Electrolytes and Interphasial Chemistry in Li Ion Devices

Electrochemistry Branch, Sensors and Electron Devices Directorate, U. S. Army Research Laboratory, Adelphi, Maryland, 20783-1197, USA
Energies 2010, 3(1), 135-154; https://doi.org/10.3390/en3010135
Submission received: 2 November 2009
/
Accepted: 15 January 2010
/
Published: 26 January 2010
(This article belongs to the Special Issue Lithium-ion Batteries)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Since its appearance in 1991, the Li ion battery has been the major power source driving the rapid digitalization of our daily life; however, much of the processes and mechanisms underpinning this newest battery chemistry remains poorly understood. As in any electrochemical device, the major challenge comes from the electrolyte/electrode interfaces, where the discontinuity in charge distribution and extreme disequality in electric forces induce diversified processes that eventually determine the kinetics of Li+ intercalation chemistry. This article will summarize the most recent efforts on the fundamental understanding of the interphases in Li ion devices. Emphasis will be placed on the formation chemistry of the so-called “SEI” on graphitic anode, the effect of solvation sheath structure of Li+ on the intercalation energy barrier, and the feasibility of tailoring a desired interphase. Biologically inspired approaches to an ideal interphase will also be briefly discussed.
1. What is Interface/Interphase?
“Interface” is where one material ends and the other begins. At interfaces, an abruptly truncated atomic array leads to discontinuity in both chemical and electric potentials, and the resultant extreme disequilibria, usually existing in a narrow region of nanometric length, lead to drastic deviations in all physicochemical properties of the bulk of the adjoining phases. Thus, the interfacial region is defined by sudden and significant changes in structure, chemistry, dynamics and reactivity. These altered properties, which remain hitherto little understood, ultimately determine most of the mass and charge transfer processes across the interfaces [1].
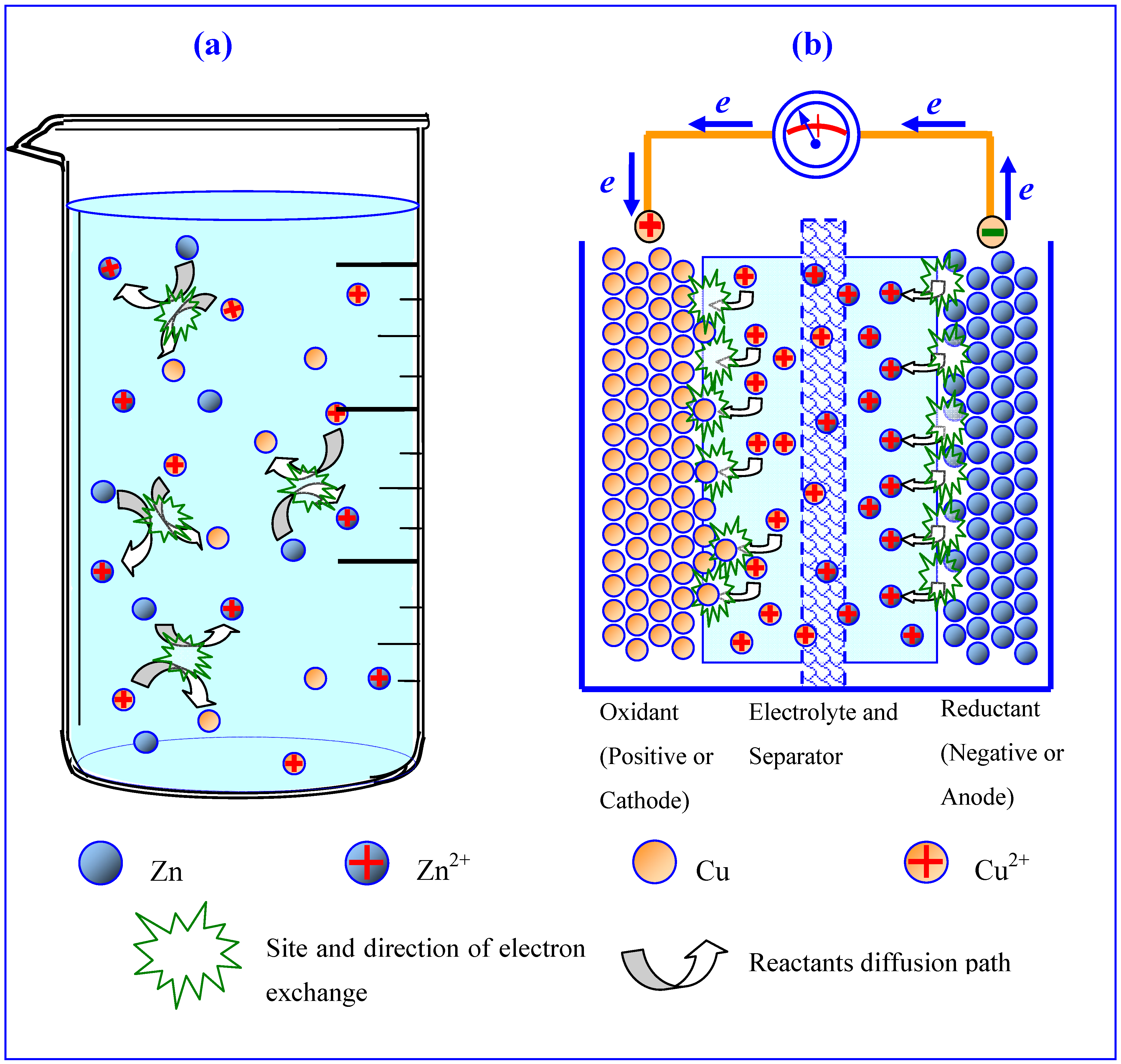
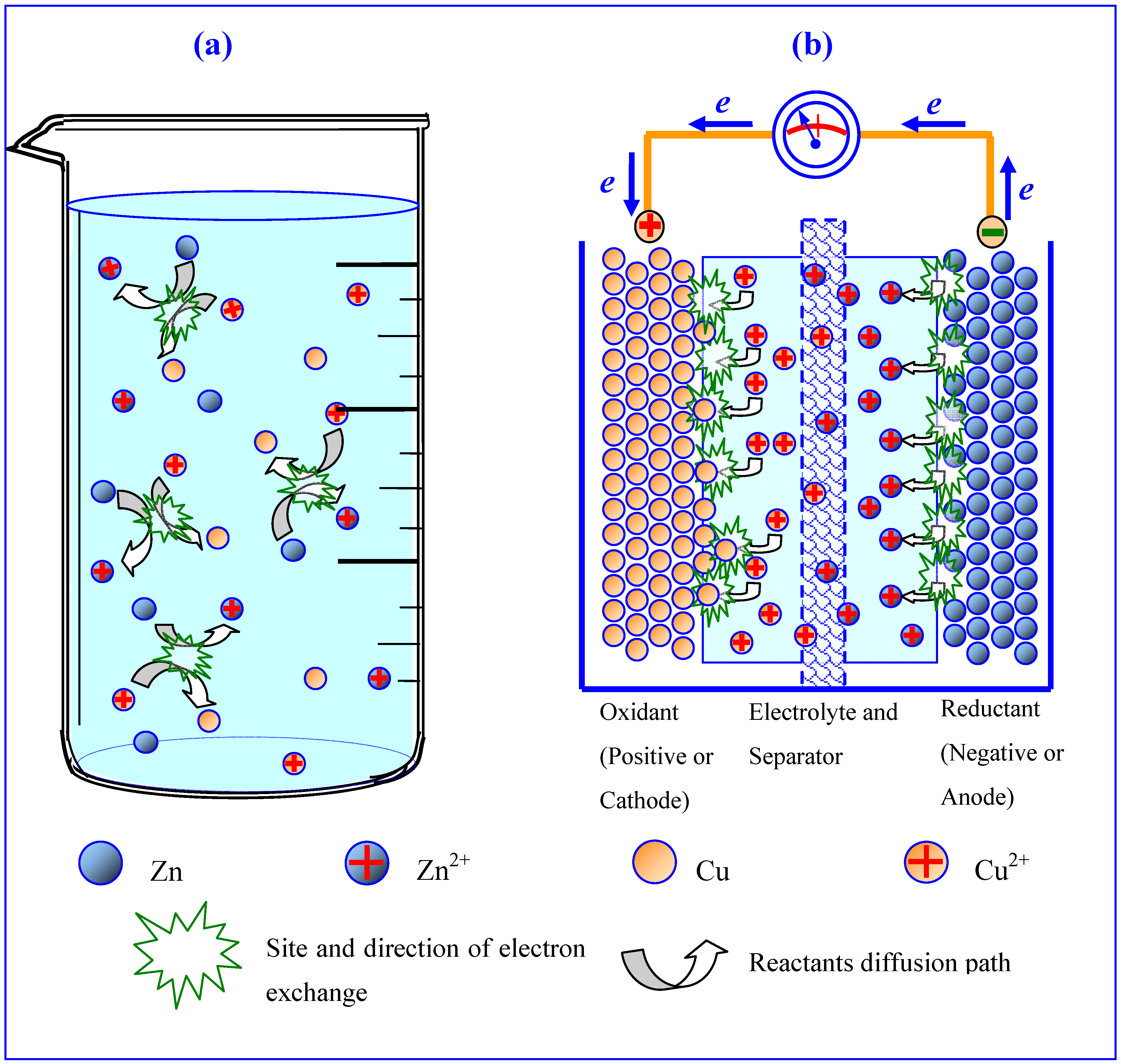
Figure 1.
Interface distinguishes electrochemistry from conventional chemistry: (a) chaotic collision of reactant particles leads to dissipation of free energy in the form of heat; (b) interfaces between electrode/electrolyte force reactions to proceed in an orderly manner, thus producing orientation movement of electrons in a Daniel cell (reproduced with permission from The Elsevier Encyclopedia of Electrochemical Power Sources, © Elsevier, November 2009).
Figure 1.
Interface distinguishes electrochemistry from conventional chemistry: (a) chaotic collision of reactant particles leads to dissipation of free energy in the form of heat; (b) interfaces between electrode/electrolyte force reactions to proceed in an orderly manner, thus producing orientation movement of electrons in a Daniel cell (reproduced with permission from The Elsevier Encyclopedia of Electrochemical Power Sources, © Elsevier, November 2009).
A typical interface is usually confined to sub-nano or slightly larger regions (e.g., 0.5~2 nm), therefore it has been traditionally treated as a 2D entity for mathematic convenience. However, as more inhomogeneous interfaces are created by the pursuit of more energetic phases, and with more advances in spectroscopic and imaging techniques, those once “2D” interfaces have become increasingly structured in our understandings. To recognize this importance of interfaces’ spatial extension, a new term “Interphase” was coined. Within this 3D entity, chemical compositions, morphology, and charge distribution are the critical properties that define the kinetics and dynamics of the charge or mass transfer processes in the region [2].
Interface is of special significance to electrochemistry, as it was the electrode/electrolyte interface that distinguished electrochemical processes from conventional chemical redox reactions. The presence of those interfaces prevents the chaotic direct electron exchange between the oxidant and reductant, hence forcing the electrons to flow through an external circuit in an orderly manner (Figure 1). In an electrochemical device, the interfaces are the only “legitimate” reaction sites where species are consumed or produced. The chemical composition and morphological structure of those two interfaces dictate the rate and efficiency of the reactions [3].
The interfaces in low-potential electrochemical devices, such as aqueous-based batteries, capacitors or fuel cells, have been primarily described as 2D in nature, and their boundaries were defined by the so-called electric double layer, or the distance from the electrode surface to the outer-Helmhotz plane (the immediate layer of counter ions in electrolyte). This simplicity is the direct benefit of the low working potentials of the electrodes, where almost all electrolyte components can remain thermodynamically stable. When more energetic electrodes are involved, however, such as with a lithium metal or lithium-based graphitic anode (0~0.20 V vs. Li+/Li), and a transition-metal oxide cathode (>4.0 V vs. Li+/Li), the interfaces separating those electrodes from electrolyte become “3D”, or interphases in our notion, mainly due to the decomposition of electrolyte components which passivate the reaction sites on the electrodes. The study on the chemistry and formation mechanism of these interphases constitutes an important part in understanding the youngest battery chemistry: Li ion batteries [4].
2. Li Ion Battery Technology: History, Glory and Challenge
The pursuit of a battery of higher energy density has been centered around how to harness the lithium metal electrode. “The Ultimate Anode” status of lithium for battery chemistry was made possible by the fortuitous combination of three factors that are irreproducible for any other element on the periodic table: (1) lithium is the metal with the lowest atomic number, hence possessing the largest theoretical specific capacity of 3,884 m·A·h·g−1; (2) it is the most electronegative metal (–3.10 V vs. Standard Hydrogen Electrode, SHE), hence generating the highest possible cell voltage against any given positive electrode, and (3) it is also the lightest metal (0.54 g·cm−3), hence contributing to the highest possible gravimetric (Wh/Kg) as well as specific energy densities (Wh/L). However, research efforts of three decades eventually diminished the feasibility of a rechargeable lithium metal electrode due to its dendritic morphology during the repeated dissolution/deposition cycles and the subsequent strong tendency to fire and explosion.
A lithium-based rechargeable chemistry was made possible only when an intercalation host was found to accommodate Li+. In this so-called “rocking chair” or “lithium ion” concept, redox reaction only occurs on the host lattice without involving the reduction of Li+, therefore issues of poor rechargibility as well as safety hazard could be circumvented [5] In the state-of-the-art Li ion battery, graphitized carbon serves as anode, whose interlayer structure accommodates Li+ at a potential ~0.20 V vs. Li+/Li, while a transition metal oxide or phosphate serves as cathode, whose layer, spinel or olivine structures accommodate Li+ at potentials in the range of 3.5~4.1 V vs. Li+/Li. Coupling of those electrodes created a secondary battery chemistry operating with energy densities between 100~180 Wh/Kg over thousands of cycles at 100% charge/discharge. Obviously, the prolonged cycle life came at the expense of energy density, which remains rather distant from the promised 500~800 Wh/Kg projected for a lithium metal electrode [6]
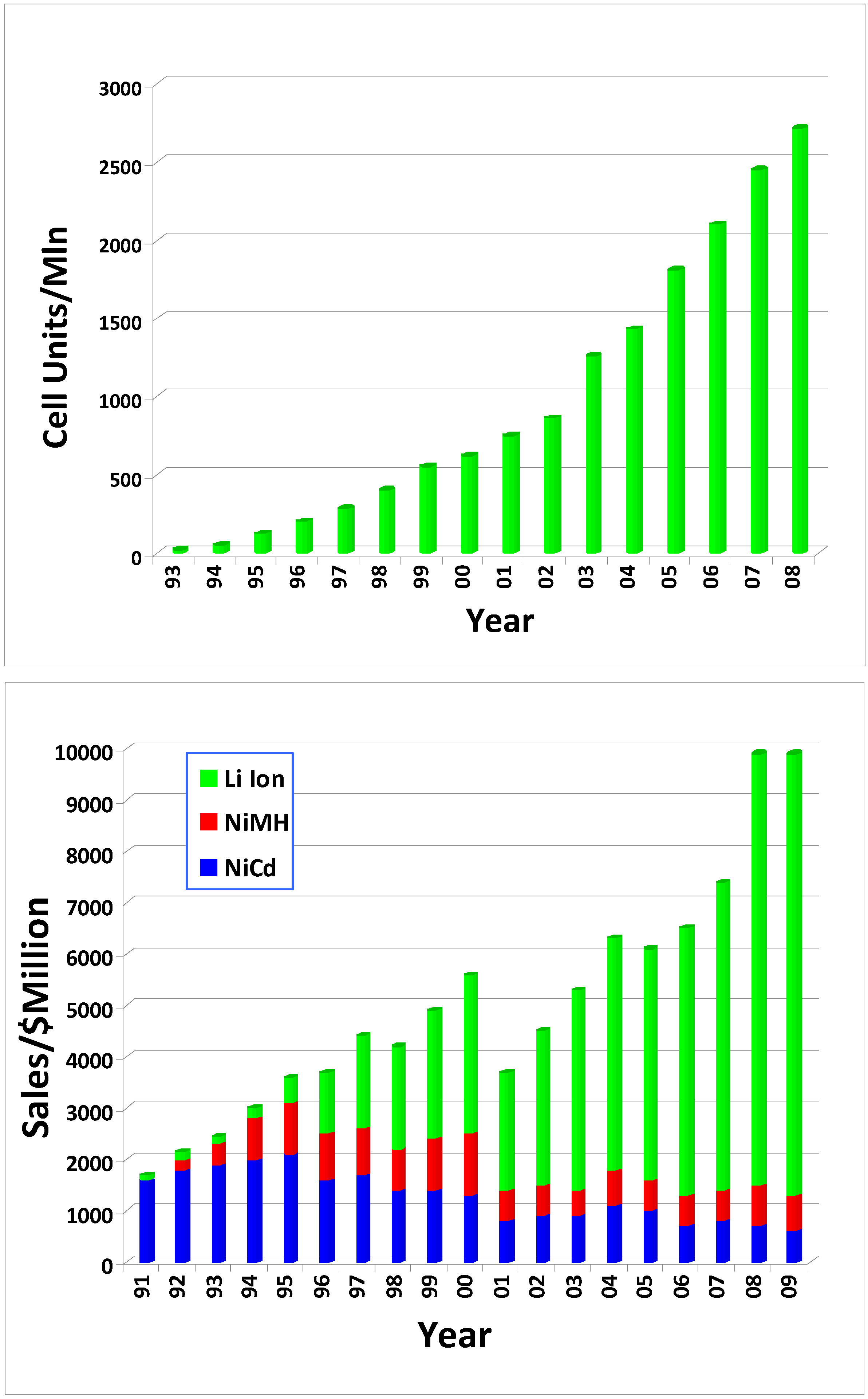
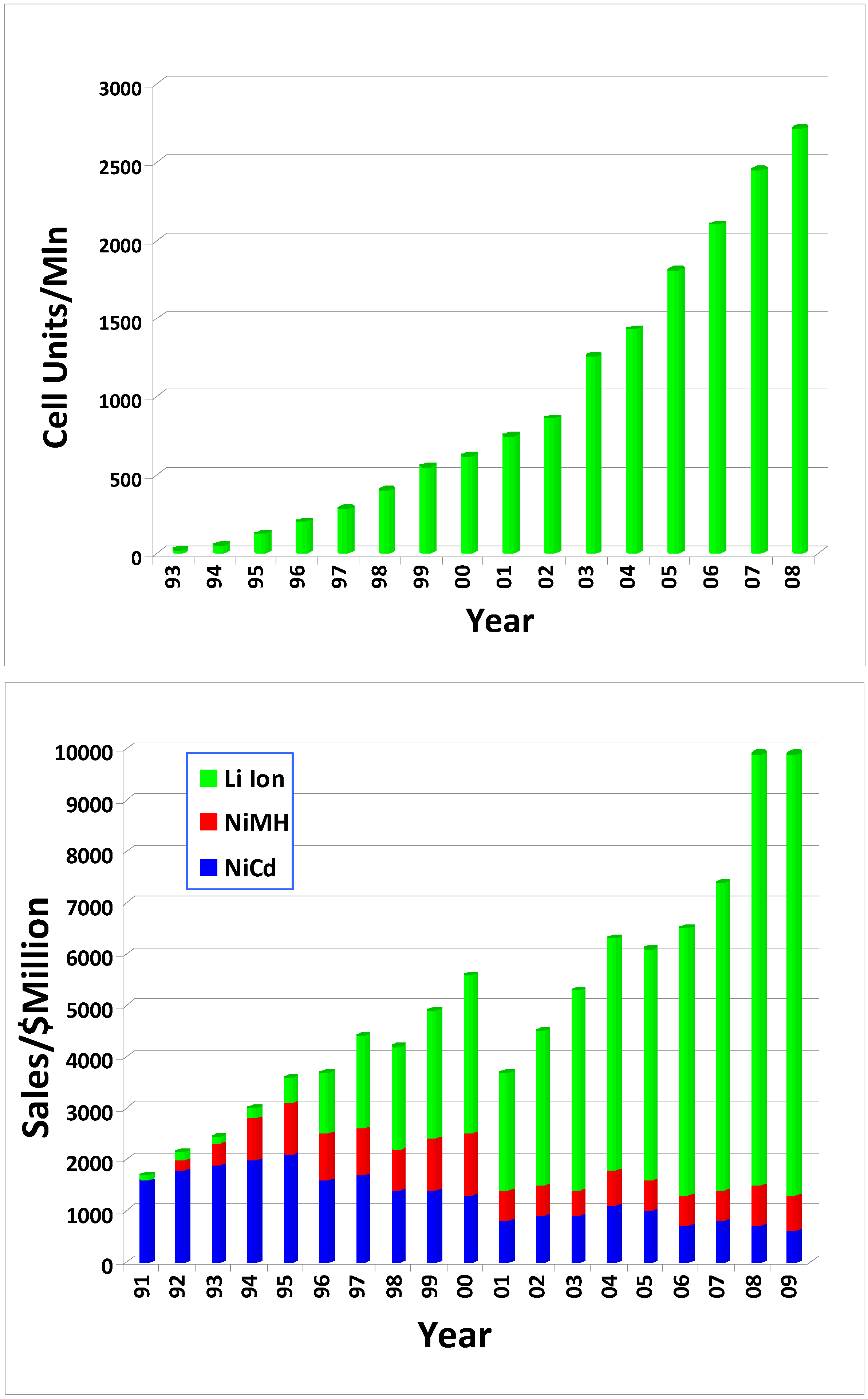
Nevertheless, this energy density is already unprecedented for a rechargeable battery chemistry, as compared with 50 Wh/Kg of Nickel Cadmium (NiCd) and 80 Wh/Kg of Nickel Metal Hydride (NiMH) batteries, the latter of which appeared on the market at almost the same time as the Li ion one. Since its birth in the early 1990s, Li ion batteries have experienced steady growth and have eventually come to dominate the rechargeable battery market for portable electronics, driving the less energy dense Ni-based chemistries into niches (Figure 2). By the end of 2008, Li ion battery industry represented an 8 billion US$ market.
The energy density of Li ion battery has been consistently improved due to combined efforts at new materials and more aggressive cell engineering. The latter probably led to the sudden surge of hazard incidents involving thermal runaway of Li ion batteries in consumer electronics around 2005, which was soon brought under media spotlight and initiated waves of recalls by major manufacturers. The highlighted safety concerns over this youngest battery chemistry indicated that the potential of the current materials has been nearly exploited to exhaustion for higher energy density, and it inevitably cast a shadow over the next possible application of Li ion batteries in vehicular electrification technologies, where battery modules composed of >100 large format Li ion cells will release multiple orders of magnitude of destruction force if similar thermal runaway occurs. In addition to safety concerns, the future challenges facing Li ion technology also include the cost and cycle life.
The 1st generation Li ion batteries were based on lithiated cobalt (Co) oxide cathodes, which still account for more than half of the cathode chemistry in cells manufactured nowadays around the world; however, Co is not an especially abundant element on the earth. Partially due to the explosive growth of Li ion industry in the past decades, the price of Co on the international market has seen dramatic rises since 2003. Both the current production and market trends indicate that Co-based chemistry will be unable to support the projected future demand from the much larger scale applications of electric (EV), hybrid electric (HEV) or plug-in hybrid electric vehicle (PHEV). This is the reason that recent research efforts have been focused on seeking a cathode intercalation chemistry that is either Co-free or at least less Co-dependent. Lithiated manganese oxide of spinel structure (LixMn2O4), doped lithiated nickel oxide (LiNi0.80Co0.15Al0.05O2), lithiated nickel manganese cobalt oxide (LiNi1/3Mn1/3Co1/3O2), and lithiated iron phosphate (LiFePO4) were all fruits of such efforts, each with its own merits and compromises [6,7].
Figure 2.
The steady growth of and rapid dominance by Li Ion batteries since its birth: (a) Li ion cell units manufactured per year; (b) sales comparison between Li ion batteries and the other two rechargeable chemistries (sources: (a) Battery Digest; (b) “Global and China's Li-ion Battery and Its Raw Materials Market Report, 2008–2009”, Market Avenue Inc., http://www.marketavenue.cn; (c) www.bccresearch.com/report/).
Figure 2.
The steady growth of and rapid dominance by Li Ion batteries since its birth: (a) Li ion cell units manufactured per year; (b) sales comparison between Li ion batteries and the other two rechargeable chemistries (sources: (a) Battery Digest; (b) “Global and China's Li-ion Battery and Its Raw Materials Market Report, 2008–2009”, Market Avenue Inc., http://www.marketavenue.cn; (c) www.bccresearch.com/report/).
The state-of-the-art small format Li ion cells used in various portable electronics can survive at least hundreds of cycles, depending on the depth of charge/discharge and user behavior. While this relatively short life duration can roughly approach or even outlive the average life expectance of most portable electronics, the battery modules designed for vehicles have to support the much longer service life of most vehicles, which are normally designed for a 10 year life or 10,000 mile driving range; Otherwise the high cost, as well as user inconvenience issues, will become the prohibitive factors preventing the marketing of EV/HEV/PHEVs. It was estimated that an advanced Li ion battery chemistry would be required to possess cycle life of at least 1,000 deep (80%) cycles to ensure that the battery modules of an electrified vehicle will need to be replaced no more than twice in its entire life.
3. Interphase in Li-based Batteries
There is perhaps no better example than the Li ion battery of the critical role of interphase in a working electrochemical device [3,8]. As the most electronegative element available, lithium has an intrinsic redox potential of –3.10 V (vs. SHE), where almost no solvent or salt can remain thermodynamically stable. The reason behind the apparent stability of lithium metal in non-aqueous electrolytes is that one or more electrolyte components decompose reductively upon contact with lithium, and the resultant decomposition products deposit on the lithium surface, thus creating a film insulating to electron conduction or tunneling but pervious to ionic diffusion. This protective film of electrolyte nature was named solid electrolyte interphase (SEI) [2,4].
In a Li ion battery, there is legitimately no presence of metallic lithium, as the anode role is served by graphitic carbon, whose interlayer structure accommodates Li+ intercalation. Since the intercalation potential is in close vicinity ( ~0.20 V vs. Li) to that of lithium metal, it had been assumed (and later turned out to be correct) that an SEI-like electron barrier must also be present between the graphitic anode and the electrolyte to stop sustaining electrolyte decomposition [9]. Presumably, a similar interphase should also exist between cathode and electrolyte in consideration of the instability of electrolyte components at high potentials of most transition metal oxides (>4.0 V vs. Li+/Li), but traditionally the term “SEI” was preserved for graphitic or metallic lithium anodes only. It was because of those two interphases that the electrolyte could remain kinetically stable against the strongly oxidative and reductive electrodes, thus supporting the reversible intercalation chemistry of Li+. In this sense, Li ion battery is a device that relies on interphases to function.
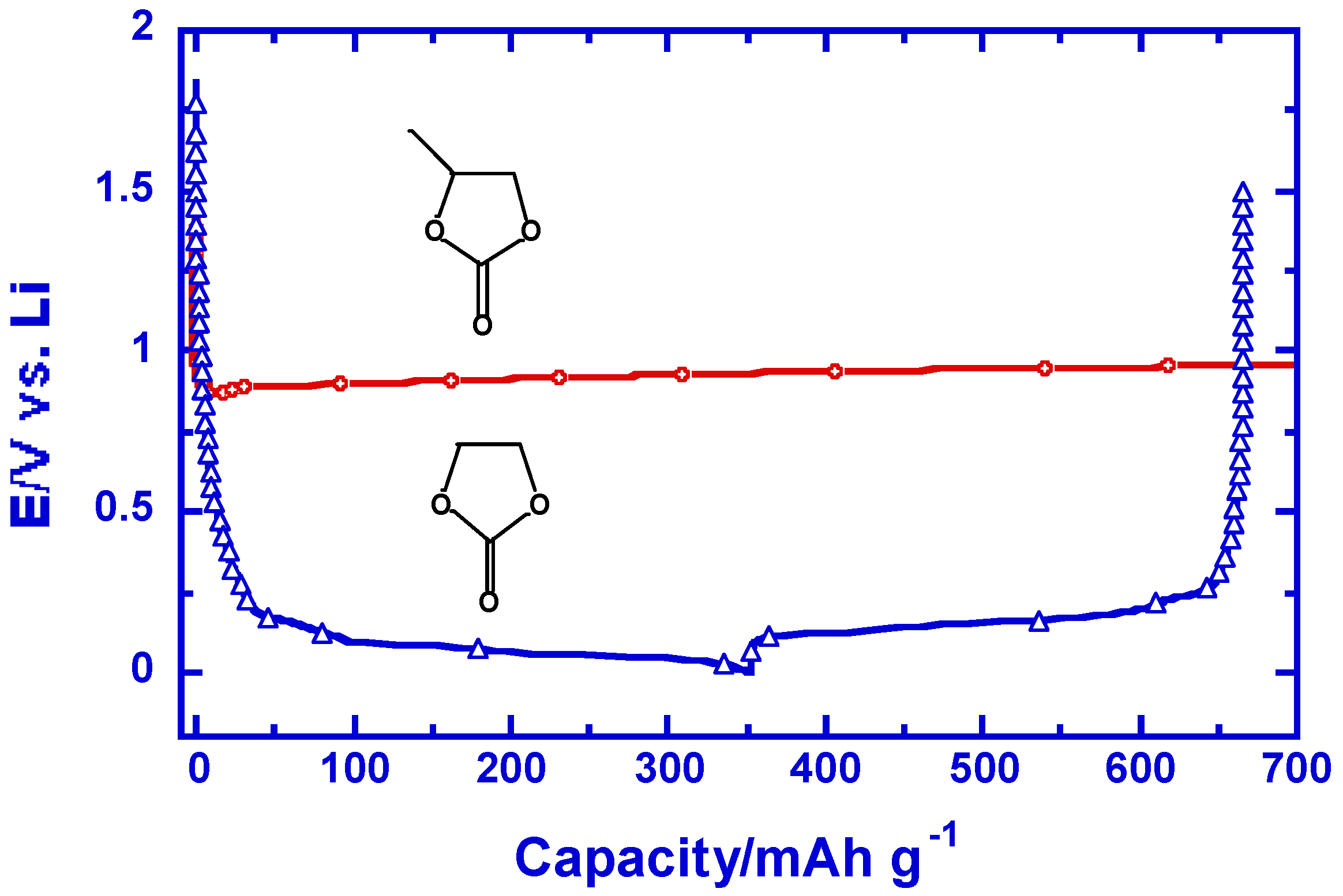
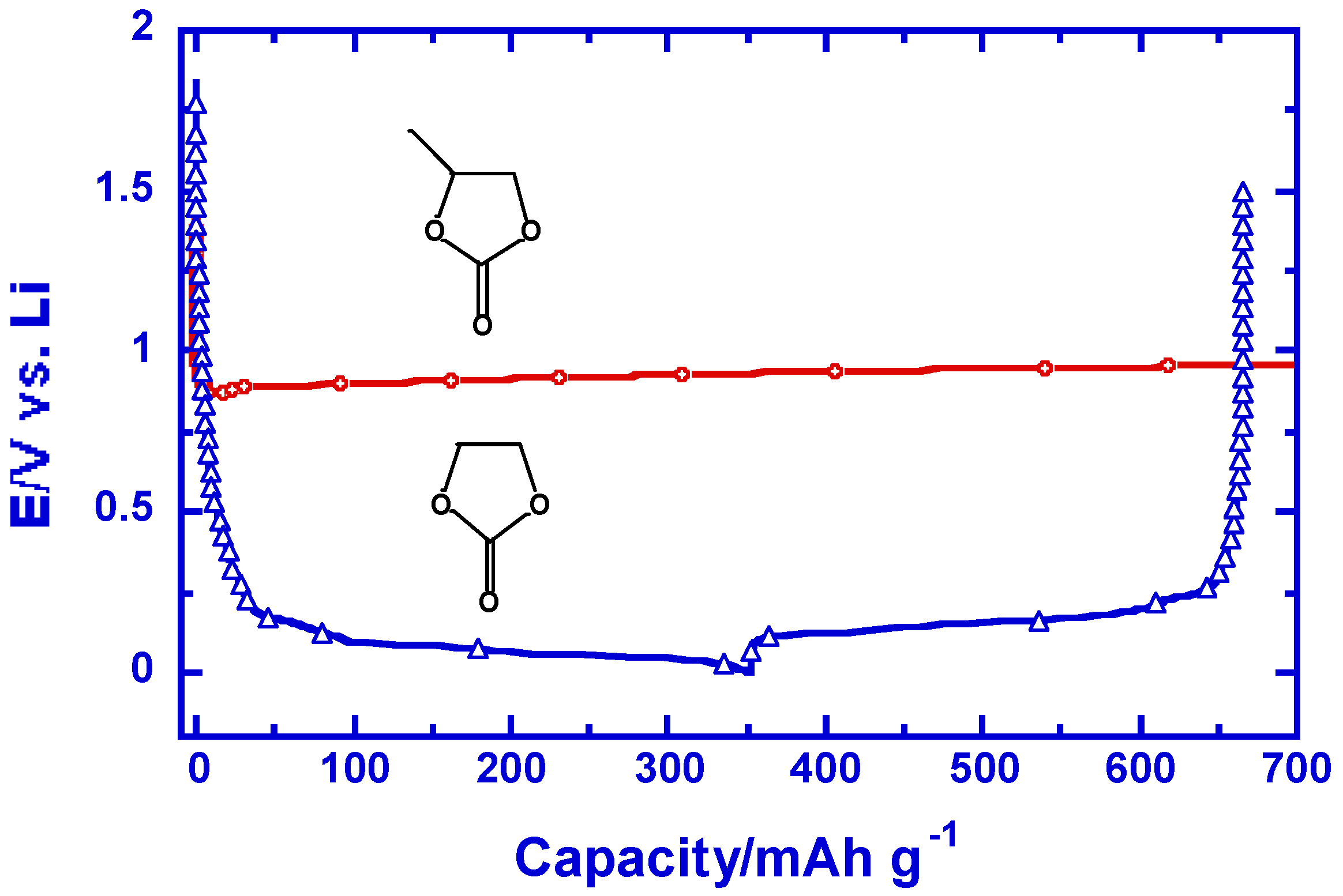
This critical importance of interphases in Li ion devices is highlighted in the so-called EC-PC disparity and the possible postponement of Li ion technology by it [10]. Early in 1950s graphite was found to form intercalation compounds with Li+ in molten lithium metal [11], but the electrochemical lithiation of graphite never succeeded during the following decades, the primary reason being that non-aqueous electrolytes during that period were almost exclusively based on propylene carbonate (PC, in Figure 3) [12]. The decomposition product of PC through single-electron pathway happens to be unable to protect the layer structure of graphite from exfoliating at ~0.70 V vs. Li+, and consequently Li+-intercalation could never occur. The lack of interphasial chemistry knowledge at the time prevented people from exploring PC’s closest homologous member, ethylene carbonate (EC, in Figure 3) until the late 1980s [13]. The interphase formed by EC, though through a similar single-electron pathway, turns out to offer a drastic surprise: it enables the reversible Li+-intercalation chemistry at potentials close to that of Li metal electrode (Figure 3), thus laying the foundation for Li ion technology [4,14]. Since then EC has become an indispensable electrolyte components in all Li ion cells manufactured. In retrospect, it is not an exaggeration to conclude that the interphasial difference of EC and PC, induced by a single methyl group, delayed the emergence of Li ion technology by nearly four decades!
Figure 3.
The difference in interphasial chemistry caused by a methyl group might be responsible for the four decade postponement of Li ion technology: voltage profiles of graphitic anodes in LiPF6/EC and LiPF6/PC during polarization to 0.002 V vs. Li (reproduced with permission from The Electrochemical Society).
Figure 3.
The difference in interphasial chemistry caused by a methyl group might be responsible for the four decade postponement of Li ion technology: voltage profiles of graphitic anodes in LiPF6/EC and LiPF6/PC during polarization to 0.002 V vs. Li (reproduced with permission from The Electrochemical Society).
SEI enables Li ion chemistry to function, but it also constitutes the most resistive component in the entire cell, as compared to electrode and electrolyte bulks (Figure 4) [3].
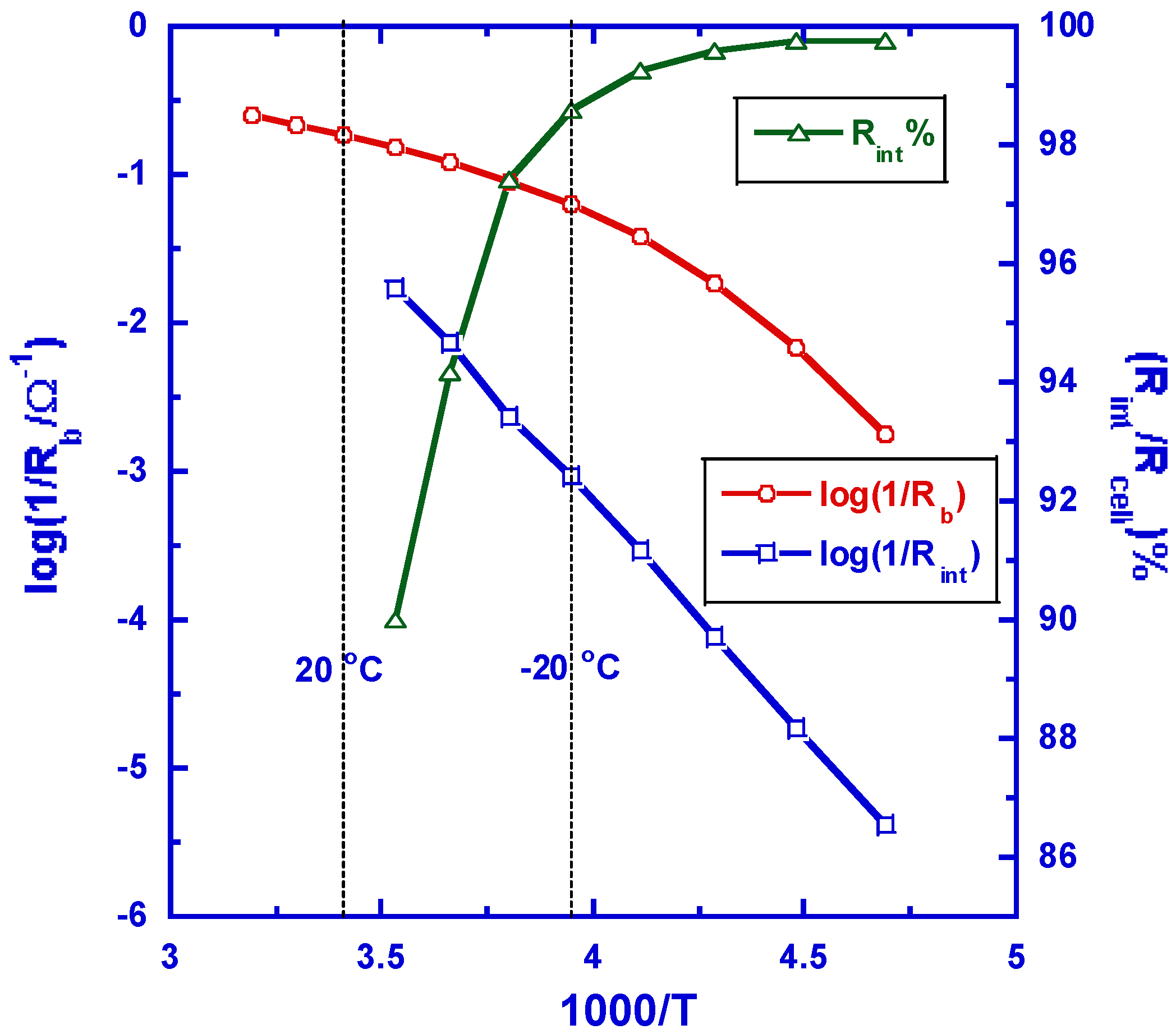
Figure 4.
The comparison of resistances generated by interphasial and the bulk electrolyte during the Li ion cell operation. Rb, Rint and Rcell represent the bulk (electrolyte), interphasial and the total cell resistance, respectively (reproduced with permission from the Elsevier Encyclopedia of Electrochemical Power Sources, ©Elsevier, November 2009).
Figure 4.
The comparison of resistances generated by interphasial and the bulk electrolyte during the Li ion cell operation. Rb, Rint and Rcell represent the bulk (electrolyte), interphasial and the total cell resistance, respectively (reproduced with permission from the Elsevier Encyclopedia of Electrochemical Power Sources, ©Elsevier, November 2009).

Since interphase is where mass/charge transfer occurs, its properties determine the kinetics of the electrochemical reactions, or the power capability of the entire cell. This bottleneck effect is more pronounced at sub-zero temperatures or in applications under high drain rate. Hitherto interphases remain the least understood component in Li ion cell, due to the fact that most surface characterization techniques must be operated under high vacuum, and hence would induce too much disturbance rather than faithfully reveal the chemical state of interphases formed in-situ during the initial charging of the Li ion cells. Nevertheless, during the past two decades, the studies on SEI have generated numerous electrochemical literatures, covering the chemistry, formation mechanism, morphology as well as physicochemical properties.
4. Chemistry and Formation Mechanism of SEI
Organic carbonates with cyclic structures, such as EC, PC , and acyclic structures, like dimethyl carbonate (DMC), diethyl carbonate (DEC), ethylmethyl carbonate (EMC), serve as the main ingredients as electrolyte solvents in state-of-the-art Li ion batteries, mainly because they can form stable interphase on transition metal oxides [8]. Occasionally unsaturated carbonates such as vinyl carbonate (VC) and vinylethyl carbonate (VEC) were also used at small concentration as additives to manipulate the chemistry of interphase formation.
Both oxidative and reductive decompositions of these solvents were investigated, but with conspicuous emphasis on the latter. The reason behind this uneven distribution of interests originated from the much higher instability of graphitic materials toward non-aqueous electrolytes than cathode ones. Since the layered structure of graphite is held together by the weak Van de Waals force (as opposed to the strong Coulombic attraction between slabs of counterions in various cathode materials), the interstitials along on c-axis are readily accessible to solvent molecules that co-intercalate with solvated Li+. Due to the large size of the solvent molecules and the average coordination number of Li+ (which is ~4), this co-intercalation almost always introduces tremendous stress to the graphite lattice. In case of organic carbonate solvents, since gaseous product is usually generated (Scheme 1), the co-intercalation almost always leads to the exfoliation of the graphene layers [15].
Aurbach undoubtedly made the most marked contribution in identifying the effective chemical ingredients in SEI. With surface spectroscopy and chemical synthesis he and coworkers determined the major products of organic carbonates from surface reduction to be semi-carbonates that are generated via a single-electron pathway [9]. More recent spectroscopic as well as diagnostic studies further confirmed Aurbach’s findings while adding an alternative two-electron pathway leading to oxalates and alkoxides (Scheme 1) [16,17].
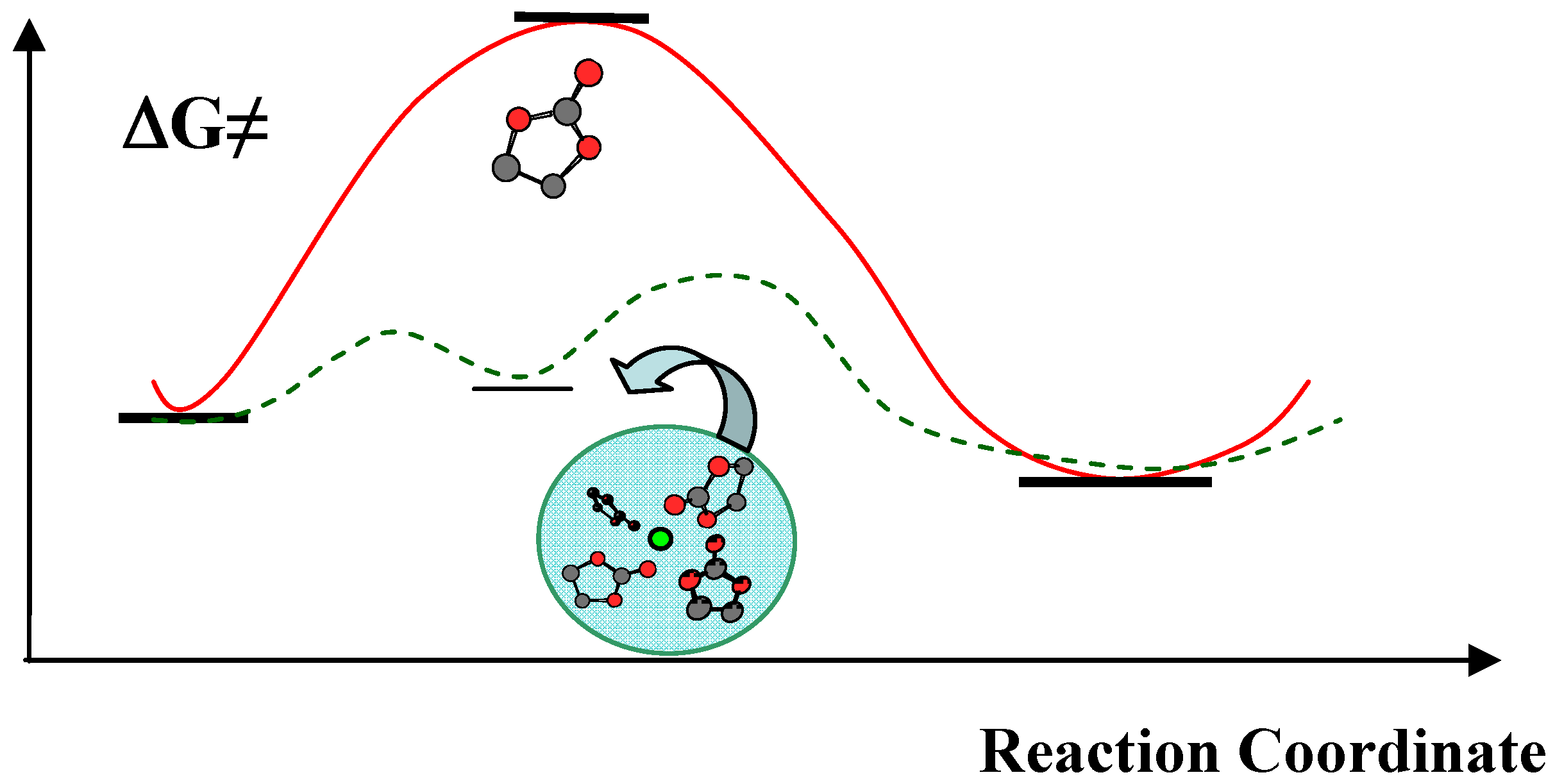
Although ab initio computations have suggested that carboxylates or semi-carbonates of higher order, such as butylene dicarbonate, would be more thermodynamically stable products of the surface reductions, NMR and FTIR spectra excluded such possibilities [16,18]. It is important to point out that the reduction of neat solvents at electrode surface should be very unlikely due to the rather high activation energy barrier; however, the presence of Li+ in the solution and the subsequent formation of its solvation sheath drastically catalyzed such reductions by lowing the energy barrier (Figure 5) [19]. Naturally the composition of this supermolecular complex Li Sn (n = 3~4) would dictate the eventual chemistry of the interphase.
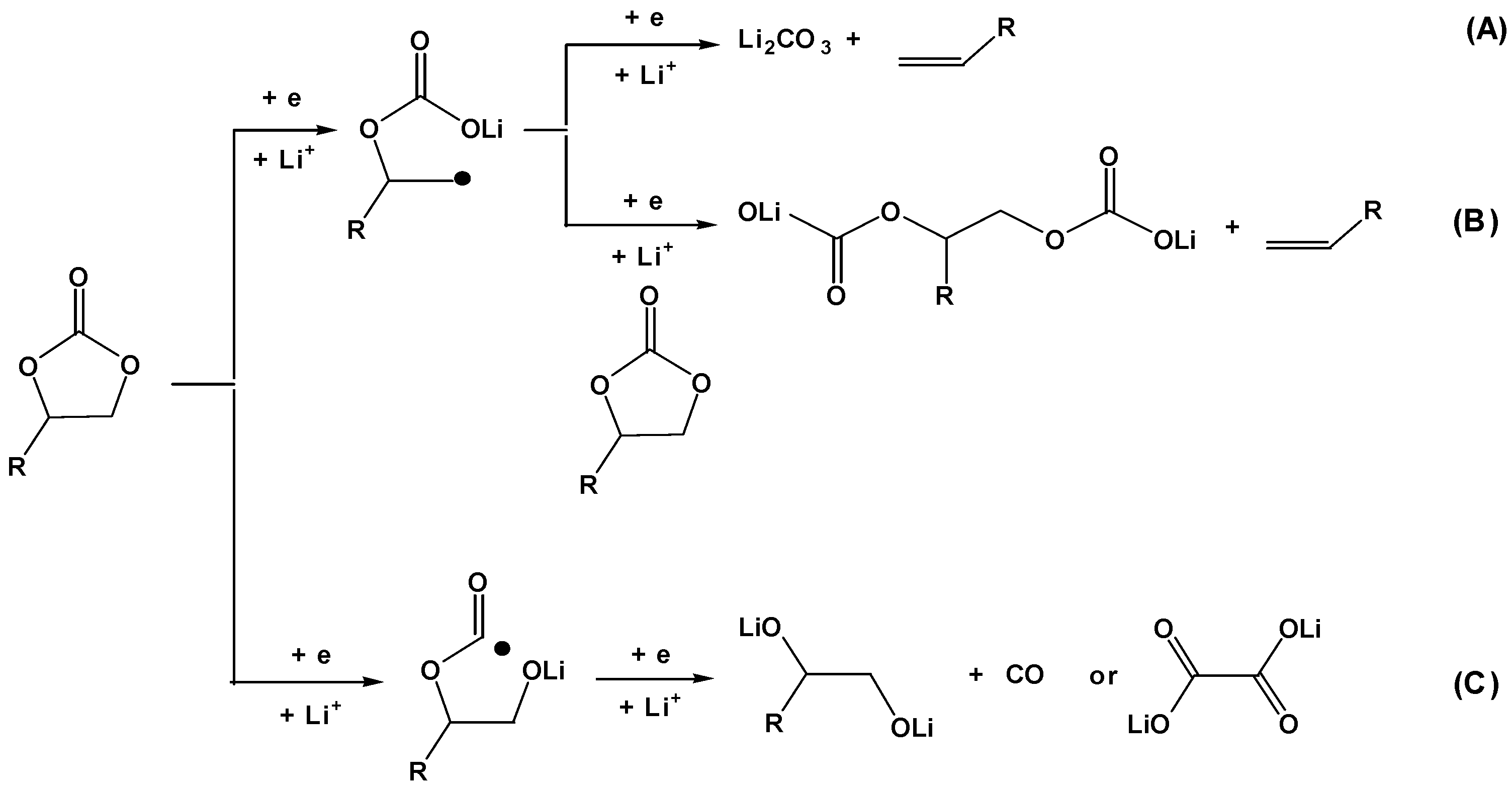
Scheme 1.
Three possible pathways for surface reductions of organic carbonates on graphitic anodes. Pathway A represents a two-electron reduction of organic carbonate, which was determined to be an unlikely route; while pathways B and C are mainly responsible for SEI ingredients.
Scheme 1.
Three possible pathways for surface reductions of organic carbonates on graphitic anodes. Pathway A represents a two-electron reduction of organic carbonate, which was determined to be an unlikely route; while pathways B and C are mainly responsible for SEI ingredients.
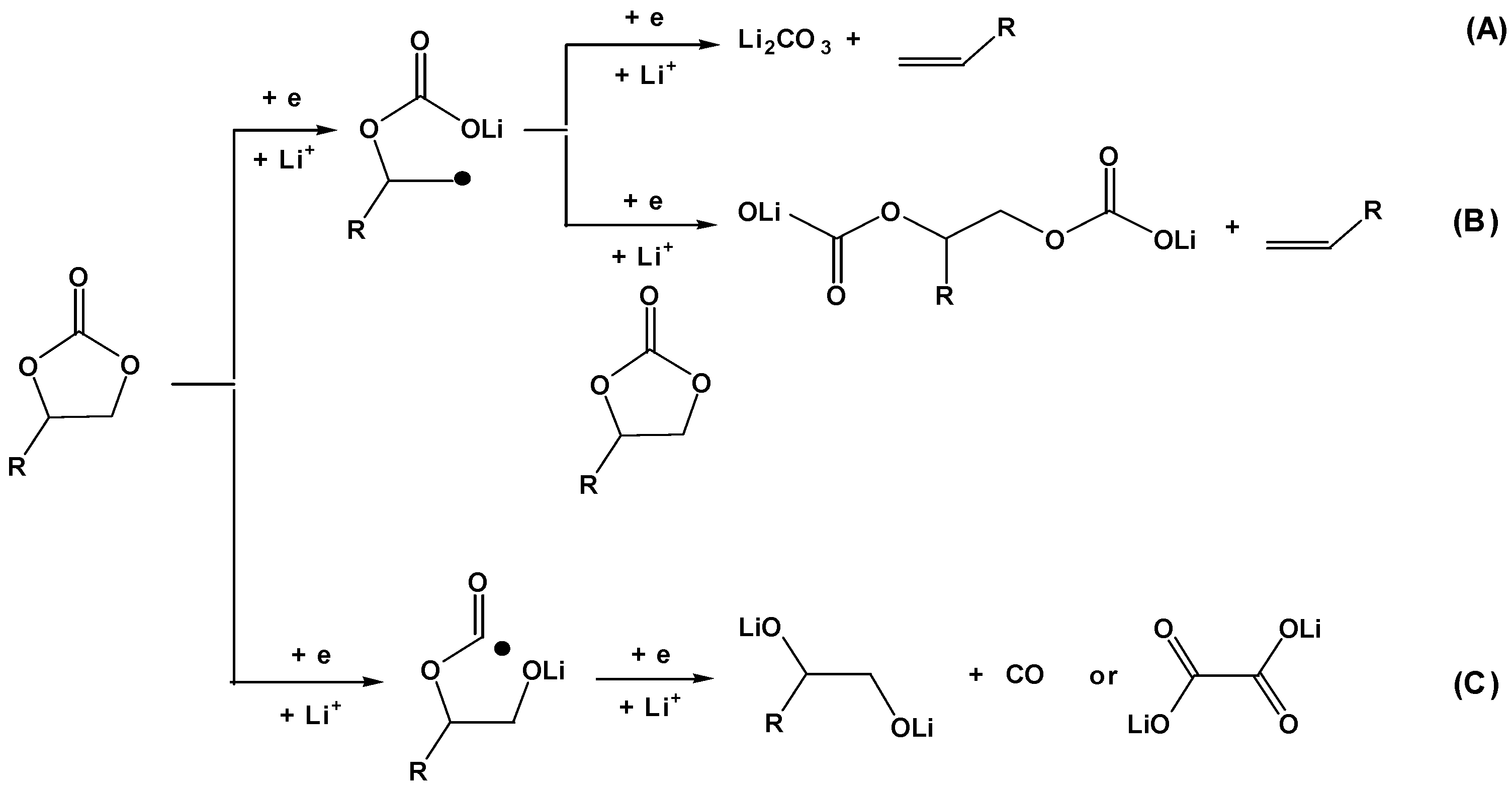
Figure 5.
Schematic illustration of the effect of Li+-solvation on the activation barrier of surface reduction for organic carbonates.
Figure 5.
Schematic illustration of the effect of Li+-solvation on the activation barrier of surface reduction for organic carbonates.
While a general agreement has been reached concerning the chemistry of interphase, controversy mainly centered around the formation mechanism with two rival models. The original SEI concept evolved from the passivation phenomenon of lithium metal electrode, which possesses a non-intercalating surface [2]. When electrolyte components are reduced on this “flat surface”, it was believed that the decomposition products should deposit on lithium electrode and form a thin film that inactivates lithium surface. Due to this manner of interphasial species accumulating on a simple surface, this formation mechanism was often referred to as “2D formation model” of SEI. It must be clarified that the nominal “2D” here denotes the mechanism only, and it differs from the dimensionless Helmhotz plane discussed above. Actually, the film-like SEI formed in 2D model still has structure and thickness of itself; for example, it has been discovered that the average thickness of SEI on lithium varies from 2.5~10 nm, and that species of more inorganic nature (such as Li2CO3, LiF etc.) are usually found in the inner region of SEI, while semi-carbonates are most likely situated further from lithium metal [20]. This latter composition gradient might have arisen from the dependence of electron tunneling probability on the distance from metallic lithium surface. When lithium ion chemistry was discovered, the 2D formation model was naturally extended to graphitic anode based on the similar operating potential (~0.20 V vs. Li+/Li).
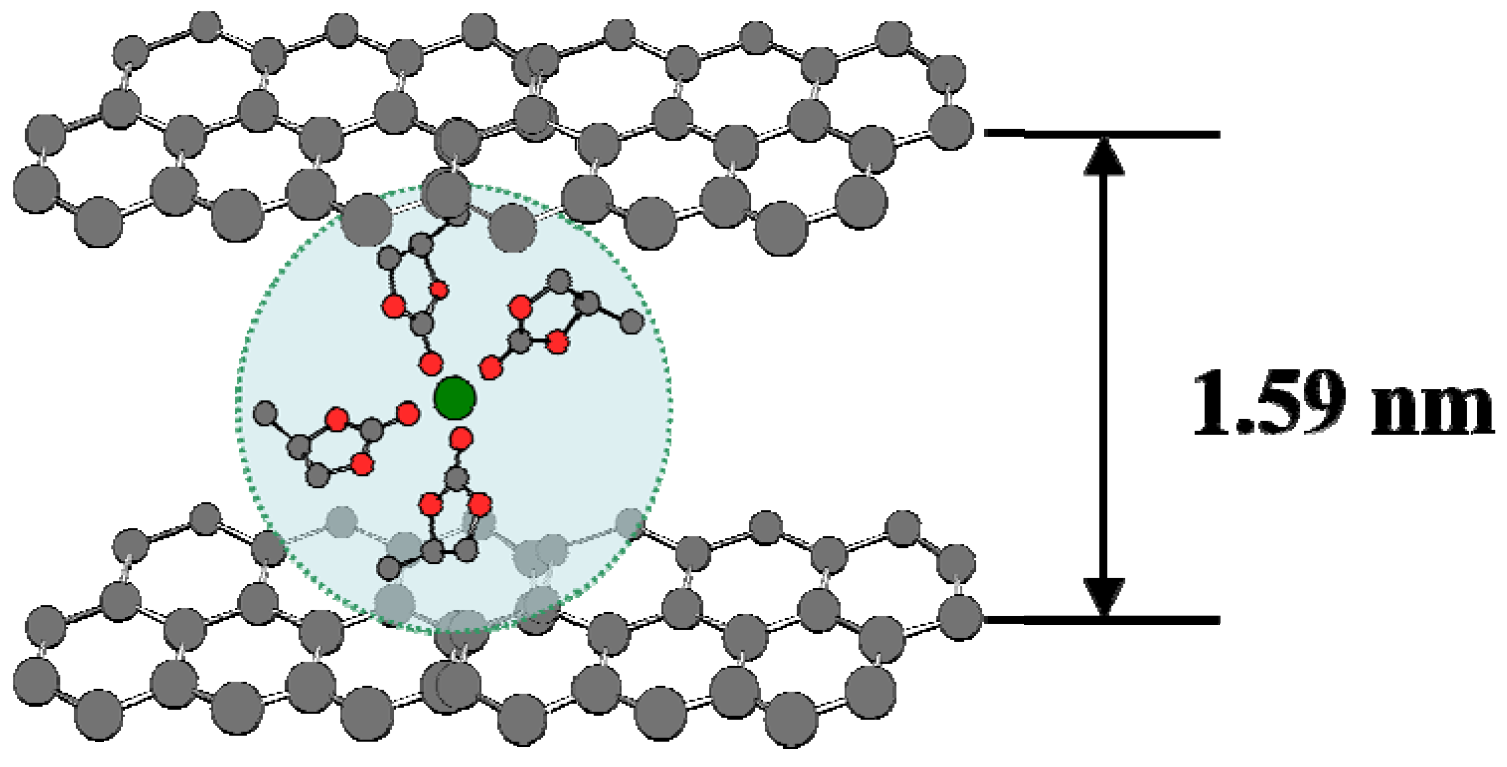
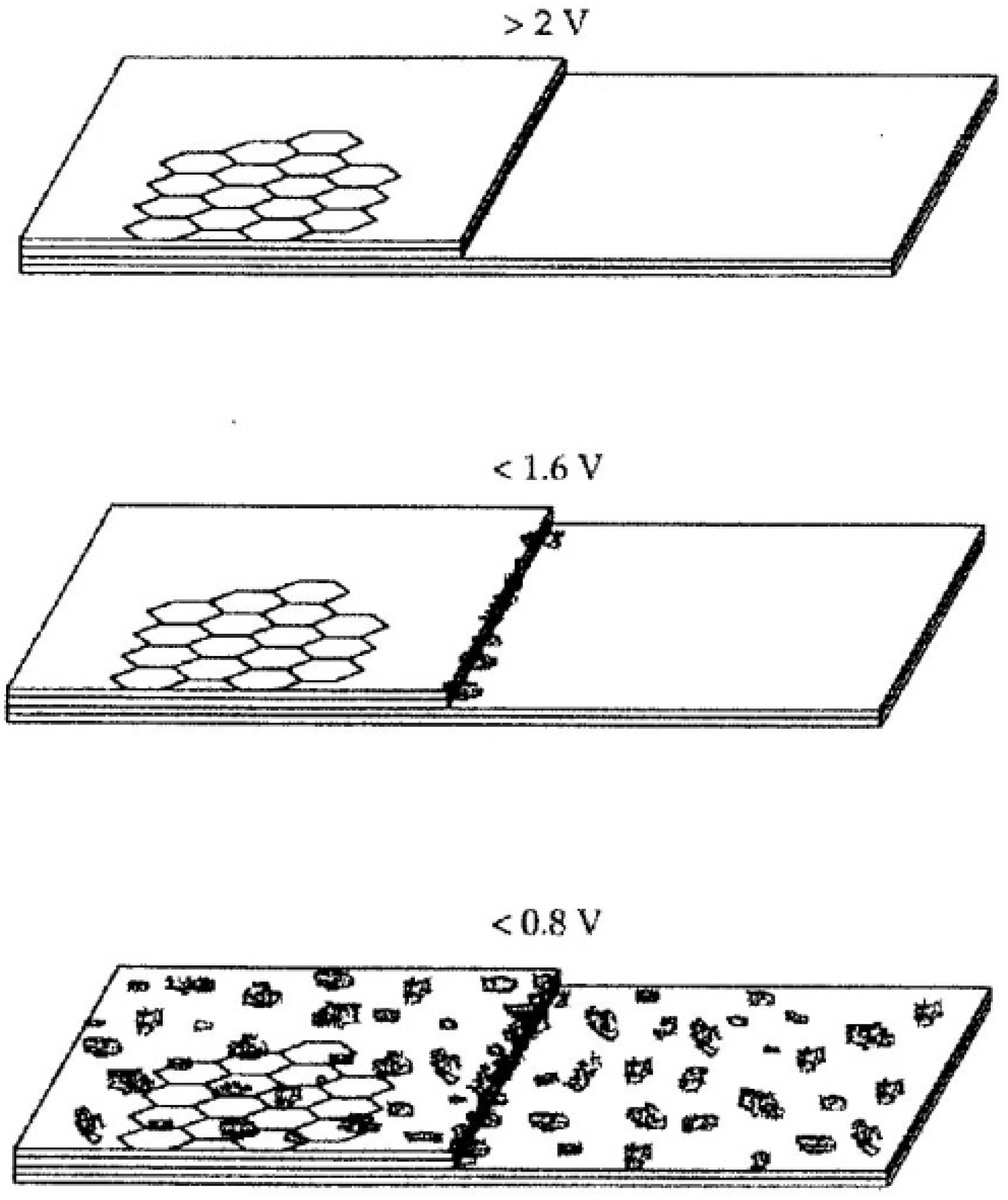
A competing model was inspired by the fact that graphitic anode surface is by no means “flat”, and that electrolyte solvents can actually be intercalated at potentials far higher than the intercalation potential of Li+, forming a so-called “Ternary Graphite Intercalation Compound (GIC)” [15]. Meticulous preparation efforts have eventually succeeded in isolating and characterizing this intercalation compounds of graphite (Figure 6), whose interlayer distance in c-axis is about five times (~1.59 nm) as large as the pristine graphite (0.33 nm) or graphite intercalation compound with naked Li+ (0.35 nm) [21]. In view of this intercalating surface, researchers believed that, before the reductive decomposition of electrolyte solvents, these very solvent molecules would have entered the intercalation sites of the graphite structure, and the subsequent SEI should be a 3D entity that bestrides the boundary between graphite electrode and electrolyte, consisting mainly from the these solvents in the solvation sheath. Evidences from more recent studies seem to support this 3D formation model of SEI, which could be universal for all intercalation-type electrodes that require an SEI.
Figure 6.
Schematic illustration of a ternary graphite intercalation compound involving a solvated Li+ accomodated between graphene layers before solvent decomposition (at ~1.0 V vs. Li+/Li); the interlayer distance was estimated from XRD from ref. [21].
Figure 6.
Schematic illustration of a ternary graphite intercalation compound involving a solvated Li+ accomodated between graphene layers before solvent decomposition (at ~1.0 V vs. Li+/Li); the interlayer distance was estimated from XRD from ref. [21].

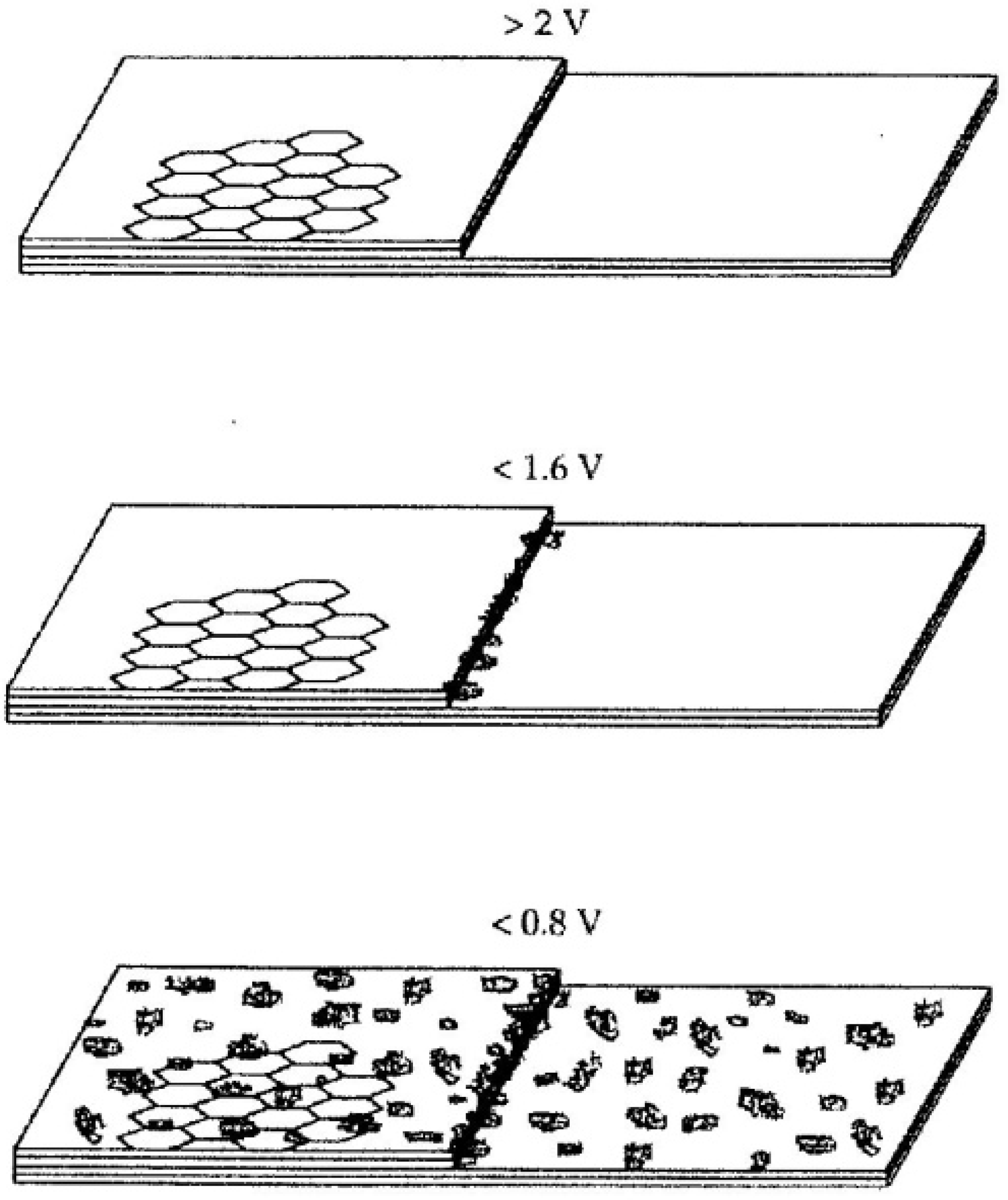
The major difference between metallic lithium (2D) and graphitic anodes (3D) is the manner in which SEI is formed, which might have implication for very different chemical compositions for the interphase. Due to the “intrinsic-potential” of metallic lithium, a SEI would form instantaneously upon its contact with electrolyte, thus there is almost no discrimination with regards to which components are reduced; however, graphite usually lies at ~3.0 V vs. Li+/Li, and the interphase would not form until the initial charging, during which the potential of graphite is brought down gradually hence allowing some electrolyte components to be preferentially reduced before others [22]. This characteristic of graphitic anode has promoted the use of electrolyte additives to tailor SEI chemistry. A further complication of graphitic electrode concerns the particular structure of graphite. Since solvated Li+ could only intercalate at the edge sites of the graphite, it is expected that chemical components of SEI would appear at edge sites instead of basal planes. AFM studies confirmed this staged and regionalized formation of SEI (Figure 7).
After the formation of SEI is completed, the interlayer distance in graphitic anode decreases from 1.59 nm to ca. 0.35 nm, and this graphene structure would be maintained nearly constant during the reversible Li+ intercalation chemistry. The so-called “breathing effect” due to repeated Li+-intercalation/de-intercalation would only cause a volume change of less than 10%. In this sense, although the ternary GIC only exists transiently upon the initial charging of graphitic anode, its significance can never be underestimated, as the graphite anode would permanently bear its chemical signature during the entire life of the device.
Figure 7.
Schematic description of the step-wise formation of SEI on graphite surface based on AFM results. (reproduced from Figure 10 in [22] with the permission from the Electrochemical Society.)
Figure 7.
Schematic description of the step-wise formation of SEI on graphite surface based on AFM results. (reproduced from Figure 10 in [22] with the permission from the Electrochemical Society.)
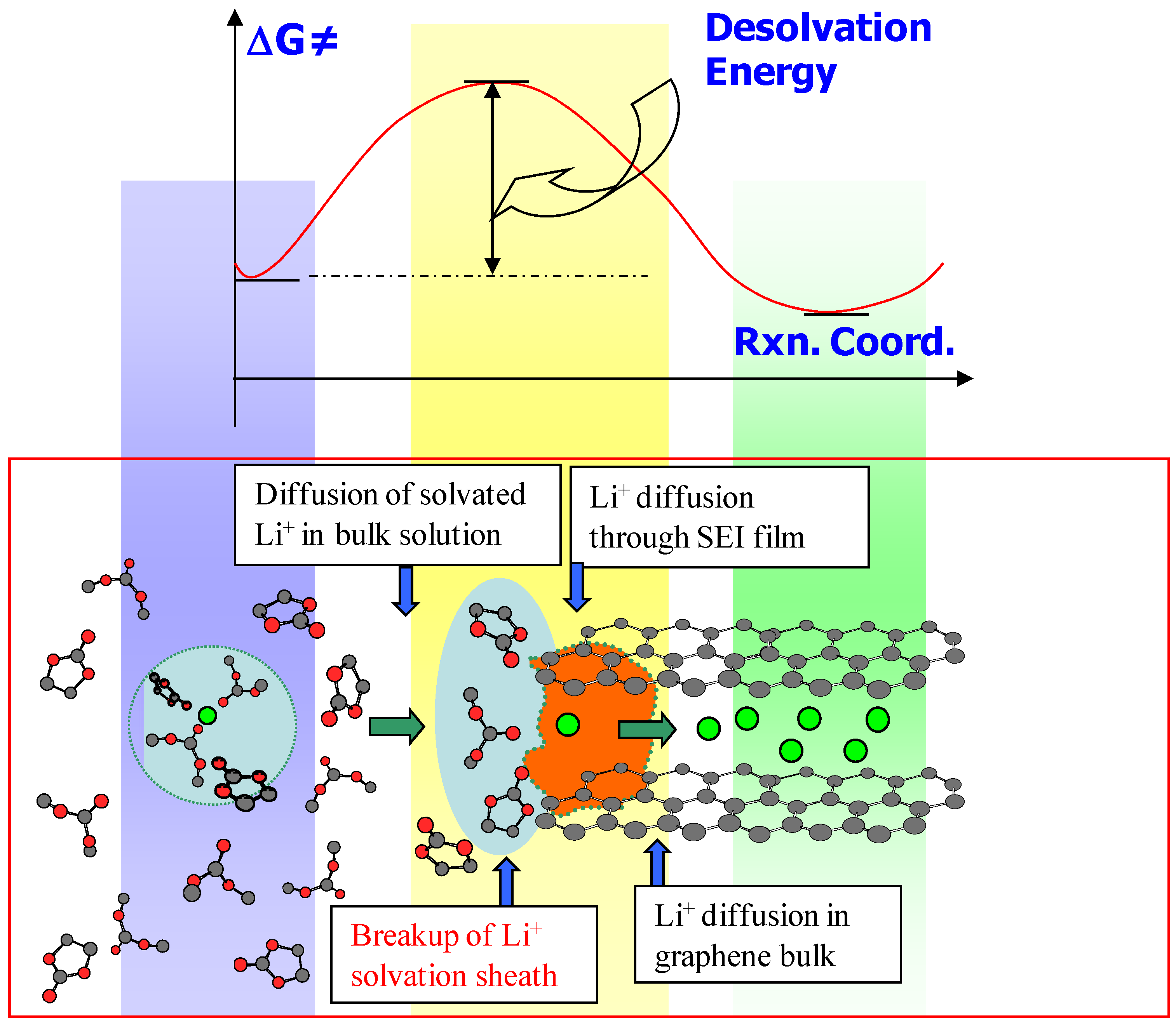
In the post-SEI formation stage, the interphase acts as filter to the migrating solvated Li+, whose solvation sheath must be stripped before a naked Li+ can be intercalated. Due to the small ionic radius of Li+, the solvent molecules in the primary solvation sheath is expected to be tightly bound, hence this desolvation process should occur with difficulty and only when the electrode provide sufficient voltage gradient to compensate the energy to be consumed by the disruption of solvation sheath (Figure 8). More recent studies by Abe et al. identified this desolvation process at electrode/electrolyte interphase as the “rate determining step” in the operation of a Li ion device, which is largely responsible for the cell resistance at low temperature or under high drain rate applications [23,24].
Figure 8.
Schematic description of a solvated Li+ diffusing through the formed SEI at electrolyte/graphite interface (below) and the associated energetic coordinates.
Figure 8.
Schematic description of a solvated Li+ diffusing through the formed SEI at electrolyte/graphite interface (below) and the associated energetic coordinates.

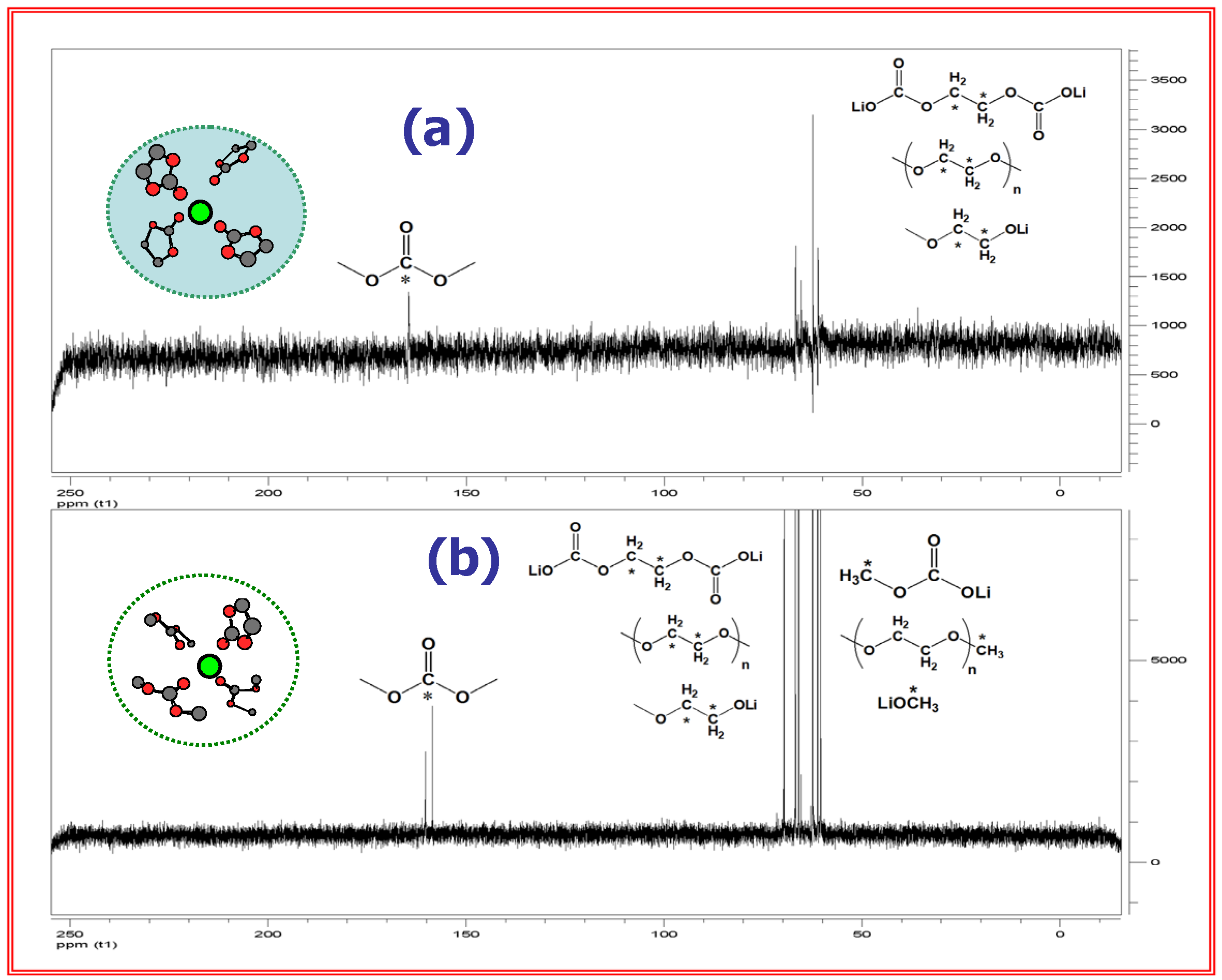
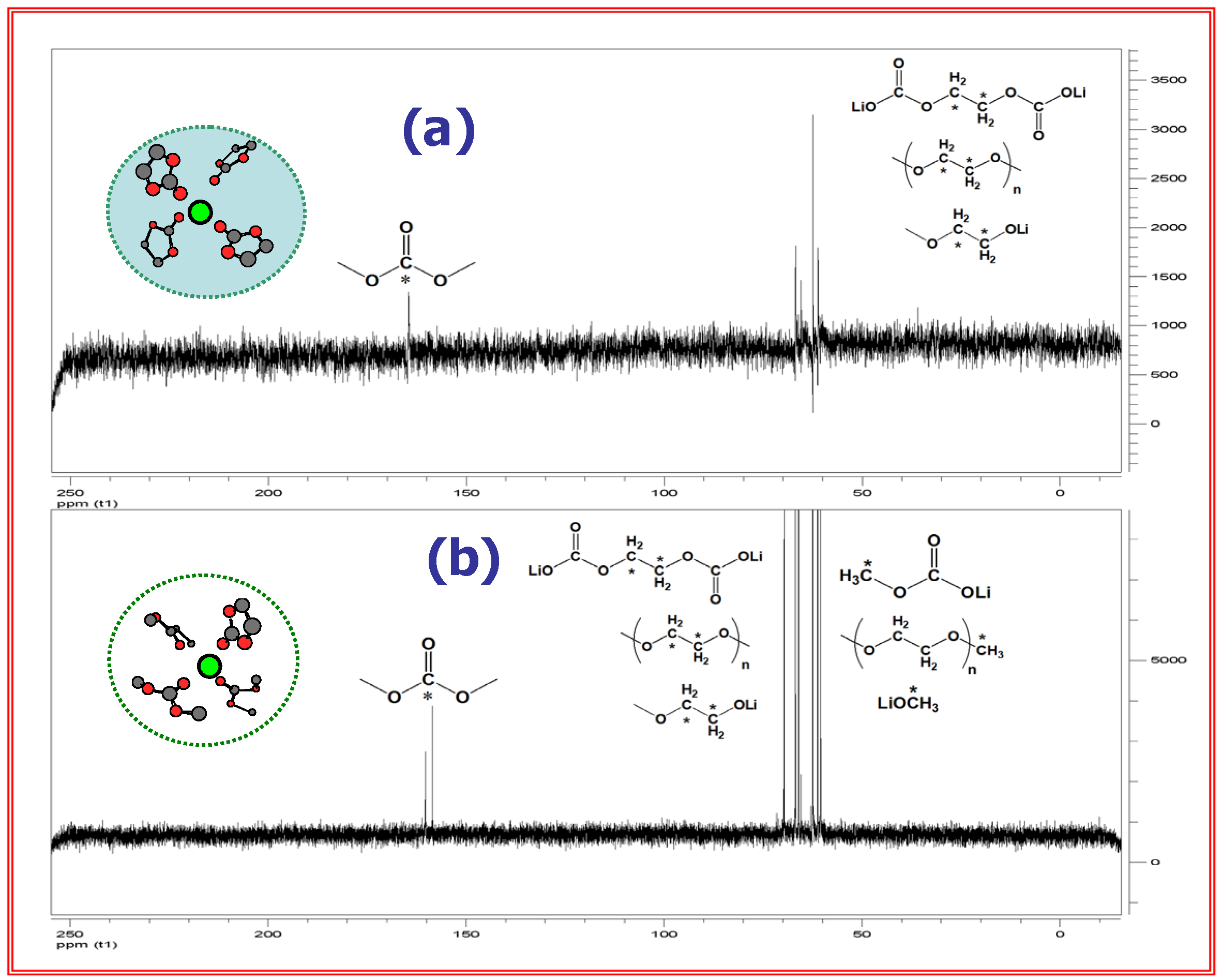
An issue of particular interest concerns how bulk electrolyte composition affects the chemical ingredients of SEI. Xu et al. attempted to establish such a correlation by using the NMR technique to analyze the SEI components collected from the cycled graphitic anodes [18,25]. By comparison with the known standard SEI ingredients that have been synthesized in vitro, it was discovered that the interphasial reduction products do not come proportionally from the bulk electrolyte composition, and that the cyclic component (EC) is the preferential ingredient of SEI as long as EC/Li ratio is more than 4, which happens to be the average solvation number of Li+ (Figure 9).
In other words, the SEI composition is not “collinear” with the bulk electrolyte. This preference of EC-originated ingredients in SEI was believed to be the direct consequence of EC-dominance in Li+-solvation sheath; thus, SEI chemistry inevitable bears the signature of the Li+-solvation sheath composition. To tailor an ideal interphase for faster Li+- intercalation kinetics, one has to consider the effect of Li+-solvation/desolvation processes.
Figure 9.
13C-NMR of graphite surfaces indicates “non-colinearity” between interphasial and bulk compositions of electrolyte. Above: SEI ingredients collected from graphitic anode in EC/DMC 5:5 (a) and 1:9 (b); Below: in EC/EMC 3:7 (a), 2:8 (b) and 1:9 (c). The contributions from acyclic reduction product are negligible until their proportion in bulk electrolyte becomes especially high (70% for DMC and 80~90% for EMC).
Figure 9.
13C-NMR of graphite surfaces indicates “non-colinearity” between interphasial and bulk compositions of electrolyte. Above: SEI ingredients collected from graphitic anode in EC/DMC 5:5 (a) and 1:9 (b); Below: in EC/EMC 3:7 (a), 2:8 (b) and 1:9 (c). The contributions from acyclic reduction product are negligible until their proportion in bulk electrolyte becomes especially high (70% for DMC and 80~90% for EMC).
5. Tailoring an Ideal Interphase
The interphase in a Li ion device is always bifunctional: it protects the electrolyte from sustaining parasitic reactions on electrodes, but also slows down the mass/charge transfer rates across the interface. Therefore the stability or reversibility of this rechargeable battery chemistry comes at the expense of its kinetics. In some application scenarios such as sub-ambient temperature discharge or high rate/pulse drain, this expense becomes too high, and researchers were induced to minimize the above negative aspect of interphase without compromising its protection of the graphene structure or the electrolyte components.
To lessen the interphasial kinetic barrier, investigators have attempted to achieve a desired interphasial chemistry by manipulating electrolyte compositions. This approach was made possible by the stepwise characteristic of interphase formation on graphitic anodes, where certain electrolyte additives, if chosen properly for their reduction potential, can be selectively decomposed on graphite surface during the initial charge cycle of a Li ion cell, before other electrolyte components could, thus leaving unique chemical signatures on the resultant interphasial chemistry (remember that this ordered reduction during the so-called “(SEI) forming cycle” is almost impossible with a metallic lithium electrode). Due to the trace presence (<5%) of those additives in electrolytes, most of them are expected to be consumed at the completion of SEI formation, hardly leaving any impact on the bulk properties of the electrolytes, such as production cost, ion conductivity, viscosity and wetting capabilities toward polyolefin-based separators [26]. It was because of these merits that the additive approach has become a favorite route of the Li ion battery manufacturers, who could adopt almost any newly-found electrolyte additives without drastically reforming their production/processing protocols.
Nowadays most of those interphase-tailoring efforts aim at forming thinnest SEI possible, taking advantage of the reduced resistance; while on a more limited scale emphases were also placed on the special functionalities that could be attained by additives, such as better protection against exfoliation so that PC could be used instead of the high melting EC, or higher stabilization that could delay the threshold trigger of possible thermal runaways, or simply as “accident warning” ingredient that activates the PTC devices built-in a Li ion cell so that cell rupture/explosion could be avoided [26].
Frequently the additives are molecular compounds that can be dissolved in electrolyte solvents, but in a few exceptional cases electrolyte solute (Li salts) were also found to be capable of impacting interphasial chemistries. Lithium bis(oxalato)borate (LiBOB) was such an example [27]. Due to the oxalate-chelate structure, the anion can be reduced at a fairly high potential into species similar to semi-carbonate, resulting in a rather robust SEI that could even protect graphene structure in neat PC electrolyte. This chemical signature of BOB anion on interphasial chemistry can be maintained even the salt was used as additive instead of bulk electrolyte solute. Lithium bis(trifluoromethane sulfonyl)imide also exhibited similar interphasial effect on PC-based electrolytes but only did so when used in high concentrations [28].
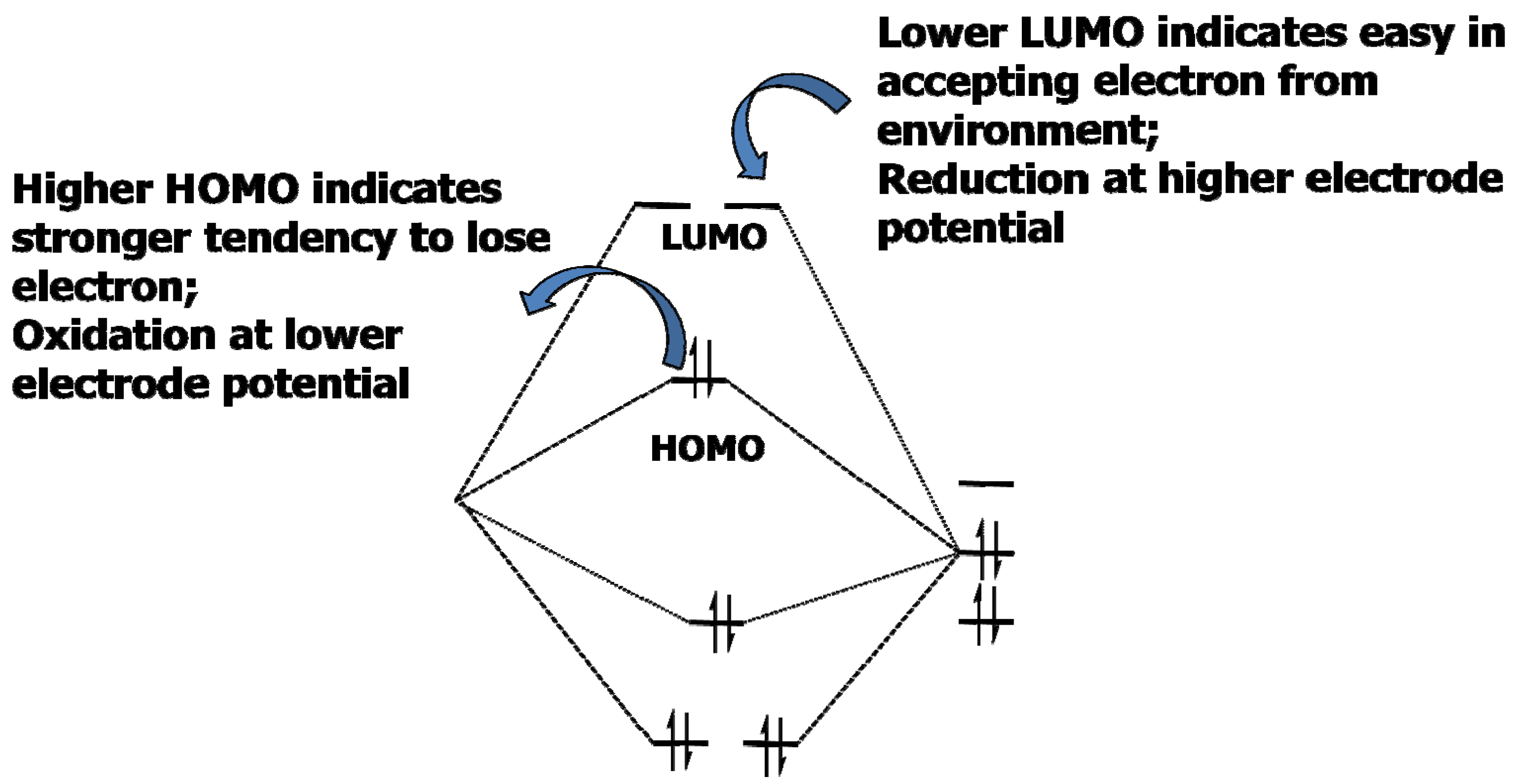
Researchers have developed models and predictive tools to screen compounds as effective additive candidates, the most well-known of which is based on the molecular orbital (MO) computations of the energy levels corresponding to highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO), assuming that a specie would be reduced preferentially with higher HOMO, or oxidized preferentially with lower LUMO (Figure 10) [29].
Figure 10.
Schematic description of the rationale behind the HOMO/LUMO model that is often used to screen SEI additive candidates.
Figure 10.
Schematic description of the rationale behind the HOMO/LUMO model that is often used to screen SEI additive candidates.

However, it must be emphasized that, notwithstanding the theoretical appearance of this model, its overall foundation remains semi-empirical, as the key physicochemical properties of the interphase, such as solubility in electrolyte solvents at elevated temperatures, adhering capability to graphite surface, impedance behavior, etc., cannot be predicted simply on the basis of their reduction or oxidation potentials. A commonality of all those tailored interphases is that the issue of Li+-desolvation was never addressed, even though it has been identified as one of the most important (if not The Most Important) kinetic barriers in the cell chemistry. One would imagine that no matter how thin or even how ionically conductive an SEI could be tailored into, the solvated Li+ must still be stripped off its solvation sheath before it can intercalate into graphene or other host lattice. At the first intuitive thought one would argue that there is little to do because the energy consumption in the desolvation process should be independent of interphasial chemistry. However, a recent reports on Cu-plated graphitic surfaces contradicts this belief and indicates that the charge-transfer process associated with Li+-desolvation exhibited much reduced activation energy [30]. The rationale behind this encouraging report remains little understood, but it is obvious that a tailored interphase could affect the Li+-desolvation. Perhaps the future efforts in electrolyte additive efforts should shift to emphasis on an SEI chemistry containing designed-in catalytic sites that assist in breaking up the solvation sheath of Li+.
6. Perspective: Inspiration from Nature
The desolvation-dictated transport at interphases in Li ion device reveals an intrinsic dilemma that has been troubling the efforts at formulating an ideal electrolyte. On one hand, the electrolyte solvents are required to have effective solvation power towards the ions of lithium salt, especially Li+, so that sufficient ion conduction could be provided; on the other, the solvents should not bind to Li+ excessively, so that Li+ could be freed at the interphase to answer the call of intercalation chemistry. Thus, a fine balance between these two conflicting requirements must be maintained, which in part leads to the current electrolyte solvents used in Li ion industry [8] Naturally, the usage of solvents at two extremes, those with limited solvation power to Li+ such as non-polar olefins, or those solvents with extra affinity to Li+ such as members from the crown-ether family, have to excluded.
On a fundamental level, the above “solvation vs. transport” dilemma was actually rooted in a single association constant that is characteristic with each solvent’s capability to solubilize Li+. An ideal solution to this dilemma would be a solvent molecule with an association parameter that can vary in different local regions of the cell, hence Li+ can be effective solvated in the bulk of electrolyte where the association parameter remains high, and be rapidly released at an interphase where the association parameter of the same solvent molecule becomes low. It can be imagined that the kinetic rate of Li+ across an interphase would be improved by orders of magnitude due to the exponential relations between diffusion rate of Li+ and the activation energy barrier it must overcome. Li ion devices with such an “active” interphase should provide rate capabilities well beyond what is restricted by an passive interphase. But is it possible for a single solvent molecule to possess varying association parameter towards an ion at the same temperature and pressure?
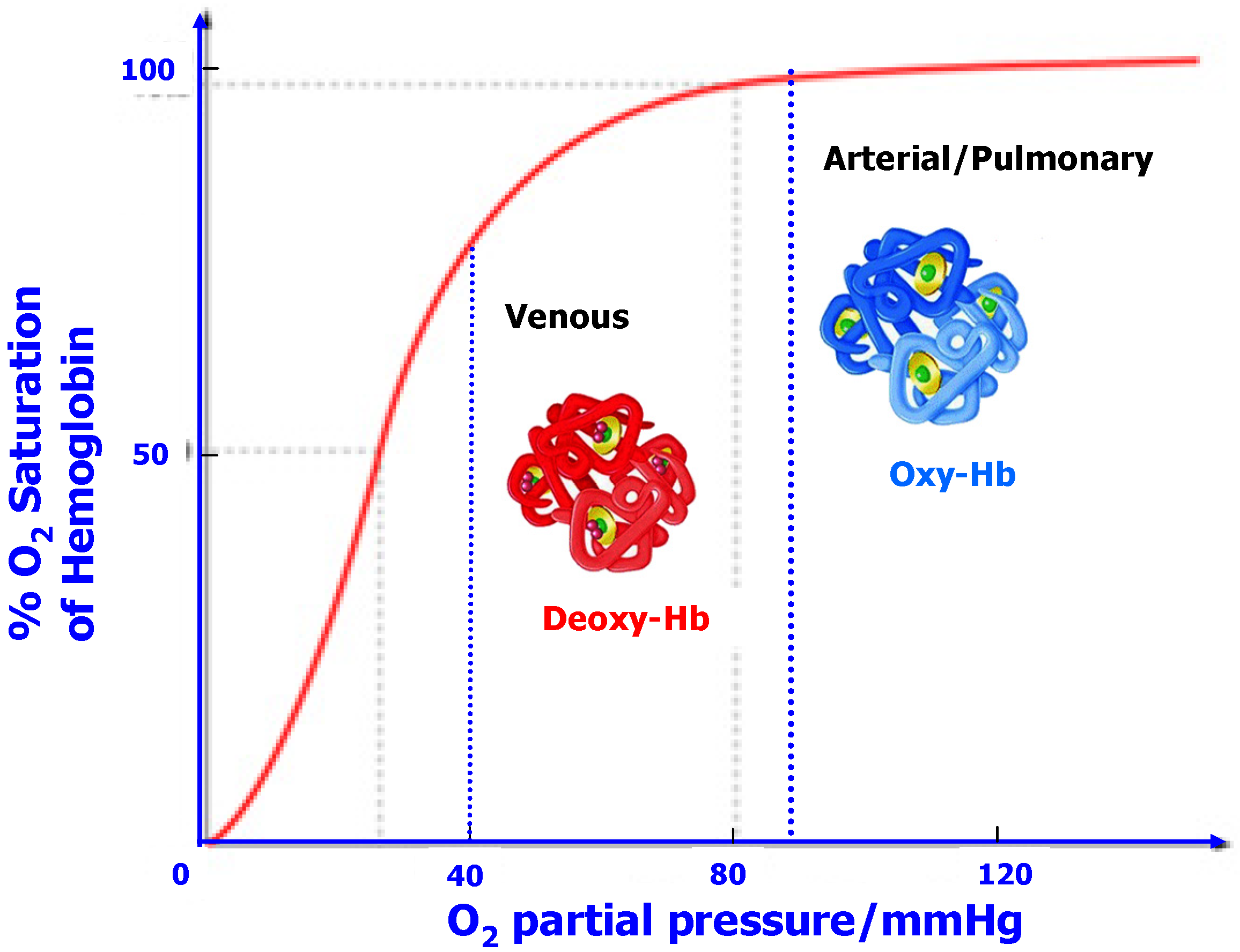
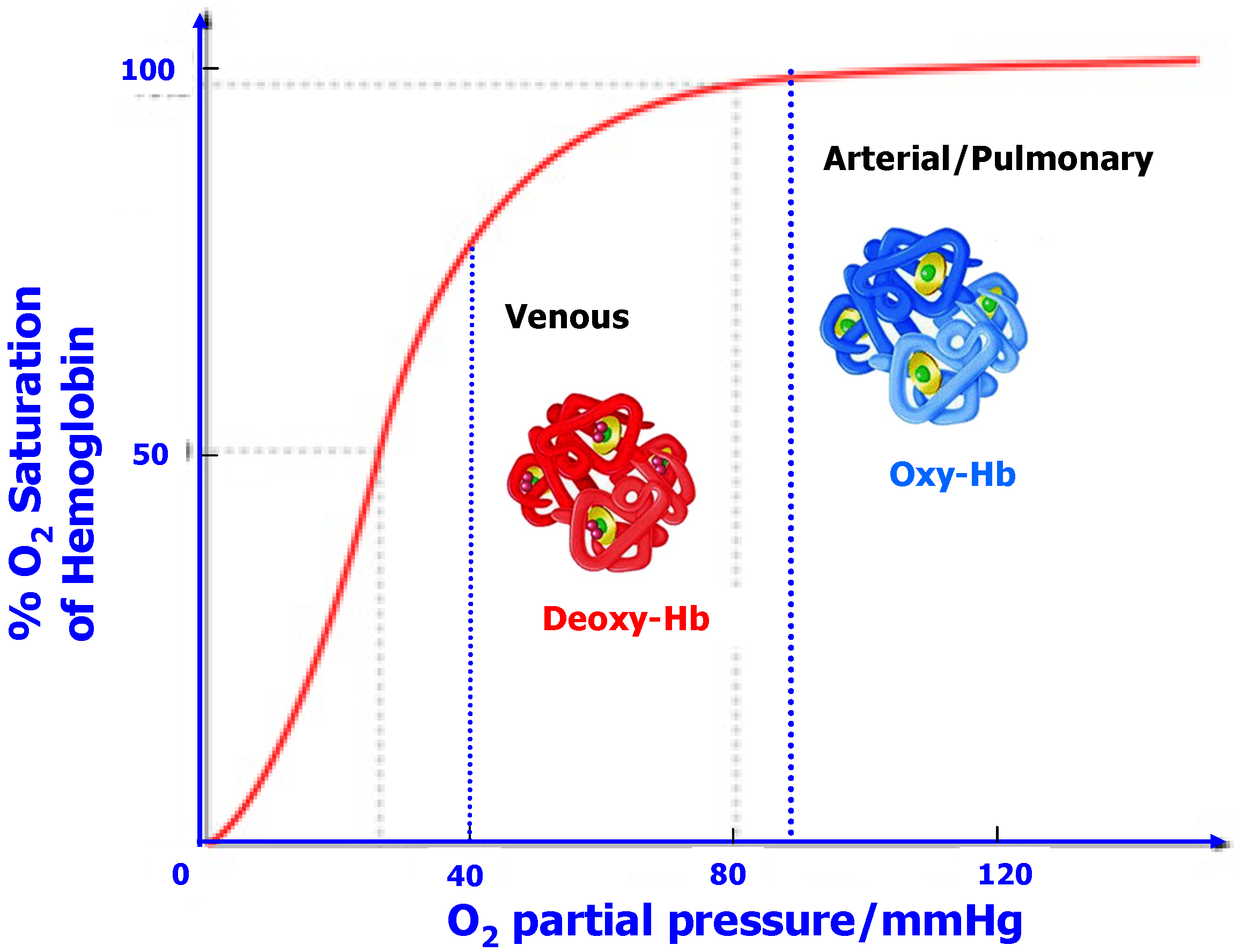
While a varying association “constant” may sound a little “exotic” in the traditional realms of physical chemistry, it is by no means an impossible stretch from reality; in fact it is a quite common phenomenon in the world of life sciences [31]. Assisted by the complicated conformations that are enabled by the large bio-molecules, the association parameter of many enzymes (polymers of α-amino acids, or proteins) would adopt different values at the request of the Nature, which is referred as the “allosteric effect”. A conspicuous example of allostery lies in the transport mechanism of oxygen within our bodies by hemoglobin (Figure 11), whose conformation in the arterial system or lungs (where O2 is in high partial pressure) encodes a rather high association parameter towards O2, thus most hemoglobin molecules are fully loaded with O2; after these O2-carrying hemoglobin molecules travel with the blood stream to venous terminals, another conformation was adopted, which encodes a much lower O2-association parameter and prompts the release of O2. Such a conformational switch is reversible and often triggered by a catalytic species present at the switch sites.
Figure 11.
In a bio-system, the O2-association parameter of hemoglobin (Hb) varies with the external O2-partial pressure by adopting two different conformations, Oxy-Hb with high affinity and deoxy-Hb with low affinity towards O2. Thus, O2 tends to be dissolved in blood where its concentration is high while released where its concentration is low. The overall result is an accelerated transport of O2 within the system, which is impossible with a simple O2-chelating agent that has a constant association parameter, like what Li ion has in conventional electrolyte.
Figure 11.
In a bio-system, the O2-association parameter of hemoglobin (Hb) varies with the external O2-partial pressure by adopting two different conformations, Oxy-Hb with high affinity and deoxy-Hb with low affinity towards O2. Thus, O2 tends to be dissolved in blood where its concentration is high while released where its concentration is low. The overall result is an accelerated transport of O2 within the system, which is impossible with a simple O2-chelating agent that has a constant association parameter, like what Li ion has in conventional electrolyte.
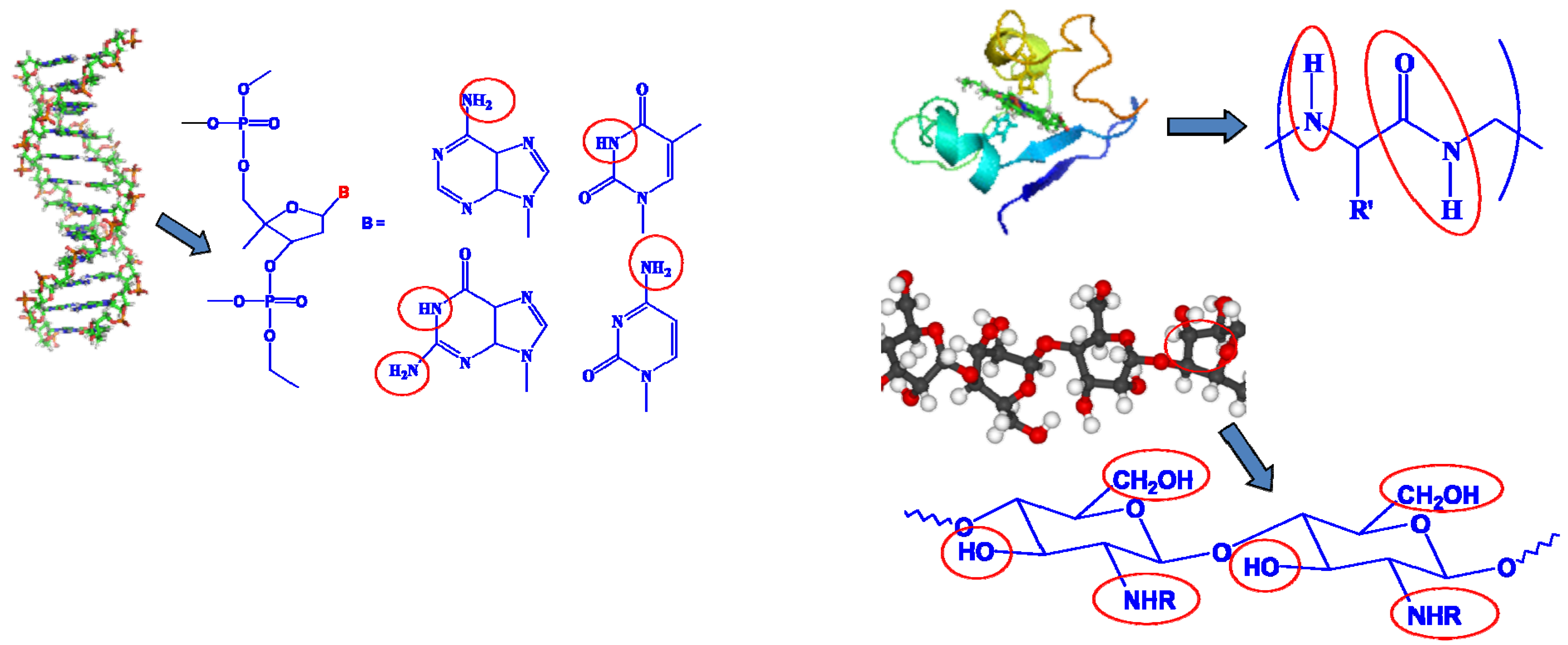
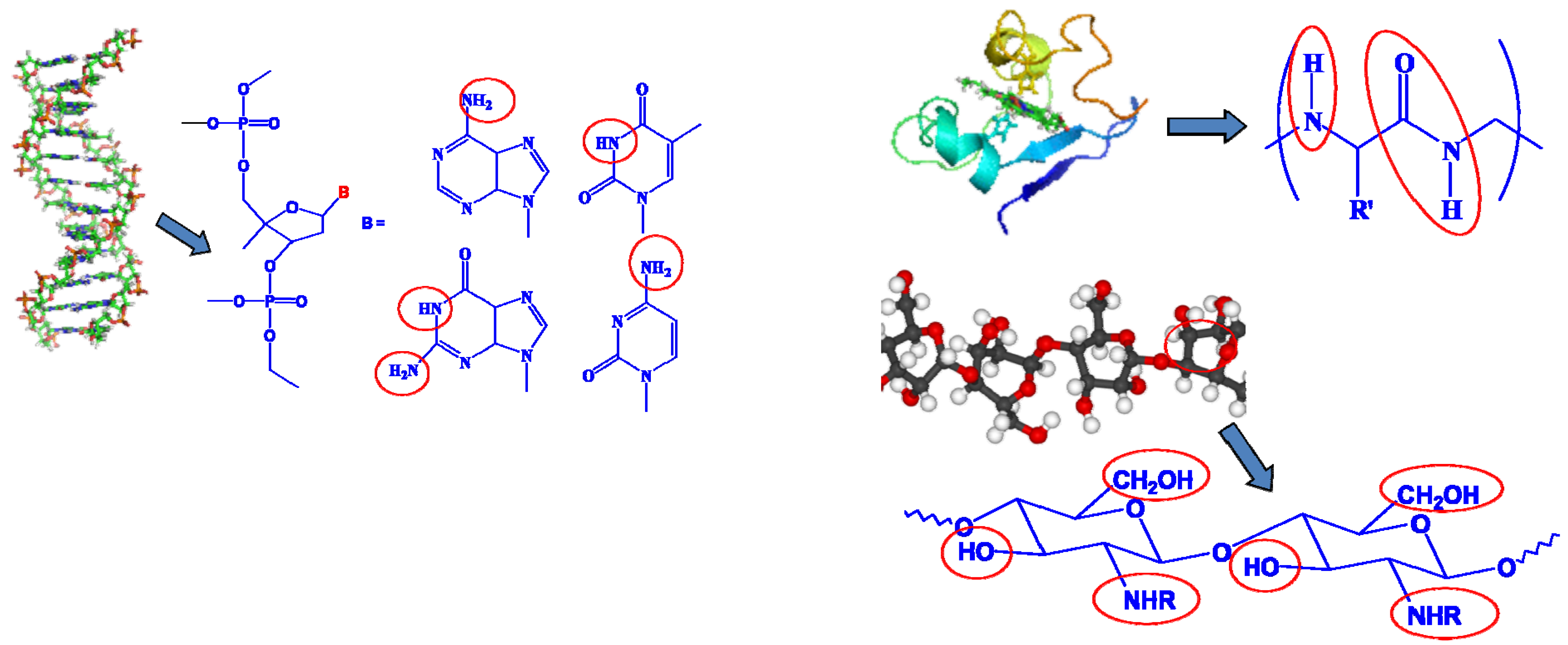
Despite the scientific validity of allostery, is it possible to design such a solvent or additive molecule that would work in similar manner in an electrochemical device? Obviously one cannot simply apply the hemoglobin or other enzymes well known in biochemistry directly to a nonaqueous electrochemical device, because (1) the presence of the active hydrogen groups in those α-amino acids cannot provide an electrochemical stability window suitable for the high potential operations of battery chemistry such as Li ion; and (2) more importantly, the non-aqueous environment would “denature” those enzymes, which can no longer maintain their native conformations to perform the desired functions (Figure 12).
Figure 12.
Unsuitability of biomolecules for nonaqueous electrochemical devices: active hydrogen that results in low oxidation potential is present universally in the building blocks for DNA/RNA on each of their nitrogenous base pairs (left); or in each monomeric unit of protein (right, above) and carbohydrates (right, below).
Figure 12.
Unsuitability of biomolecules for nonaqueous electrochemical devices: active hydrogen that results in low oxidation potential is present universally in the building blocks for DNA/RNA on each of their nitrogenous base pairs (left); or in each monomeric unit of protein (right, above) and carbohydrates (right, below).
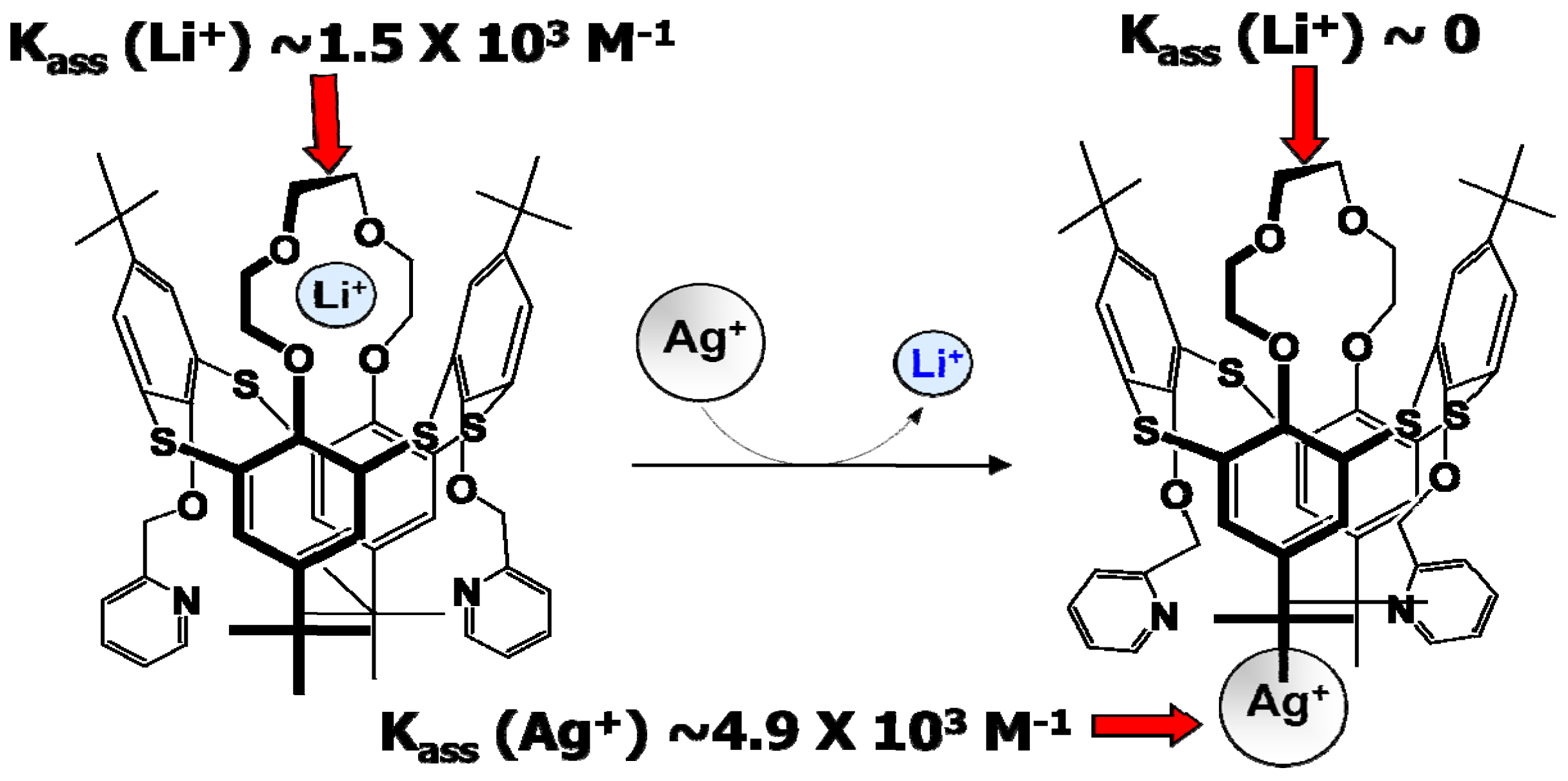
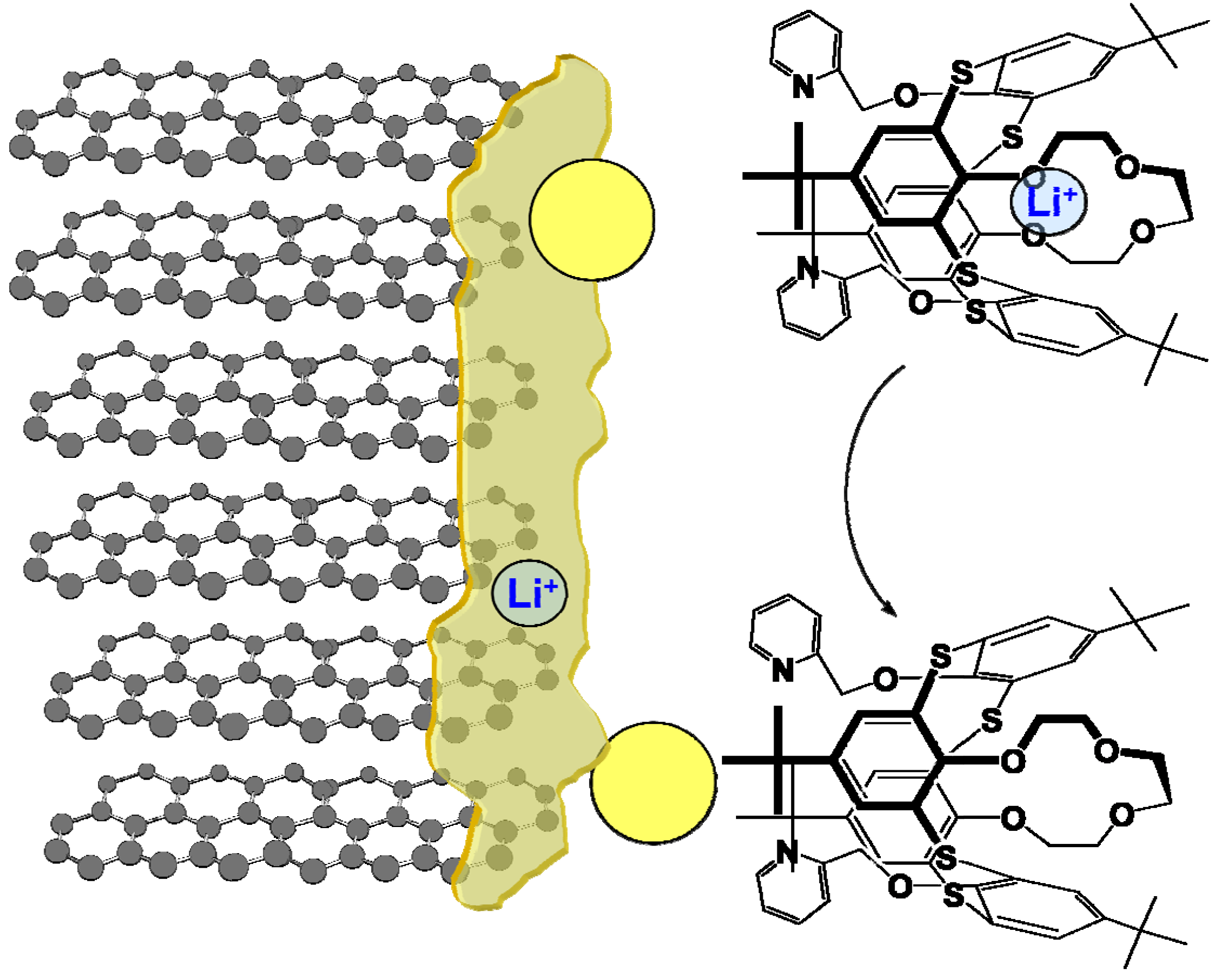
However, there is always a way to design molecules that mimic the allostery structure using building blocks that are insensitive to the non-aqueous environment and electrochemically stable in a wide potential range. Recent work by Perez-Casas et al. proved that such an artificial allosteric effect could be achieved through a thiacalixarene molecule that is insensitive to non-aqueous environments and should be stable in a wide electrochemical window as judged by its chemical composition (Figure 13) [32]. The crown-ether moieties in this supermolecule, which is known to coordinate with Li+ preferentially, acts as the site for Li+-recognition, while the bipyridine moieties in combination with Ag+ serve as switch to turn off the Li+-recognition. This allosteric effect results into a difference by three orders of magnitude in the Li+-association parameter. With proper interphasial chemistry engineering, it is possible to immobilize the trigger species on the site where Li+ is desired to be released and harvest the advantage of this active interphasial transport process (Figure 14).
Figure 13.
Artificial allostery effect forged: switch on-off of Li+-recognition by a regulator (Ag+) in 1,3-alternate-thia[4]calixarene with crown ether and bipyridine moieties results in change in Li+ association constant by orders of magnitude.
Figure 13.
Artificial allostery effect forged: switch on-off of Li+-recognition by a regulator (Ag+) in 1,3-alternate-thia[4]calixarene with crown ether and bipyridine moieties results in change in Li+ association constant by orders of magnitude.
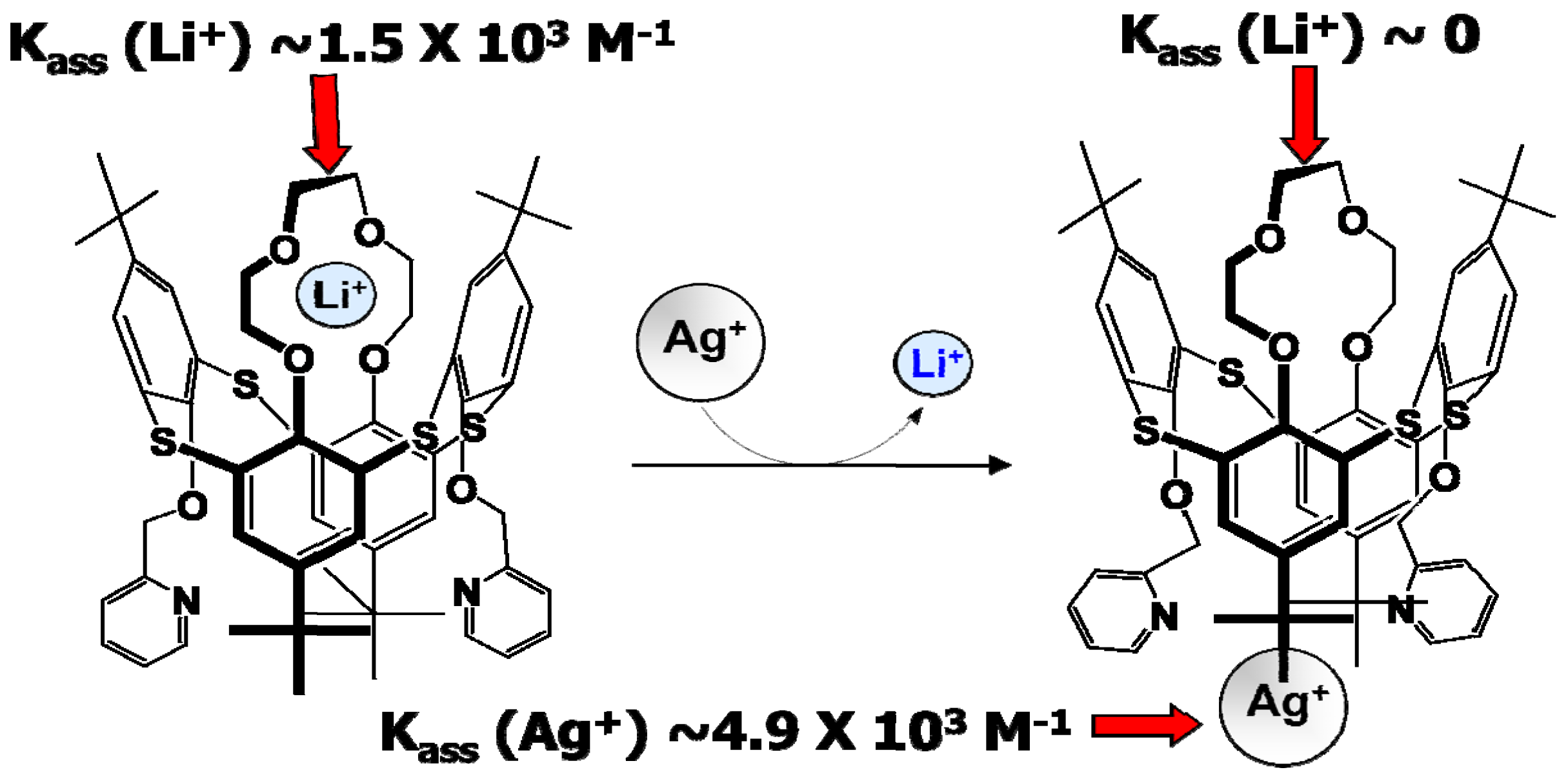
Figure 14.
An imagined active transport of Li+ across an interphase by an allosteric electrolyte additive, which intends to coordinate with Li+ in the electrolyte bulk but releases Li+ at the interphase.
Figure 14.
An imagined active transport of Li+ across an interphase by an allosteric electrolyte additive, which intends to coordinate with Li+ in the electrolyte bulk but releases Li+ at the interphase.
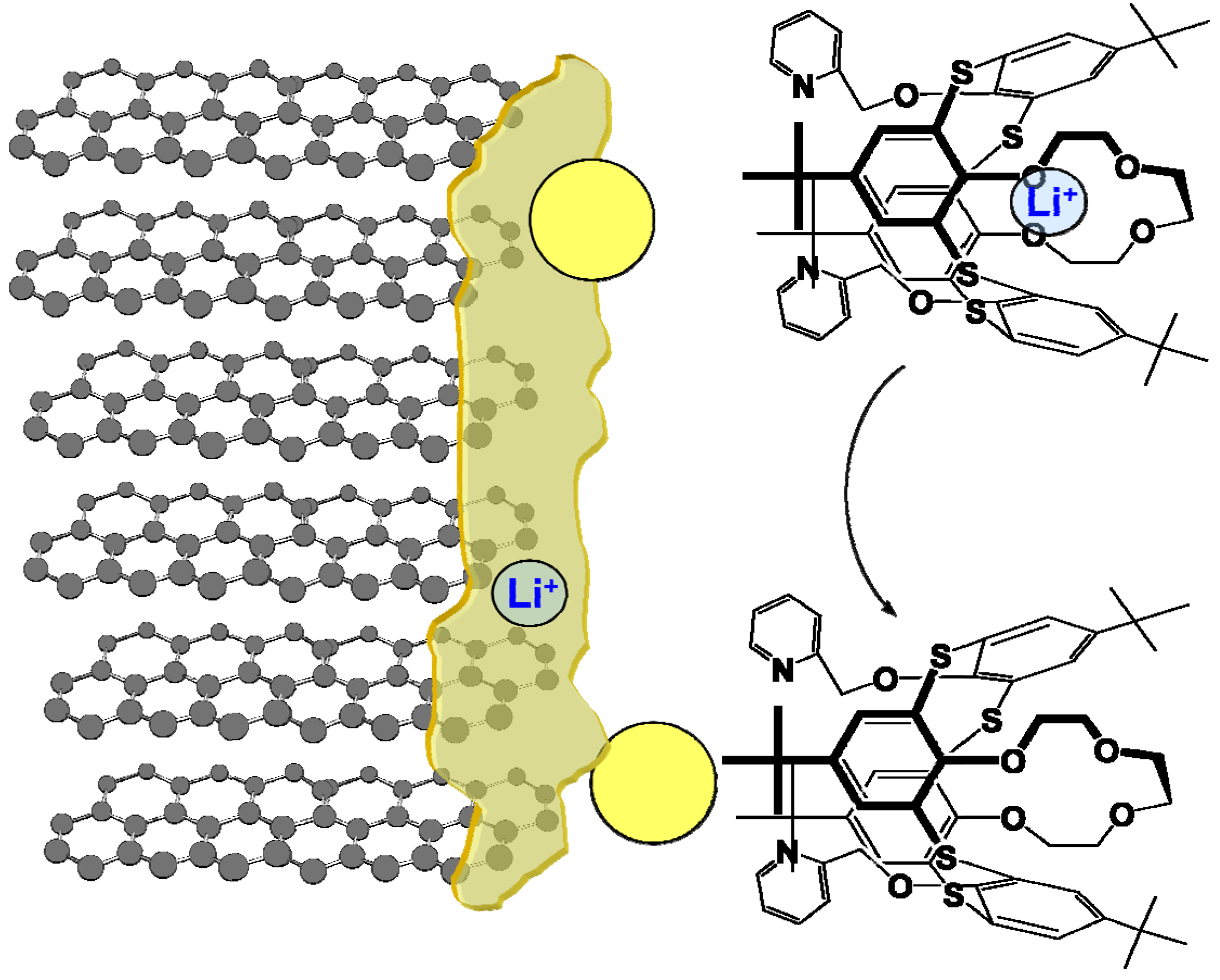
It must be pointed out that at the current stage the above thiacalixarene molecule is only proof-of-concept, and it might yet take years to finally ready it for any practical applications. However, such an active interphase that assists in transporting ion/molecular species is expected to be a major breakthrough for not only Li ion batteries or other electrochemical devices, but also for a wider spectrum of applications that involves surface reactions such as catalysis. Therefore, it is well worthwhile encouraging active efforts in this new direction.
7. Concluding Remarks
Interphase is a key subcomponent in Li ion batteries that dictates the rate capability of Li intercalation chemistry but remains elusive despite extensive research efforts to understand its formation mechanism and chemistry. Recent progress has identified the factors that would determine its chemical nature and manner to tailor its physicochemical properties. Since the Li+ desolvation process remains a major energy consuming step during its migration from electrolyte bulk to interphase, an active interphasial transport mechanism should be sought after if transformational improvement is desired. Inspiration from the allosteric effects that widely exist in bio-molecular realms provide a possible pathway.
References
- Bockris, J.O’M.; Reddy, A.K.N. Modern Electrochemistry, 2nd Ed ed; Plenum Press: New York, NY, USA, 2000; Volume 2. [Google Scholar]
- Peled, E. The Electrochemical Behavior of Alkali and Alkaline Earth Metals in Nonaqueous Battery Systems—The Solid Electrolyte Interphase Model. J. Electrochem. Soc. 1979, 126, 2047–2051. [Google Scholar] [CrossRef]
- Xu, K. Electrolytes: Overview. In Encyclopedia of Electrochemical Power Sources; Garche, J., Dyer, C., Moseley, P., Ogumi, Z., Rand, D., Scrosati, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; Volume 5, pp. 51–70. [Google Scholar]
- Fong, R.; Sacken, U.; Dahn, J.R. Studies of Lithium Intercalation into Carbons Using Nonaqueous Electrochemical Cells. J. Electrochem. Soc. 1990, 137, 2009–2013. [Google Scholar] [CrossRef]
- Lazzari, M.; Scrosati, B. A Cyclable Lithium Organic Electrolyte Cell Based on Two Intercalation Electrodes. J. Electrochem. Soc. 1980, 127, 773–774. [Google Scholar] [CrossRef]
- Whittingham, M.S. Lithium Batteries and Cathode Materials. Chem. Rev. 2004, 104, 4271–4302. [Google Scholar]
- Chung, S.Y.; Bloking, J.T.; Chiang, Y.M. Electronically Conductive Phospho-Olivines as Lithium Storage Electrodes. Nature Mater. 2002, 1, 123. [Google Scholar]
- Xu, K. Nonaqueous Liquid Electrolytes for Lithium-Based Rechargeable Batteries. Chem. Rev. 2004, 104, 4303–4418. [Google Scholar] [CrossRef] [PubMed]
- Aurbach, D.; Gofer, Y. The Behavior of Lithium Electrodes in Mixtures of Alkyl Carbonates and Ethers. J. Electrochem. Soc. 1991, 138, 3529–3536. [Google Scholar] [CrossRef]
- Xu, K. Whether EC and PC Differ in Interphasial Chemistry on Graphitic Anode and How. J. Electrochem. Soc. 2009, 156, A751–A755. [Google Scholar] [CrossRef]
- Hérold, A. Recherches sur les Composés d’Insertion du Graphite. Bull. Soc. Chim. Fr. 1955, 187, 999–1012. [Google Scholar]
- Dey, A.N; Sullivan, B.P. The Electrochemical Decomposition of Propylene Carbonate on Graphite. J. Electrochem. Soc. 1970, 117, 222–224. [Google Scholar] [CrossRef]
- Pistoia, G.; de Rossi, M.; Scrosati, B. Study of the Behavior of Ethylene Carbonate as A Nonaqueous Battery Solvent. J. Electrochem. Soc. 1970, 117, 500–502. [Google Scholar] [CrossRef]
- Aurbach, D.; Daroux, M.L.; Faguy, P.W.; Yeager, E. Identification of Surface Films Formed on Lithium in Propylene Carbonate Solutions. J. Electrochem. Soc. 1987, 134, 1611–1620. [Google Scholar] [CrossRef]
- Besenhard, J.O.; Winter, M.; Yang, J.; Biberacher, W. Filming Mechanism of Lithium-Carbon Anodes in Organic and Inorganic Electrolytes. J. Power Sources 1993, 54, 228–231. [Google Scholar] [CrossRef]
- Zhuang, G.V.; Ross, P.N. Analysis of the Chemical Composition of the Passive Film on Li-Ion Battery Anodes Using Attentuated Total Reflection Infrared Spectroscopy. Electrochem. Solid State Lett. 2003, 6, A136. [Google Scholar] [CrossRef]
- Onuki, M.; Kinoshita, S.; Sakata, Y.; Yanagidate, M.; Otake, Y.; Ue, M. Identification of the Source of Evolved Gas in Li-Ion Batteries Using 13C-labeled Solvents. J. Electrochem. Soc. 2008, 155, A794–A797. [Google Scholar] [CrossRef]
- Xu, K.; Zhuang, G.V.; Allen, J.L.; Lee, U.; Zhang, S.; Ross, P.N.; Jow, T.R. Syntheses and Characterization of Lithium Alkyl Mono- and Dicarbonates as Components of Surface Films in Li-Ion Batteries. J. Phys. Chem.: B 2006, 110, 7708–7719. [Google Scholar] [CrossRef]
- Wang, Y.; Nakamura, S.; Ue, M.; Balbuena, P.B. Theoretical Studies to Understand Surface Chemistry on Carbon Anodes for Lithium-Ion Batteries: Reduction Mechanisms of Ethylene Carbonate. J. Am. Chem. Soc. 2001, 123, 11708–11718. [Google Scholar] [CrossRef] [PubMed]
- Peled, E.; Golodnitsky, D.; Ardel, G. Advanced Model for Solid Electrolyte Interphase Electrodes in Liquid and Polymer Electrolytes. J. Electrochem. Soc. 1997, 144, L208–L210. [Google Scholar] [CrossRef]
- Wagner, M.R.; Albering, J.H.; Moeller, K.C.; Besenhard, J.O.; Winter, M. XRD Evidence for the Electrochemical Formation of Li+(PC)yCn− in PC-based Electrolytes. Electrochem. Commun. 2005, 7, 947–952. [Google Scholar] [CrossRef]
- Chu, A.C.; Josefowicz, J.Y.; Farrington, G.C. Electrochemistry of Highly Ordered Pyrolytic Graphite Surface Film Formation Observed by Atomic Force Microscopy. J. Electrochem. Soc. 1997, 144, 4161–4169. [Google Scholar] [CrossRef]
- Abe, T.; Fukuda, H.; Iriyama, Y.; Ogumi, Z. Solvated Li-Ion Transfer at Interface Between Graphite and Electrolyte. J. Electrochem. Soc. 2004, 151, A1120–1123. [Google Scholar] [CrossRef]
- Yamada, Y.; Iriyama, Y.; Abe, T.; Ogumi, Z. Kinetics of Lithium Ion Transfer at the Interface between Graphite and Liquid Electrolytes: Effects of Solvent and Surface Film. Langmuir 2009, 25, 12766–12770. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Lam, Y.; Zhang, S.S.; Jow, T.R.; Curtis, T.B. Solvation Sheath of Li+ in Nonaqueous Electrolytes and Its Implication of Graphite/Electrolyte Interface Chemistry. J. Phys. Chem. C 2007, 111, 7411–7421. [Google Scholar] [CrossRef]
- Zhang, S. A review on electrolyte additives for lithium-ion batteries. J. Power Sources 2006, 162, 1379–1394. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, S.S.; Jow, T.R.; Xu, W.; Angell, C.A. LiBOB as Salt for Lithium-Ion Batteries: A Possible Solution for High Temperature Operation. Electrochem. Solid-State Lett. 2002, 5, A26–A29. [Google Scholar] [CrossRef]
- Jeong, S.; Inaba, M.; Iriyama, Y.; Abe, T.; Ogumi, Z. Electrochemical Intercalation of Lithium Ion within Graphite from Propylene Carbonate Solutions. Electrochem. Solid-State Lett. 2003, 6, A13–A15. [Google Scholar] [CrossRef]
- Morita, M.; Ishikawa, M.; Matsuda, Y. Organic Electrolytes for Rechargeable Lithium Ion Batteries. In Lithium Ion Batteries: Fundamentals and Performance; Wakihara, M., Yamamoto, O., Eds.; Wiley-VCH: New York, NY, USA, 1999; p. 156. [Google Scholar]
- Nobili, F.; Dsoke, S.; Mancini, M.; Tossici, R.; Marassi, R. Electrochemical Investigation of Polarization Phenomena and Intercalation Kinetics of Oxidized Graphite Electrodes Coated with Evaporated Metal Layers. J. Power Sources 2008, 180, 845–851. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J.G. Biochemistry, 3rd Ed. ed; John Wiley & Sons: New York, NY, USA, 2004; Chapter 10; pp. 154–196. [Google Scholar]
- Perez-Casas, C.; Rahman, S.; Begum, N.; Xi, Z.; Yamato, T. Allosteric Bindings of Thiacalix[4]arene-Based Receptors with 1,3- Alternate Conformation Having Two Different Side Arms. J. Incl. Phenom. Macrocycl. Chem. 2008, 60, 173–185. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Xu, K. Electrolytes and Interphasial Chemistry in Li Ion Devices. Energies 2010, 3, 135-154. https://doi.org/10.3390/en3010135
AMA Style
Xu K. Electrolytes and Interphasial Chemistry in Li Ion Devices. Energies. 2010; 3(1):135-154. https://doi.org/10.3390/en3010135
Chicago/Turabian StyleXu, Kang. 2010. "Electrolytes and Interphasial Chemistry in Li Ion Devices" Energies 3, no. 1: 135-154. https://doi.org/10.3390/en3010135