New Approaches for the Production of Hydrocarbons from Hydrate Bearing Sediments

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
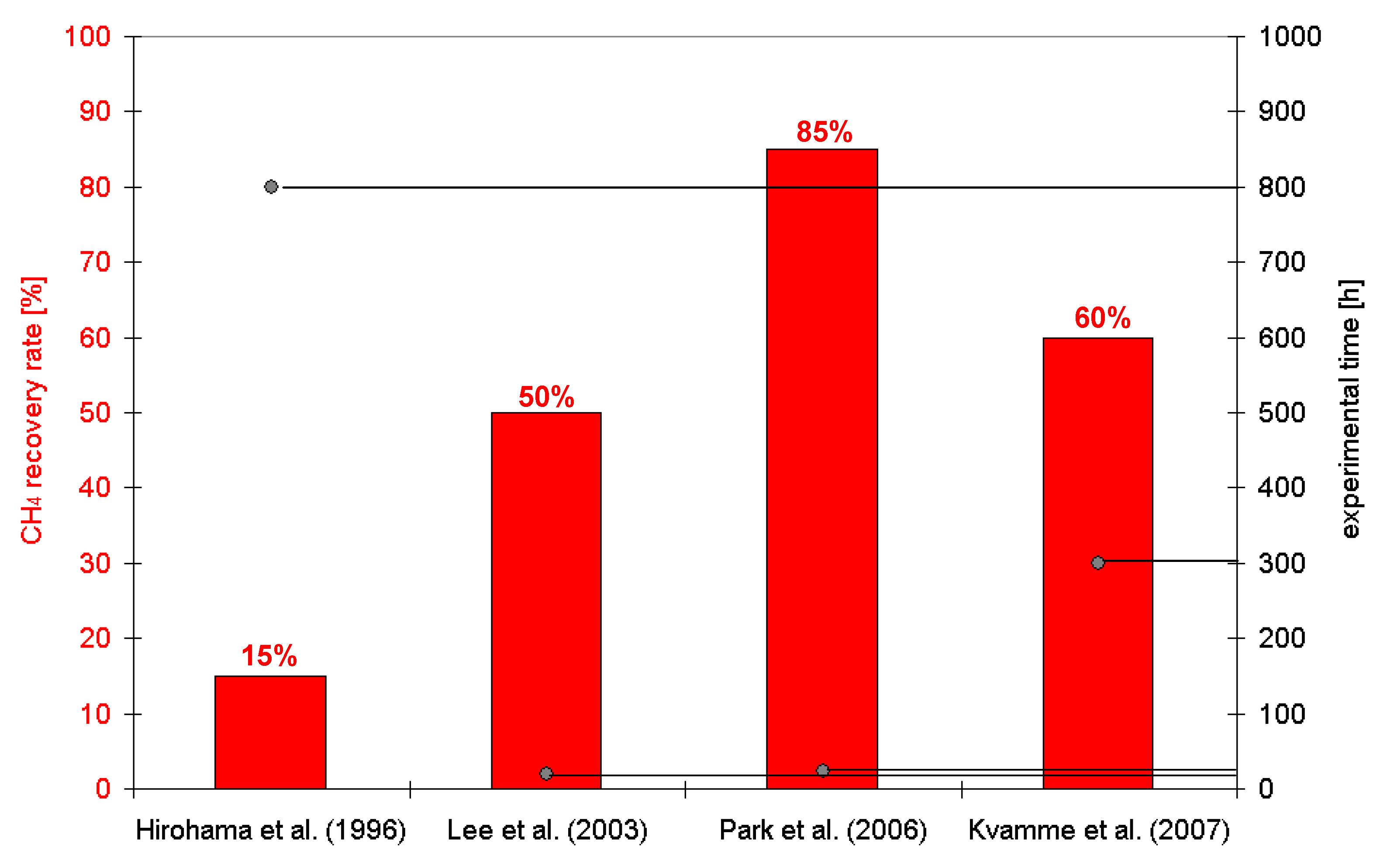
:1. Introduction

2. Results and Discussion
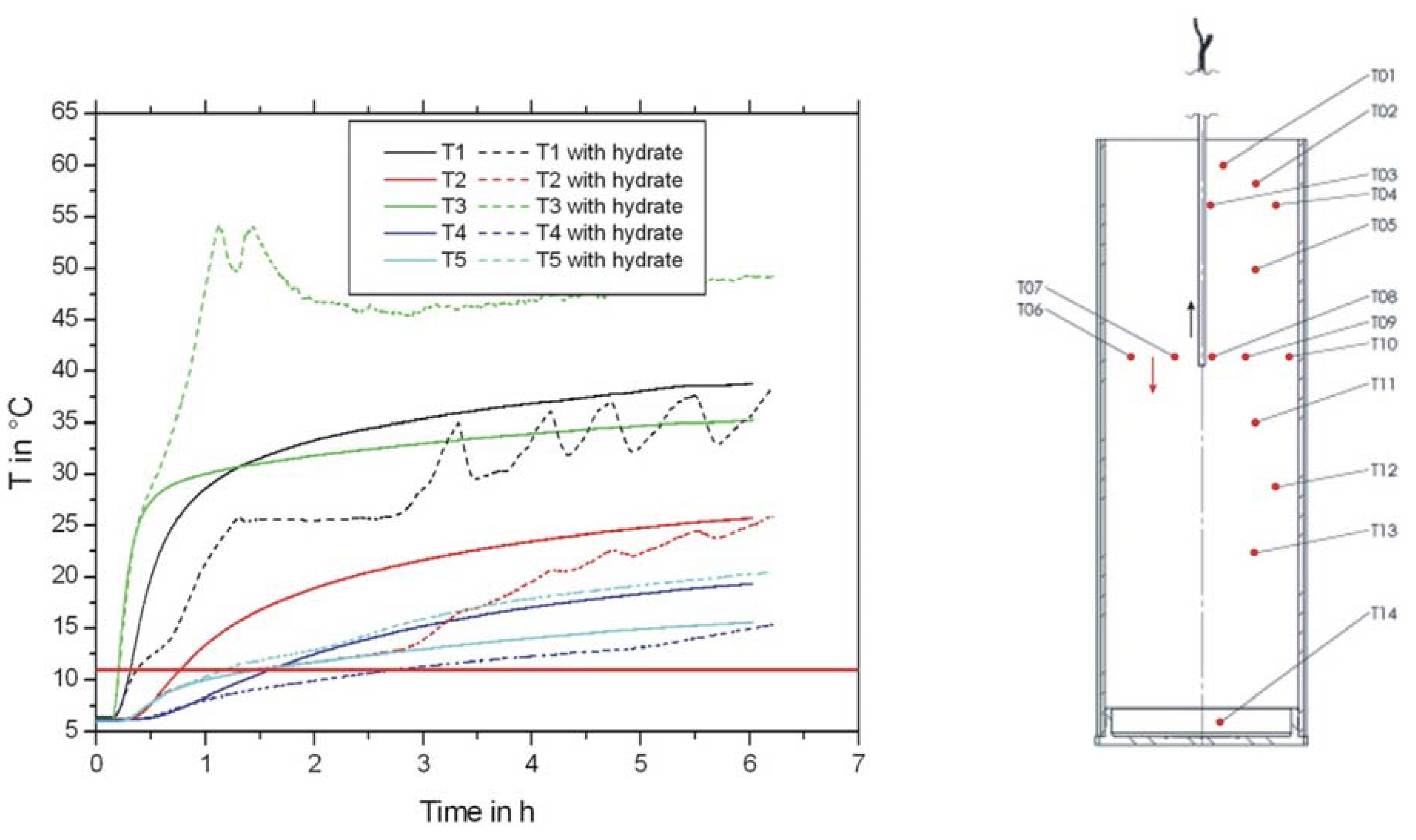
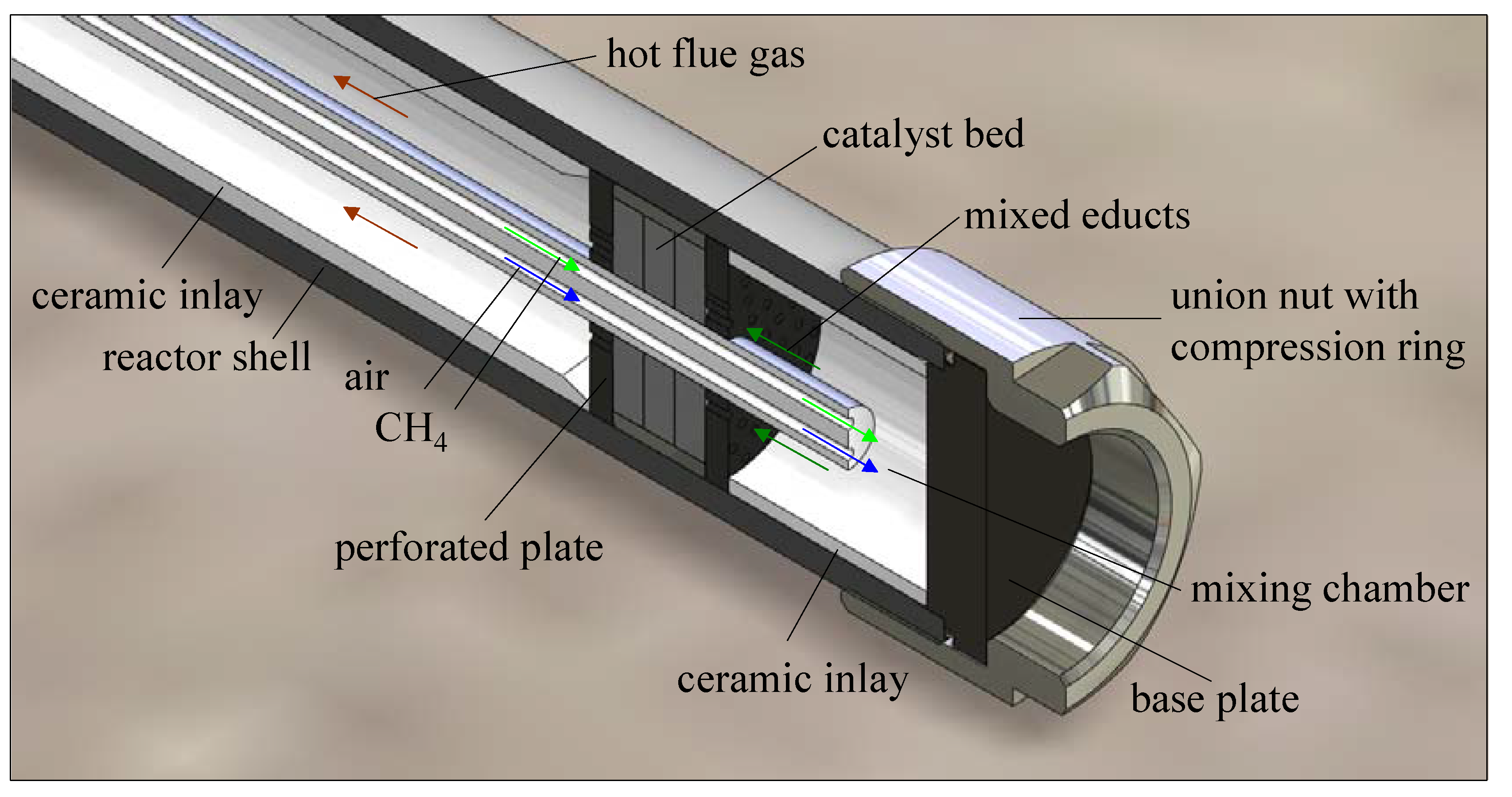
2.1. Development and Test of a Heater for the Thermal Stimulation of Hydrate Bearing Sediments
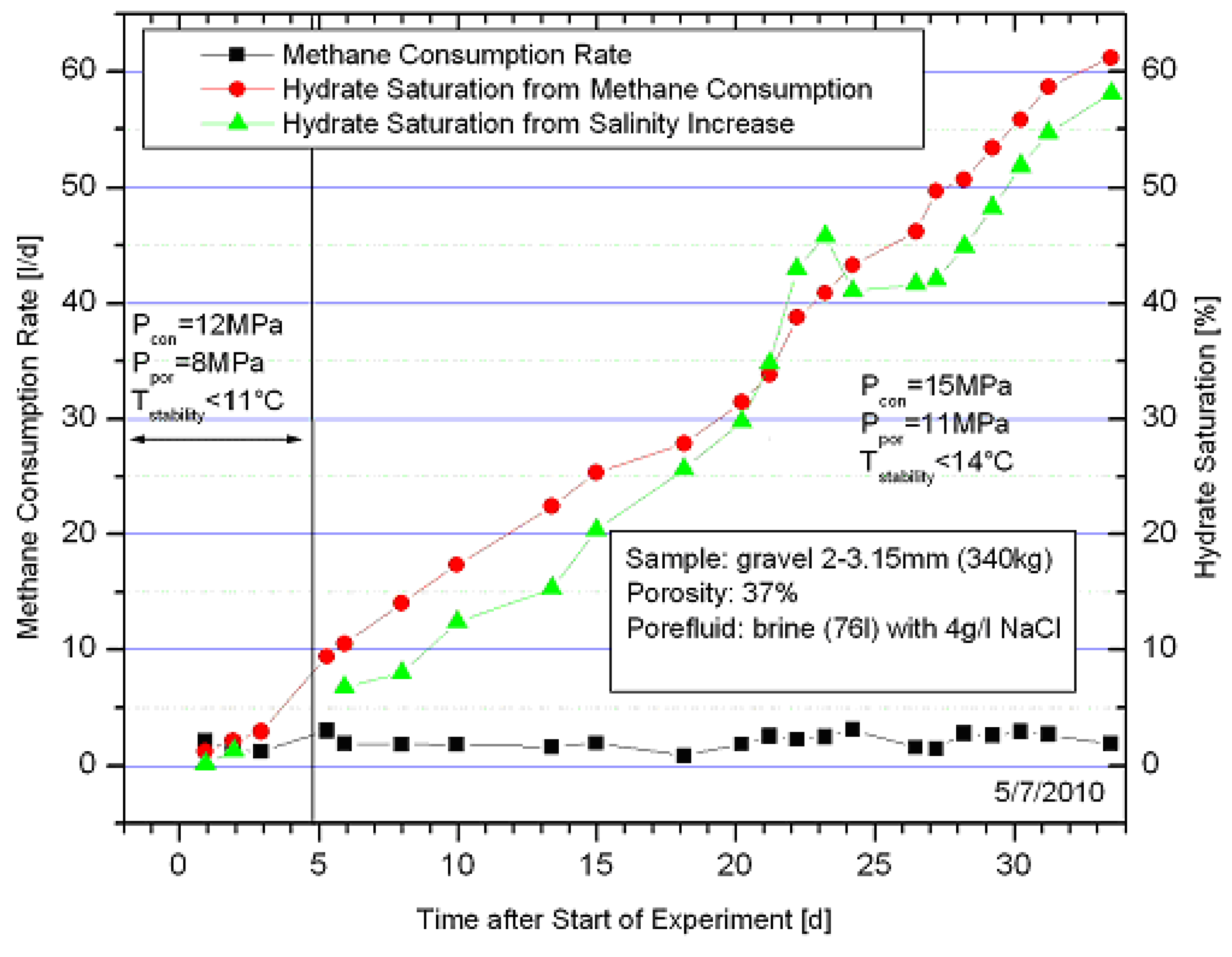
2.2. Thermal Stimulation of a Hydrate Bearing Sediment Using An Electrical Heater

- (1)
- The hydrate formation from CH4 dissolved in water to a sufficient hydrate saturation in a limited time frame was successful, and
- (2)
- Thermal stimulation and CH4 hydrate decomposition were successfully induced applying a heater.

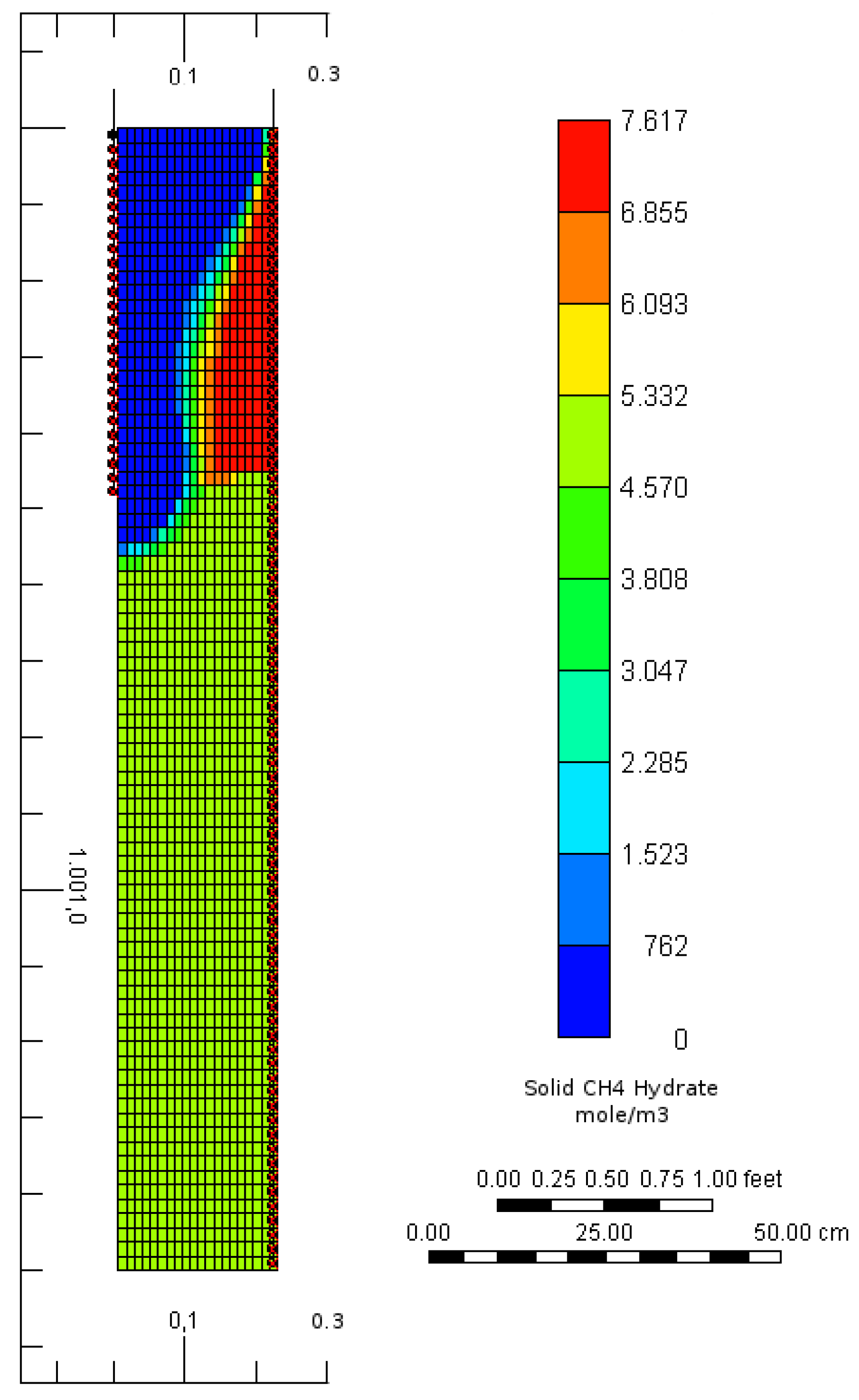
2.3. Numerical Simulation

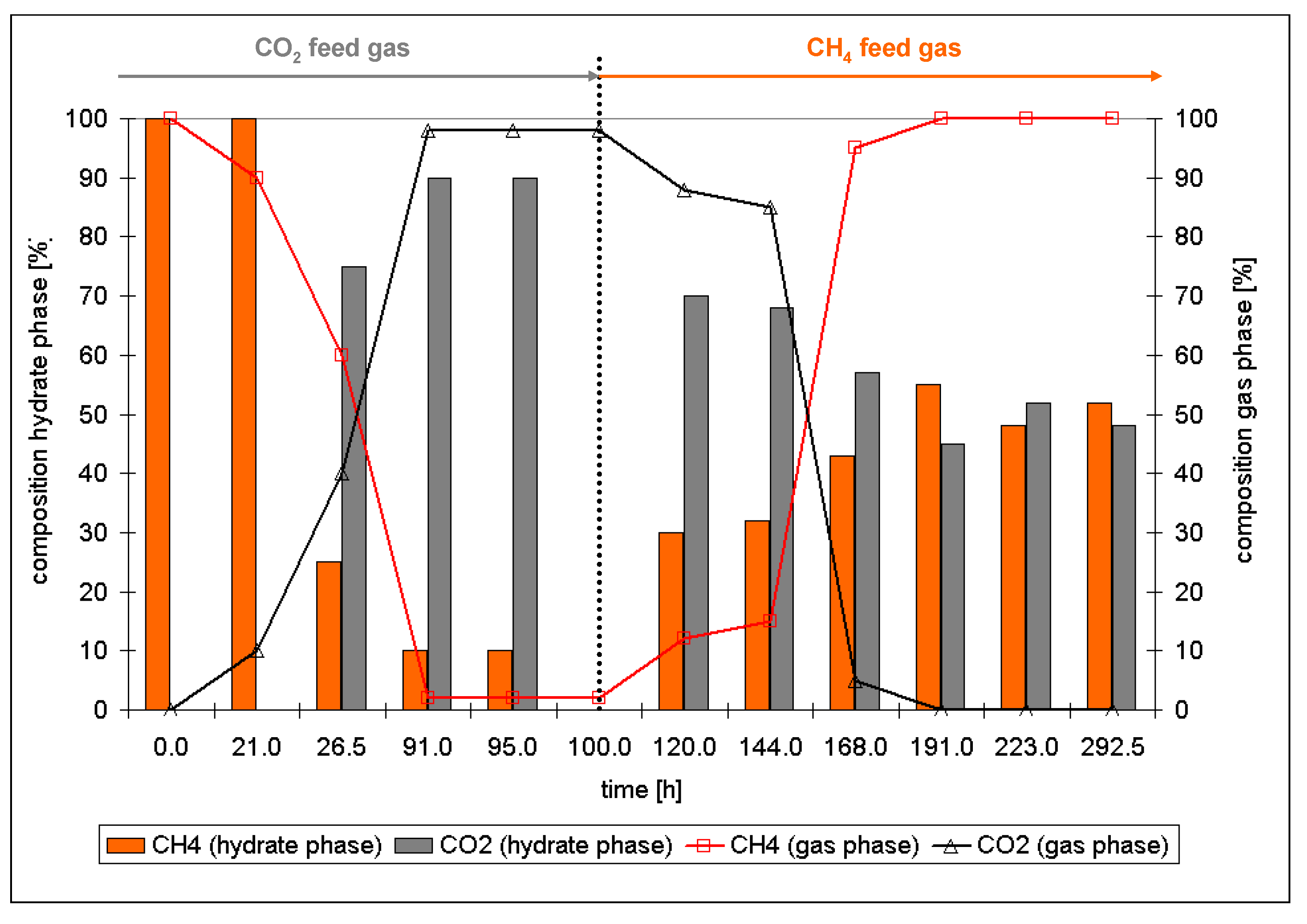
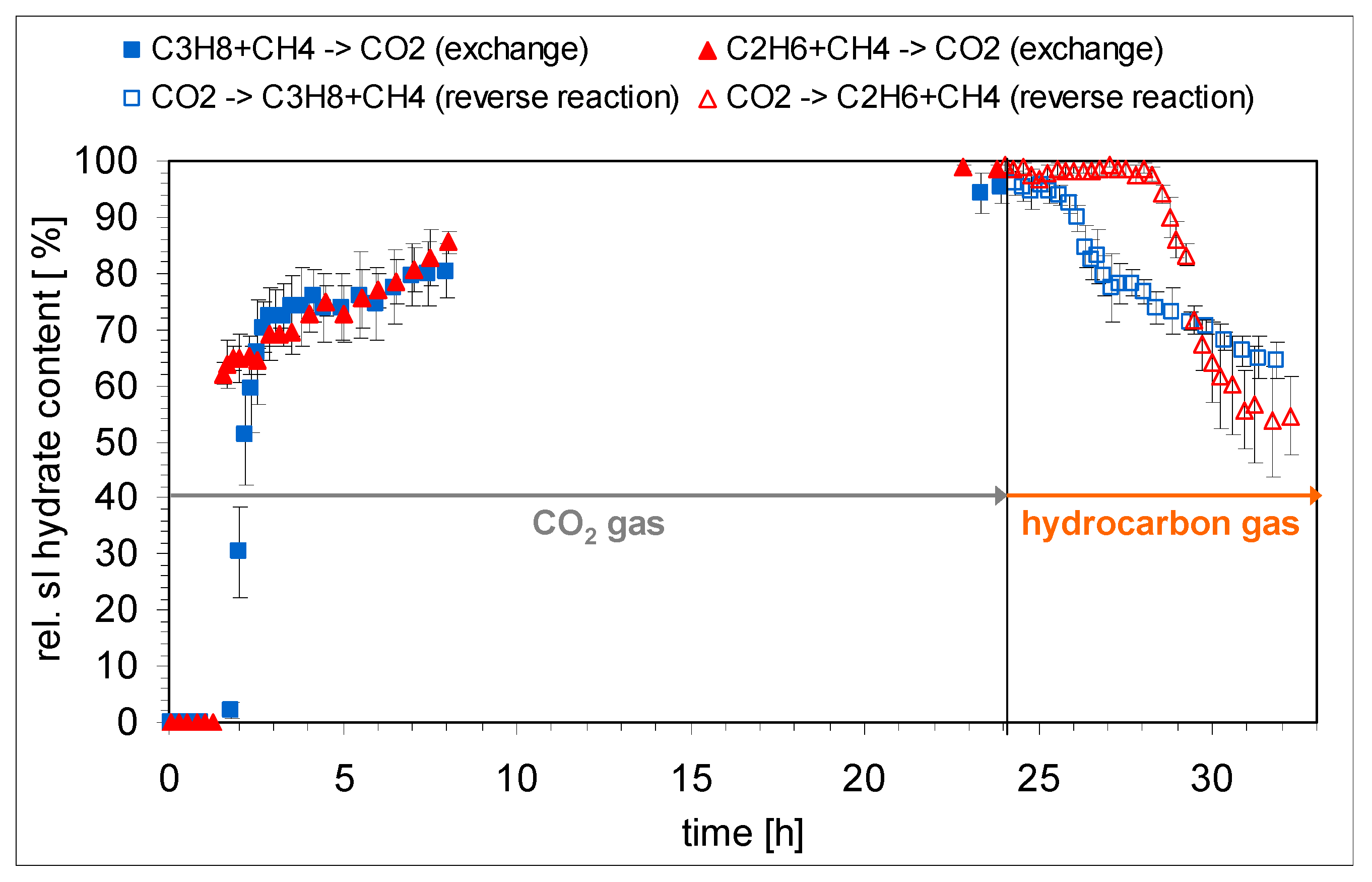
2.4. Conversion of CH4/Hydrocarbon Hydrates to CO2 Hydrates
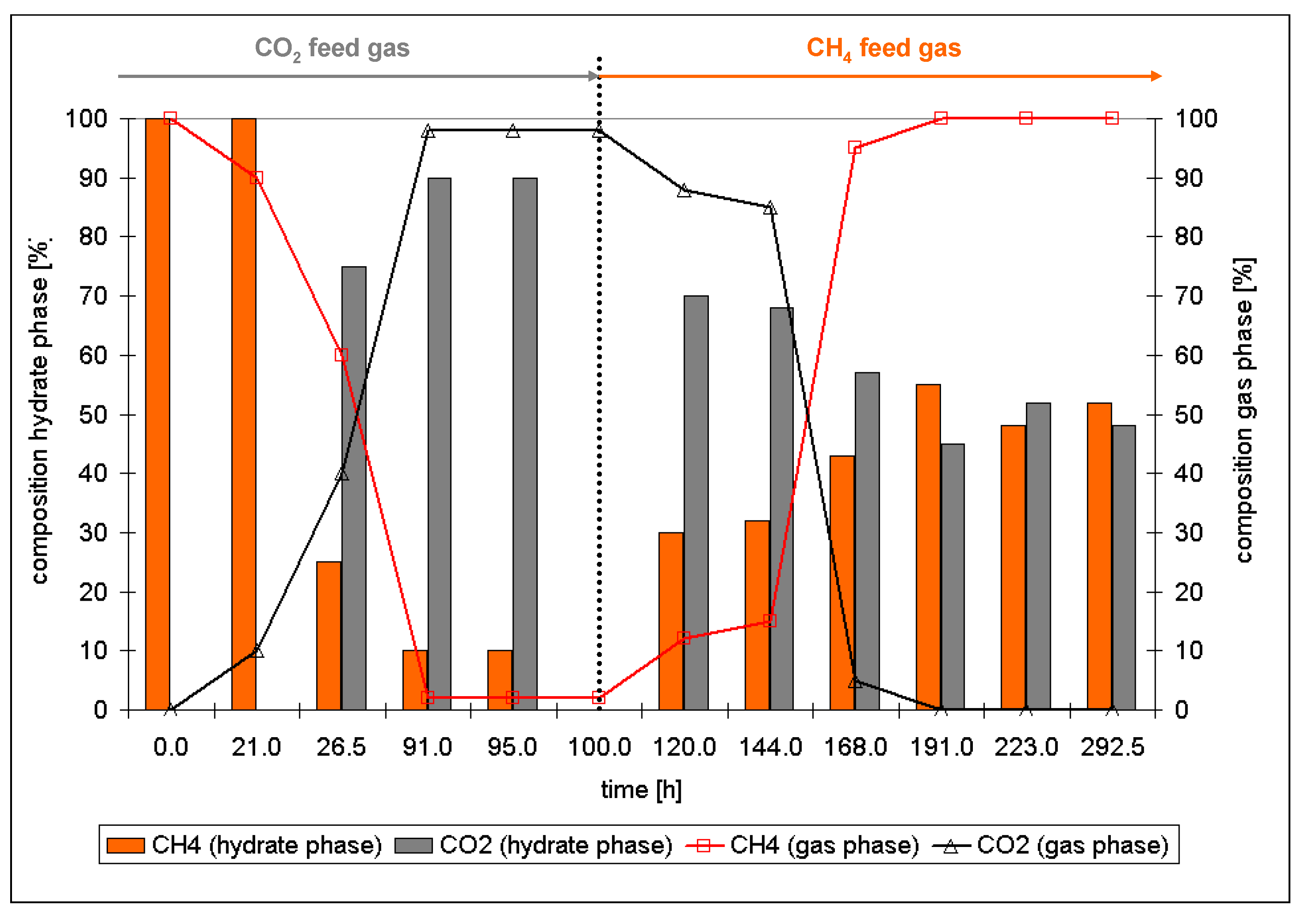

- The ratio of large to small cavities in structure I (6:2) and structure II hydrates (8:16) differ. Also, for the formation of a large cavity of structure II hydrate (hexacaidecaeder 51264) from a large cavity of structure I (tetracaidecaeder 51262) four more water molecules have to be incorporated. Thus, for the structural changes the hydrate phase has to decompose at least in terms of a partial decomposition of the water cavities, a release of the gas molecules and a rearrangement of the water molecules.
- This rearrangement goes along with a reformation of the hydrate phase with a composition in chemical equilibrium with the environmental gas phase.
3. Experimental
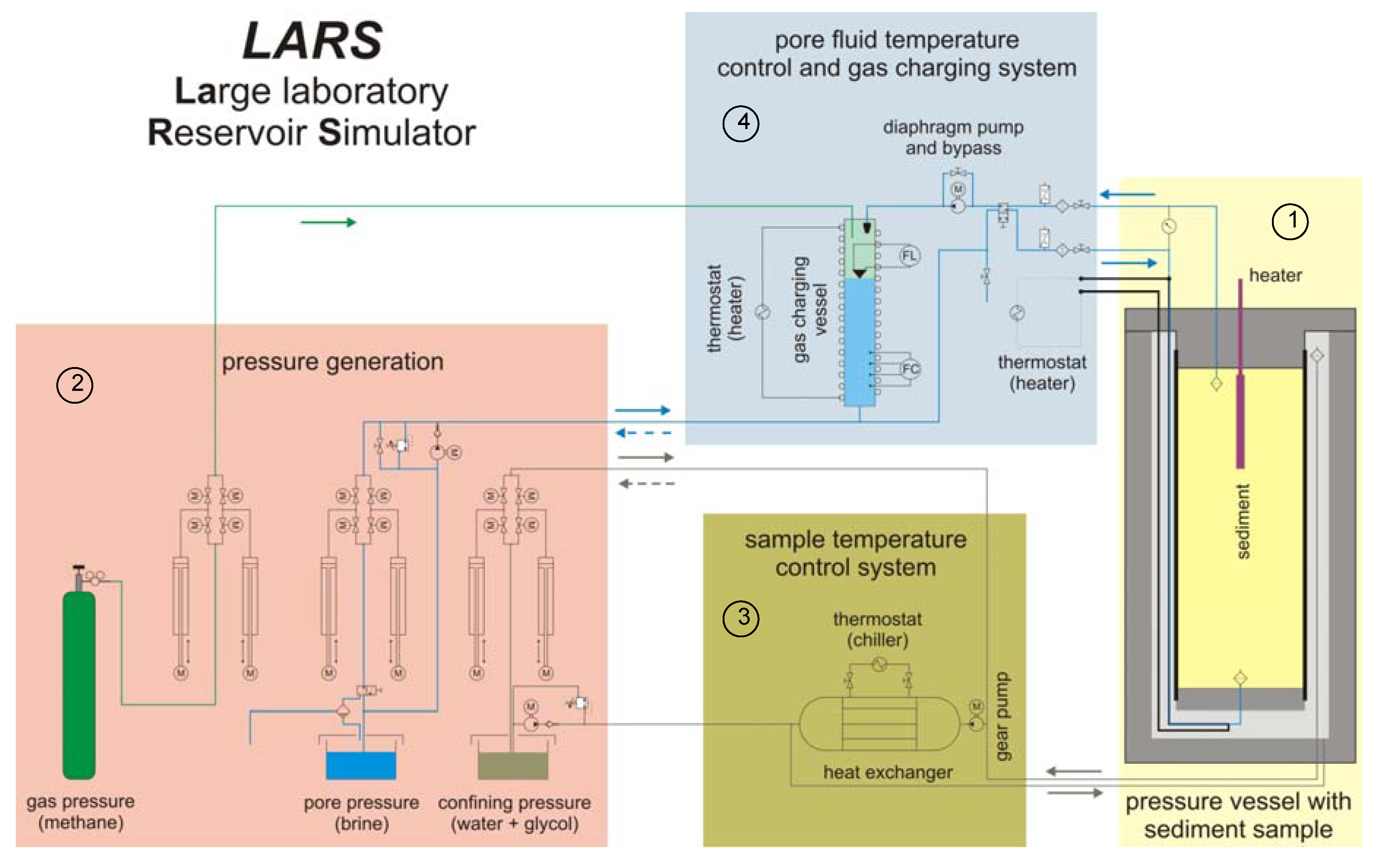
3.1. Autothermal Catalytic Reactor and LArge Scale Reservoir Simulator (LARS)


3.2. Raman Spectroscopy and Powder X-ray Diffraction
4. Conclusions
- •
- The conversion process is induced by a chemical disequilibrium state between the hydrate phase and the environmental gas phase.
- •
- On a molecular level the conversion process can be described as a decomposition and reformation process, in terms of a rearrangement of molecules.
Acknowledgements
References
- Sloan, E.D.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press Taylor and Francis Group: Boca Raton, FL, USA, 2008. [Google Scholar]
- Kvenvolden, K.A.; Lorenson, T.D. The Global Occurrence of Natural Gas Hydrates. In Natural Gas Hydrates—Occurrences, Distribution, and Detection; Paull, C.K., Dillon, W.P., Eds.; American Geophysical Union: Washington, DC, USA, 2001; pp. 3–18. [Google Scholar]
- Milkov, A.V. Molecular and stable isotope compositions of natural gas hydrates: A revised global dataset and basic interpretations in the context of geological settings. Org. Geochem. 2005, 36, 681–702. [Google Scholar] [CrossRef]
- Milkov, A.V.; Claypool, G.E.; Lee, Y.-J.; Sassen, R. Gas hydrate systems at Hydrate Ridge offshore Oregon inferred from molecular and isotopic properties of hydrate-bound and void gases. Geochim. Cosmochim. Acta 2005, 69, 1007–1026. [Google Scholar] [CrossRef]
- Trofimuk, A.A.; Cherskiy, N.V.; Tsarev, V.P. Accumulation of natural gases in zones of hydrate—Formation in the hydrosphere. Doklady Akademii Nauk SSSR 1973, 212, 931–934. [Google Scholar]
- Kvenvolden, K.A.; Grantz, A. Gas hydrates of the Arctic Ocean region. In The Arctic Ocean Region. The Geology of North America; Grantz, A., Johnson, L., Sweeney, J.F., Eds.; Geological Society of America: Boulder, CO, USA, 1990; pp. 539–549. [Google Scholar]
- Klauda, J.B.; Sandler, S.I. Global Distribution of Methane Hydrate in Ocean Sediment. Energy Fuels 2005, 19, 459–470. [Google Scholar] [CrossRef]
- Yasuda, M.; Dallimore, S. Summery of the Methane Hydrate Second Mallik Production Test. J. Jpn. Assoc. Pet. Technol. 2007, 72, 603–607. [Google Scholar] [CrossRef]
- Moridis, G.J.; Collett, T.S.; Boswell, R.; Kurihara, M.; Reagan, M.T.; Koh, C.; Sloan, E.D. Toward Production from Gas Hydrates: Current Status, Assessment of Resources, and Simulation-Based Evaluation of Technology and Potential. Reservoir Eval. Eng. 2009, 12, 745–771. [Google Scholar] [CrossRef]
- Yamamoto, K.; Dallimore, S. Aurora-JOGMEC_NRCan Mallik 2006-2008 Gas Hydrates Research Project Progress. Fire in the Ice Methane Hydrate Newsletter, Summer 2008. Available online: http://www.netl.doe.gov/technologies/oil-gas/futuresupply/methanehydrates/newsletter/newsletter.htm (accessed on 11 January 2011).
- Cranganu, C. In-situ stimulation of gas hydrates. J. Pet. Sci. Technol. 2009, 65, 76–80. [Google Scholar]
- Yang, X.; Gates, I.D. Design of Hybrid Steam-In Situ Combustion Bitumen Recovery Processes. Nat. Resour. Res. 2009, 18, 213–233. [Google Scholar] [CrossRef]
- Cranganu, C. Method for Producing Natural Gas from Gas Hydrate Deposits. In Proceedings of the AAPG Annual Convention, Calgary, Canada, June 2005.
- Zwinkels, M.F.M.; Järås, S.G.; Menon, P.G.; Griffin, T.A. Catalytic Materials for High-Temperature Combustion. Catal. Rev. Sci. Eng. 1993, 35, 319–358. [Google Scholar] [CrossRef]
- Air Liquide. Downloadable Material Safety Data Sheet (MSDS) for CH4; Air Liquide: Paris, France, 1966. [Google Scholar]
- Rydzy, M.B.; Schicks, J.M.; Naumann, R.; Erzinger, J. Dissociation Enthalpies of Synthesized Multicomponent Gas Hydrates with Respect to the Guest Composition and Cage Occupancy. J. Phys. Chem. B 2007, 111, 9539–9545. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Trimm, D.L. Catalytic combustion of methane. Fuel Process. Technol. 1995, 42, 339–359. [Google Scholar] [CrossRef]
- Cranganu, C.; Nitzov, B. The Outlook for Gas Hydrates in the Black Sea: Technology and Economics. In Proceedings of the AAPG Annual Convention, Dallas, TX, USA, April 2004.
- Tsang, S.C.; Claridge, J.B.; Green, M.L.H. Recent advances in the conversion of methane to synthesis gas. Catal. Today 1995, 23, 3–15. [Google Scholar] [CrossRef]
- Pérez-Fortes, M.; Bojarski, A.D.; Velo, E.; Nougués, J.M.; Puigjaner, L. Conceptual model and evaluation of generated power and emissions in an IGCC plant. Energy 2009, 34, 1721–1732. [Google Scholar] [CrossRef]
- Sánchez, D.; Chacartegui, R.; Muñoz, J.M.; Muñoz, A.; Sánchez, T. Performance analysis of a heavy duty combined cycle power plant burning various syngas fuels. Int. J. Hydrogen Energy 2010, 35, 337–345. [Google Scholar] [CrossRef]
- Ormerod, R.M. Solid oxide fuel cells. Chem. Soc. Rev. 2003, 32, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.R.; Luo, J.L.; Chuang, K.T. The study of Au/MoS2 anode catalyst for solid oxide fuel cell (SOFC) using H2S-containing syngas fuel. J. Power Sources 2009, 188, 458–462. [Google Scholar] [CrossRef]
- Keim, W. Industrial Chemicals via C1 Processes. ACS Symp. Ser. 1987, 328, 1–16. [Google Scholar]
- Kamara, B.I.; Coetzee, J. Overview of High-Temperature Fischer-Tropsch Gasoline and Diesel Quality. Energy Fuels 2009, 23, 2242–2247. [Google Scholar] [CrossRef]
- King, D.L.; Cusumano, J.A.; Garten, R.L. A Technological Perspective for Catalytic Processes Based on Synthesis Gas. Catal. Rev. Sci. Eng. 1981, 23, 233–263. [Google Scholar] [CrossRef]
- Riedel, E. Anorganische Chemie; de Gruyter: Berlin, Germany, 1988; p. 352. [Google Scholar]
- Park, Y.; Kim, D.-Y.; Lee, J.-W.; Huh, D.-G.; Park, K.-P.; Lee, J.; Lee, H. Sequestering carbon dioxide into complex structures of naturally occurring gas hydrates. Proc. Natl. Acad. Sci. 2006, 103, 12690–12694. [Google Scholar] [CrossRef] [PubMed]
- Kvamme, B.; Graue, A.; Buanes, T.; Kuznetsova, T.; Ersland, G. Storage of CO2 in natural gas hydrate reservoirs and the effect of hydrate as an extra sealing in cold aquifers. Int. J. Greenhouse Gas Control 2007, 1, 236–246. [Google Scholar] [CrossRef]
- Hirohama, S.; Shimoyama, Y.; Wakabayashi, A.; Tatsuta, S.; Nishida, N.J. Conversion of CH4-hydrate to CO2-hydrate in liquid CO2. J. Chem. Eng. Jpn. 1996, 29, 1014–1020. [Google Scholar] [CrossRef]
- Lee, H.; Seo, Y.; Seo, Y.-T.; Moudrakovski, I.L.; Ripmeester, J. Recovering Methane from Solid Methane Hydrate with Carbon Dioxide. Angew. Chem. Int. Ed. 2003, 42, 5048–5051. [Google Scholar] [CrossRef]
- Steinhauer, B.; Schicks, J.M.; Giese, R. Application of an autothermal reactor for the catalytic partial oxidation of methane to synthesis gas on a Pd-ZrO2 catalyst. Energy Fuels 2010. Submitted. [Google Scholar]
- Stern, L.A.; Kirby, S.H.; Durham, W.B.; Circone, S.; Waite, W.F. Laboratory synthesis of pure methane hydrate suitable for measurement of physical properties and decomposition behaviour. In Natural Gas Hydrate in Oceanic and Permafrost Environments; Max, M.D., Ed.; Springer: New York, NY, USA, 2000; pp. 323–348. [Google Scholar]
- Spangenberg, E.; Kulenkampff, J.; Naumann, R.; Erzinger, J. Pore Space Hydrate Formation from Methane dissolved in Water. Geophys. Res. Lett. 2005, 32, 1–4. [Google Scholar]
- Spangenberg, E.; Kulenkampff, J. Influence of methane hydrate content on the electrical sediment properties. Geophys. Res. Lett. 2006, 33, 1–5. [Google Scholar]
- Wilder, J.W.; Moridis, G.J.; Wilson, S.J.; Kurihara, M.; White, M.D.; Masuda, Y.; Anderson, B.J.; Collett, T.S.; Hunter, R.B.; Narita, H.; Pooladi-Darvish, M.; Rose, K.; Boswell, R. An international effort to compare gas hydrate reservoir simulators. In Proceedings of the 6th International Conference on Gas Hydrates, Vancouver, Canada, 6–10 July 2008.
- Waite, W.F.; Santamarina, J.C.; Cortes, D.D.; Dugan, B.; Espinoza, D.N.; Germaine, J.; Jang, J.; Jung, J.W.; Kneafsey, T.J.; Shin, H.; Soga, K.; Winters, W.J.; Yun, T.-S. Physical properties of hydrate-bearing sediments. Rev. Geophys. 2009, 47, RG4003. [Google Scholar] [CrossRef]
- Schick, J.M.; Ripmeester, J.A. The Coexistence of Two Different Methane Hydrate Phases under Moderate Pressure and Temperature Conditions: Kinetic versus Thermodynamic Products. Angew. Chem. Int. Ed. 2004, 43, 3310–3313. [Google Scholar] [CrossRef]
- Schicks, J.M.; Naumann, R.; Erzinger, J.; Hester, K.C.; Koh, C.A.; Sloan, E.D. Phase Transitions in Mixed Gas Hydrates: Experimental Observations versus Calculated Data. J. Phys. Chem. B 2006, 110, 11468–11474. [Google Scholar] [CrossRef] [PubMed]
- Burke, E.A.J. Raman microspectrometry of fluid inclusions. Lithos 2001, 55, 139–158. [Google Scholar] [CrossRef]
- Luzi, M.; Girod, M.; Naumann, R.; Schicks, J.M.; Erzinger, J. A high pressure cell for kinetic studies on gas hydrates by powder X-ray diffraction. Rev. Sci. Instrum. 2010, 81, 125105. [Google Scholar] [CrossRef] [PubMed]
- TOPAS, Version 3.0; Bruker AXS GmbH: Karlsruhe, Germany, 2005.
- Priest, J.A.; Best, A.I.; Clayton, C.R.I. A laboratory investigation into the seismic velocities of methane gas hydrate-bearing sand. J. Geophys. Res. 2005, 110, B04102. [Google Scholar]
- Winters, W.J.; Pecher, I.A.; Waite, W.F.; Mason, D. Physical properties and rock physics models of sediment containing natural and laboratory-formed methane hydrate. Am. Mineral. 2004, 89, 1221–1227. [Google Scholar]
- Priest, J.A.; Rees, E.V.L.; Clayton, C.R.I. Influence of gas hydrate morphology on the seismic velocities of sands. J. Geophys. Res. 2009, 114, B11205. [Google Scholar]
- Spangenberg, E.; Beeskow-Strauch, B.; Luzi, M.; Naumann, R.; Schicks, J.M. The process of hydrate formation in clastic sediments and its impact on their physical properties. In Proceedings of the 6th International Conference on Gas Hydrates, Vancouver, Canada, 6–10 July 2008.
- Schicks, J.M.; Luzi, M.; Beskow-Strauch, B. The conversion process of hydrocarbon hydrates into CO2 hydrates and vice versa: thermodynamic consideration. J. Phys. Chem. A 2010. Submitted. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schicks, J.M.; Spangenberg, E.; Giese, R.; Steinhauer, B.; Klump, J.; Luzi, M. New Approaches for the Production of Hydrocarbons from Hydrate Bearing Sediments. Energies 2011, 4, 151-172. https://doi.org/10.3390/en4010151
Schicks JM, Spangenberg E, Giese R, Steinhauer B, Klump J, Luzi M. New Approaches for the Production of Hydrocarbons from Hydrate Bearing Sediments. Energies. 2011; 4(1):151-172. https://doi.org/10.3390/en4010151
Chicago/Turabian StyleSchicks, Judith M., Erik Spangenberg, Ronny Giese, Bernd Steinhauer, Jens Klump, and Manja Luzi. 2011. "New Approaches for the Production of Hydrocarbons from Hydrate Bearing Sediments" Energies 4, no. 1: 151-172. https://doi.org/10.3390/en4010151