Comparison of Lithium-Ion Anode Materials Using an Experimentally Verified Physics-Based Electrochemical Model

 and
and
Abstract
:1. Introduction
2. Results
2.1. Parameter Identification
2.1.1. Battery Manufacturing and Physical Property Characterization
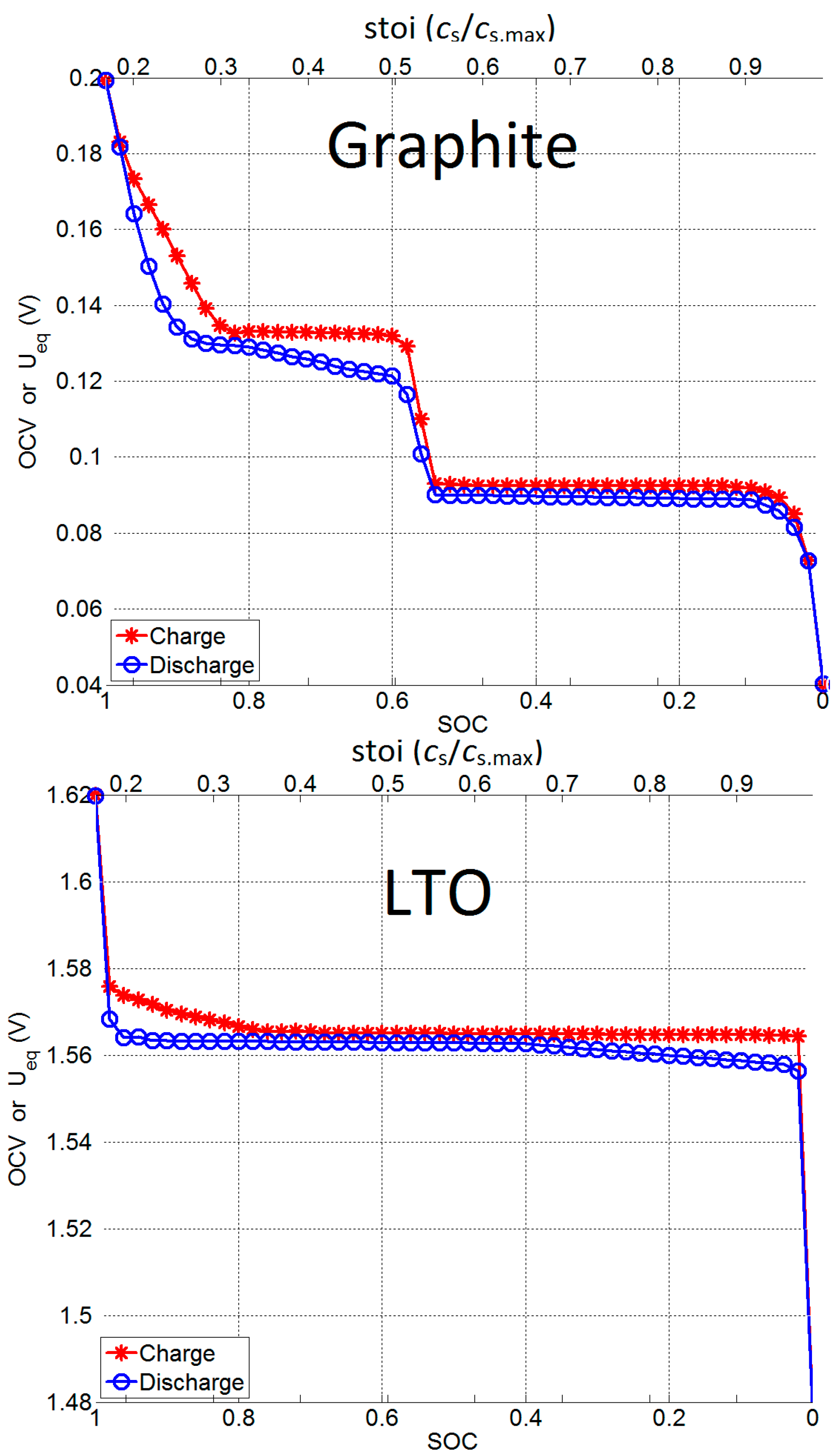
2.1.2. Open Circuit Voltage (OCV) Measurement
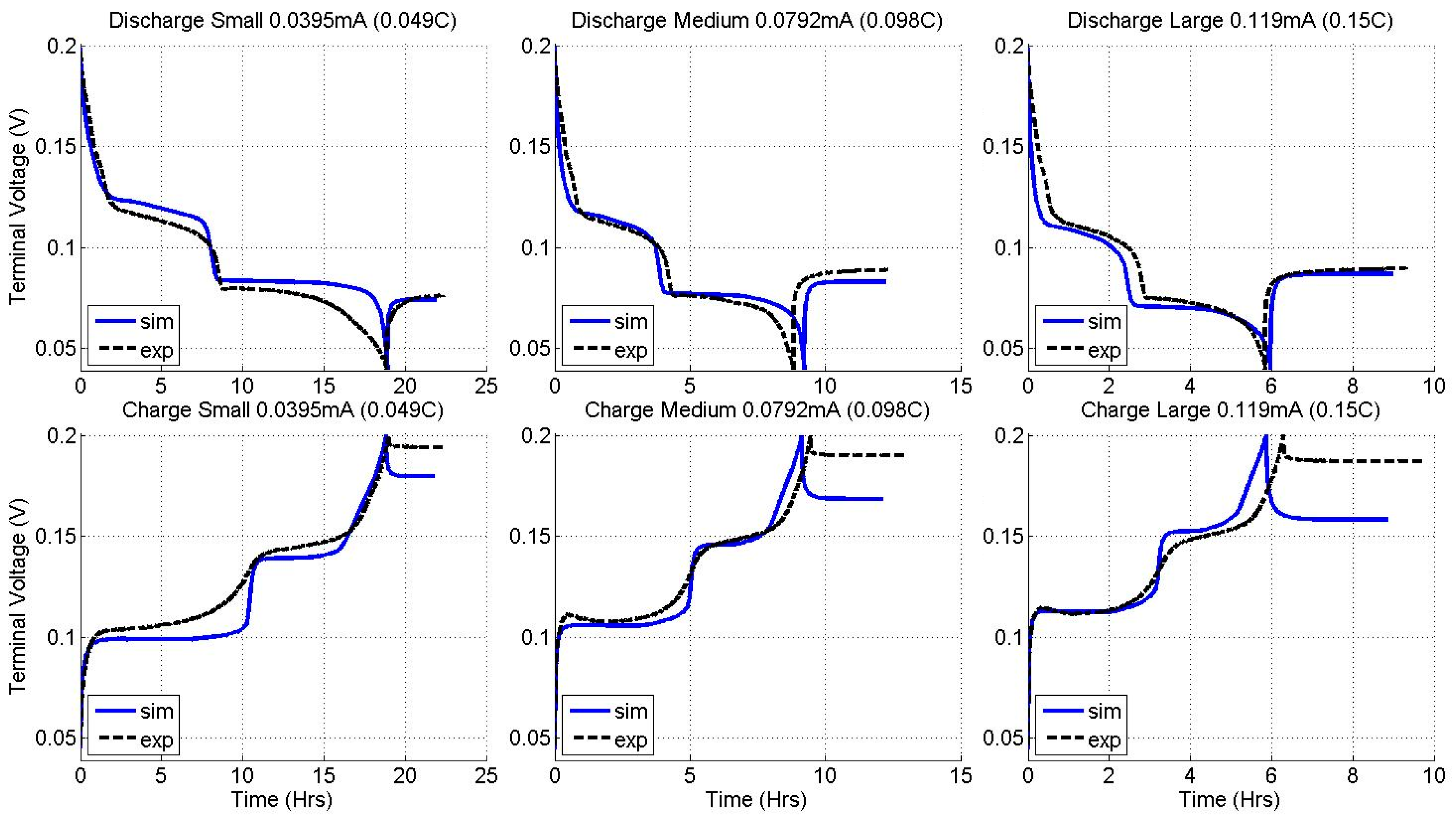
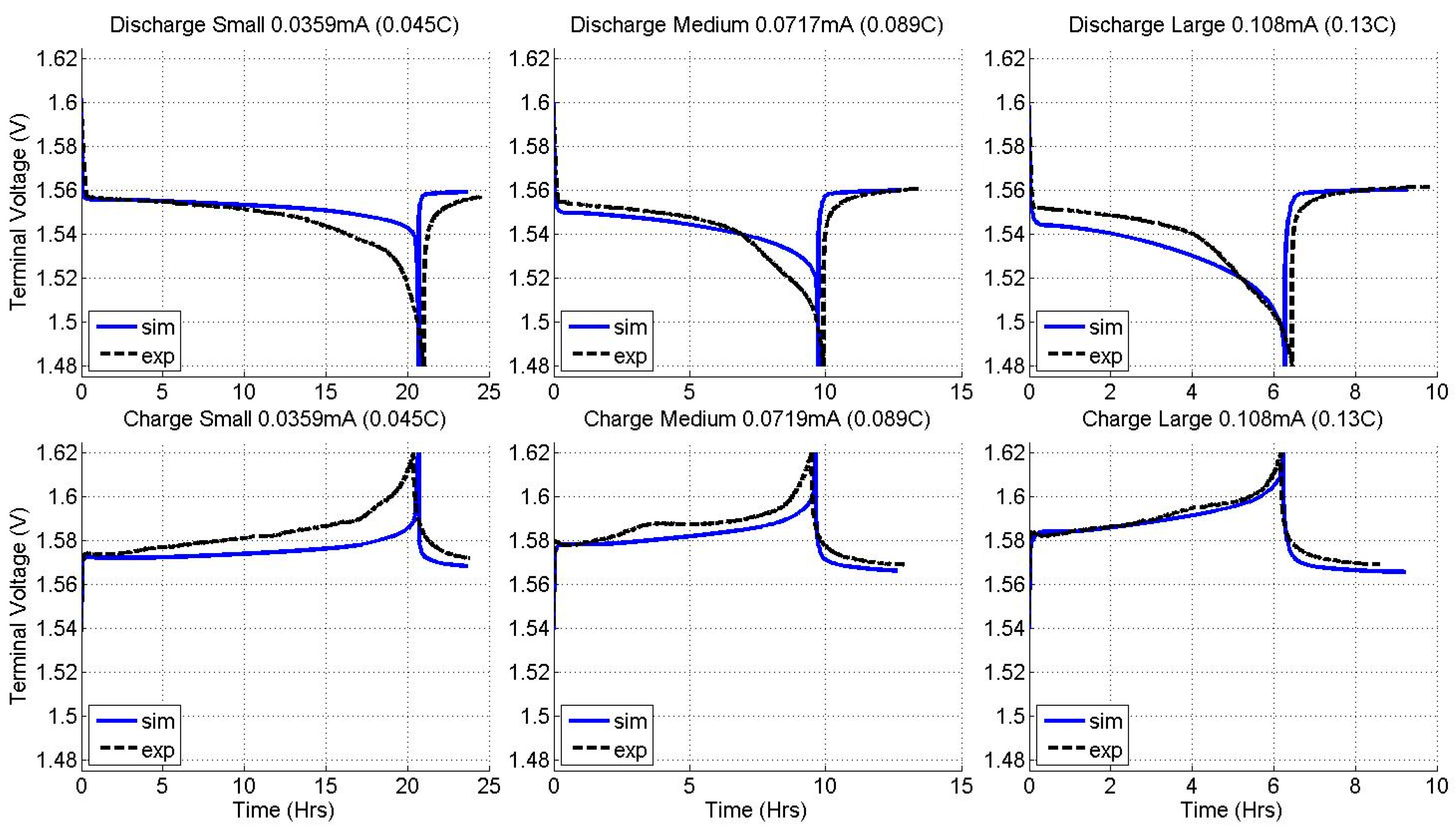
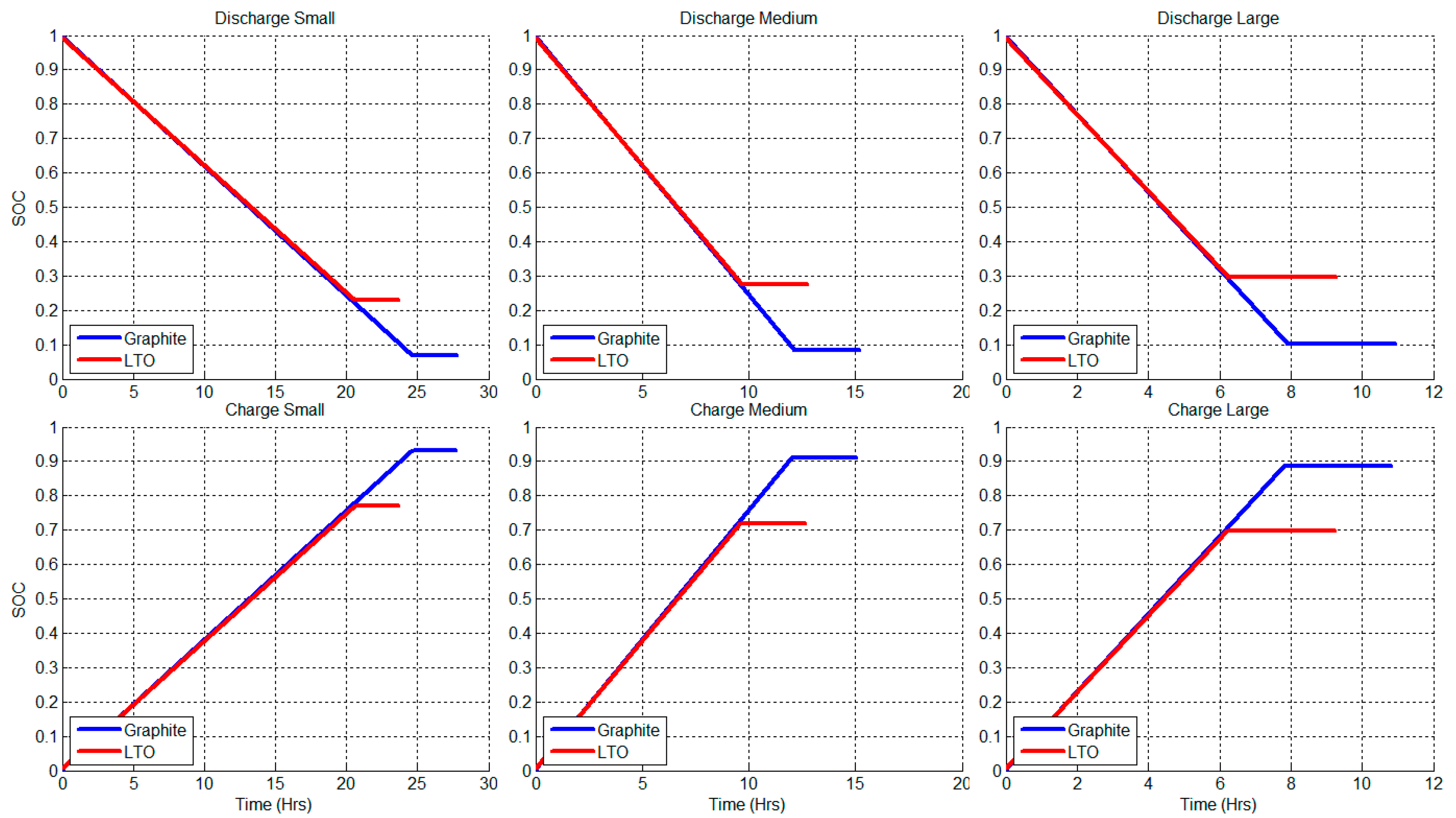
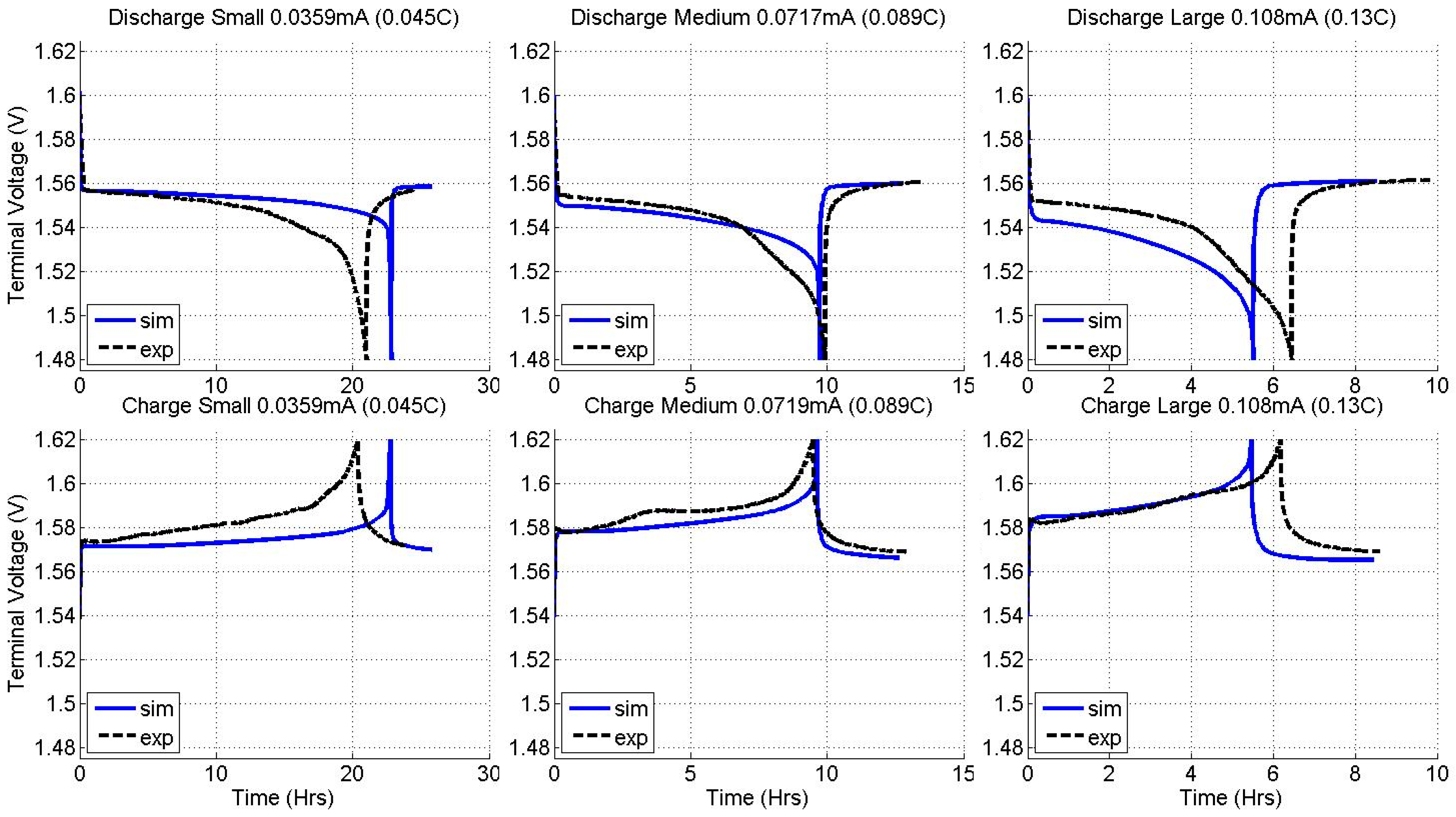
2.2. Constant Current Charge/Discharge and Model Validation
3. Discussion
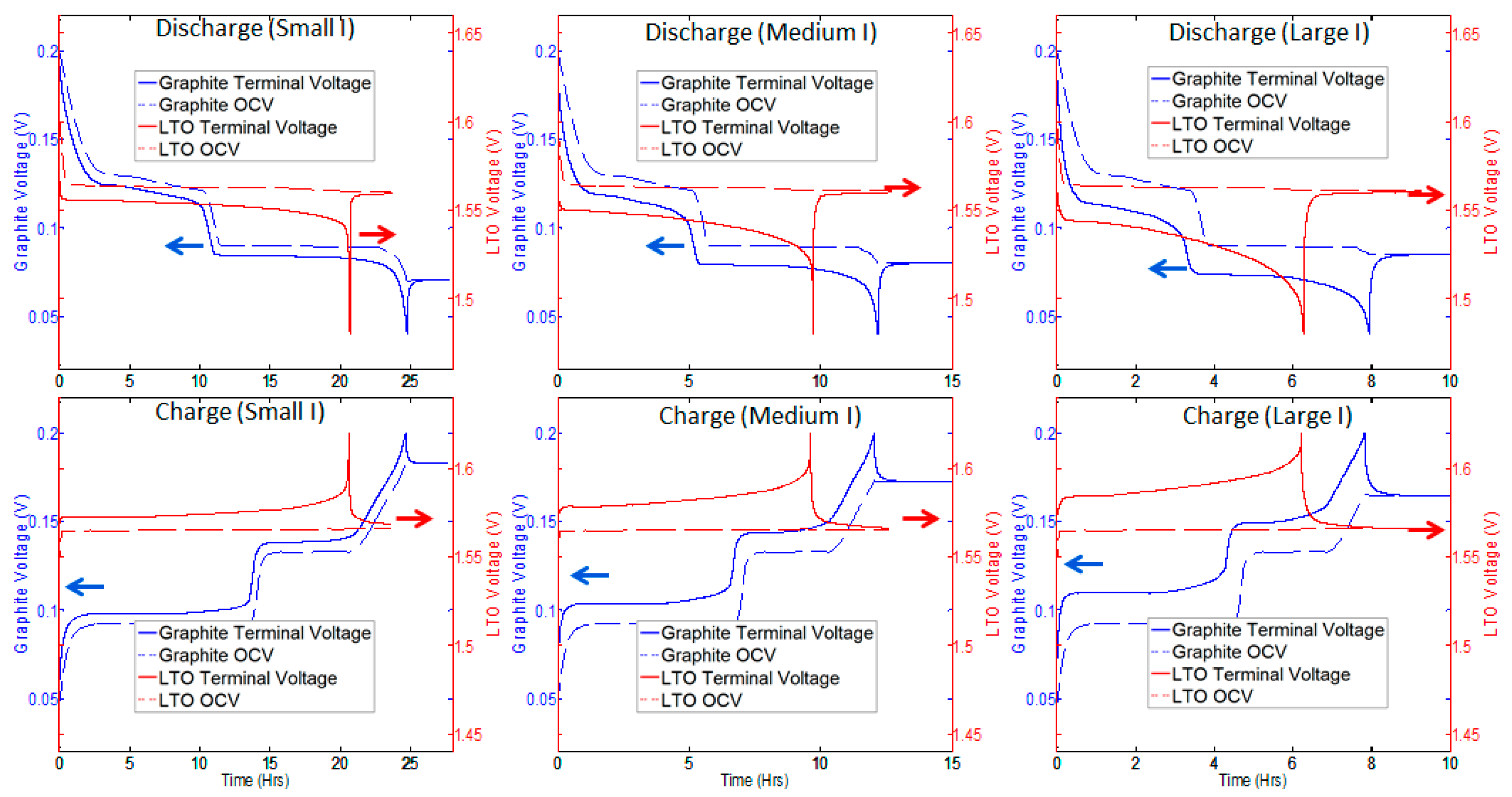
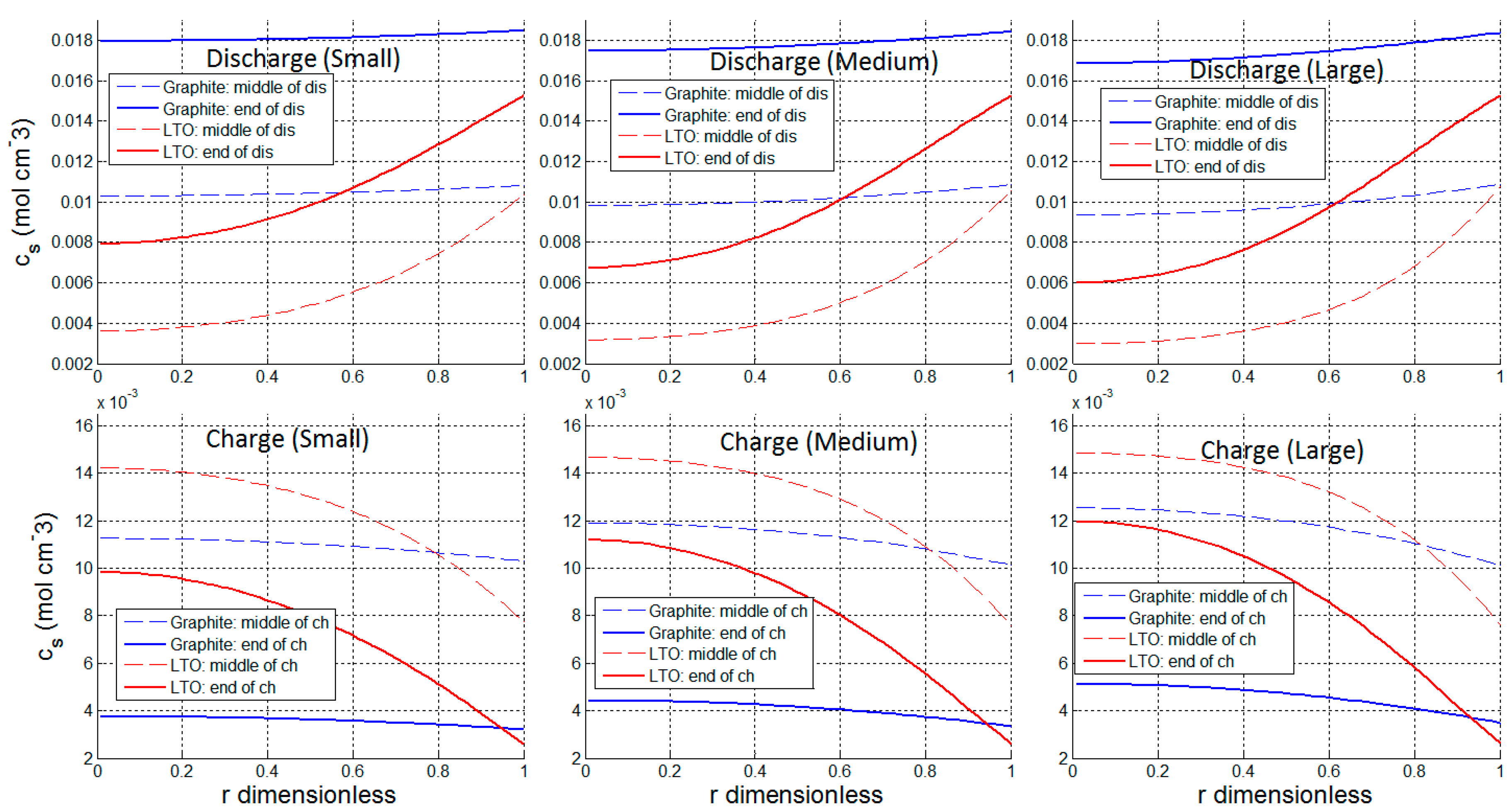
3.1. Utilization and Ion Diffusion in Solid Phase
3.2. Current Dependence and Large Current Applications
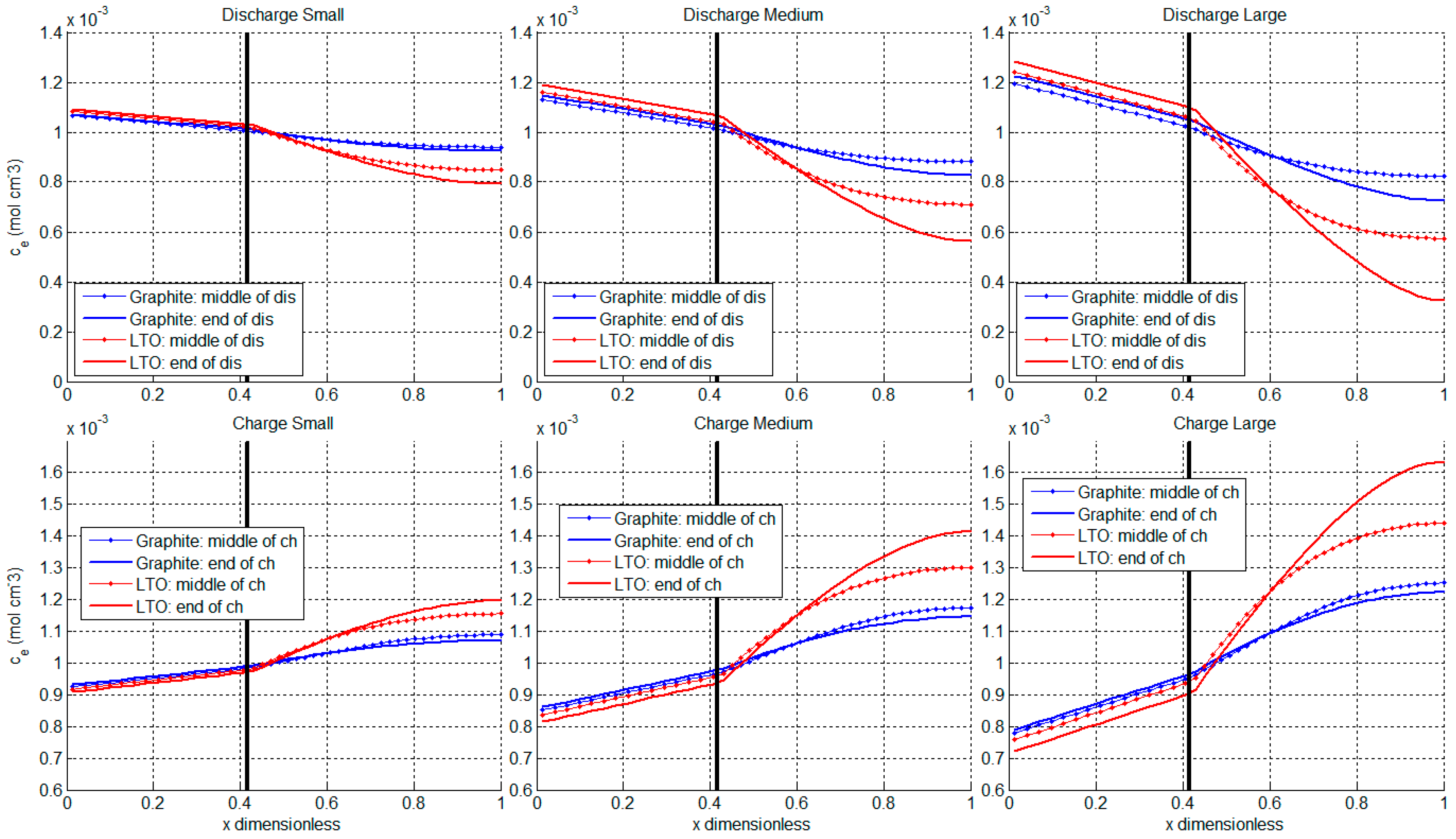
3.3. Ion Diffusion in Electrolyte
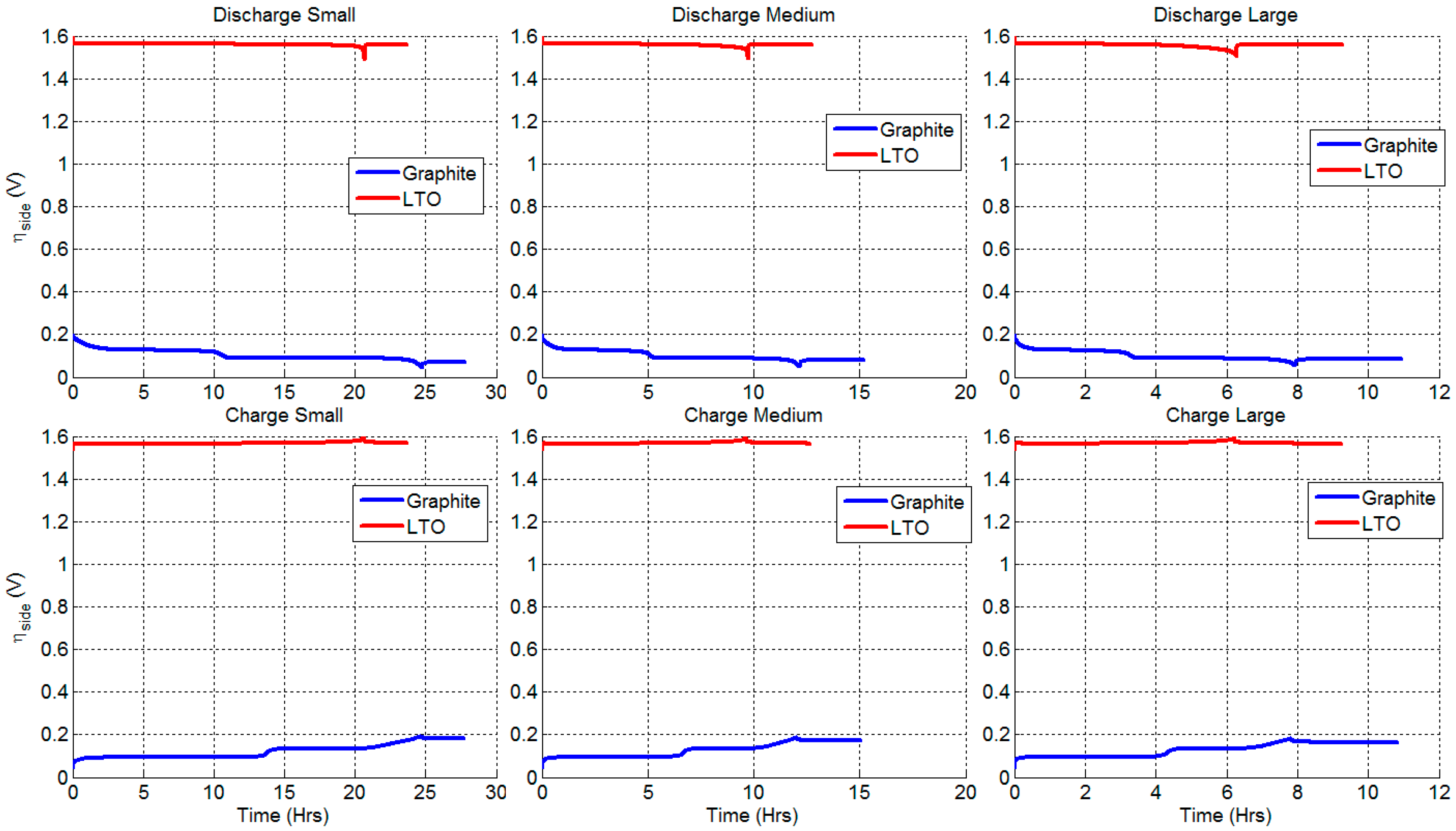
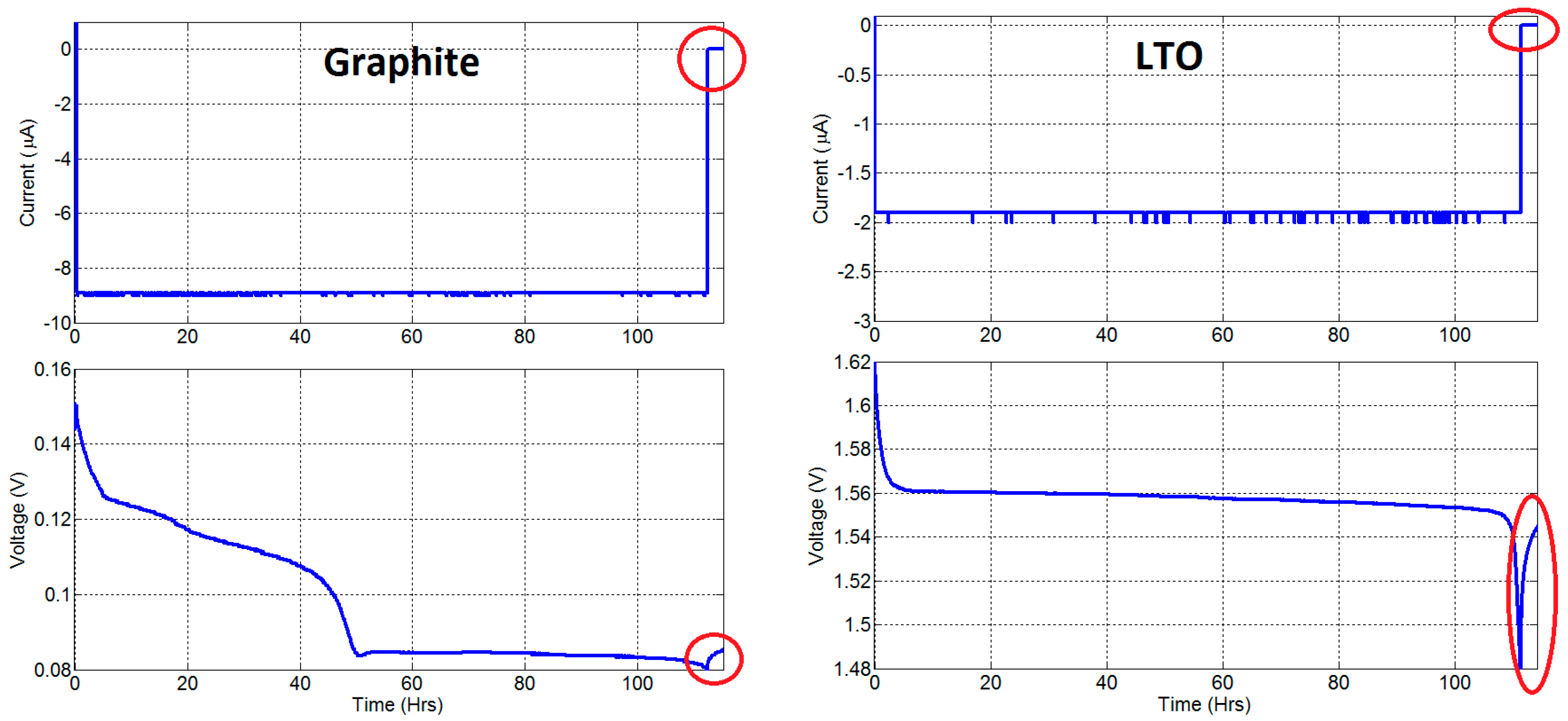
3.4. Degradation Caused by Side Reactions
4. Materials and Methods
4.1. Anode Material Selection
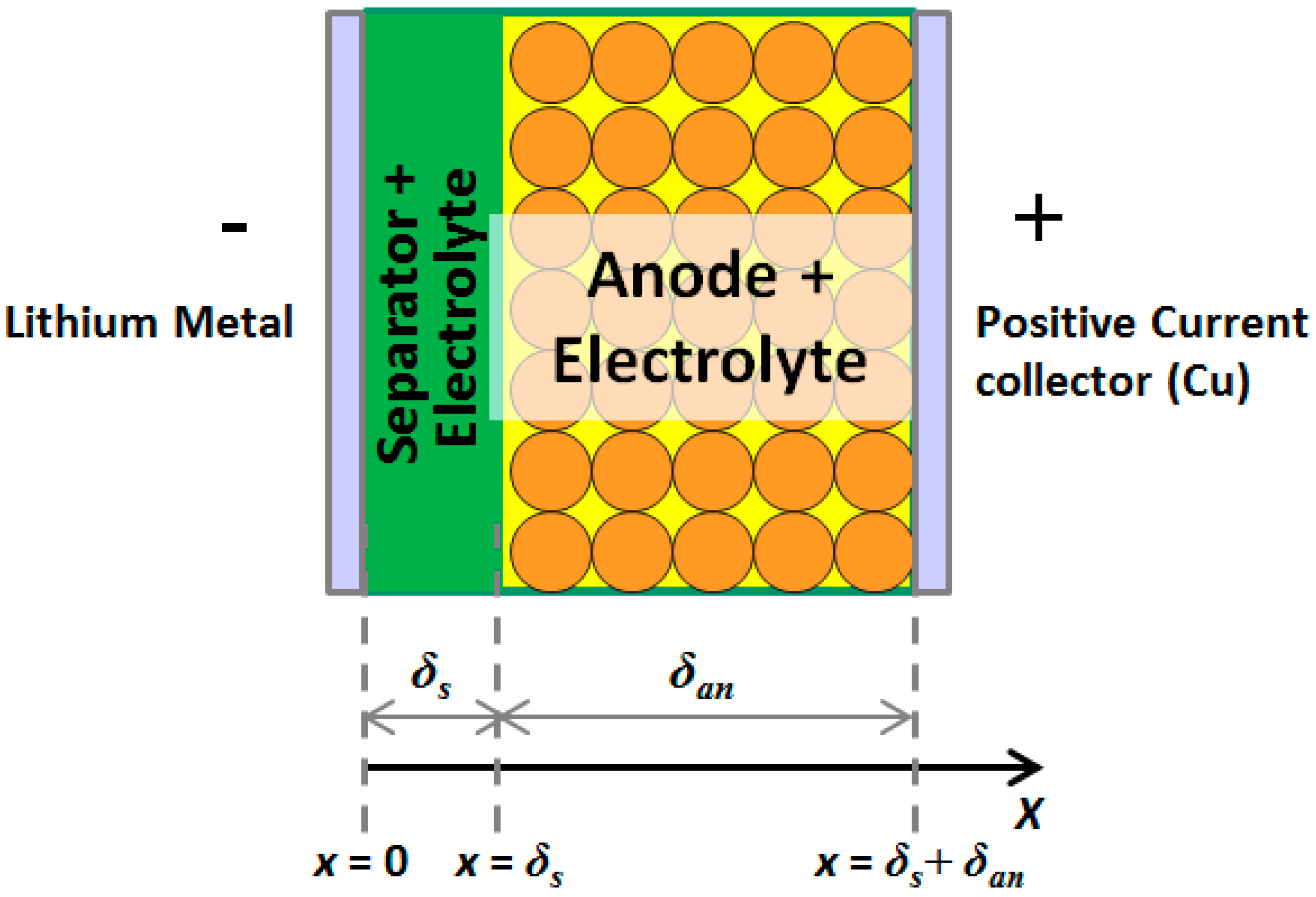
4.2. Model Development
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. SEM of Anode Materials

Appendix B. An Alternative OCV Test Method

Appendix C

References
- Tarascon, J.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, J.B.; Park, K. The Li-Ion Rechargeable Battery: A perspective. J. Am. Chem. Soc. 2013, 135, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xu, J.; Cao, B.; Zhou, X. A novel multimode hybrid energy storage system and its energy management strategy for electric vehicles. J. Power Source 2015, 281, 432–443. [Google Scholar]
- Chaturvedi, N.A.; Klein, R.; Christensen, J.; Ahmed, J.; Kojic, A. Algorithms for Advanced Battery-Management Systems. IEEE Control Syst. Mag. 2010, 30, 49–68. [Google Scholar]
- Zhang, X.; Lu, J.; Yuan, S.; Zhou, X. A novel method for identification of Lithium-ion battery equivalent circuit model parameters considering electrochemical properties. J. Power Sources 2017, 345, 21–29. [Google Scholar] [CrossRef]
- Xiong, R.; Yu, Q.; Wang, L.Y.; Li, C. A novel method to obtain the open circuit voltage for the state of charge of lithium ion batteries in electric vehicles by using H infinity filter. Appl. Energy 2017, 207, 341–348. [Google Scholar] [CrossRef]
- Xiong, R.; Sun, F.; Gong, X.; Gao, C. A data-driven based adaptive state of charge estimator of lithium-ion polymer battery used in electric vehicles. Appl. Energy 2014, 113, 1421–1433. [Google Scholar] [CrossRef]
- Xiong, R.; Tian, J.P.; Mu, H.; Wang, C. A systematic model-based degradation behavior recognition and health monitor method of lithium-ion batteries. Appl. Energy 2017, 207, 367–378. [Google Scholar]
- Xiong, R.; Zhang, Y.; He, H.; Zhou, X.; Pecht, M. A double-scale, particle-filtering, energy state prediction algorithm for lithium-ion batteries. IEEE Trans. Ind. Electron. 2018, 65, 1526–1538. [Google Scholar]
- Sung, W.; Shin, C.B. Electrochemical model of a lithium-ion battery implemented into an automotive battery management system. Comput. Chem. Eng. 2015, 76, 87–97. [Google Scholar] [CrossRef]
- Doyle, M.; Fuller, T.F.; Newman, J. Modeling of Galvanostatic Charge and Discharge of the Lithium/Polymer/Insertion Cell. J. Electrochem. Soc. 1993, 140, 1526–1533. [Google Scholar] [CrossRef]
- Pals, C.R.; Newman, J. Thermal Modeling of the Lithium/Polymer Battery. J. Electrochem. Soc. 1995, 142, 3282–3288. [Google Scholar] [CrossRef]
- Doyle, M.; Newman, J. Comparison of Modeling Predictions with Experimental Data from Plastic Lithium Ion Cells. J. Electrochem. Soc. 1996, 143, 1890–1903. [Google Scholar] [CrossRef]
- Gu, W.B.; Wang, C.Y.; Liaw, B.Y. Numerical Modeling of Coupled Electrochemical and Transport Processes in Lead-Acid Batteries. J. Electrochem. Soc. 1997, 144, 2053–2061. [Google Scholar] [CrossRef]
- Srinivasan, V.; Wang, G.Q.; Wang, C.Y. Mathematical Modeling of Current-Interrupt and Pulse Operation of Valve-Regulated Lead Acid Cells. J. Electrochem. Soc. 2003, 150, A316–A325. [Google Scholar] [CrossRef]
- Gu, W.B.; Wang, C.Y.; Liaw, B.Y. Micro-Macroscopic Coupled Modeling of Batteries and Fuel Cells. J. Electrochem. Soc. 1998, 145, 3418–3427. [Google Scholar] [CrossRef]
- Gu, W.B.; Wang, C.Y.; Li, S.M.; Geng, M.M.; Liaw, B.Y. Modeling discharge and charge characteristics of nickel-metal hydride batteries. Electrochim. Acta 1999, 44, 4525–4541. [Google Scholar] [CrossRef]
- Smith, K.A.; Rahn, C.D.; Wang, C.Y. Control oriented 1D electrochemical model of lithium ion battery. Energy Convers. Manag. 2007, 48, 2565–2578. [Google Scholar] [CrossRef]
- Smith, K.A.; Rahn, C.D.; Wang, C.Y. Model-Based Electrochemical Estimation and Constraint Management for Pulse Operation of Lithium Ion batteries. IEEE Trans. Control Syst. Technol. 2010, 18, 654–663. [Google Scholar] [CrossRef]
- Tanim, T.R.; Rahn, C.D.; Wang, C.Y. A Temperature Dependent Single Particle, Lithium Ion Cell Model Including Electrolyte Diffusion. J. Dyn. Syst. Meas. Control 2015, 137, 011005. [Google Scholar] [CrossRef]
- Subramanian, V.R.; Boovaragavan, V.; Diwakar, V.D. Toward Real-Time Simulation of Physics Based Lithium-Ion Battery Models. Electrochem. Solid-State Lett. 2007, 10, A255–A260. [Google Scholar] [CrossRef]
- Boovaragavan, V.; Harinipriya, S.; Subramanian, V.R. Toward real-time (milliseconds) parameters estimation of Lithium-Ion Batteries using reformulated physics-based models. J. Power Sources 2008, 183, 361–365. [Google Scholar] [CrossRef]
- Kim, G.; Smith, K.; Lee, K.; Santhanagopalan, S.; Pesaran, A. Multi-Domain Modeling of Lithium-Ion Batteries Encompassing Multi-Physics in Varied Length Scales. J. Electrochem. Soc. 2011, 158, A955–A969. [Google Scholar] [CrossRef]
- Zhang, Q.; White, R.E. Capacity fade analysis of a lithium ion cell. J. Power Sources 2008, 179, 793–798. [Google Scholar] [CrossRef]
- Ramasamy, R.P.; White, R.E.; Popov, B.N. Calendar life performance of pouch lithium-ion cells. J. Power Sources 2005, 141, 298–306. [Google Scholar] [CrossRef]
- Arora, P.; White, R.E.; Doyle, M. Capacity Fade Mechanisms and Side Reactions in Lithium-Ion Batteries. J. Electrochem. Soc. 1998, 145, 3647–3667. [Google Scholar] [CrossRef]
- Coman, P.; Rayman, S.; White, R.E. A lumped model of venting during thermal runaway in a cylindrical lithium cobalt oxide lithium-ion cell. J. Power Sources 2016, 307, 56–62. [Google Scholar] [CrossRef]
- Marongiu, A.; Nubbaum, F.; Waag, W.; Garmendia, M.; Sauer, D. Comprehensive study of the influence of aging on the hysteresis behavior of a lithium iron phosphate. Appl. Energy 2016, 171, 629–645. [Google Scholar] [CrossRef]
- Dreyer, W.; Jamnik, J.; Guhlke, C.; Huth, R.; Moskon, J.; Gaberscek, M. The thermodynamic origin of hysteresis in insertion batteries. Nat. Mater. 2010, 9, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Choe, S.Y.; Agubra, V.; Fergus, J. Development of a physics-based degradation model for lithium ion polymer batteries considering side reactions. J. Power Sources 2015, 278, 506–521. [Google Scholar] [CrossRef]
- Fu, R.; Choe, S.Y.; Agubra, V.; Fergus, J. Modeling of degradation effects considering side reactions for a pouch type Li-ion polymer battery with carbon anode. J. Power Sources 2014, 261, 120–135. [Google Scholar] [CrossRef]
- Smith, K.; Wang, C. Solid-state diffusion limitations on pulse operation of a lithium ion cell for hybrid electric vehicles. J. Power Sources 2006, 161, 628–639. [Google Scholar] [CrossRef]
- Smith, K.; Wang, C. Power and thermal characterization of a lithium-ion battery pack for hybrid-electric vehicles. J. Power Sources 2006, 160, 662–673. [Google Scholar] [CrossRef]
- Xiao, M.; Choe, S.Y. Dynamic modeling and analysis of a pouch type LiMn2O4/Carbon high power Li-polymer battery based on electrochemical-thermal principles. J. Power Sources 2012, 218, 357–367. [Google Scholar] [CrossRef]
- Arora, P.; Doyle, M.; White, R. Mathematical modeling of the lithium deposition overcharge reaction in lithium−ion batteries using carbon−based negative electrodes. J. Electrochem. Soc. 1999, 146, 3543–3553. [Google Scholar] [CrossRef]
- Christensen, J.; Srinivasan, V.; Newman, J. Optimization of lithium titanate electrodes for high-power cells. J. Electrochem. Soc. 2006, 153, A560–A565. [Google Scholar] [CrossRef]
- Stewart, S.; Srinivasan, V.; Newman, J. Modeling the performance of lithium-ion batteries and capacitors during hybrid-electric-vehicle operation. J. Electrochem. Soc. 2008, 155, A664–A671. [Google Scholar] [CrossRef]
- Srinivasan, V.; Newman, J. Discharge Model for the Lithium Iron-Phosphate Electrode. J. Electrochem. Chem. 2004, 151, A1517–A1529. [Google Scholar] [CrossRef]
- Delacourt, C.; Safari, M. Analysis of lithium deinsertion/insertion in LiyFePO4 with a simple mathematical model. Electrochim. Acta 2011, 56, 5222–5229. [Google Scholar] [CrossRef]
- Maheshwari, A.; Dumitrescu, M.A.; Destro, M.; Santarelli, M. Inverse parameter determination in the development of an optimized lithium iron phosphate e Graphite battery discharge model. J. Power Sources 2016, 307, 160–172. [Google Scholar] [CrossRef]
- Ramadass, P.; Haran, B.; Gomadam, P.; White, R.; Popov, B. Development of First Principles Capacity Fade Model for Li-Ion Cells. J. Electrochem. Soc. 2004, 151, A196–A203. [Google Scholar] [CrossRef]
- Santhanagopalan, S.; Zhang, Q.; Kumaresan, K.; White, R. Parameter Estimation and Life Modeling of Lithium-Ion Cells. J. Electrochem. Soc. 2008, 155, A345–A353. [Google Scholar] [CrossRef]
- Sikha, G.; Popov, B.; White, R. Effect of Porosity on the Capacity Fade of a Lithium-Ion Battery: Theory. J. Electrochem. Soc. 2004, 151, A1104–A1114. [Google Scholar] [CrossRef]
- Ploehn, H.; Ramadass, P.; White, R. Solvent Diffusion Model for Aging of Lithium-Ion Battery Cells. J. Electrochem. Soc. 2004, 151, A456–A462. [Google Scholar] [CrossRef]
- Aurbach, D.; Levi, M.; Levi, E.; Schechter, A. Failure and Stabilization Mechanisms of Graphite Electrodes. J. Phys. Chem. B 1997, 101, 2195–2206. [Google Scholar] [CrossRef]
- Verma, P.; Maire, P.; Novak, P. A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries. Electrochim. Acta 2010, 55, 6332–6341. [Google Scholar] [CrossRef]
- Goriparti, S.; Miele, E.; Angelis, F.D.; Fabrizo, E.D.; Zaccaria, R.P.; Capiglia, C. Review on recent progress of nanostructured anode materials for Li-ion batteries. J. Power Sources 2014, 257, 421–443. [Google Scholar] [CrossRef]
- Zaghib, K.; Simoneau, M.; Armand, M.; Gauthier, M. Electrochemical study of Li4Ti5O12 as negative electrode for Li-ion polymer rechargeable batteries. J. Power Sources 1999, 81–82, 300–305. [Google Scholar] [CrossRef]
- Sun, X.; Radovanovic, P.V.; Cui, B. Advances in spinel Li4Ti5O12 anode materials for lithium-ion batteries. New J. Chem. 2015, 39, 38–63. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Graphite Half-Cell | LTO Half-Cell | ||
|---|---|---|---|
| 100% SOC | OCV (OCVmax) | 0.2 V | 1.62 V |
| Stoichiometric number (stoimin) | 0.168 | 0.165 | |
| 0% SOC | OCV (OCVmin) | 0.04 V | 1.48 V |
| Stoichiometric number (stoimax) | 0.989 | 0.987 | |
| Parameter | Half-Cell with Graphite | Half-Cell with LTO | Unit | Source |
|---|---|---|---|---|
| Thickness of composite anode, δan | 31.9 × 10−4 | 35.0 × 10−4 | cm | Measured |
| Thickness of separator, δs | 25 × 10−4 | 25 × 10−4 | mg | Measured |
| Mass of composite anode | 2.11 | 4.23 | mg | Measured |
| Stoichiometry at 0% SOC (anode lithiated): xmax | 0.989 | 0.987 | Assumed | |
| Stoichiometry at 100% SOC (anode de-lithiated): xmin | 0.168 | 0.165 | Assumed | |
| Capacity from xmax to xmin | 0.80 | 0.97 | mAh | Identified from OCV tests |
| Area of composite anode | 1.27 | 1.27 | cm2 | Measured |
| Volume fraction of active material in composite anode, εs,an | 0.50 | 0.64 | Identified from charge/discharge tests | |
| Volume fraction of inactive material in composite anode, εin,an | 0.17 | 0.17 | Identified from charge/discharge tests | |
| Volume fraction of electrolyte in composite anode, εe,an | 0.33 | 0.19 | Calculated by 1 − εs,an − εin,an | |
| Volume fraction of electrolyte in separator region, εe,sep | 0.5 | 0.5 | Literature [30,31,32,33,34] | |
| Volume fraction of inactive material in separator region, εin,sep | 0.5 | 0.5 | Literature [30,31,32,33,34] | |
| Anode particle radius, rs | 10 × 10−4 | Refer to Table 3 | cm | Rough estimate from SEM and Identified from charge/discharge tests |
| Averaged ion concentration in electrolyte, ce,0 | 1.0 × 10−3 | 1.0 × 10−3 | mol cm−3 | Controlled in cell fabrication |
| Exchange current density coefficient, ki0 | 1 | 1 | A cm−2 | Literature [32] |
| Charge-transfer coefficient, αa, αc | 0.5, 0.5 | 0.5, 0.5 | Literature [32,34] | |
| Li+ diffusion coefficient in solid phase, Ds | 50 × 10−12 | 5 × 10−12 | cm2 s−1 | Identified from charge/discharge tests |
| Solid phase conductivity, σ | 1 | 1 | S cm−1 | Literature [32,33,35,36,37] |
| Li+ diffusion coefficient in electrolyte, De | 2.08 × 10−8 | 2.08 × 10−8 | cm2 s−1 | Identified from charge/discharge tests |
| Bruggeman’s porosity exponent, p | 1.5 | 1.5 | Literature [30,31,32,33,34] | |
| Electrolyte phase ionic conductivity, κ | 15.8ce⋅ | 15.8ce⋅ | S cm−1 | Literature [30,31,32,33] |
| exp(−13,472ce1.4) | exp(−13,472ce1.4) | |||
| SEI resistance (multiplied by reaction area) | 300 | 300 | Ω cm2 | Assumed (similar to [35]) |
| Li+ transference number, t+0 | 0.363 | 0.363 | Literature [30,31,32,33,34] | |
| Ohmic resistance (multiplied by cell area, including lithium metal, current collector and coin cell casing) | 8.08 × 10−6 | 8.08 × 10−6 | Ω cm2 | Calculated from cell dimensions |
| Current (LTO) | 0.045 C | 0.089 C | 0.13 C |
| Equivalent Rs (LTO) | 13 × 10−4 cm | 10 × 10−4 cm | 8.6 × 10−4 cm |
| Parameter | Half-Cell with Graphite | Half-Cell with LTO | Unit |
|---|---|---|---|
| Thickness of composite anode, δan | 35.0 × 10−4 | 35.0 × 10−4 | cm |
| Mass of composite anode | 2.32 | 4.23 | mg |
| Capacity from xmax to xmin | 0.87 | 0.97 | mAh |
| Δcs at Small Current (0.045 C) | Δcs at Large Current (0.13 C) | Increase of Δcs from Small Current to Charge Current | ||
|---|---|---|---|---|
| Graphite | Discharge | 0.955 × 10−3 mol cm−3 | 2.41 × 10−3 mol cm−3 | 152% |
| Charge | 0.513 × 10−3 mol cm−3 | 1.52 × 10−3 mol cm−3 | 196% | |
| LTO | Discharge | 6.48 × 10−3 mol cm−3 | 7.26 × 10−3 mol cm−3 | 12% |
| Charge | 6.74 × 10−3 mol cm−3 | 7.77 × 10−3 mol cm−3 | 15% | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, R.; Zhou, X.; Fan, H.; Blaisdell, D.; Jagadale, A.; Zhang, X.; Xiong, R. Comparison of Lithium-Ion Anode Materials Using an Experimentally Verified Physics-Based Electrochemical Model. Energies 2017, 10, 2174. https://doi.org/10.3390/en10122174
Fu R, Zhou X, Fan H, Blaisdell D, Jagadale A, Zhang X, Xiong R. Comparison of Lithium-Ion Anode Materials Using an Experimentally Verified Physics-Based Electrochemical Model. Energies. 2017; 10(12):2174. https://doi.org/10.3390/en10122174
Chicago/Turabian StyleFu, Rujian, Xuan Zhou, Hengbin Fan, Douglas Blaisdell, Ajay Jagadale, Xi Zhang, and Rui Xiong. 2017. "Comparison of Lithium-Ion Anode Materials Using an Experimentally Verified Physics-Based Electrochemical Model" Energies 10, no. 12: 2174. https://doi.org/10.3390/en10122174