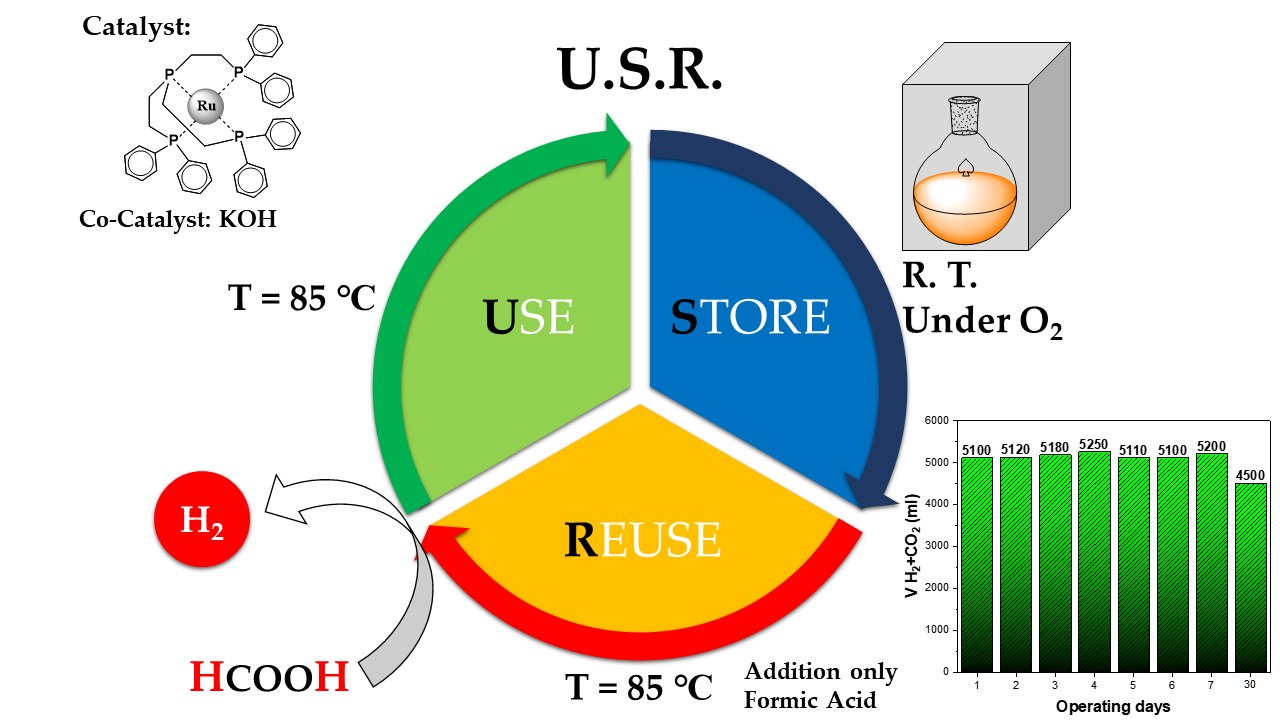
A Use-Store-Reuse (USR) Concept in Catalytic HCOOH Dehydrogenation: Case-Study of a Ru-Based Catalytic System for Long-Term USR under Ambient O2

 , ,
, , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Catalytic Procedures
2.3. Characterization Techniques
3. Results and Discussion
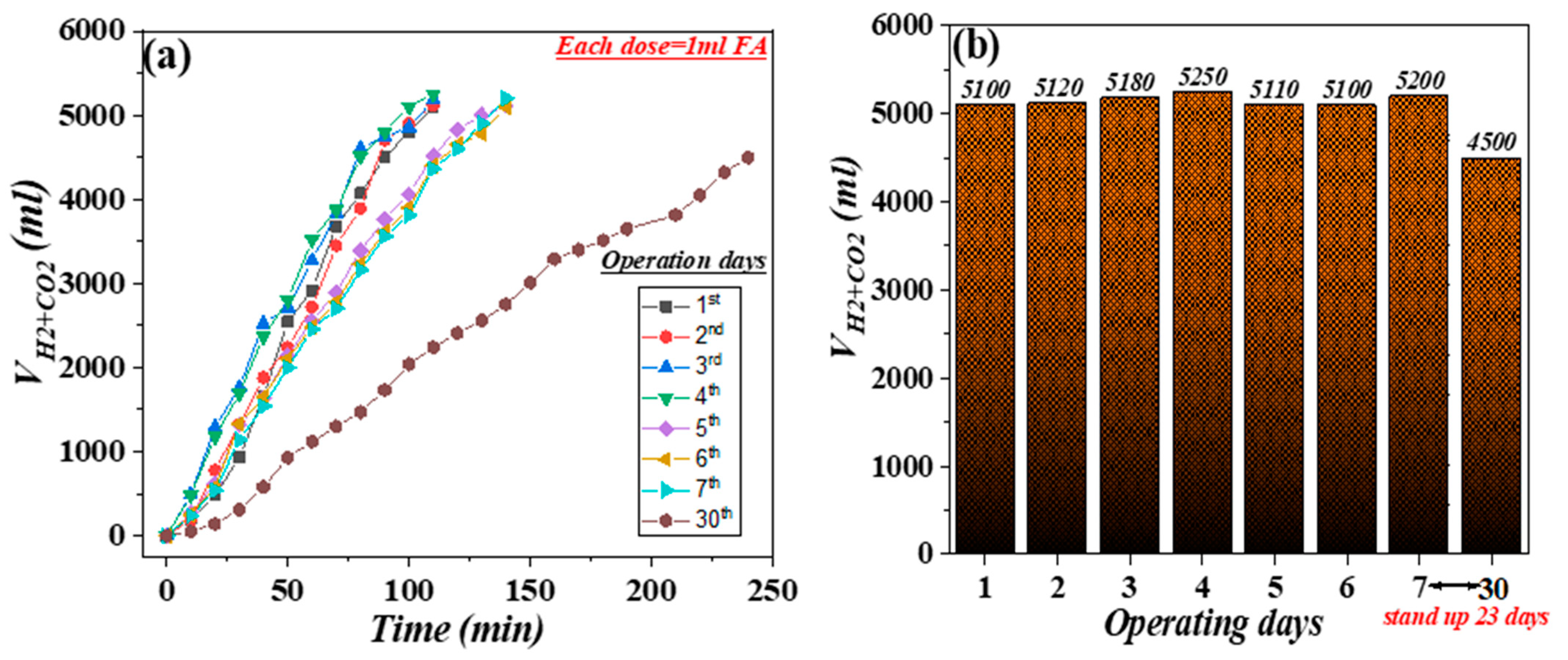
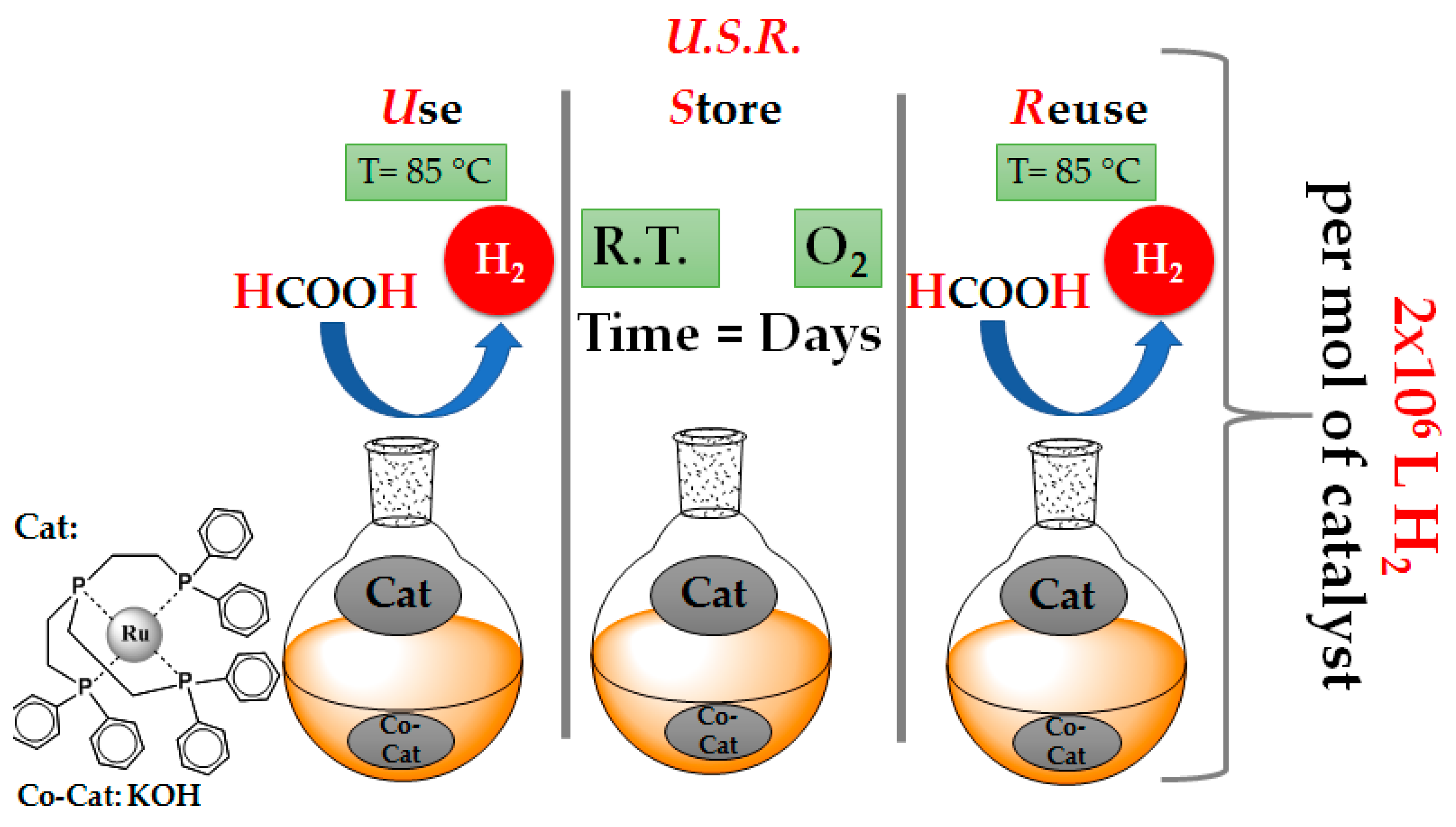
3.1. Operation Mode and Catalytic Data
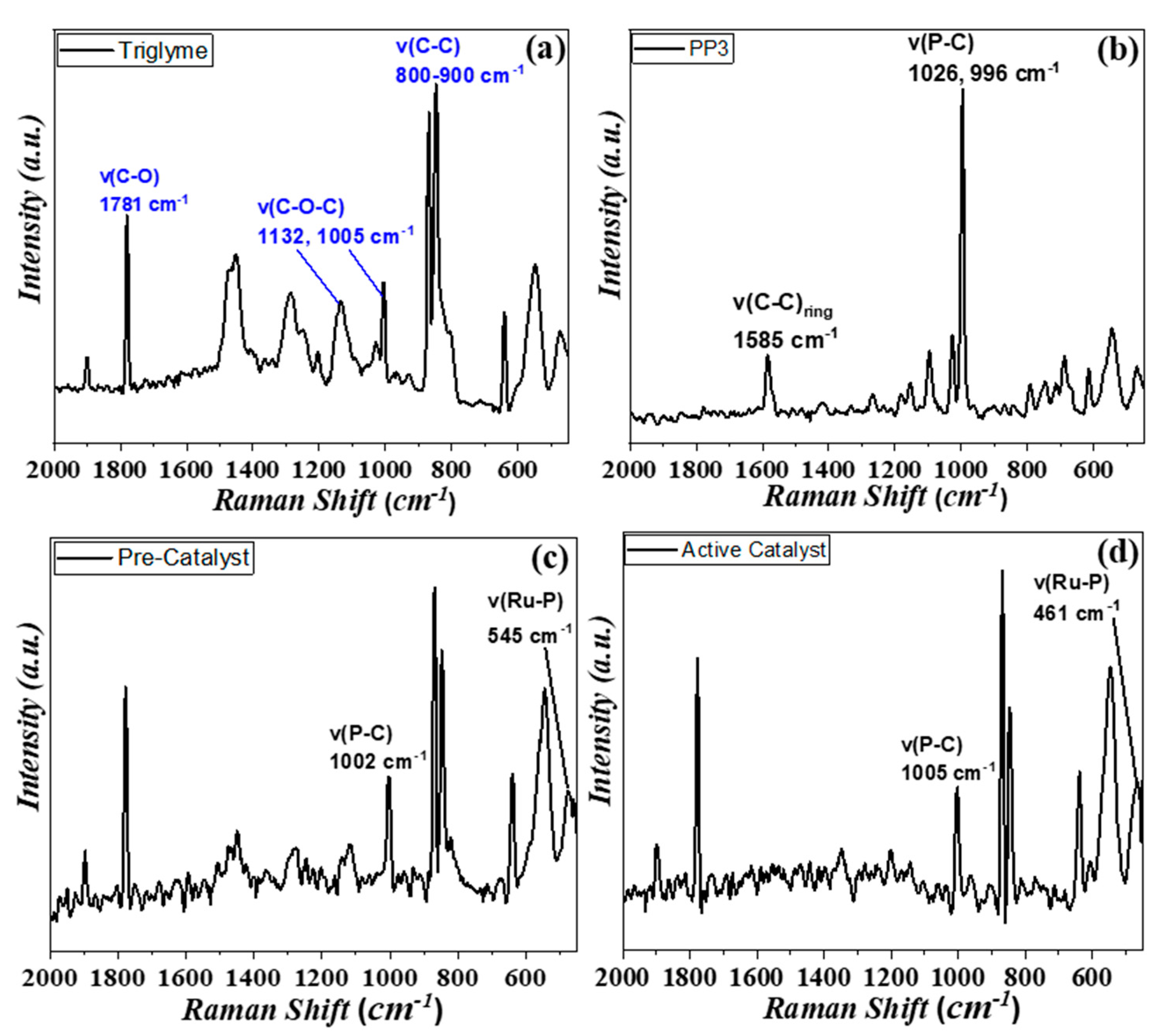
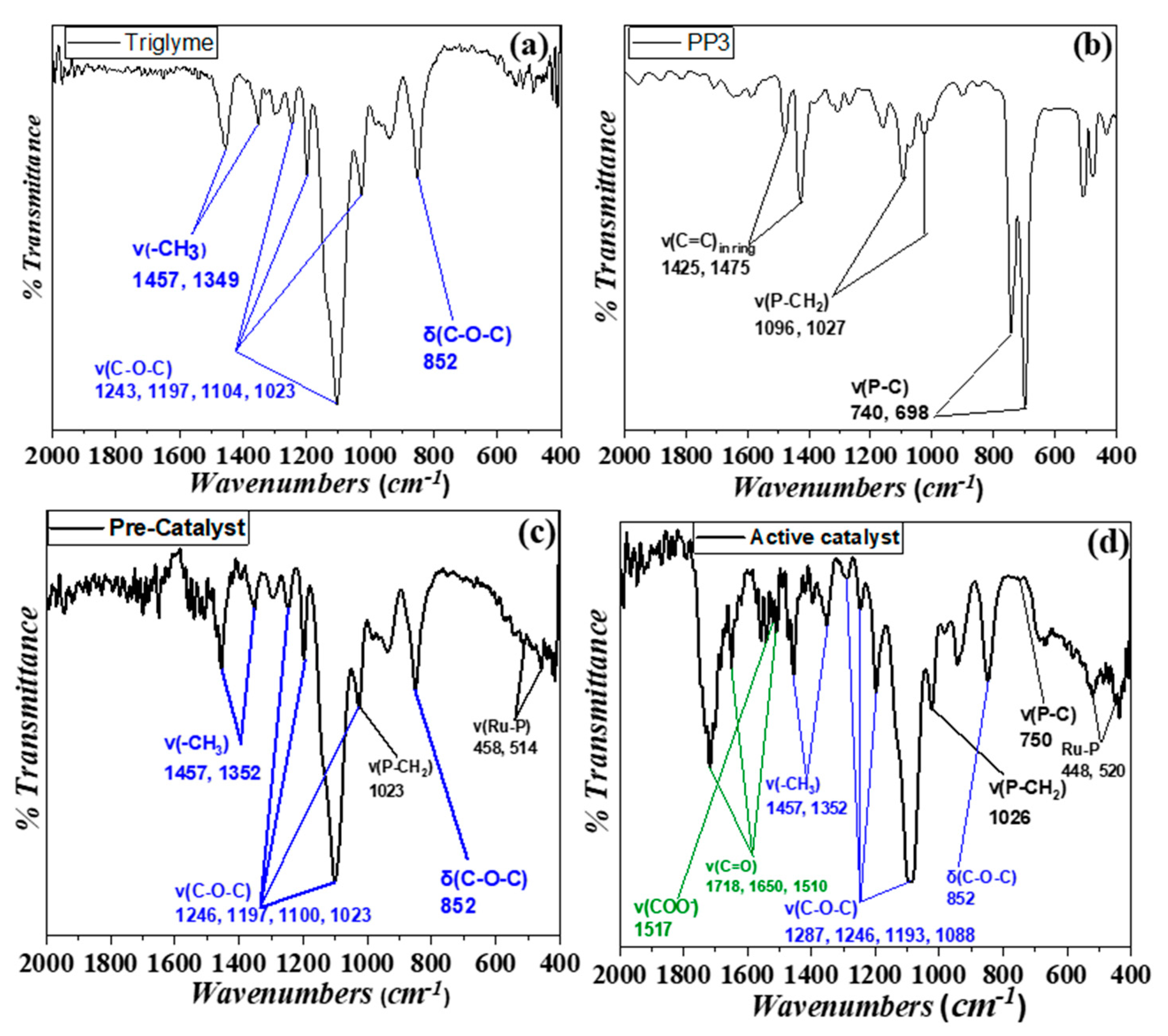
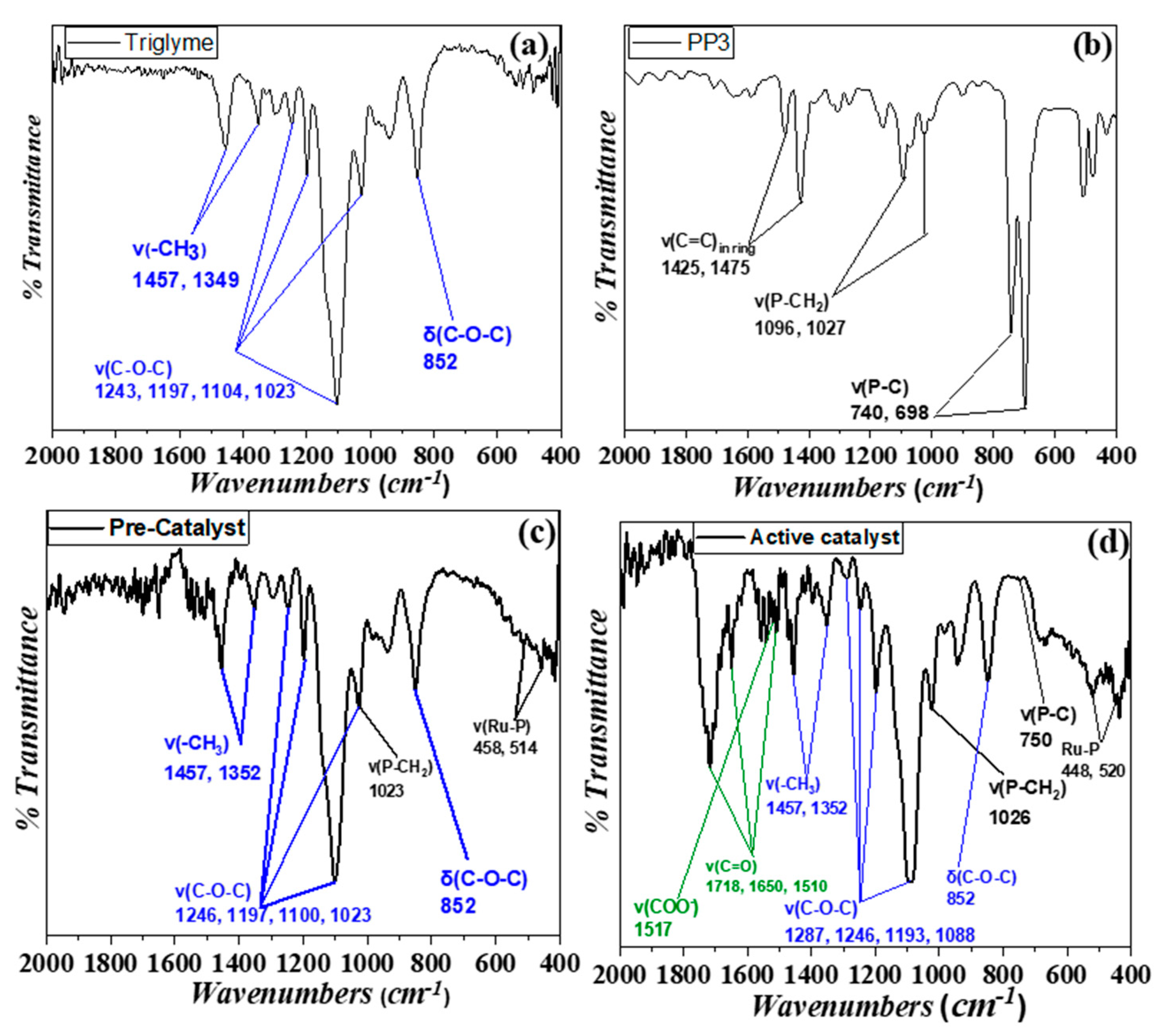
3.2. Spectroscopic Study of Catalysis In Situ
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jacobson, M.Z. Review of solutions to global warming, air pollution, and energy security. Energy Environ. Sci. 2009, 2, 148–173. [Google Scholar] [CrossRef]
- Hansen, J. Climate sensitivity: Analysis of feedback mechanisms. Clim. Process. Clim. Sensit. 1984, 5, 130–163. [Google Scholar]
- Andreas, Z.; Borgschulte, A. Handbook of Fuels Beyond Oil and Gas: The Methanol Economy Molten Carbonate Fuel Cells, 1st ed.; Wiley-VCH: Weinheim, Germany, 2007; ISBN 9783527307401. [Google Scholar]
- Elam, C.C.; Padró, C.E.G.; Sandrock, G.; Luzzi, A.; Lindblad, P.; Hagen, E.F. Realizing the hydrogen future: The International Energy Agency’s efforts to advance hydrogen energy technologies. Int. J. Hydrogen Energy 2003, 28, 601–607. [Google Scholar] [CrossRef]
- Amende, M.; Kaftan, A.; Bachmann, P.; Brehmer, R.; Preuster, P.; Koch, M.; Wasserscheid, P.; Libuda, J. Regeneration of LOHC dehydrogenation catalysts: In-situ IR spectroscopy on single crystals, model catalysts, and real catalysts from UHV to near ambient pressure. Appl. Surf. Sci. 2016, 360, 671–683. [Google Scholar] [CrossRef]
- Jörissen, L. Hydrogen and Fuel Cells. Fundamentals, Technologies and Applications. Edited by Detlev Stolten. Angew. Chem. Int. Ed. 2011, 50, 9787. [Google Scholar] [CrossRef]
- Aakko-Saksa, P.T.; Cook, C.; Kiviaho, J.; Repo, T. Liquid organic hydrogen carriers for transportation and storing of renewable energy–Review and discussion. J. Power Sources 2018, 396, 803–823. [Google Scholar] [CrossRef]
- Crabtree, R.H. Hydrogen storage in liquid organic heterocycles. Energy Environ. Sci. 2008, 1, 134–138. [Google Scholar] [CrossRef]
- Joó, F. Breakthroughs in hydrogen storage\-formic acid as a sustainable storage material for hydrogen. ChemSusChem 2008, 1, 805–808. [Google Scholar] [CrossRef]
- Sheet, S.D. Formic acid Formic acid. ICIS Chem. Bus. 2017, 1, 1–12. [Google Scholar]
- Carra, S.; Santacesaria, E. Engineering Aspects of Gas-Liquid Catalytic Reactions. Catal. Rev. 1980, 22, 75–140. [Google Scholar] [CrossRef]
- Stathi, P.; Solakidou, M.; Louloudi, M.; Deligiannakis, Y. From homogeneous to heterogenized molecular catalysts for H2 production by formic acid dehydrogenation: Mechanistic aspects, role of additives, and co-catalysts. Energies 2020, 13, 733. [Google Scholar] [CrossRef] [Green Version]
- Onishi, N.; Laurenczy, G.; Beller, M.; Himeda, Y. Recent progress for reversible homogeneous catalytic hydrogen storage in formic acid and in methanol. Coord. Chem. Rev. 2018, 373, 317–332. [Google Scholar] [CrossRef]
- Coffey, R.S. The decomposition of formic acid catalysed by soluble metal complexes. Chem. Commun. 1967, 18, 923–924. [Google Scholar] [CrossRef]
- Loges, B.; Boddien, A.; Junge, H.; Beller, M. Controlled generation of hydrogen from formic acid amine adducts at room temperature and application in H2/O2 fuel cells. Angew. Chemie Int. Ed. 2008, 47, 3962–3965. [Google Scholar] [CrossRef] [PubMed]
- Fellay, C.; Yan, N.; Dyson, P.J.; Laurenczy, G. Selective formic acid decomposition for high-pressure hydrogen generation: A mechanistic study. Chem. A Eur. J. 2009, 15, 3752–3760. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Pan, C.L.; Zhang, Y.; Li, H.; Min, S.; Guo, X.; Zheng, B.; Chen, H.; Anders, A.; Lai, Z.; et al. Selective hydrogen generation from formic acid with well-defined complexes of ruthenium and phosphorus-nitrogen PN3-pincer ligand. Chem. Asian J. 2016, 11, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Boddien, A.; Mellmann, D.; Gärtner, F.; Jackstell, R.; Junge, H.; Dyson, P.J.; Laurenczy, G.; Ludwig, R.; Beller, M. Efficient dehydrogenation of formic acid using an iron catalyst. Science 2011, 333, 1733–1736. [Google Scholar] [CrossRef] [Green Version]
- Stathi, P.; Deligiannakis, Y.; Louloudi, M. Co-catalytic enhancement of H2 production by SiO2 nanoparticles. Catal. Today 2015, 242, 146–152. [Google Scholar] [CrossRef]
- Solakidou, M.; Deligiannakis, Y.; Louloudi, M. Heterogeneous amino-functionalized particles boost hydrogen production from Formic Acid by a ruthenium complex. Int. J. Hydrogen Energy 2018, 21386–21397. [Google Scholar] [CrossRef]
- Solakidou, M.; Theodorakopoulos, M.; Deligiannakis, Y.; Louloudi, M. Double-ligand Fe, Ru catalysts: A novel route for enhanced H2 production from Formic Acid. Int. J. Hydrogen Energy 2020, 45, 17367–17377. [Google Scholar] [CrossRef]
- Boddien, A.; Loges, B.; Junge, H.; Gärtner, F.; Noyes, J.R.; Beller, M. Continuous hydrogen generation from formic acid: Highly active and stable ruthenium catalysts. Adv. Synth. Catal. 2009, 351, 2517–2520. [Google Scholar] [CrossRef]
- Mellone, I.; Gorgas, N.; Bertini, F.; Peruzzini, M.; Kirchner, K.; Gonsalvi, L. Selective Formic Acid Dehydrogenation Catalyzed by Fe-PNP Pincer Complexes Based on the 2,6-Diaminopyridine Scaffold. Organometallics 2016, 35, 3344–3349. [Google Scholar] [CrossRef]
- Guan, C.; Zhang, D.D.; Pan, Y.; Iguchi, M.; Ajitha, M.J.; Hu, J.; Li, H.; Yao, C.; Huang, M.H.; Min, S.; et al. Dehydrogenation of Formic Acid Catalyzed by a Ruthenium Complex with an N,N′-Diimine Ligand. Inorg. Chem. 2017, 56, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Agapova, A.; Alberico, E.; Kammer, A.; Junge, H.; Beller, M. Catalytic Dehydrogenation of Formic Acid with Ruthenium-PNP-Pincer Complexes: Comparing N-Methylated and NH-Ligands. ChemCatChem 2019, 11, 1910–1914. [Google Scholar] [CrossRef] [Green Version]
- Stathi, P.; Deligiannakis, Y.; Avgouropoulos, G.; Louloudi, M. Efficient H2 production from formic acid by a supported iron catalyst on silica. Appl. Catal. A Gen. 2015, 498, 176–184. [Google Scholar] [CrossRef]
- Mellmann, D.; Barsch, E.; Bauer, M.; Grabow, K.; Boddien, A.; Kammer, A.; Sponholz, P.; Bentrup, U.; Jackstell, R.; Junge, H.; et al. Base-free non-noble-metal-catalyzed hydrogen generation from formic acid: Scope and mechanistic insights. Chem. A Eur. J. 2014, 20, 13589–13602. [Google Scholar] [CrossRef]
- Boddien, A.; Loges, B.; Gärtner, F.; Torborg, C.; Fumino, K.; Junge, H.; Ludwig, R.; Beller, M. Iron-catalyzed hydrogen production from formic acid. J. Am. Chem. Soc. 2010, 132, 8924–8934. [Google Scholar] [CrossRef]
- Sánchez-De-Armas, R.; Xue, L.; Ahlquist, M.S.G. One site is enough: A theoretical investigation of iron-catalyzed dehydrogenation of formic acid. Chem. A Eur. J. 2013, 19, 11869–11873. [Google Scholar] [CrossRef]
- Yang, X. Mechanistic insights into iron catalyzed dehydrogenation of formic acid: β-hydride elimination vs. direct hydride transfer. Dalt. Trans. 2013, 42, 11987–11991. [Google Scholar] [CrossRef]
- Léval, A.; Agapova, A.; Steinlechner, C.; Alberico, E.; Junge, H.; Beller, M. Hydrogen production from formic acid catalyzed by a phosphine free manganese complex: Investigation and mechanistic insights. Green Chem. 2020, 22, 913–920. [Google Scholar] [CrossRef]
- Mellone, I.; Bertini, F.; Peruzzini, M.; Gonsalvi, L. An active, stable and recyclable Ru(II) tetraphosphine-based catalytic system for hydrogen production by selective formic acid dehydrogenation. Catal. Sci. Technol. 2016, 6, 6504–6512. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
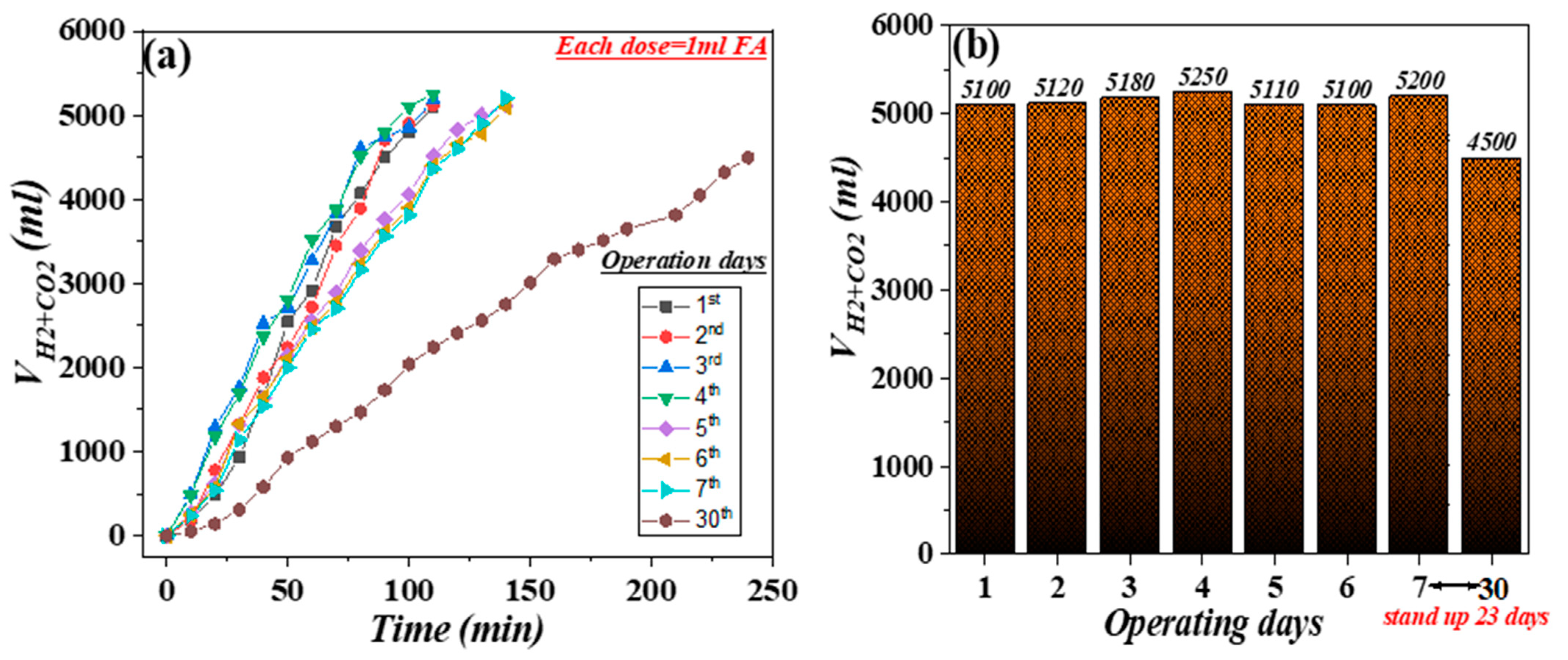
| Entry * | FA Added (mL) | Produced Gas Volume (L) | TONs | TOFs (h−1) | Reaction Time (min) |
|---|---|---|---|---|---|
| 1 | 5 | 5.10 | 11,468 | 6267 | 110 |
| 2 | 5 | 5.12 | 11,513 | 6291 | 110 |
| 3 | 5 | 5.18 | 11,648 | 6365 | 110 |
| 4 | 5 | 5.25 | 11,806 | 6451 | 110 |
| 5 | 5 | 5.11 | 11,491 | 4931 | 140 |
| 6 | 5 | 5.10 | 11,468 | 4922 | 140 |
| 7 | 5 | 5.20 | 11,693 | 5018 | 140 |
| 30 | 5 | 4.50 | 10,119 | 2529 | 240 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theodorakopoulos, M.; Solakidou, M.; Deligiannakis, Y.; Louloudi, M. A Use-Store-Reuse (USR) Concept in Catalytic HCOOH Dehydrogenation: Case-Study of a Ru-Based Catalytic System for Long-Term USR under Ambient O2. Energies 2021, 14, 481. https://doi.org/10.3390/en14020481
Theodorakopoulos M, Solakidou M, Deligiannakis Y, Louloudi M. A Use-Store-Reuse (USR) Concept in Catalytic HCOOH Dehydrogenation: Case-Study of a Ru-Based Catalytic System for Long-Term USR under Ambient O2. Energies. 2021; 14(2):481. https://doi.org/10.3390/en14020481
Chicago/Turabian StyleTheodorakopoulos, Marinos, Maria Solakidou, Yiannis Deligiannakis, and Maria Louloudi. 2021. "A Use-Store-Reuse (USR) Concept in Catalytic HCOOH Dehydrogenation: Case-Study of a Ru-Based Catalytic System for Long-Term USR under Ambient O2" Energies 14, no. 2: 481. https://doi.org/10.3390/en14020481