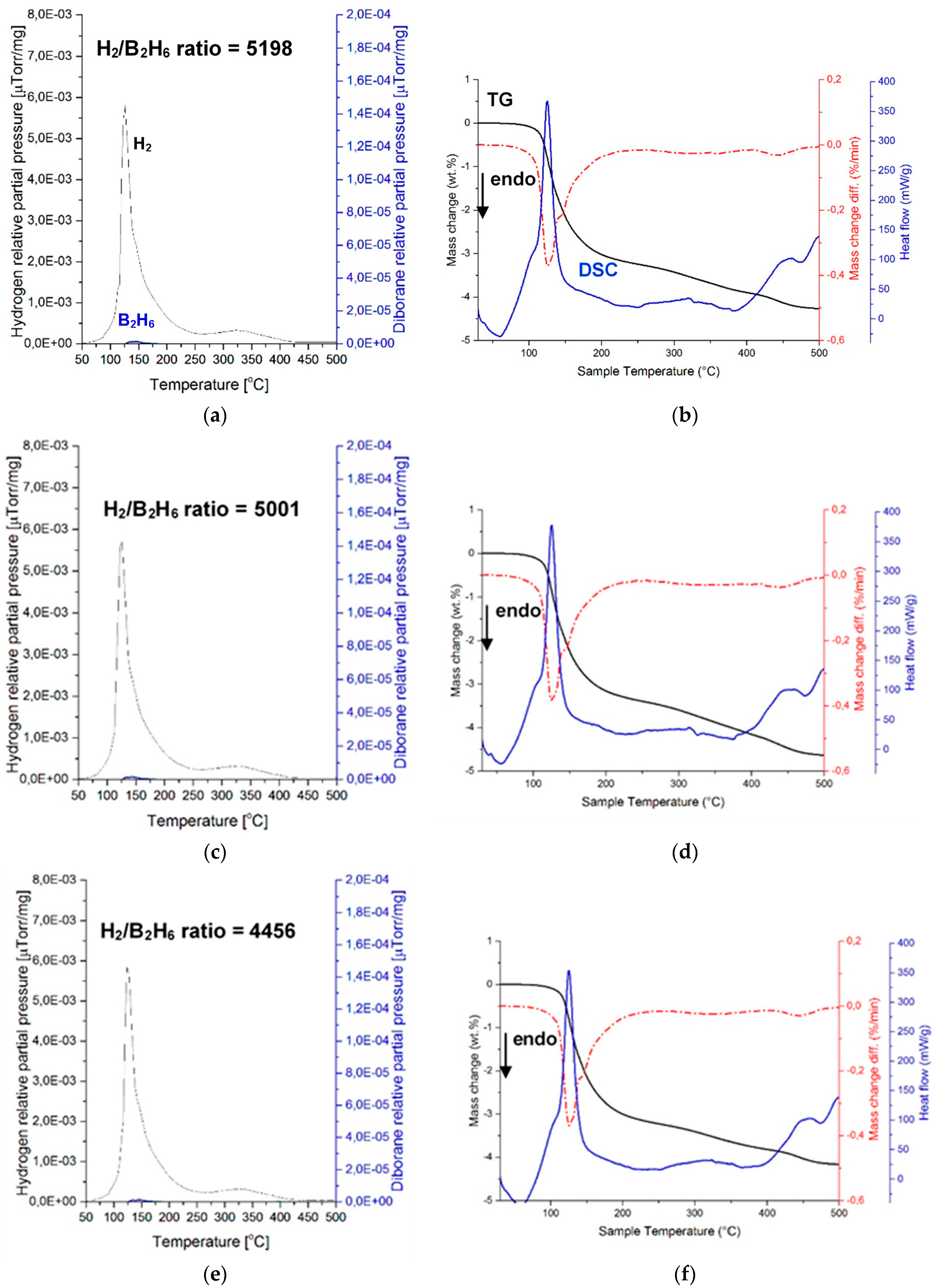
Figure 6 shows the mass spectrometry (MS) results of a (H
2 + B
2H
6) gas mixture and the corresponding TGA and DSC curves during gas desorption for three samples of a ((2NaBH
4 + MnCl
2) + 5 wt. % LiNH
2) composition, ball milled for 0.5 h (Q
TR = 36.4 kJ/g). MS of gas desorption is quite remarkable, since it clearly shows nearly no presence of B
2H
6. The intensity ratios of the MS H
2/B
2H
6 signal up to 200 °C are 5198, 5001, and 4456 for samples 1, 2, and 3, respectively. Apparently, the LiNH
2 additive is a very potent suppressor of B
2H
6, as already reported by Song et al. [
12] for a LiBH
4/MnF
2 mixture upon its thermal decomposition. The same effect of LiNH
2 is confirmed for the first time for a mechanochemically reacted system of (2NaBH
4 + MnCl
2).
2.2. Crystalline c-Mn(BH4)2 after Solvent Filtration/Extraction
The NaCl-type salt that is formed as a by-product of the mechanochemical reaction during ball milling of the additive-free (2NaBH
4 + MnCl
2) [
11], or the samples containing the additives, as in this work, is completely useless because it is a “dead-weight” that adds up to the total molecular weight of the system.
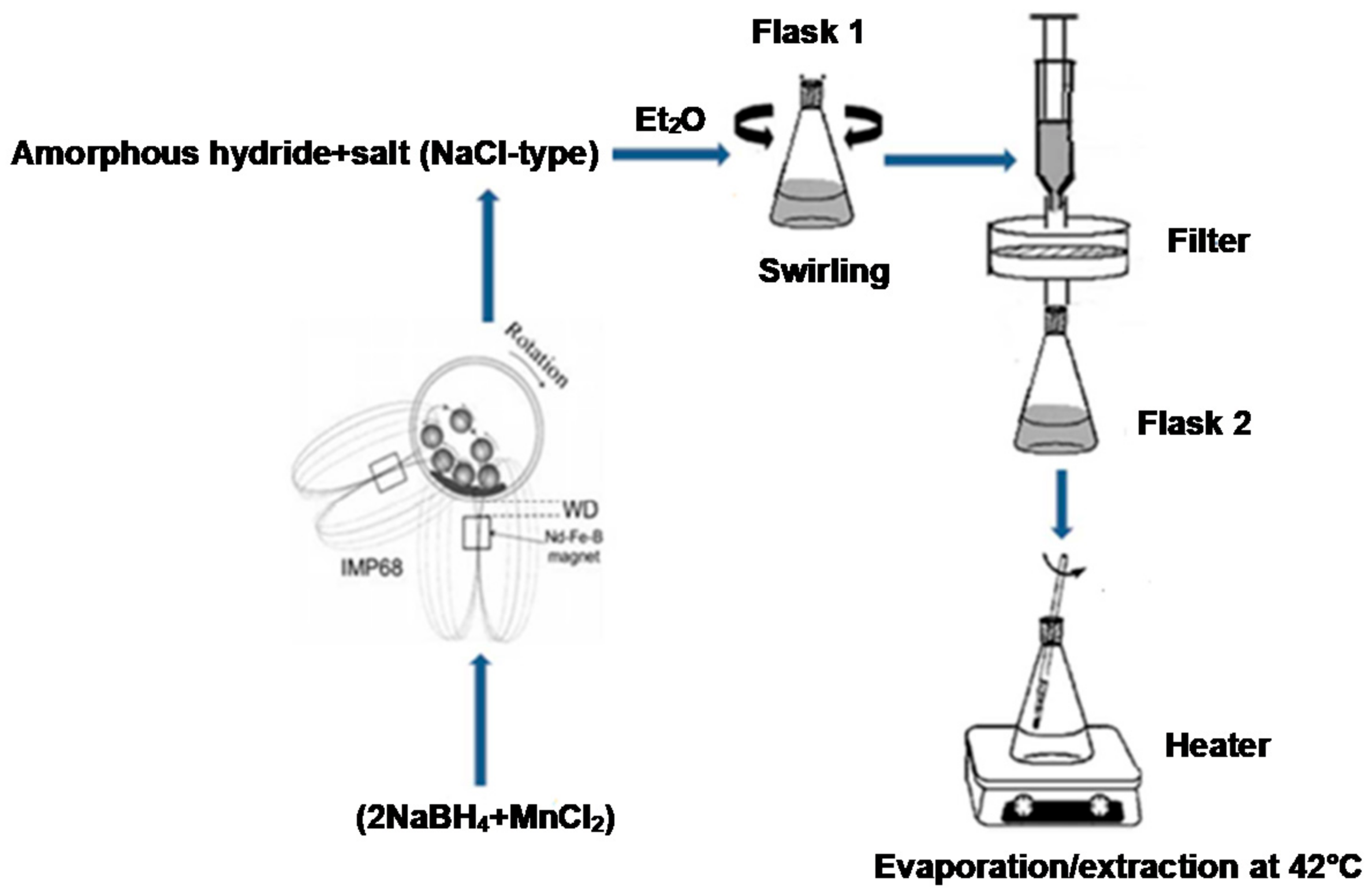
In contrast to the pale brown color of the ball milled mixture, the dry powder obtained after solvent filtration extraction (see Materials and Methods) had a light grey color, which is a completely different color from the pale yellow of the Mn(BH
4)
2-NaCl mixture obtained in the final drying step of the solvent synthesis route [
14].
Figure 7 shows the scanning electron micrographs of the extracted, additive-free (2NaBH
4 + MnCl
2) sample. Very characteristic thin “slabs” resembling highly compacted powder can be seen at both magnifications.
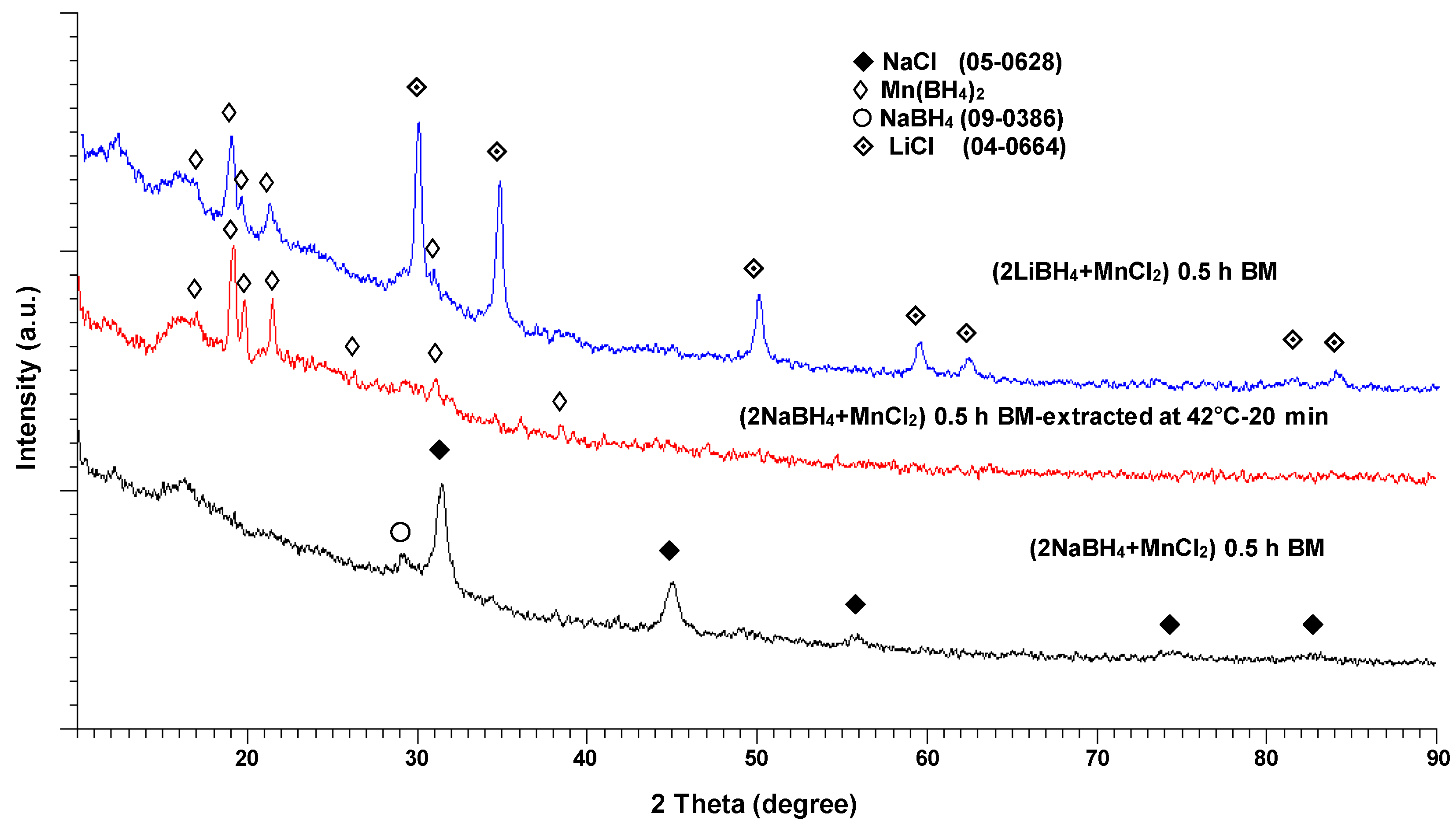
The XRD pattern for the ball milled (2NaBH
4 + MnCl
2) sample is shown at the bottom of
Figure 8. The pattern in the center is for the same sample after solvent filtration/extraction at 42 °C (as reported by Varin et al. [
11]). For comparison, the XRD pattern for the ball milled (2LiBH
4 + MnCl
2) mixture is also included at the top of
Figure 8.
Solely the NaCl-type diffraction peaks accompanied by a weak, singular NaBH
4 peak can be seen in the BM (2NaBH
4 + MnCl
2) XRD pattern (bottom), as already reported by Varin et al. [
11]. The center XRD pattern in
Figure 8, which belongs to the same sample after solvent filtration/extraction, is very similar to the XRD pattern of the BM (2LiBH
4 + MnCl
2) mixture containing the
crystalline c-Mn(BH
4)2 hydride co-existing with a LiCl salt [
3,
4,
5,
6,
7,
8,
9,
10] (a top XRD pattern). The NaCl-type salt was effectively filtered during the filtration stage, and therefore, no NaCl-type diffraction peaks are discernible in
Figure 8 in the XRD pattern for the solvent extracted sample (center). These findings clearly show that an
amorphous a-Mn(BH
4)
2-type hydride, that was formed through ball milling, was transformed into a
crystalline c-Mn(BH
4)
2 hydride during solvent filtration/extraction. When the 1 h ball milled (2NaBH
4 + MnCl
2) sample was used, we observed identical results (the XRD patterns are not shown here).
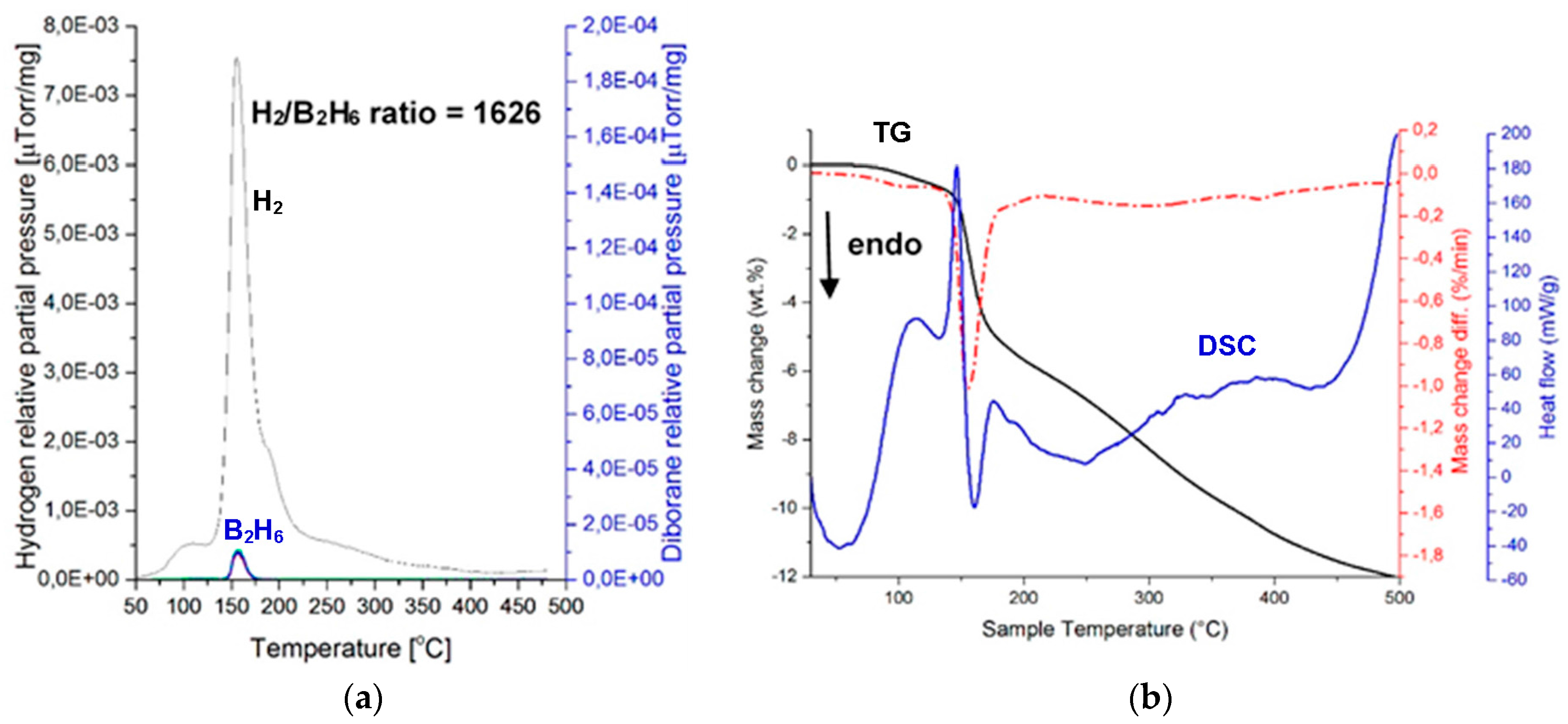
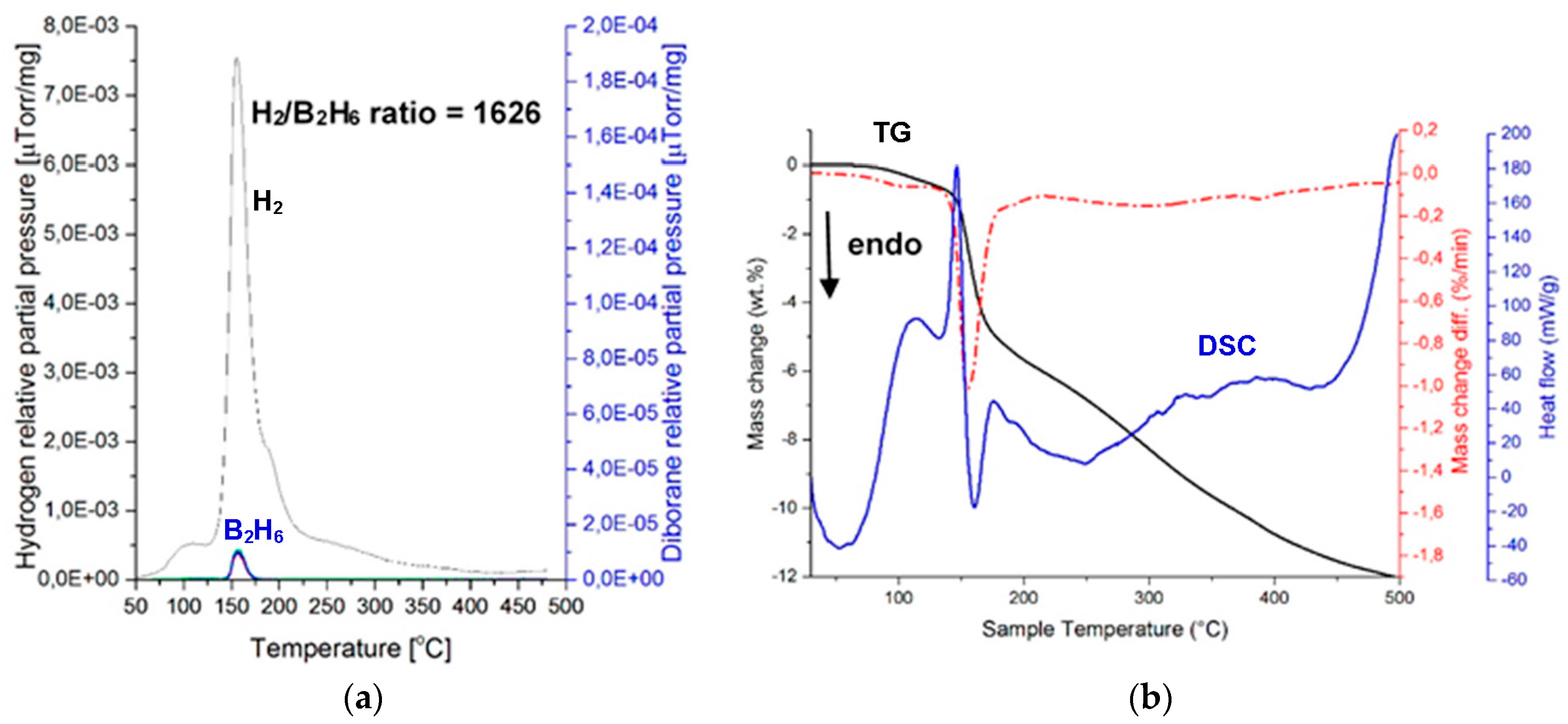
Figure 9 shows MS (gas desorption spectra) of a (H
2 + B
2H
6) mixture (
Figure 9a) and the corresponding TGA and DSC thermal decomposition curves (
Figure 9b) for the (2NaBH
4 + MnCl
2) sample ball milled for 1 h (Q
TR = 72.8 kJ/g), and then extracted at 42 °C. The intensity ratio of the MS H
2/B
2H
6 signal up to 200 °C equals 1626, which is much higher than 948 for a ball milled (2NaBH
4 + MnCl
2) counterpart [
11], and also much higher than the ratio of 200–600 for a ball milled (2LiBH
4 + MnCl
2)[
8]. The second extracted sample, which was investigated by MS, exhibited the MS ratio H
2/B
2H
6 = 1546, which confirms a very low quantity of released B
2H
6 from the extracted
crystalline c-Mn(BH
4)
2. Apparently, the
crystalline c-Mn(BH
4)
2 hydride that was extracted from the ball milled, additive-free (2NaBH
4 + MnCl
2), has a very low release of B
2H
6 during thermal decomposition, which is nearly on par with the ((2NaBH
4 + MnCl
2) + 5 wt. % graphene) in
Figure 3a. Furthermore, as shown in
Figure 9b, the DSC decomposition curve for the extracted
crystalline c-Mn(BH
4)
2 hydride still has an endothermic character. The TG curve is also quite peculiar, as it shows a rapid mass loss of about 5.7 wt. % until 200 °C, and then a slower second stage with the total mass loss of about 8 wt. % up to 300 °C. This is rather consistent with the MS curve in
Figure 9a, which shows a rapid H
2 loss up to ~200 °C and then a slower H
2 release up to 300 °C. However, the mass loss still continuous up to 500 °C with a total of 12 wt. % mass loss. The MS curve in
Figure 9b also shows slight H
2 release between 300 and 500 °C. It is possible that the H
2 release/mass loss between 300 and 500 °C is due to decomposition of a small amount of residual NaBH
4 whose remnant diffraction peak is observed after ball milling in
Figure 8. The residual NaBH
4 could have been extracted together with c-Mn(BH
4)
2 during solvent filtration/extraction.
A similar result was observed when the ball milled ((2NaBH4 + MnCl2) + 5 wt. % Ni) was filtered/extracted (the XRD patterns are not shown here). The same types of peaks associated with crystalline c-Mn(BH4)2 were observed. Interestingly, no Ni peaks were observed in the XRD patterns, which strongly suggests that the ultrafine filamentary Ni did not pass through the syringe filter during filtration. Thus, the catalytic effect of Ni was not observed in the thermal desorption process of the sample containing filamentary Ni.
It must be pointed out that from the Full Width at Half Maximum (FWHM) for the Bragg diffraction peaks for crystalline c-Mn(BH
4)
2, obtained from three filtered/extracted powder samples, like those in
Figure 8, we calculated corresponding crystallite (grain) sizes residing in the powder particles. The results are shown in
Table 1.
Experimentally obtained lattice spacings, d
(hkl), for
crystalline c-Mn(BH
4)
2, that was solvent extracted from the 0.5 and 1 h ball milled (2NaBH
4 + MnCl
2) samples, are listed in
Table 2. For comparison, the d
(hkl) values calculated from synchrotron radiation as reported by Varin et al. [
8], and those obtained from selected area electron diffraction pattern (SAEDP) [
10], are also included for comparison. The agreement between the d
(hkl) values in this work and those reported by Varin et al. [
8] and Shirani Bidabadi [
10] is very good. Since different precursors and methods for the Mn(BH
4)
2 synthesis were used in the present work than those reported by Varin et al. [
8] and Shirani Bidabadi [
10], some small differences in the lattice spacings are expected.
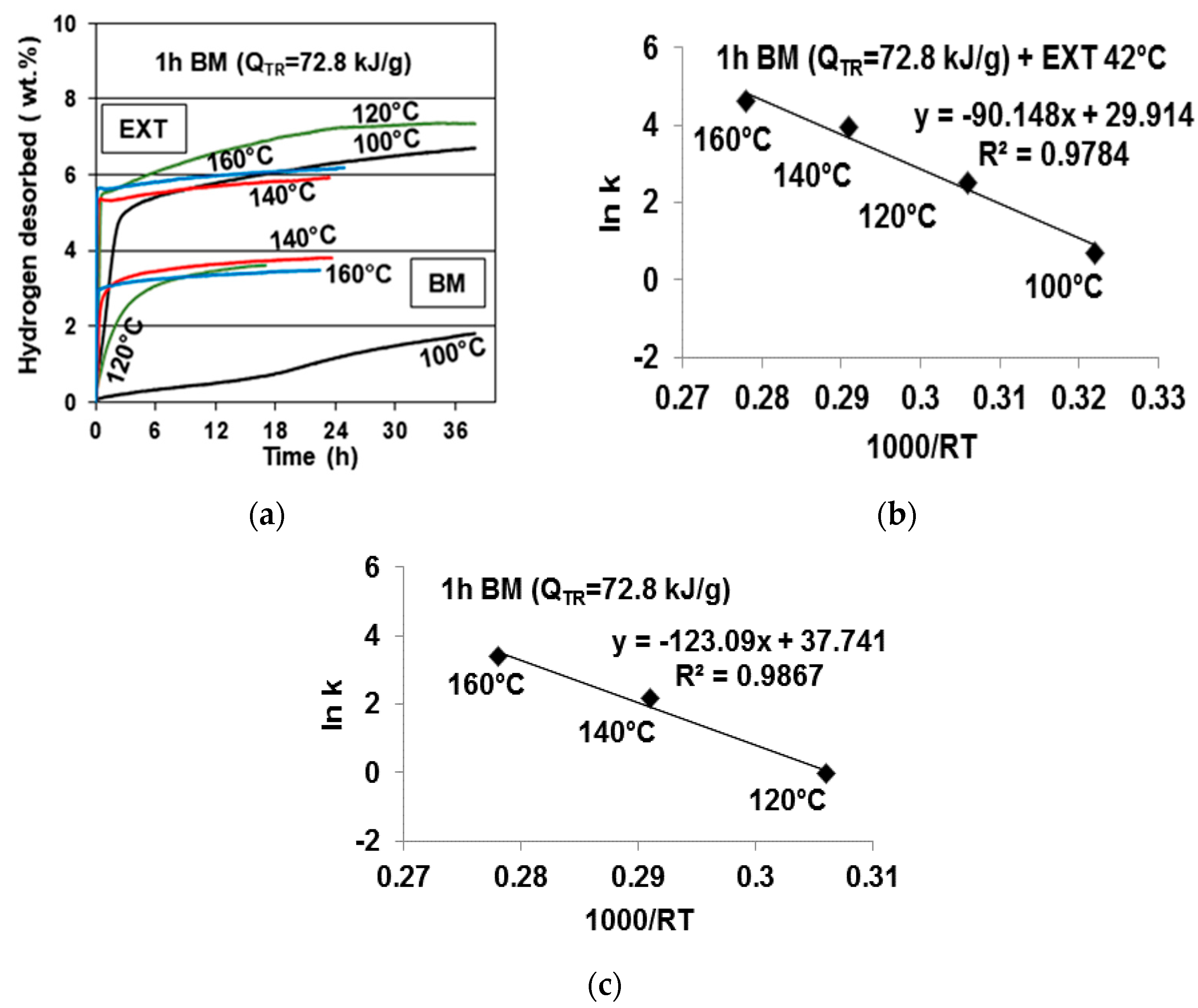
Figure 10a shows a comparison of the thermal desorption curves at four temperatures for the filtered/extracted sample (EXT), and the same (2NaBH
4 + MnCl
2) sample ball milled (BM) for 1 h before filtration/extraction. It is apparent that all of the desorption curves for each temperature for the filtered/extracted sample (EXT) are located much above their BM counterparts, which indicates a much higher H
2 capacity released from the filtered/extracted sample. Within an experimental scatter, the desorption curves approach about 7–7.5 wt. % H
2 (as mentioned earlier, due to a very small quantity of released B
2H
6 (
Figure 9a), the
y axis for the gas desorption curves in
Figure 10a is designated as desorbing solely H
2). Desorption at 140 °C and 160 °C seems to be suppressed, for some reasons, after an initial rapid stage as compared with that at 100 °C and 120 °C. Also, the H
2 desorption at 100 °C is much faster than that for their ball milled counterpart.
Figure 10b shows the Arrhenius plot for the filtered/extracted sample with the estimated AAE of 90.15 kJ/mol. This can be compared with a higher AAE of 123.09 kJ/mol for its ball milled counterpart (
Figure 10c [
11]). It is quite obvious that the c-Mn(BH
4)
2 hydride formed after filtration/extraction has much better thermal desorption characteristics than its
amorphous a-Mn(BH
4)
2-type precursor, which was synthesized during ball milling.
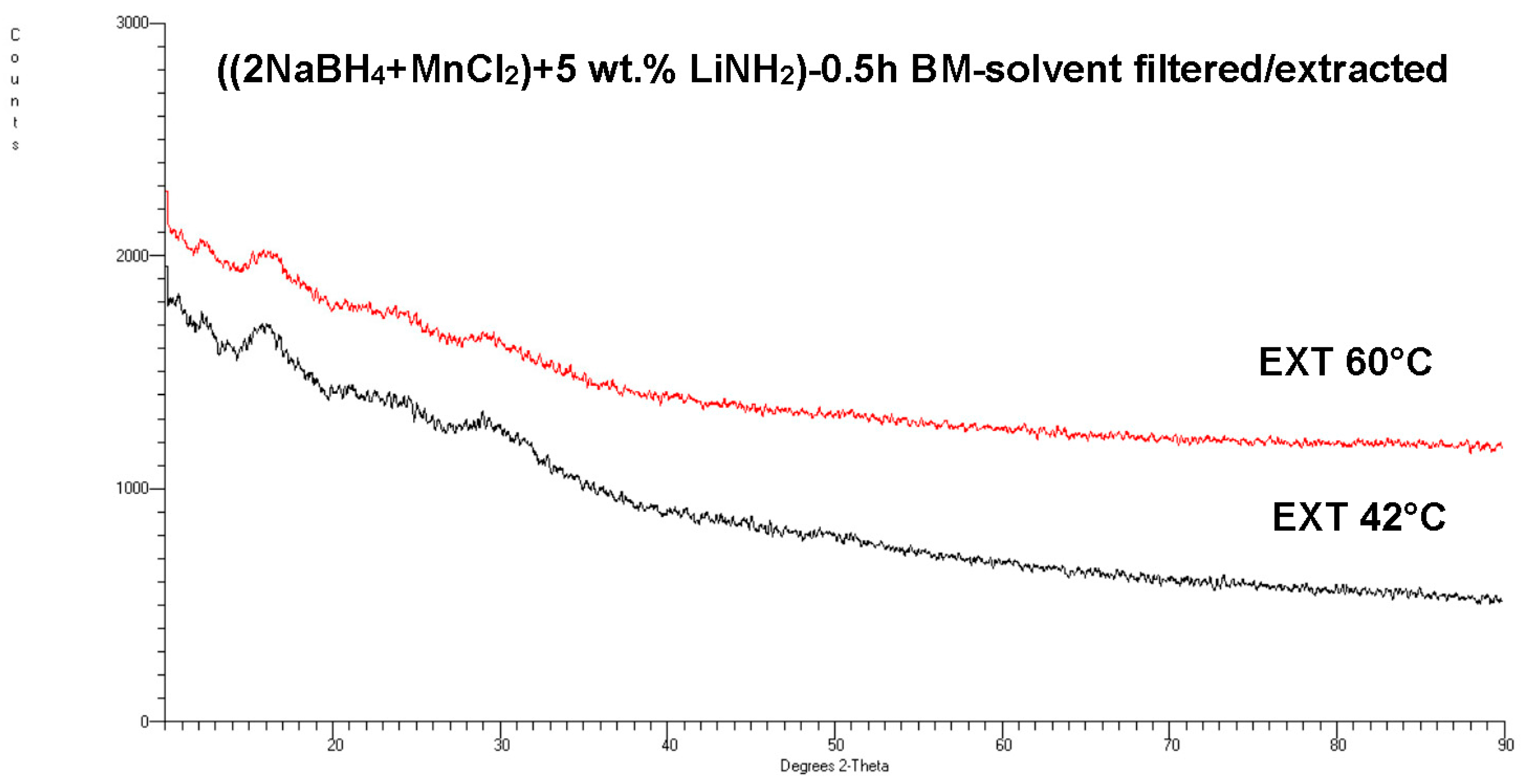
Figure 11 shows an XRD pattern for the 0.5 h BM ((2NaBH
4 + MnCl
2) + 5 wt. % LiNH
2) sample (Q
TR = 36.4 kJ/g), after filtration/extraction at 42 °C and 60 °C. No diffraction peaks for
crystalline c-Mn(BH
4)
2 are present in
Figure 11, which is similar to
Figure 8 for an additive-free sample. This result is very striking, since it strongly suggests that the LiNH
2 additive suppressed the transformation of an
amorphous Mn(BH
4)
2-type hydride into a
crystalline Mn(BH
4)
2 hydride during solvent extraction.
Figure 12 shows the MS (
Figure 12a) and TGA/DSC (
Figure 12b) thermal behavior for the same ((2NaBH
4 + MnCl
2) + 5 wt. % LiNH
2) sample, whose XRD pattern is shown in
Figure 11. MS shows no presence of desorbed diborane (B
2H
6), although simultaneously, the H
2 capacity is also reduced for reasons that are not clear. However, a complete elimination of B
2H
6 from the released gas is striking.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}