
In Situ Surface-Enhanced Raman Spectroscopy Study of the Electrocatalytic Effect of PtFe/C Nanocatalyst on Ethanol Electro-Oxidation in Alkaline Medium

Abstract
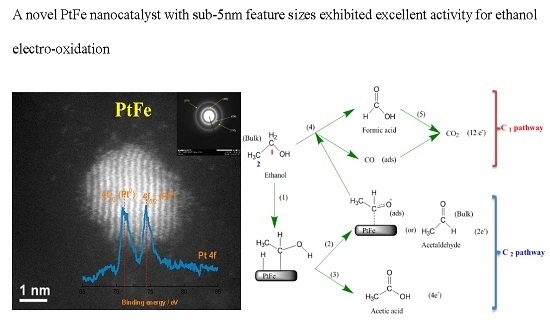
:
1. Introduction
2. Results and Discussion
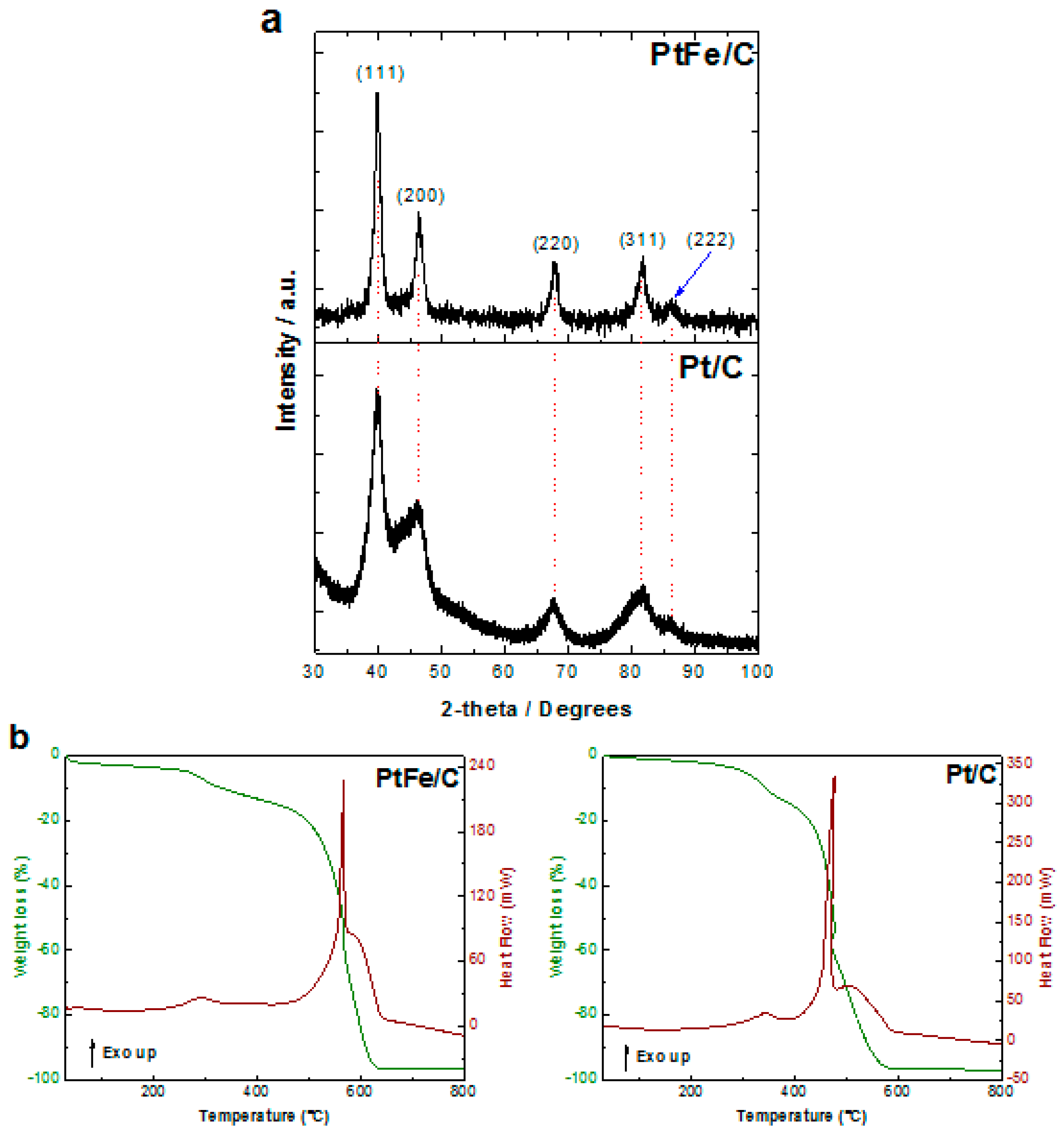
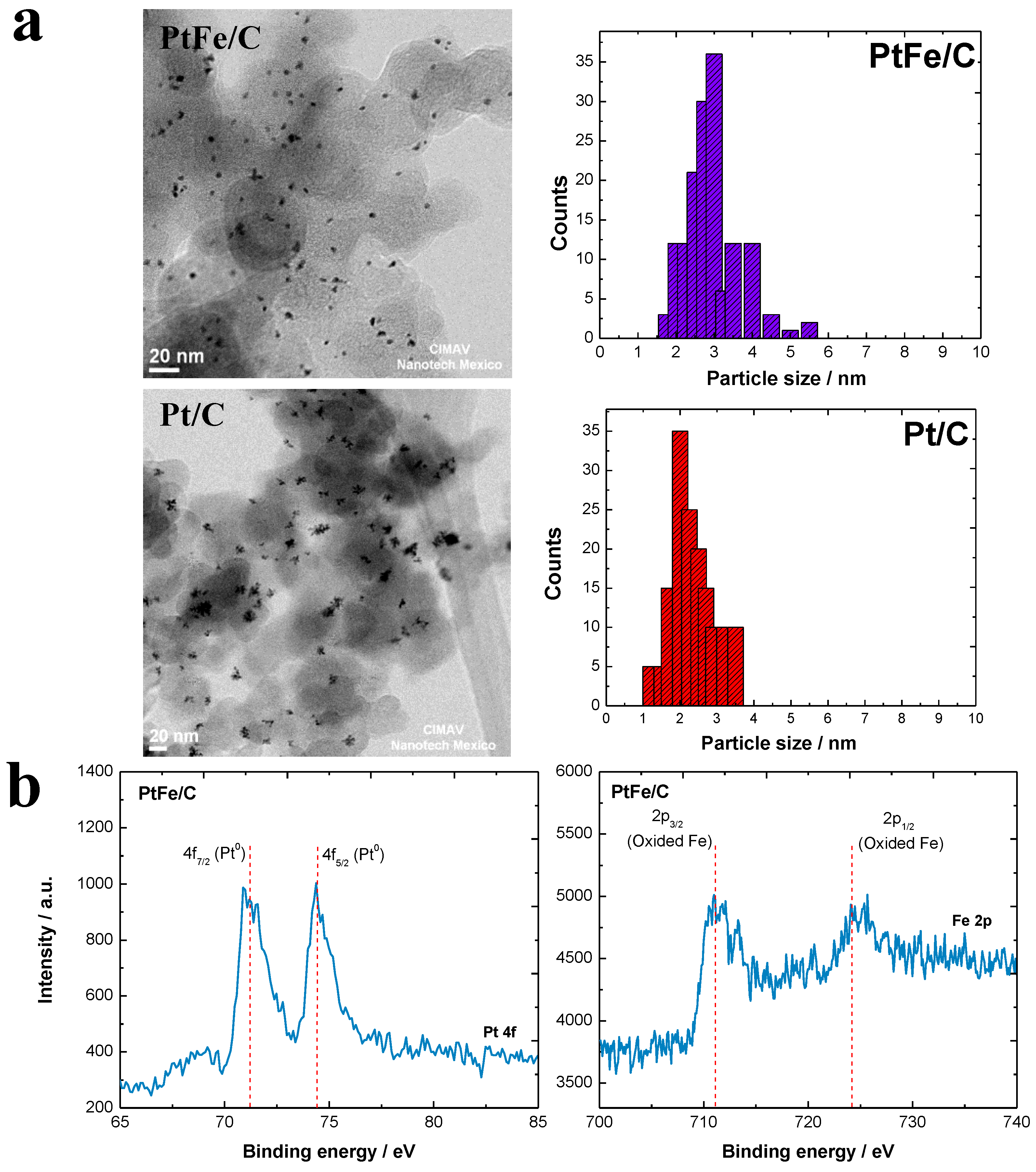
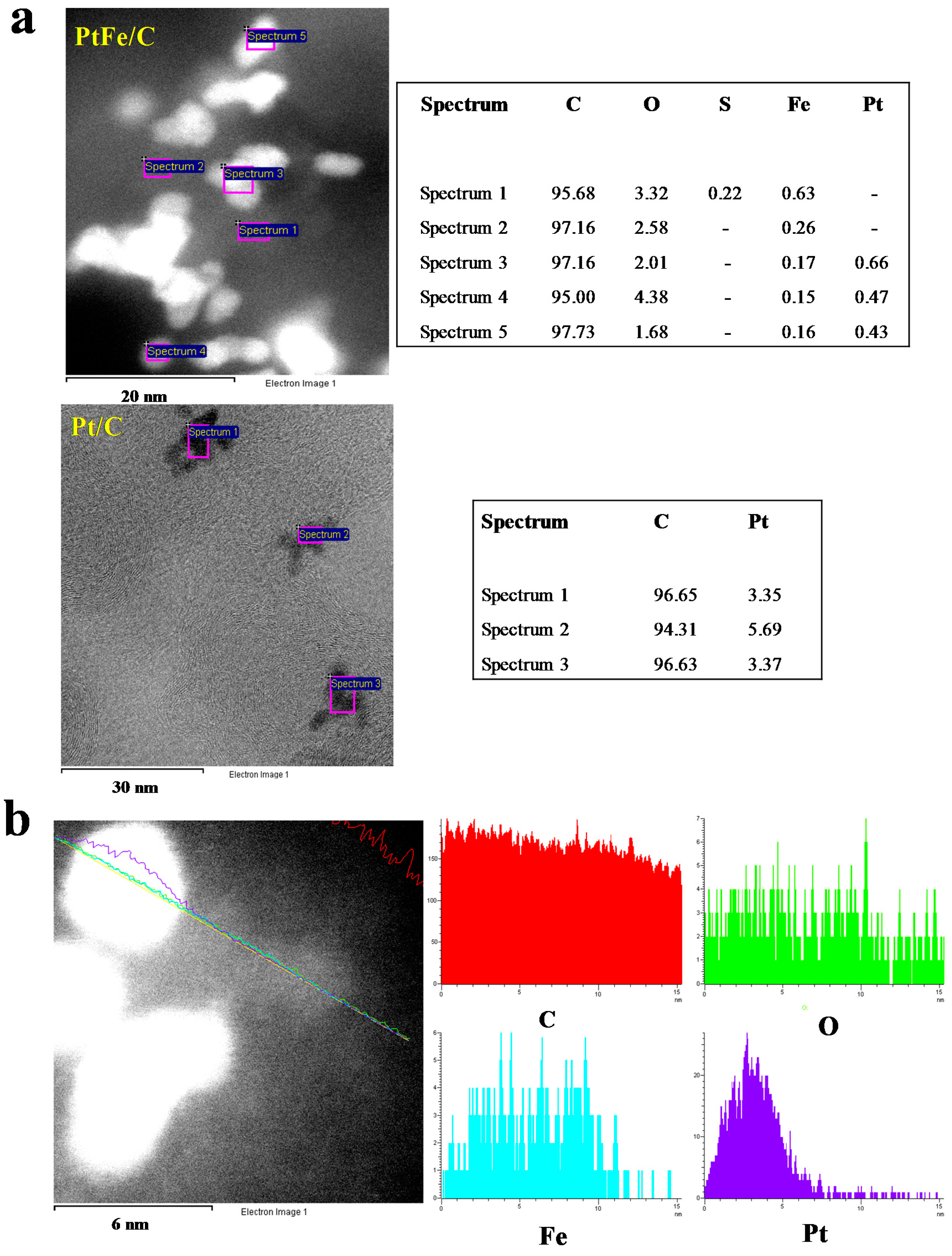
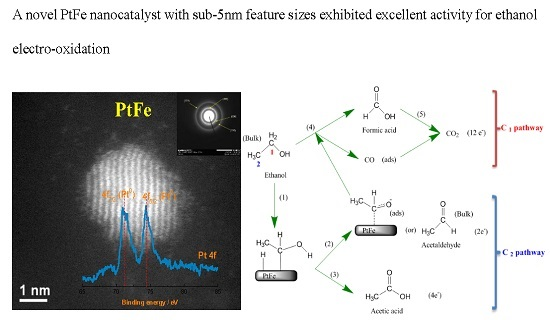
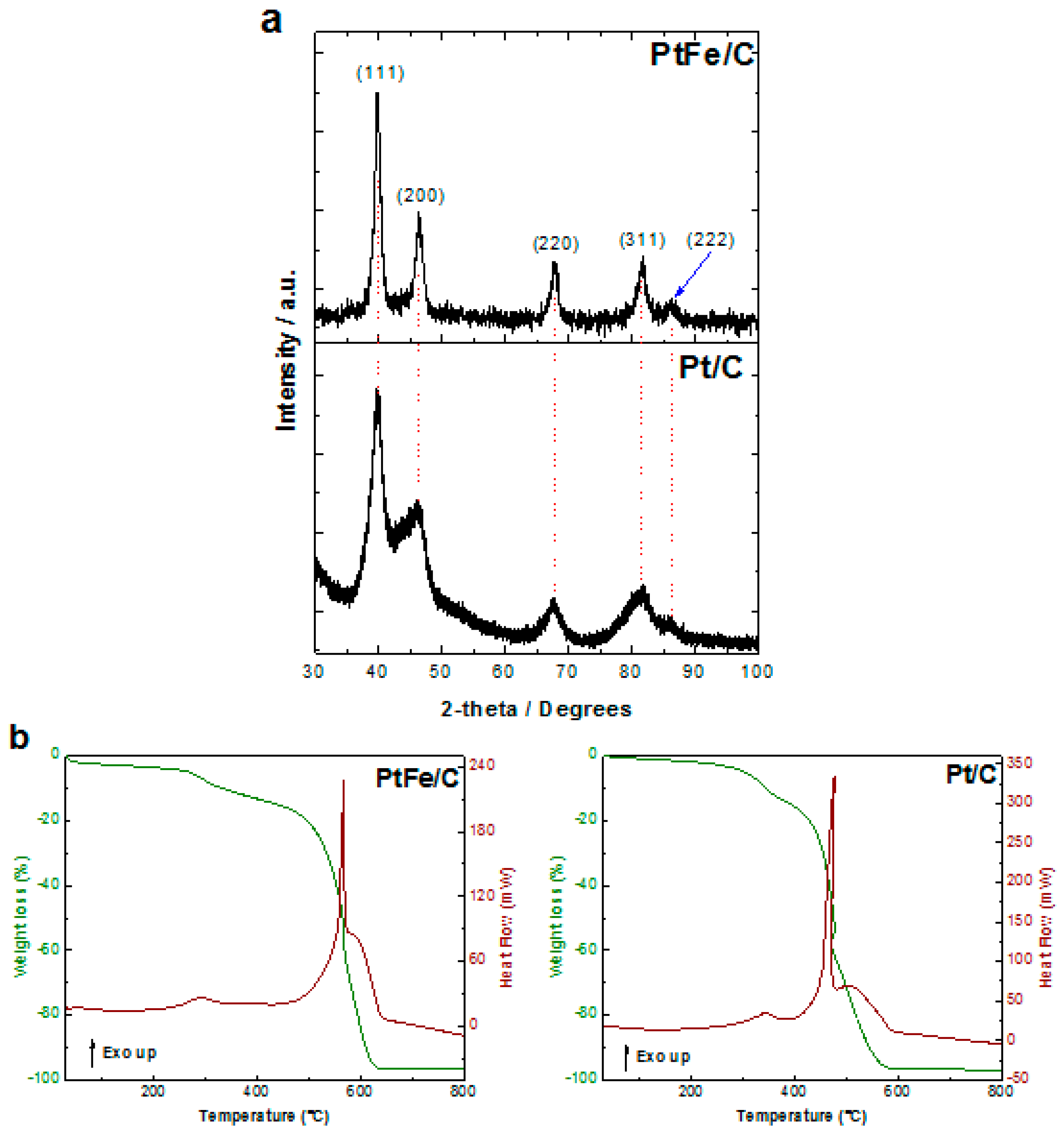
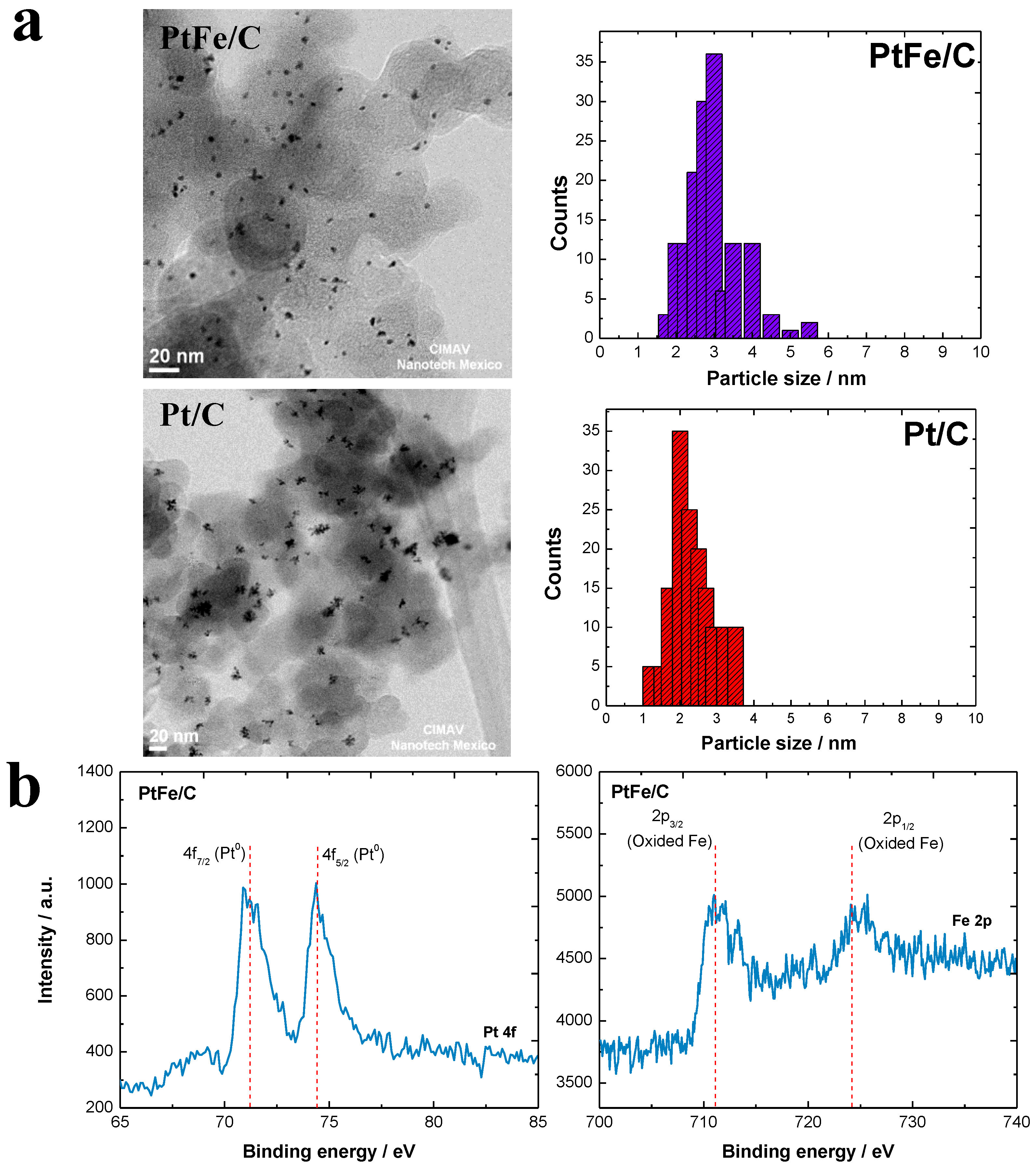
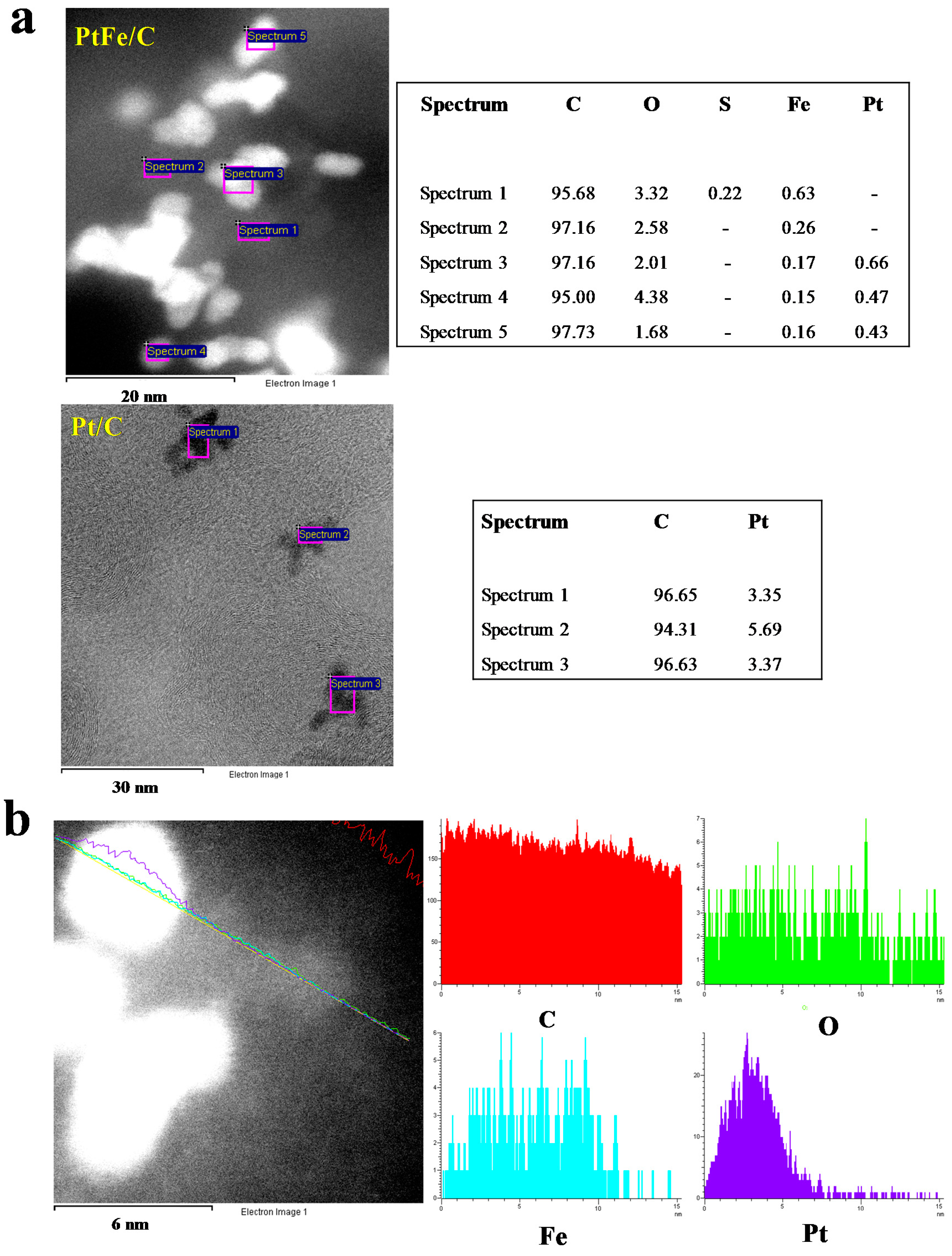
2.1. Physicochemical Characterization
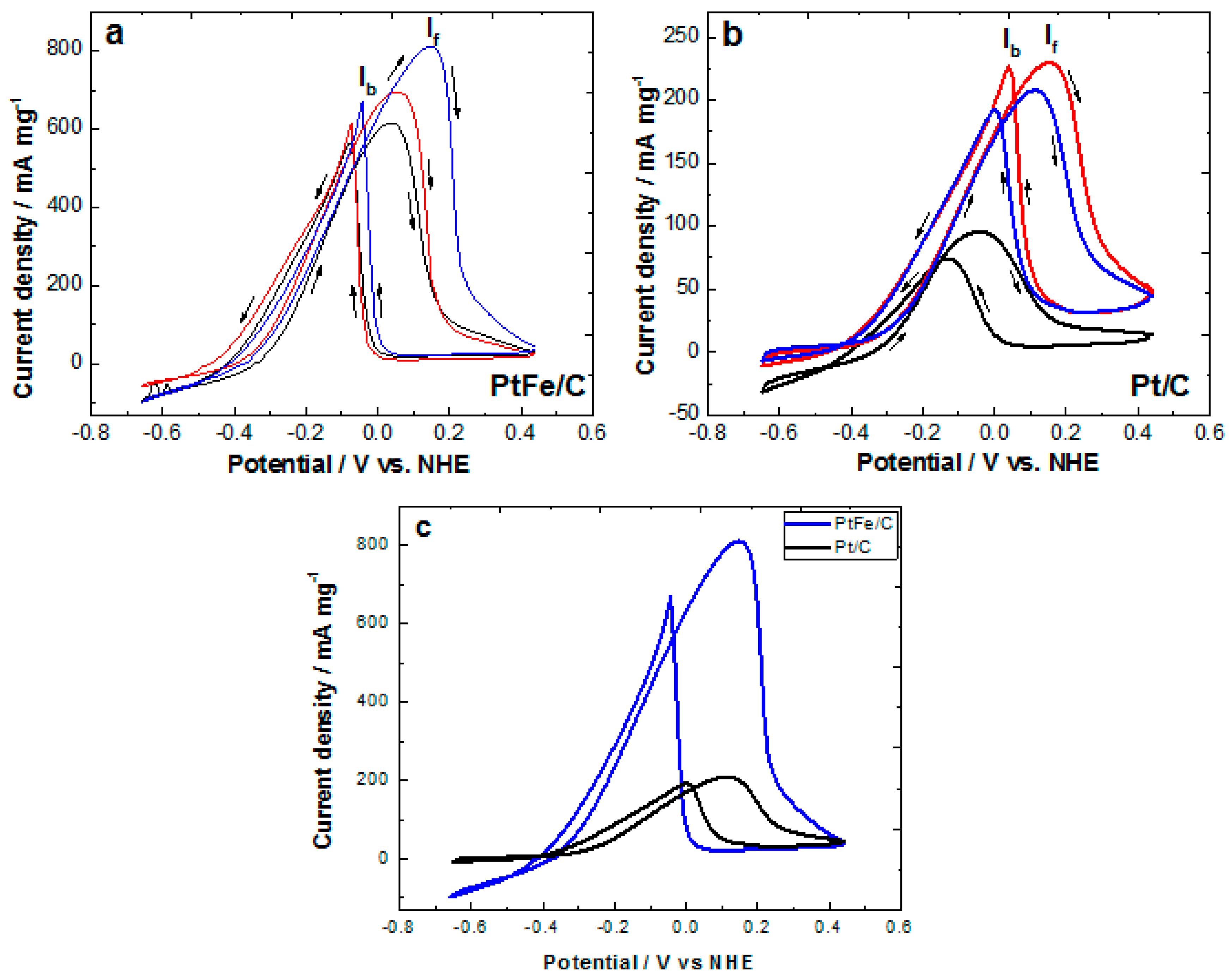
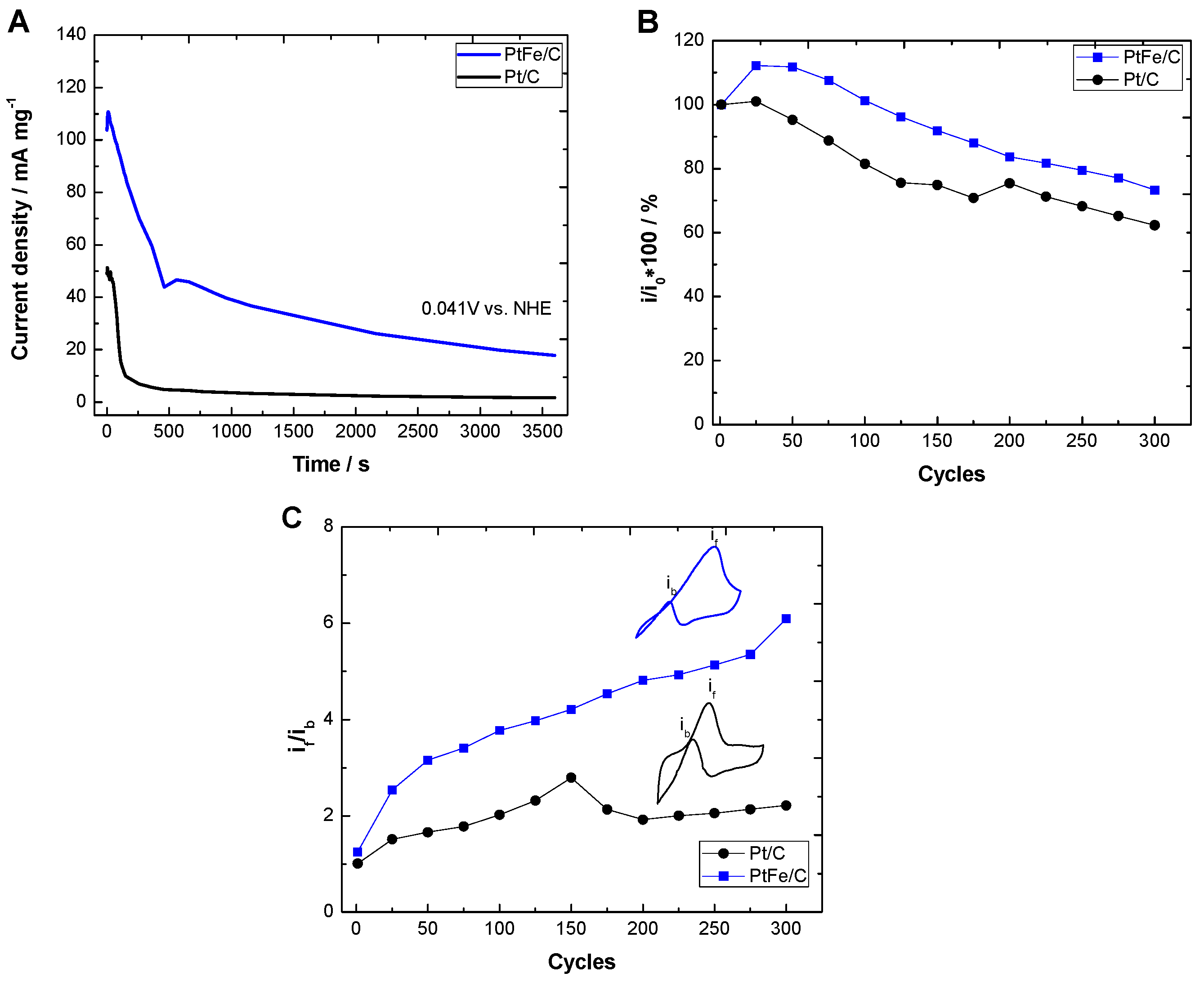
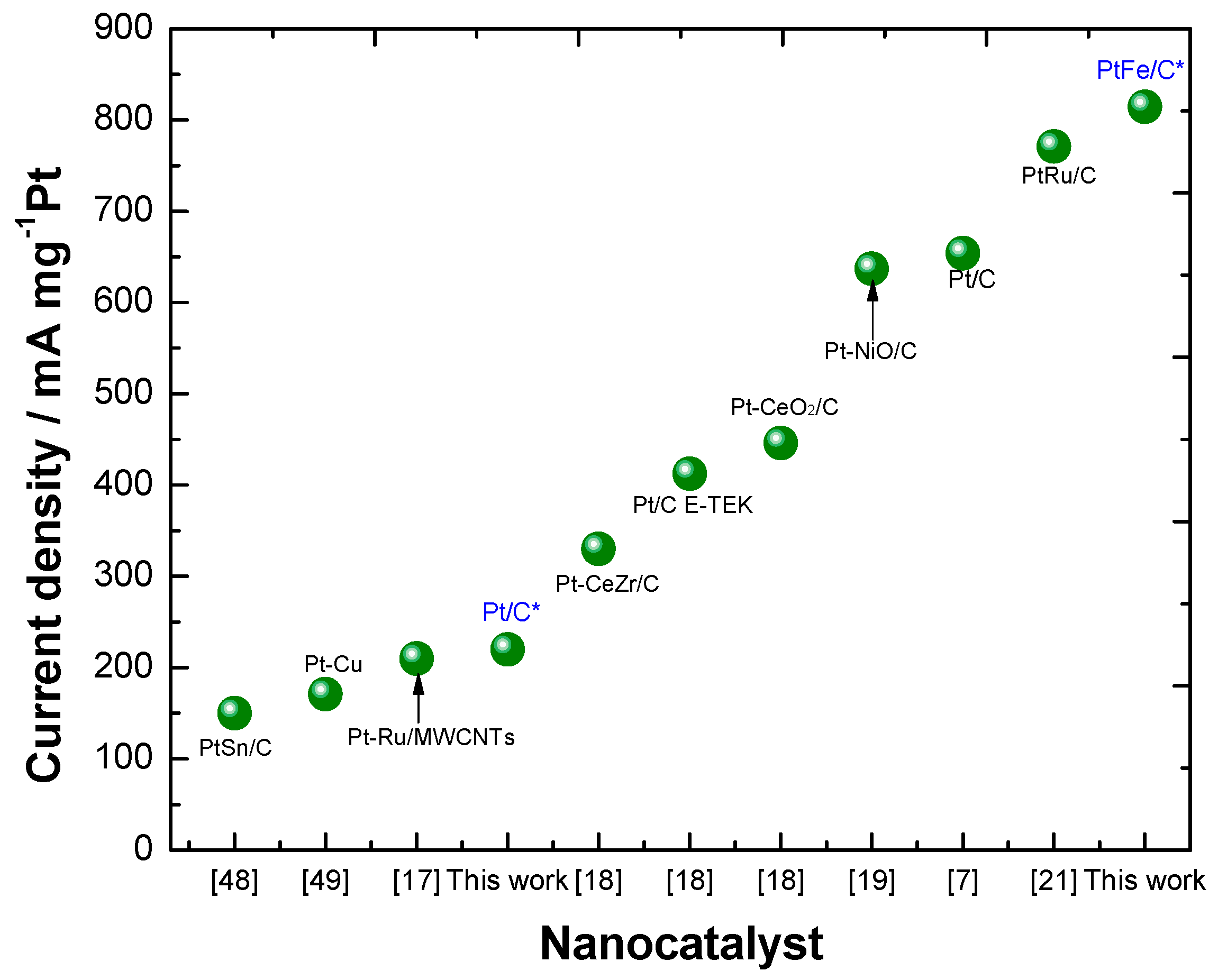
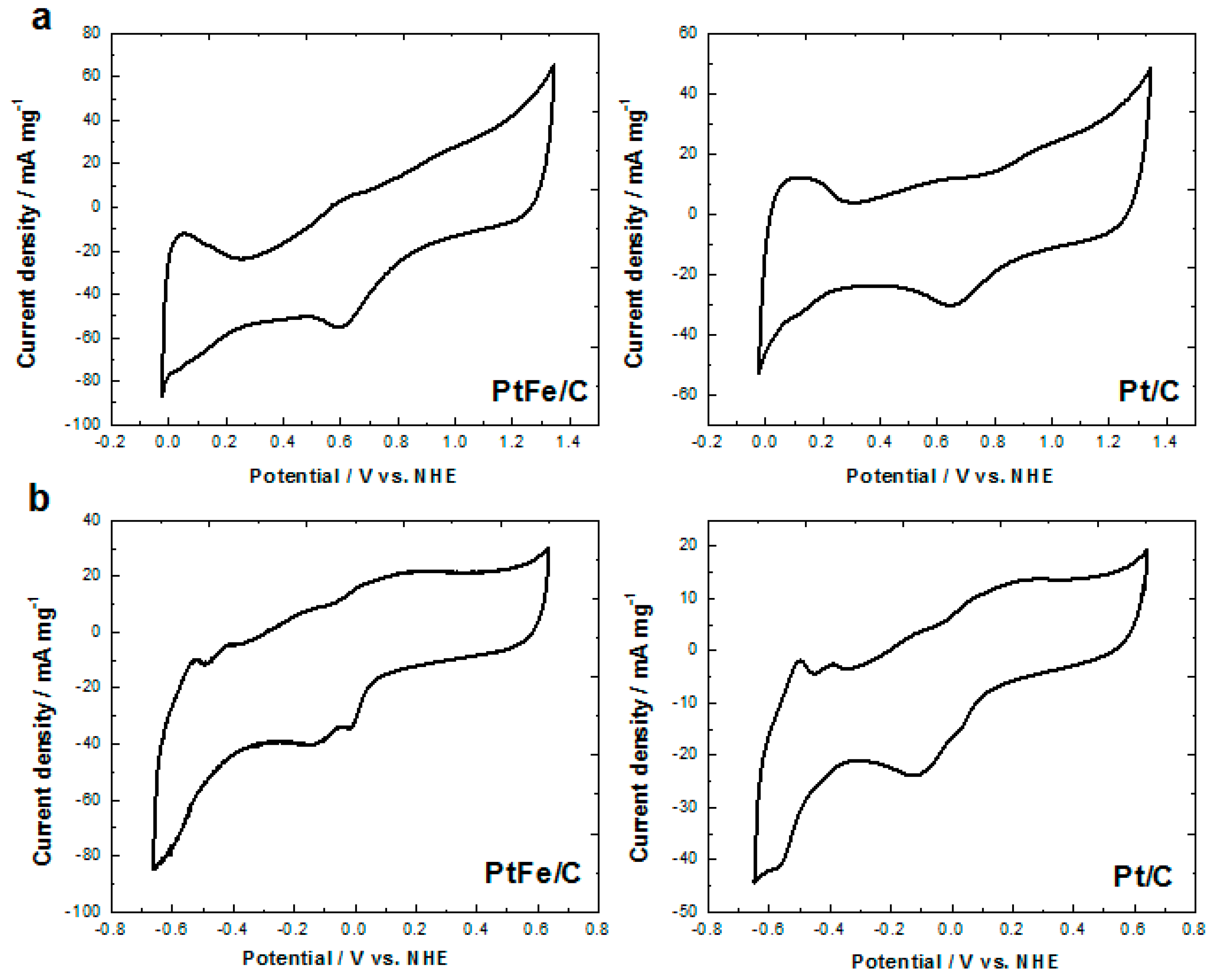
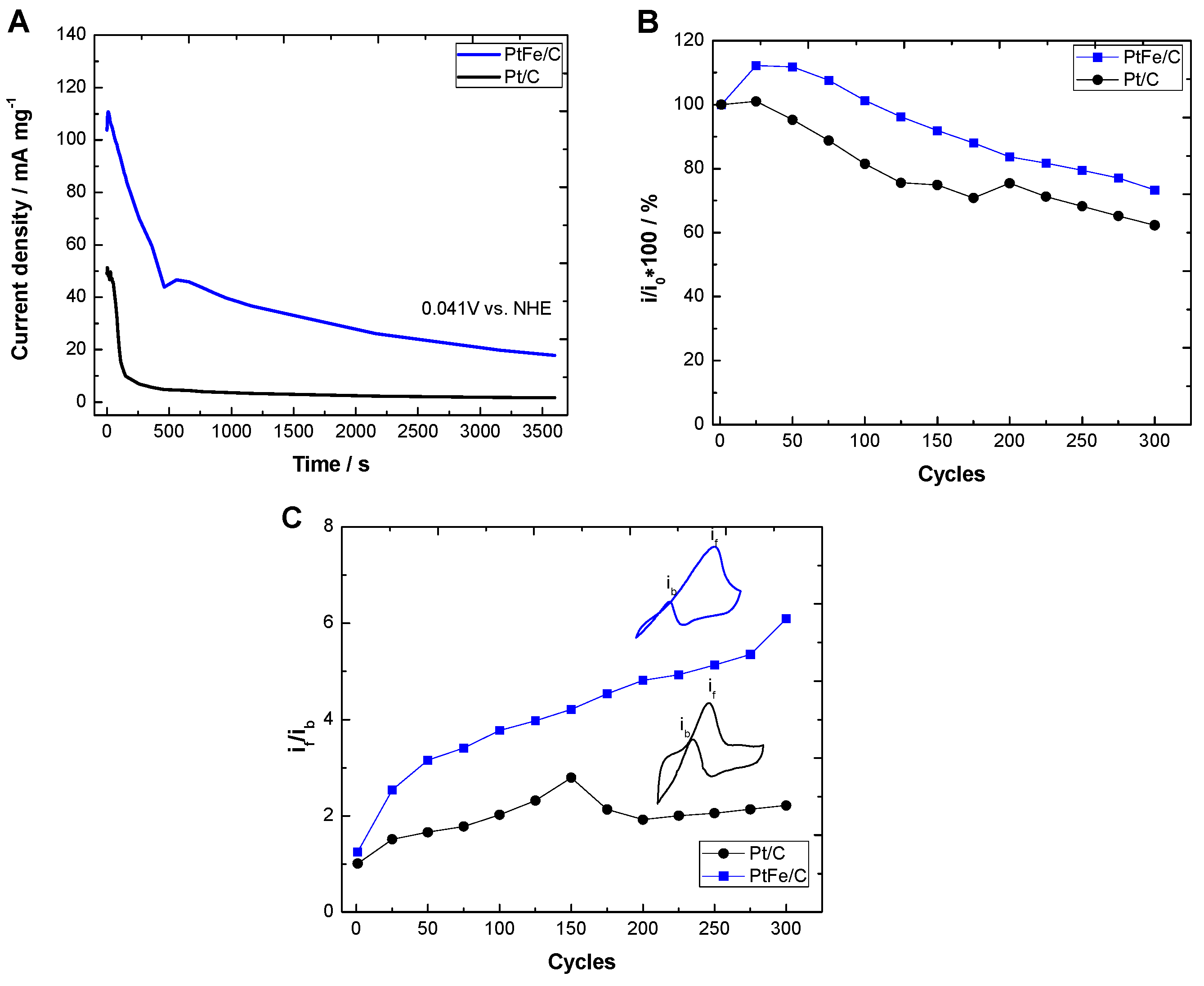
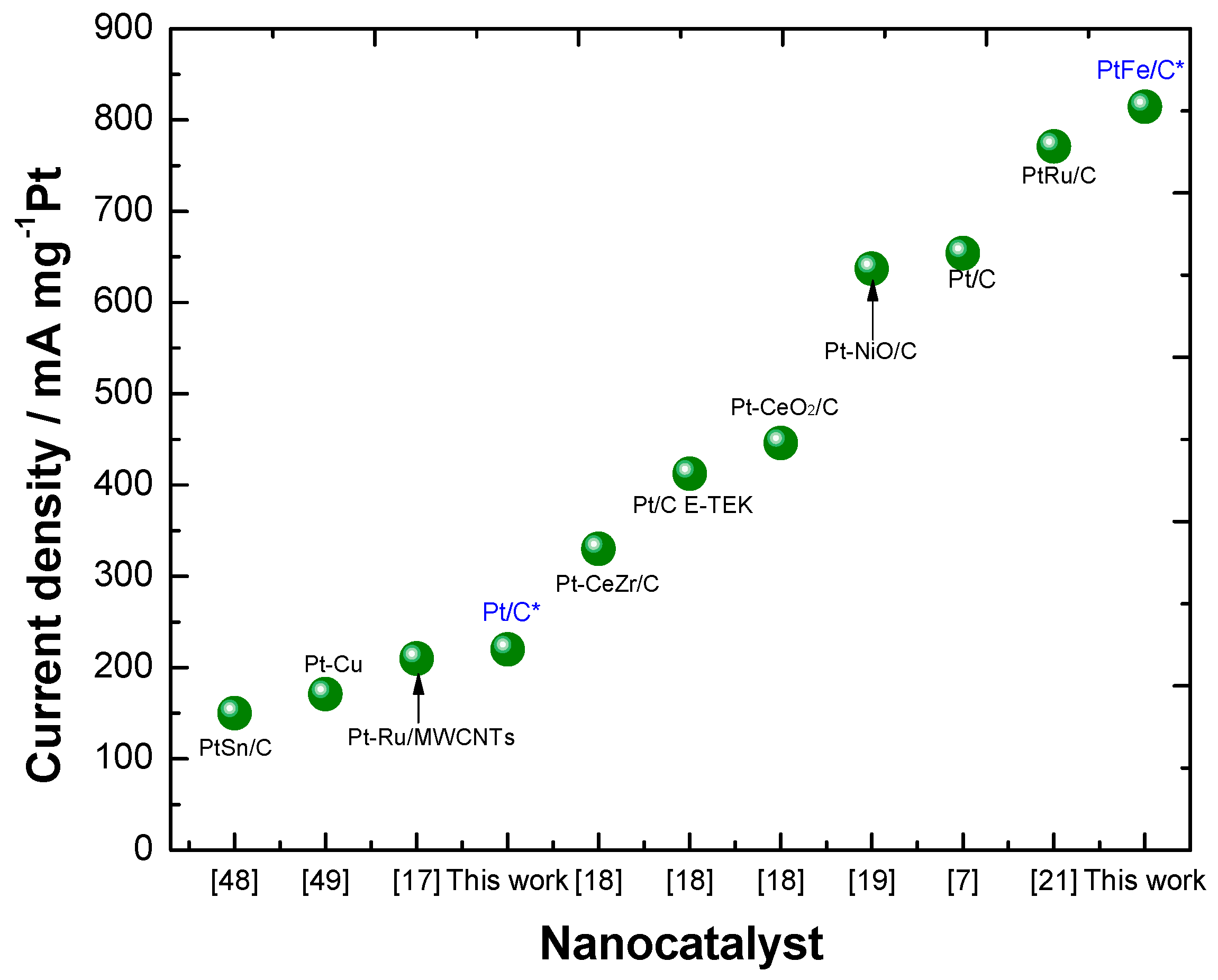
2.2. Electrochemical Characterization
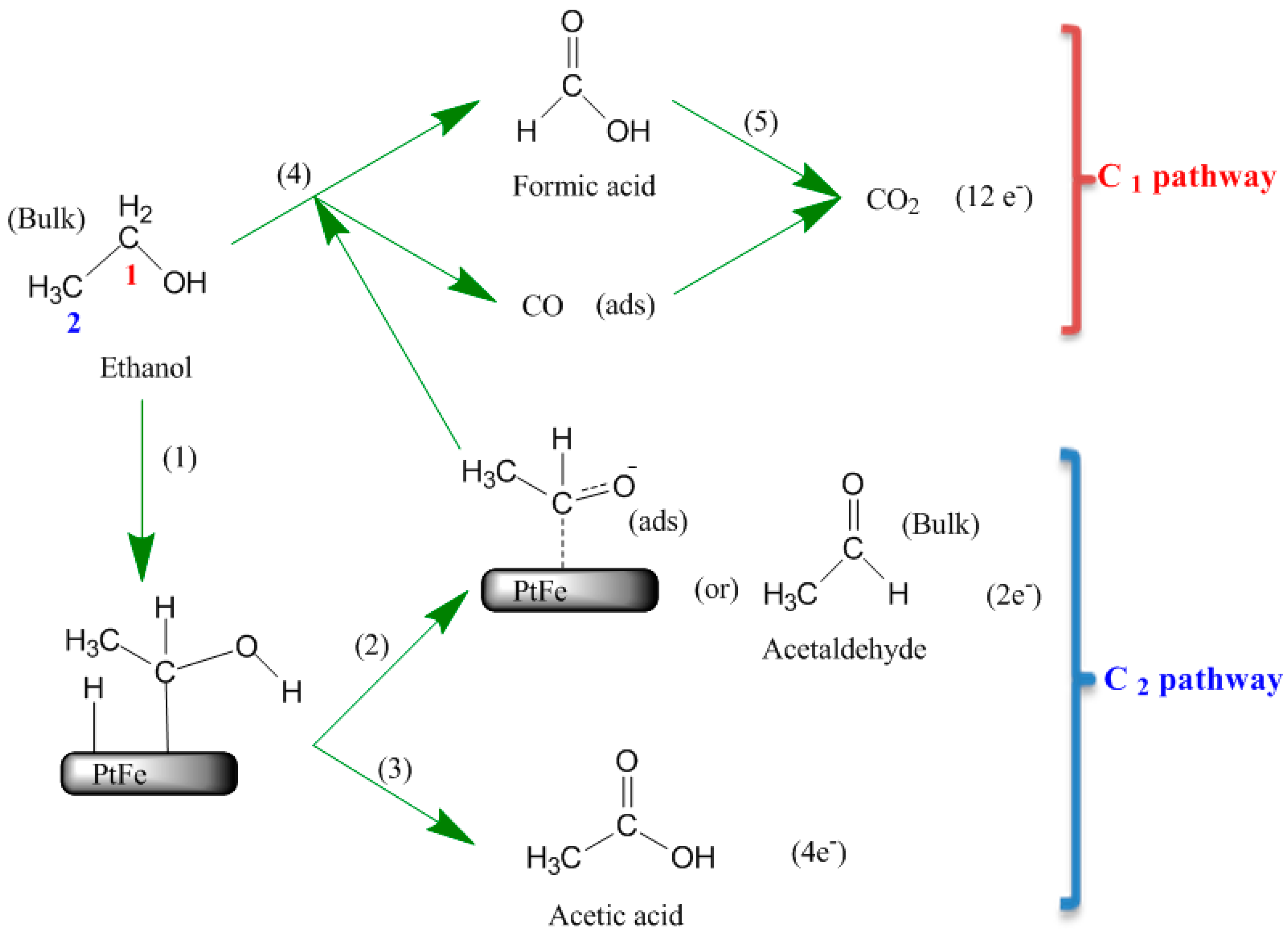
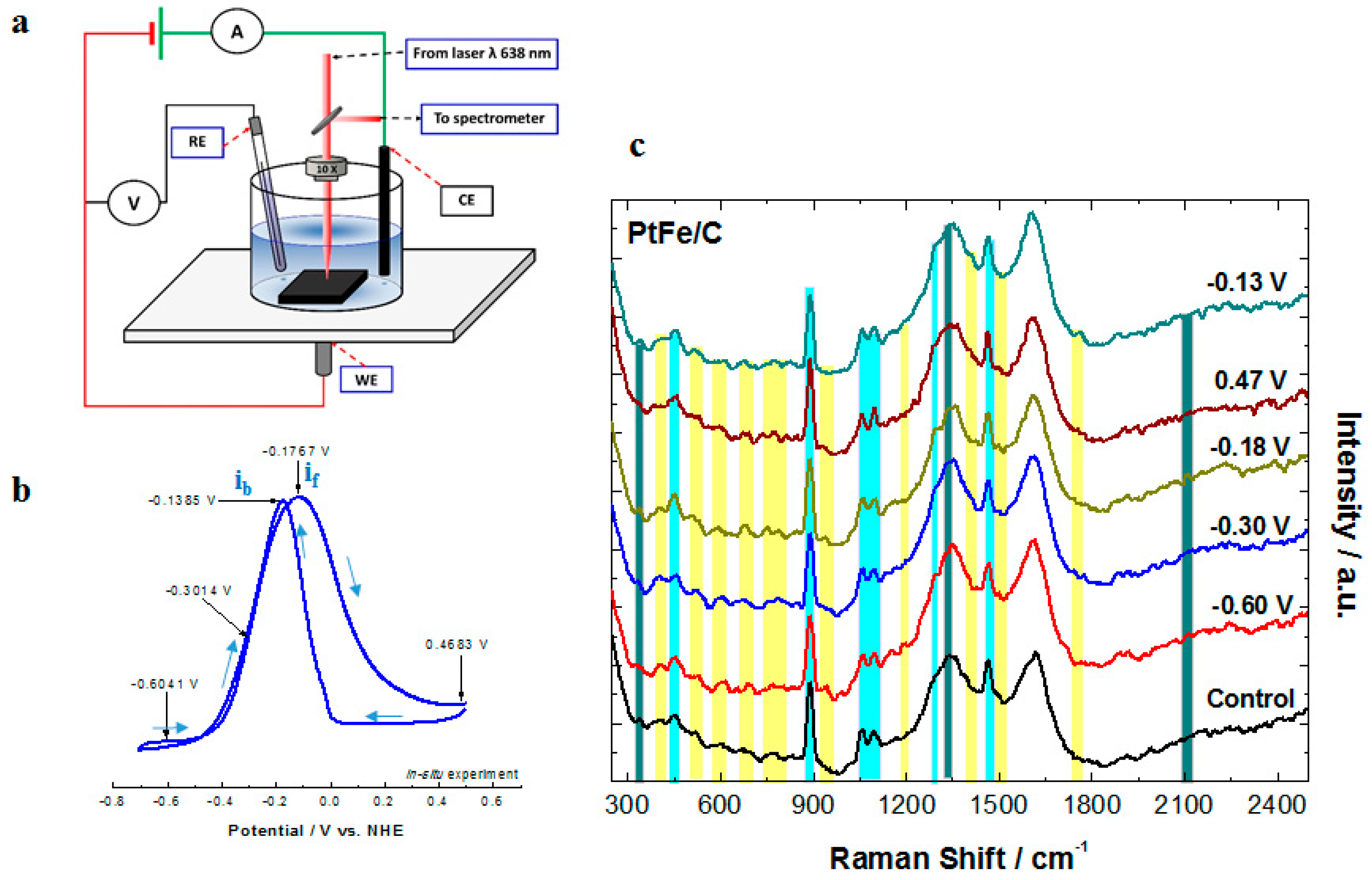
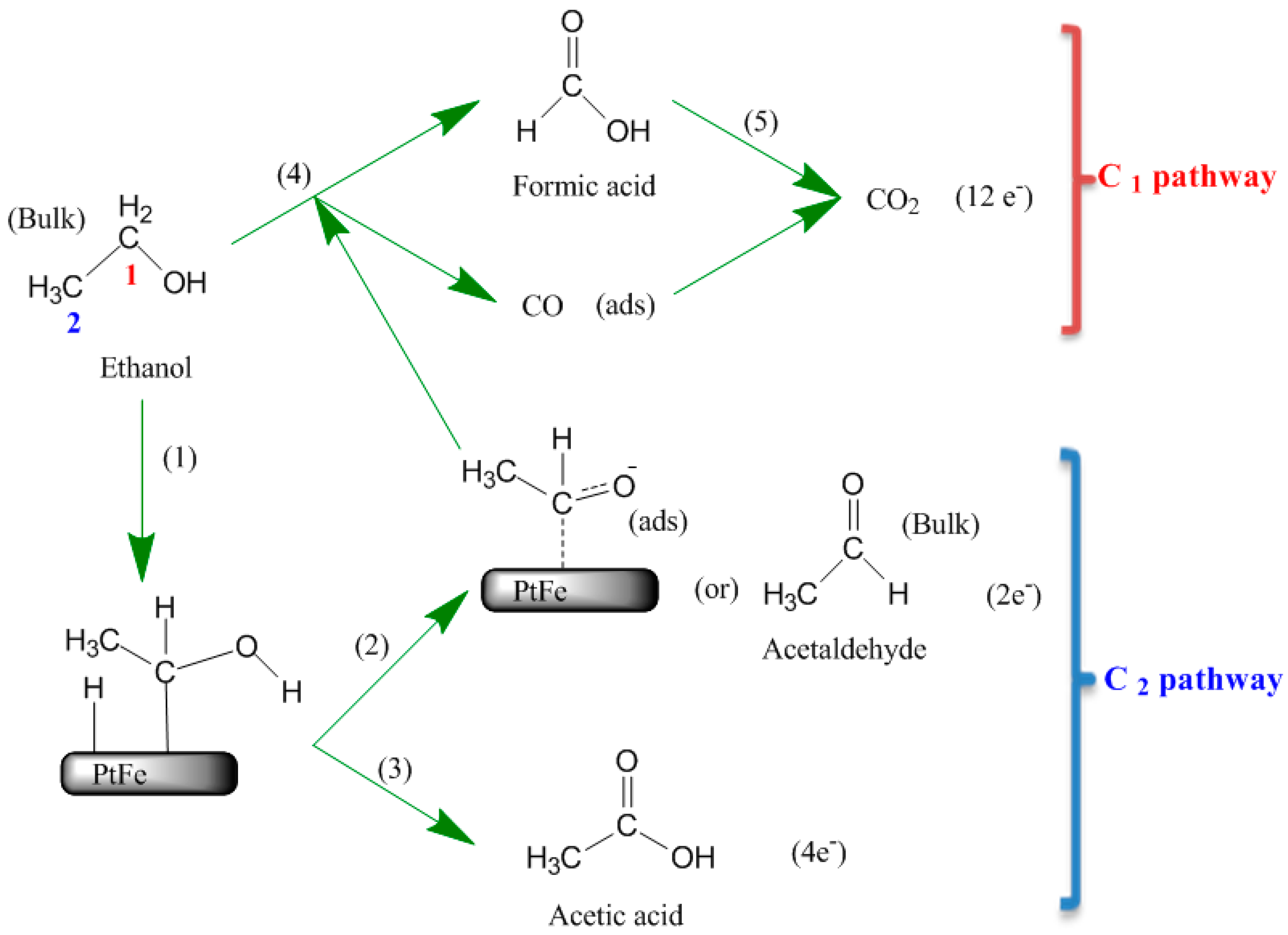
2.3. Surface Enhanced Raman Spectroscopy (SERS) Analysis
3. Materials and Methods
3.1. Synthesis of Pt/C
3.2. Synthesis of PtFe/C
3.3. Physicochemical Characterization
3.4. Electrochemical Characterization
3.5. Surface-Enhanced Raman Spectroscopy Experiments
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Otomo, J.; Nishida, S.; Takahashi, H.; Nagamoto, H. Electro-oxidation of methanol and ethanol on carbon-supported Pt catalyst at intermediate temperature. J. Electroanal. Chem. 2008, 615, 84–90. [Google Scholar] [CrossRef]
- Giz, M.J.; Camara, G.A. The ethanol electrooxidation reaction at Pt (111): The effect of ethanol concentration. J. Electroanal. Chem. 2009, 625, 117–122. [Google Scholar] [CrossRef]
- Li, Y.S.; Zhao, T.S.; Liang, Z.X. Performance of alkaline electrolyte-membrane-based direct ethanol fuel cells. J. Power Sources 2009, 187, 387–392. [Google Scholar] [CrossRef]
- An, L.; Zhao, T.S.; Chen, R.; Wu, Q.X. A novel direct ethanol fuel cell with high power density. J. Power Sources 2011, 196, 6219–6222. [Google Scholar] [CrossRef]
- Andreadis, G.; Tsiakaras, P. Ethanol crossover and direct ethanol PEM fuel cell performance modeling and experimental validation. Chem. Eng. Sci. 2006, 61, 7497–7508. [Google Scholar] [CrossRef]
- Merle, G.; Wssling, M.; Nijmeijer, K. Anion exchange membranes for alkaline fuel cells: A review. J. Membr. Sci. 2011, 377, 1–35. [Google Scholar] [CrossRef]
- Ma, L.; Chu, D.; Chen, R. Comparison of ethanol electro-oxidation on Pt/C and Pd/C catalysts in alkaline media. Int. J. Hydrog. Energy 2012, 37, 11185–11194. [Google Scholar] [CrossRef]
- Akhairi, M.A.F.; Kamarudin, S.K. Catalysts in direct ethanol fuel cell (DEFC): An overview. Int. J. Hydrog. Energy 2016, 41, 4214–4228. [Google Scholar] [CrossRef]
- Kamarudin, M.Z.F.; Kamarudin, S.K.; Masdar, M.S.; Daud, W.R.W. Review: Direct ethanol fuel cells. Int. J. Hydrog. Energy 2013, 38, 9438–9453. [Google Scholar] [CrossRef]
- Antolini, E. Catalysts for direct ethanol fuel cells. J. Power Sources 2007, 170, 1–12. [Google Scholar] [CrossRef]
- Zignani, S.C.; Baglio, V.; Linares, J.J.; Monforte, G.; Gonzalez, E.R.; Aricò, A.S. Performance and selectivity of PtxSn/C electro-catalysts for ethanol oxidation prepared by reduction with different formic acid concentrations. Electrochim. Acta 2012, 70, 255–265. [Google Scholar] [CrossRef]
- Camara, G.A.; de Lima, R.B.; Iwasita, T. Catalysis of ethanol electrooxidation by PtRu: The influence of catalyst composition. Electrochem. Commun. 2004, 6, 812–815. [Google Scholar] [CrossRef]
- Kannan, P.; Maiyalagan, T.; Opallo, M. One-pot synthesis of chain-like palladium nanocubes and their enhanced electrocatalytic activity for fuel-cell applications. Nano Energy 2013, 2, 677–687. [Google Scholar] [CrossRef]
- Liu, Y.; Li, D.; Sun, S. Pt-based composite nanoparticles for magnetic, catalytic, and biomedical applications. J. Mater. Chem. 2011, 21, 12579–12587. [Google Scholar] [CrossRef]
- Dai, Y.; Ou, L.; Liang, W.; Yang, F.; Liu, Y.; Chen, S. Efficient and superiorly durable Pt-lean electrocatalysts of Pt−W alloys for the oxygen reduction reaction. J. Phys. Chem. C 2011, 115, 2162–2168. [Google Scholar] [CrossRef]
- Stamenkovic, V.R.; Fowler, B.; Mun, B.S.; Wang, G.; Ross, P.N.; Lucas, C.A.; Markovic, N.M. Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability. Science 2007, 315, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Byhan, S.; Coutanceau, C.; Léger, J.-M.; Napporn, T.W.; Kadirgan, F. Promising anode candidates for direct ethanol fuel cell: Carbon supported PtSn-based trimetallic catalysts prepared by Bönnemann method. Int. J. Hydrog. Energy 2013, 38, 6830–6841. [Google Scholar] [CrossRef]
- Fan, Y.; Liu, P.-F.; Zhang, Z.-W.; Cui, Y.; Zhang, Y. Three-dimensional hierarchical porous platinum–copper alloy networks with enhanced catalytic activity towards methanol and ethanol electro-oxidation. J. Power Sources 2015, 296, 282–289. [Google Scholar] [CrossRef]
- Sieben, J.M.; Duarte, M.M.E. Methanol, ethanol and ethylene glycol electro-oxidation at Pt and Pt–Ru catalysts electrodeposited over oxidized carbon nanotubes. Int. J. Hydrog. Energy 2012, 37, 9941–9947. [Google Scholar] [CrossRef]
- Du, W.; Su, D.; Wang, Q.; Frenkel, A.I.; Teng, X. Promotional effects of bismuth on the formation of platinum-bismuth nanowires network and the electrocatalytic activity toward ethanol oxidation. Cryst. Growth Des. 2011, 11, 594–599. [Google Scholar] [CrossRef]
- Bai, Y.; Wu, J.; Qiu, X.; Xi, J.; Wang, J.; Li, J.; Zhu, W.; Chen, L. Electrochemical characterization of Pt-CeO2/C and Pt-CexZr1−xO2/C catalysts for ethanol electro-oxidation. Appl. Catal. B 2007, 73, 144–149. [Google Scholar] [CrossRef]
- Comignani, V.; Sieben, J.M.; Brigante, M.E.; Duarte, M.M.E. Carbon supported Pt–NiO nanoparticles for ethanol electro-oxidation in acid media. J. Power Sources 2015, 278, 119–127. [Google Scholar] [CrossRef]
- Zhou, W.J.; Li, W.Z.; Song, S.Q.; Zhou, Z.H.; Jiang, L.H.; Sun, G.Q.; Xin, Q.; Poulianitis, K.; Kontou, S.; Tsiakaras, P. Bi- and tri-metallic Pt-based anode catalysts for direct ethanol fuel cells. J. Power Sources 2004, 131, 217–223. [Google Scholar] [CrossRef]
- Antolini, E. Iron-containing platinum-based catalysts as cathode and anode materials for low-temperature acidic fuel cells: A review. RSC Adv. 2016, 6, 3307–3325. [Google Scholar] [CrossRef]
- Huang, T.; Liu, J.; Li, R.; Cai, W.; Yu, A. A novel route for preparation of PtRuMe (Me=Fe, Co, Ni) and their catalytic performance for methanol electrooxidation. Electrochem. Commun. 2009, 11, 643–646. [Google Scholar] [CrossRef]
- Malheiro, A.R.; Perez, J.; Villullas, H.M. Surface structure and electronic properties of Pt–Fe/C nanocatalysts and their relation with catalytic activity for oxygen reduction. J. Power Sources 2010, 195, 3111–3118. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Wang, P.; Huang, R.; Li, M.; Su, S. Preparation of FePt/GC nanocatalysts and their electrocatalytic activities for ethanol oxidation. CIESC J. 2010, 61, 101–105. [Google Scholar]
- Dong, L.; Dong, H.; Yu, L.; Zhang, Q.; Bai, J.; Sui, J.; Ma, B. Comparative study of different carbon nanotube-supported platinum bimetallic and trimetallic catalysts for methanol and ethanol oxidation. ECS Trans. 2011, 41, 1317–1321. [Google Scholar]
- Wang, R.; Wang, H.; Li, H.; Wang, W.; Key, J.; Khotseng, L.; Ji, S. An Fe@Fe3C-inserted carbon nanotube/graphite composite support providing highly dispersed Pt nanoparticles for ethanol oxidation. Electrochim. Acta 2014, 132, 251–257. [Google Scholar] [CrossRef]
- Xu, Z.; Hu, J.; Yan, Z.; Yang, S.; Zhou, J.; Lu, W. Potassium ferrate (VI) and decomposed K2FeO4 assisted methanol electro-oxidation in alkaline media. Electrochim. Acta 2009, 54, 3548–3552. [Google Scholar] [CrossRef]
- Léger, J.M.; Rousseau, S.; Coutanceau, C.; Hahn, F.; Lamy, C. How bimetallic electrocatalysts does work for reactions involved in fuel cells? Example of ethanol oxidation and comparison to methanol. Electrochim. Acta 2005, 50, 5118–5125. [Google Scholar] [CrossRef]
- Zhang, Y.; Weaver, M.J. Application of surface-enhanced Raman spectroscopy to organic electrocatalytic systems: Decomposition and electrooxidation of methanol and formic acid on gold and platinum-film electrodes. Langmuir 1993, 9, 1397–1403. [Google Scholar] [CrossRef]
- Lubarda, V.A. On the effective lattice parameter of binary alloys. Mech. Mater. 2003, 35, 53–68. [Google Scholar] [CrossRef]
- Merte, L.R.; Grabow, L.C.; Peng, G.; Knudsen, J.; Zeuthen, H.; Kudernatsch, W.; Porsgaard, S.; Lægsgaard, E.; Mavrikakis, M.; Besenbacher, F. Tip-dependent scanning tunneling microscopy imaging of ultrathin FeO films on Pt(111). J. Phys. Chem. C 2011, 115, 2089–2099. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, M.; Wu, Z.; Li, W. Ultra-thin PtFe-nanowires as durable electrocatalysts for fuel cells. Nanotechnology 2011, 22, 015602. [Google Scholar] [CrossRef] [PubMed]
- Jingyan, S.; Yuwen, L.; Zhiyong, W.; Cunxin, W. Investigation of thermal decomposition of ascorbic acid by TG-FTIR and thermal kinetics analysis. J. Pharm. Biomed. Anal. 2013, 77, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Baturina, O.A.; Aubuchon, S.R.; Wynne, K.J. Thermal stability in air of Pt/C catalysts and PEM fuel cell catalyst layers. Chem. Mater. 2006, 18, 1498–1504. [Google Scholar] [CrossRef]
- Silva, M.F.; da Silva, F.C.; Fogo, F.C.; Pineda, E.A.G.; Hechenleitner, A.A.W. Thermal and FTIR study of polyvinylpyrrolidone/lignin blends. J. Therm. Anal. Calorim. 2005, 79, 367–370. [Google Scholar] [CrossRef]
- Kim, M.-C.; Kim, T.-W.; Kim, H.J.; Kim, C.-U.; Bae, J.W. Aqueous phase reforming of polyols for hydrogen production using supported Pt-Fe bimetallic catalysts. Renew. Energy 2016, 95, 396–403. [Google Scholar] [CrossRef]
- Mei, H.; Wu, W.; Yu, B.; Wu, H.; Wang, S.; Xia, Q. Nonenzymatic electrochemical sensor based on Fe@Pt core–shell nanoparticles for hydrogen peroxide, glucose and formaldehyde. Sens. Actuators B 2016, 223, 68–75. [Google Scholar] [CrossRef]
- Arjona, N.; Guerra-Balcázar, M.; Trejo, G.; Álvarez-Contreras, L.; Ledesma-García, J.; Arriaga, L.G. Staircase and pulse potential electrochemical techniques for the facile and rapid synthesis of Pt and PtAg materials. Electrochim. Acta 2014, 115, 46–55. [Google Scholar] [CrossRef]
- Lee, Y.W.; Kim, M.; Kim, Y.; Kang, S.W.; Lee, J.-H.; Han, S.W. Synthesis and electrocatalytic activity of Au−Pd alloy nanodendrites for ethanol oxidation. J. Phys. Chem. C 2010, 114, 7689–7693. [Google Scholar] [CrossRef]
- Hong, J.W.; Kim, D.; Lee, Y.W.; Kim, M.; Kang, S.W.; Han, S.W. Atomic-distribution-dependent electrocatalytic activity of Au–Pd bimetallic nanocrystals. Angew. Chem. 2011, 123, 9038–9042. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, T.; Pan, Q.; Zhang, X.; Guo, J. PtFe/nitrogen-doped graphene for high-performance electrooxidation of formic acid with composition sensitive electrocatalytic activity. RSC Adv. 2015, 5, 60237–60245. [Google Scholar] [CrossRef]
- Arjona, N.; Guerra-Balcázar, M.; Ortiz-Frade, L.; Osorio-Monreal, G.; Álvarez-Contreras, L.; Ledesma-García, J.; Arriaga, L.G. Electrocatalytic activity of well-defined and homogeneous cubic-shaped Pd nanoparticles. J. Mater. Chem. A 2013, 1, 15524–15529. [Google Scholar] [CrossRef]
- Pereira, L.G.S.; Paganin, V.A.; Ticianelli, E.A. Investigation of the CO tolerance mechanism at several Pt-based bimetallic anode electrocatalysts in a PEM fuel cell. Electrochim. Acta 2009, 54, 1992–1998. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Jeon, S. Highly active graphene-supported NixPd100–x Binary alloyed catalysts for electro-oxidation of ethanol in an alkaline media. ACS Catal. 2014, 4, 1830–1837. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Yin, J.; Liu, M.; Dong, Q.; Su, Y. Synthesis and electrocatalytic alcohol oxidation performance of Pd–Co bimetallic nanoparticles supported on graphene. Int. J. Hydrog. Energy 2014, 39, 1325–1335. [Google Scholar] [CrossRef]
- Feng, Y.-Y.; Liu, Z.-H.; Kong, W.-Q.; Lin, Q.-Y.; Du, L.-X. Promotion of palladium catalysis by silver for ethanol electro-oxidation in alkaline electrolyte. Int. J. Hydrog. Energy 2014, 39, 2497–2504. [Google Scholar] [CrossRef]
- Vié, R.; Drahi, E.; Baudino, O.; Blayac, S.; Berthon-Fabry, S. Synthesis of carbon nanospheres for the development of inkjet-printed resistive layers and sensors. Flex. Print. Electron. 2016, 1. [Google Scholar] [CrossRef]
- Lai, S.C.S.; Koper, M.T.M. Ethanol electro-oxidation on platinum in alkaline media. Phys. Chem. Chem. Phys. 2009, 11, 10446–10456. [Google Scholar] [CrossRef] [PubMed]
- She, C.-X.; Xiang, J.; Ren, B.; Zhong, Q.-L.; Wang, X.-C.; Tian, Z.-Q. The Investigation of electro-oxidation of methanol on Pt-Ru electrode surfaces by in-situ Raman spectroscopy. J. Korean Electrochem. Soc. 2002, 5, 221–225. [Google Scholar] [CrossRef]
- De Souza, R.F.B.; Neto, É.T.; Calegaro, M.L.; Santos, E.A.; Martinho, H.S.; dos Santos, M.C. Ethanol electro-oxidation on Pt/C electrocatalysts: An “in situ” Raman spectroelectrochemical study. Electrocatalysis 2011, 2, 28–34. [Google Scholar] [CrossRef]
- Himmelblau, D.M. Diffusion of dissolved gases in liquids. Chem. Rev. 1964, 64, 527–550. [Google Scholar] [CrossRef]
- Wang, Y.; Zou, S.; Cai, W.-B. Recent advances on electro-oxidation of ethanol on Pt- and Pd-based catalysts: From reaction mechanisms to catalytic materials. Catalysis 2015, 5, 1507–1534. [Google Scholar] [CrossRef]
- Pletcher, D. Electrocatalysis: Present and future. J. Appl. Electrochem. 1984, 14, 403–415. [Google Scholar] [CrossRef]
- Brouzgou, A.; Podias, A.; Tsiakaras, P. PEMFCs and AEMFCs directly fed with ethanol: A current status comparative review. J. Appl. Electrochem. 2013, 43, 119–136. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Material | ||
|---|---|---|---|
| Pt/C | PtFe/C | ||
| Pt | Fe | ||
| Crystallite size (nm) | 3.31 | 3.94 | |
| Lattice constant (Å) | 3.9333 | 3.9119 | |
| Mass content by XRF (%) | 100 | 89 | 11 |
| Average atomic by EDX (%) | 4.13 | 0.52 | 0.16 |
| Residual metallic content by TGA (%) | 3.443 | 3.669 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Monsiváis, A.C.; Velázquez-Hernández, I.; Álvarez-Contreras, L.; Guerra-Balcázar, M.; Arriaga, L.G.; Arjona, N.; Ledesma-García, J. In Situ Surface-Enhanced Raman Spectroscopy Study of the Electrocatalytic Effect of PtFe/C Nanocatalyst on Ethanol Electro-Oxidation in Alkaline Medium. Energies 2017, 10, 290. https://doi.org/10.3390/en10030290
Gómez-Monsiváis AC, Velázquez-Hernández I, Álvarez-Contreras L, Guerra-Balcázar M, Arriaga LG, Arjona N, Ledesma-García J. In Situ Surface-Enhanced Raman Spectroscopy Study of the Electrocatalytic Effect of PtFe/C Nanocatalyst on Ethanol Electro-Oxidation in Alkaline Medium. Energies. 2017; 10(3):290. https://doi.org/10.3390/en10030290
Chicago/Turabian StyleGómez-Monsiváis, A. C., I. Velázquez-Hernández, L. Álvarez-Contreras, M. Guerra-Balcázar, L. G. Arriaga, N. Arjona, and J. Ledesma-García. 2017. "In Situ Surface-Enhanced Raman Spectroscopy Study of the Electrocatalytic Effect of PtFe/C Nanocatalyst on Ethanol Electro-Oxidation in Alkaline Medium" Energies 10, no. 3: 290. https://doi.org/10.3390/en10030290