Results from a Novel Method for Corrosion Studies of Electroplated Lithium Metal Based on Measurements with an Impedance Scanning Electrochemical Quartz Crystal Microbalance

Abstract
:1. Introduction
2. Results and Discussion
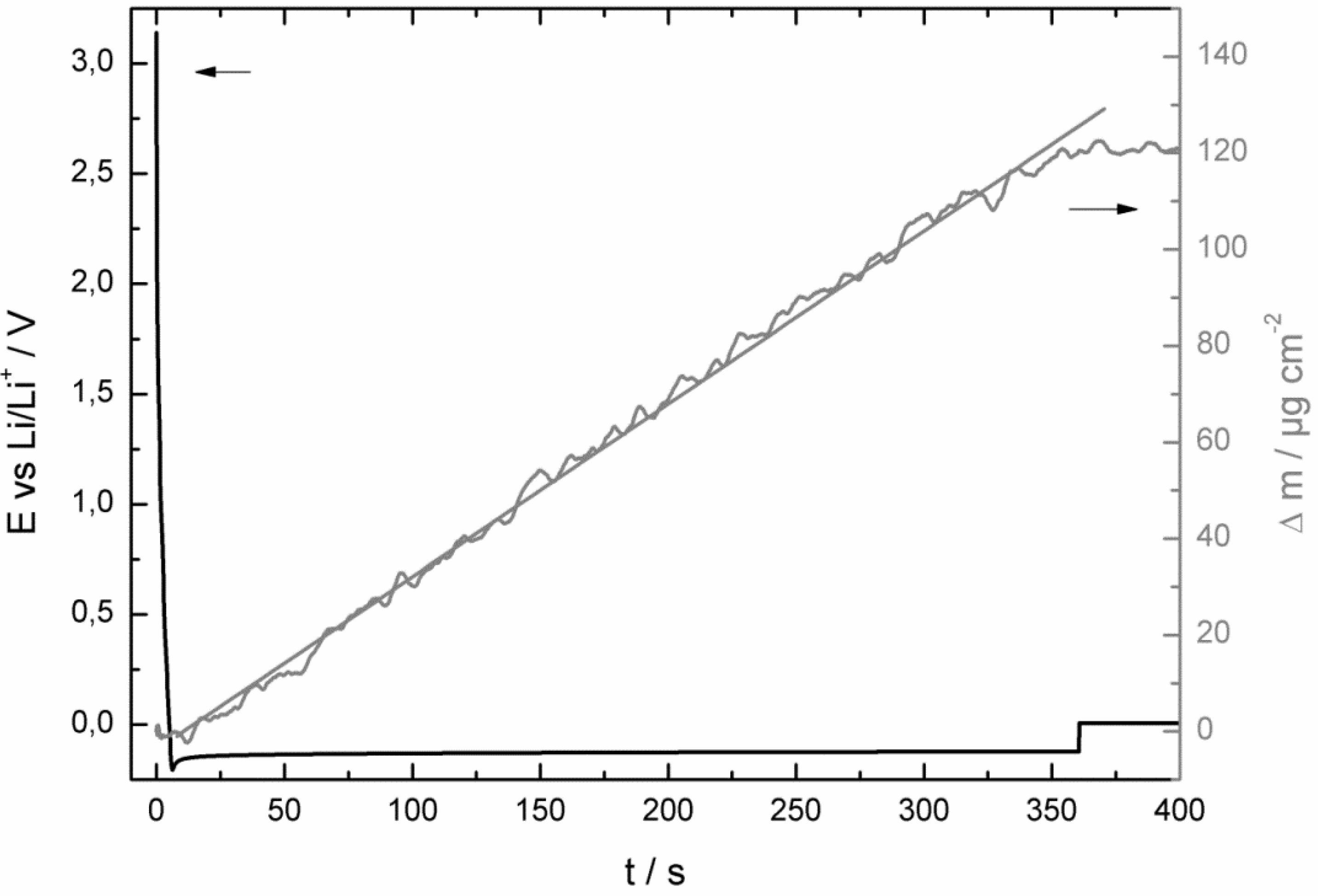
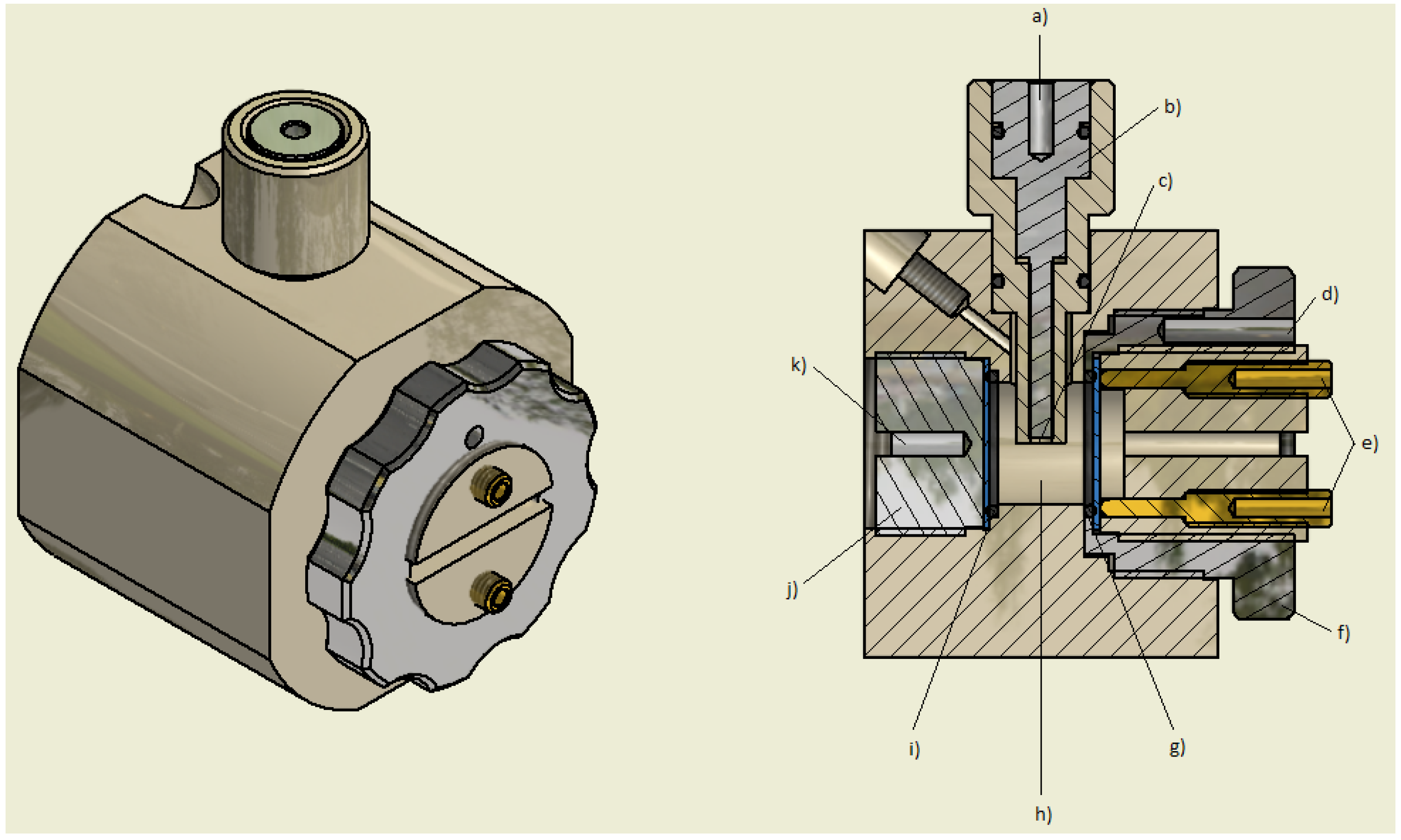
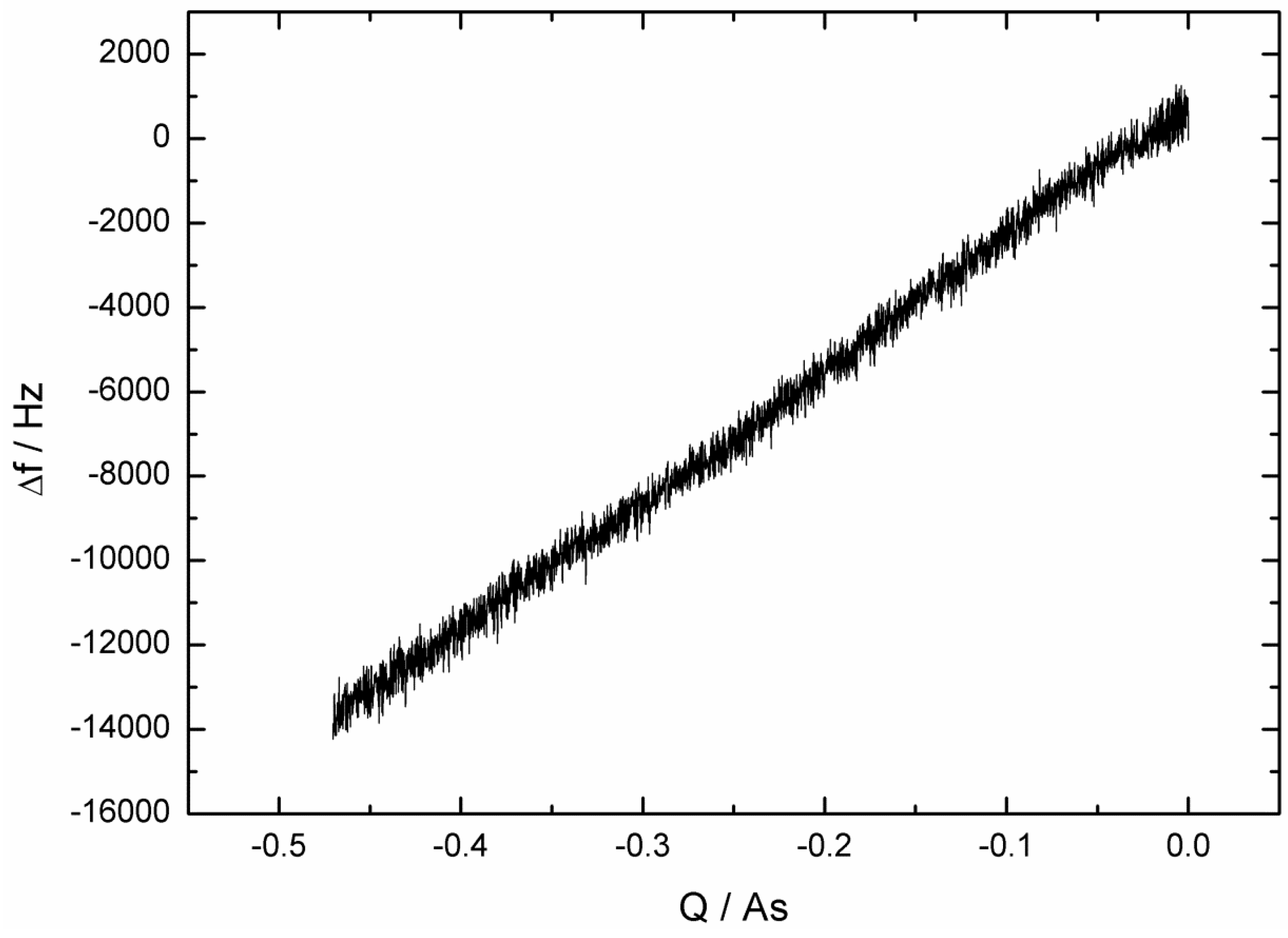
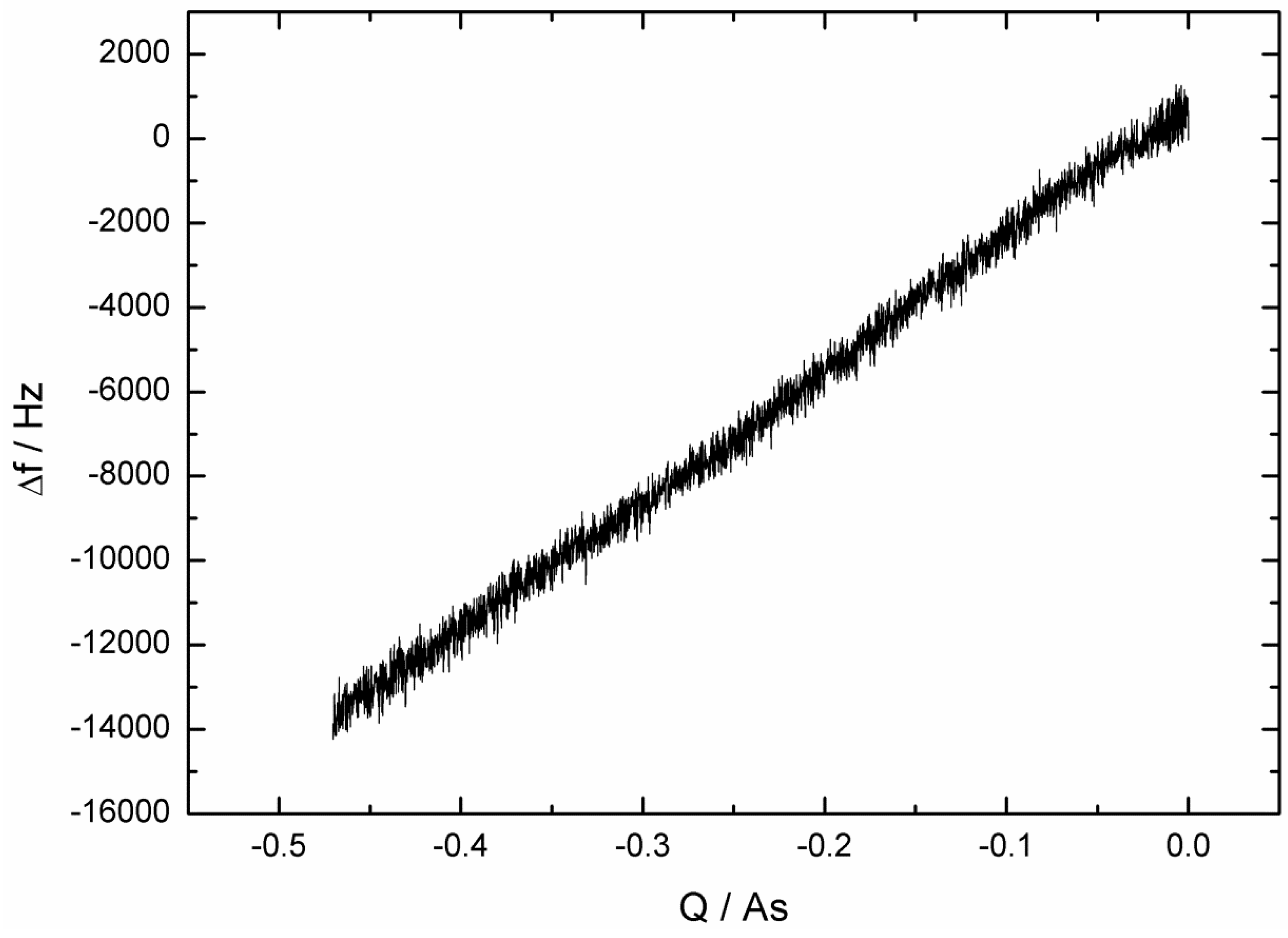
2.1. Fast Impedance Scanning Electrochemical Quartz Crystal Microbalance (fis-EQCM)

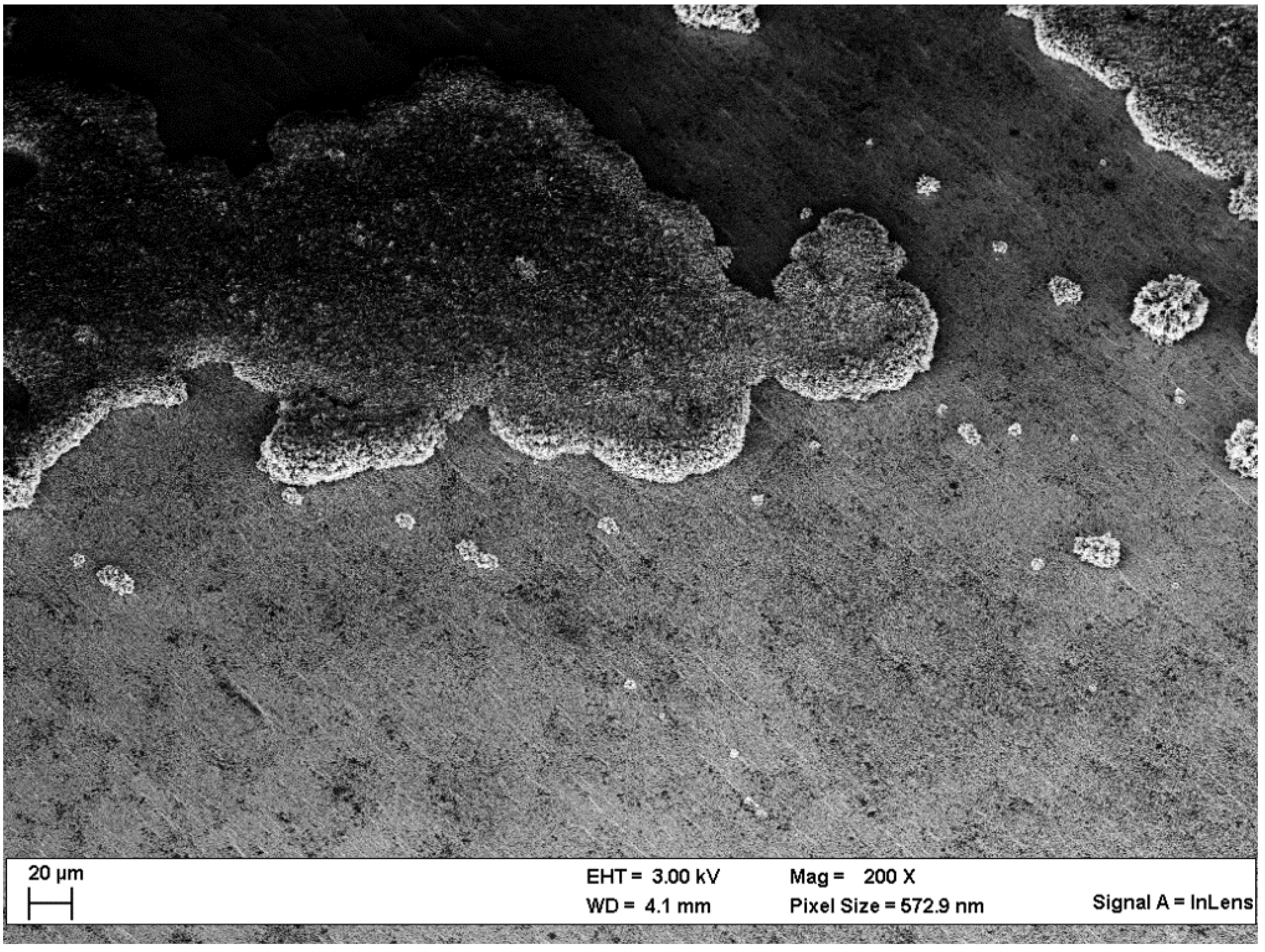
Scanning Electron Microscopy (SEM)





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lithium metal deposits analysis of | 7Li | 11B | 12C | |||
| x/cps | rmsd/cps | x/cps | rmsd/cps | x/cps | rmsd/cps | |
| 1 M LiDFOB in EC:DEC (3:7, by wt.) | 1.61 × 107 | 5.38 × 106 | 2.47 × 105 | 8.53 × 104 | 2.45 × 106 | 6.93 × 105 |
| - | 7Li | 31P | 12C | |||
| x/cps | rmsd/cps | x/cps | rmsd/cps | x/cps | rmsd/cps | |
| 1 M LiPF6 in EC:DEC (3:7, by wt.) | 6.17 × 107 | 1.83 × 107 | 2.58 × 105 | 7.63 × 104 | 2.88 × 106 | 1.44 × 105 |

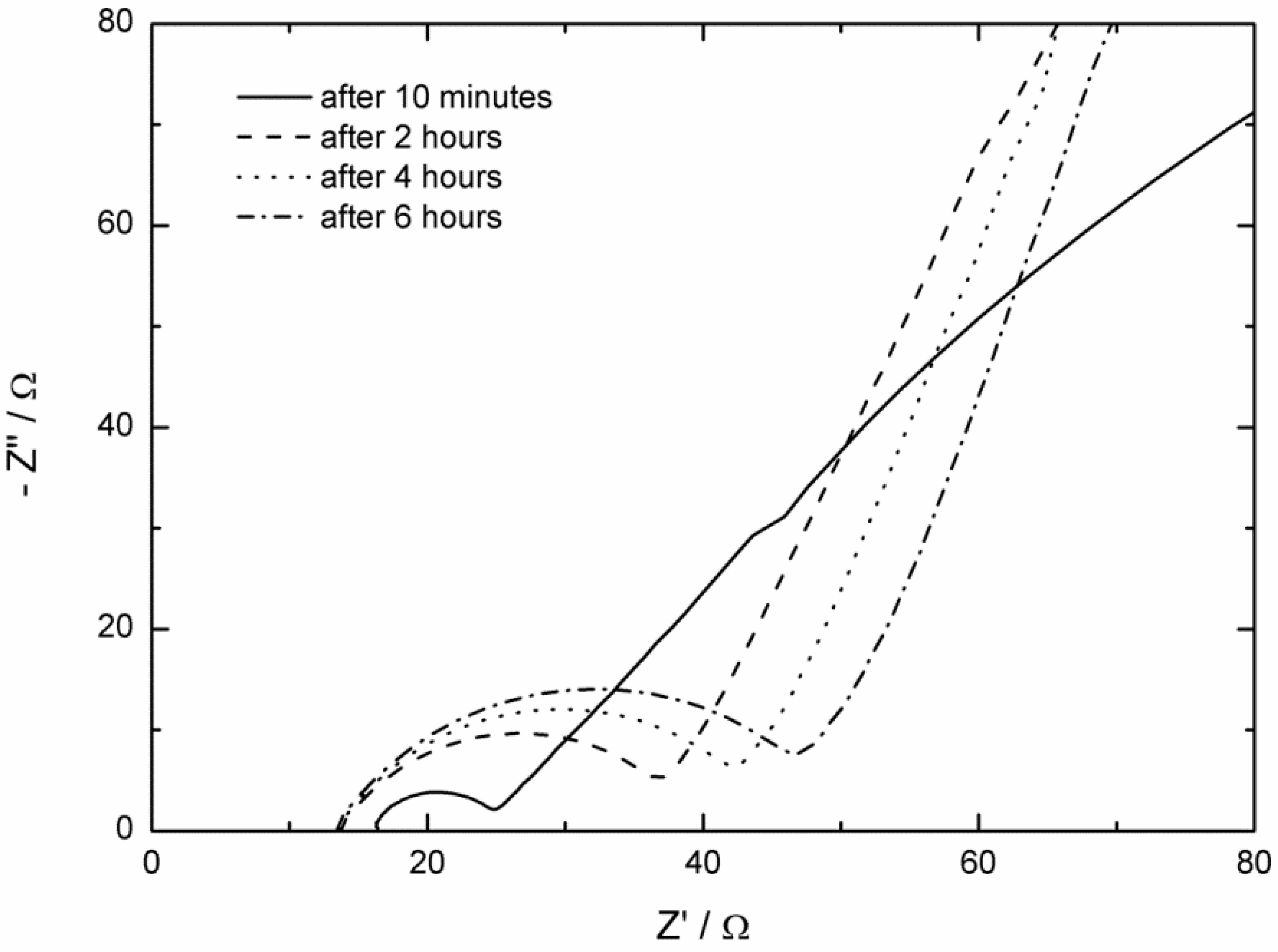
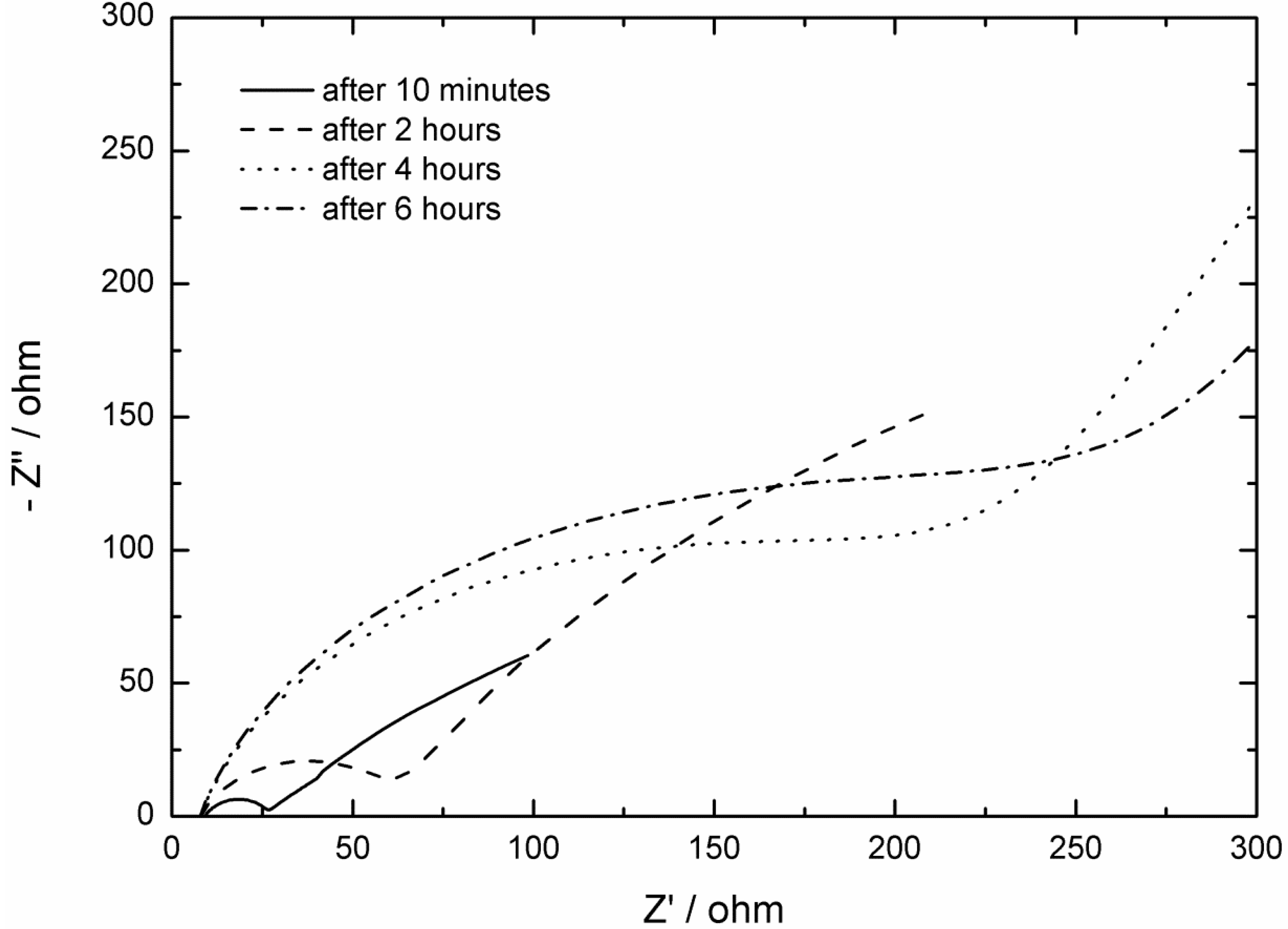
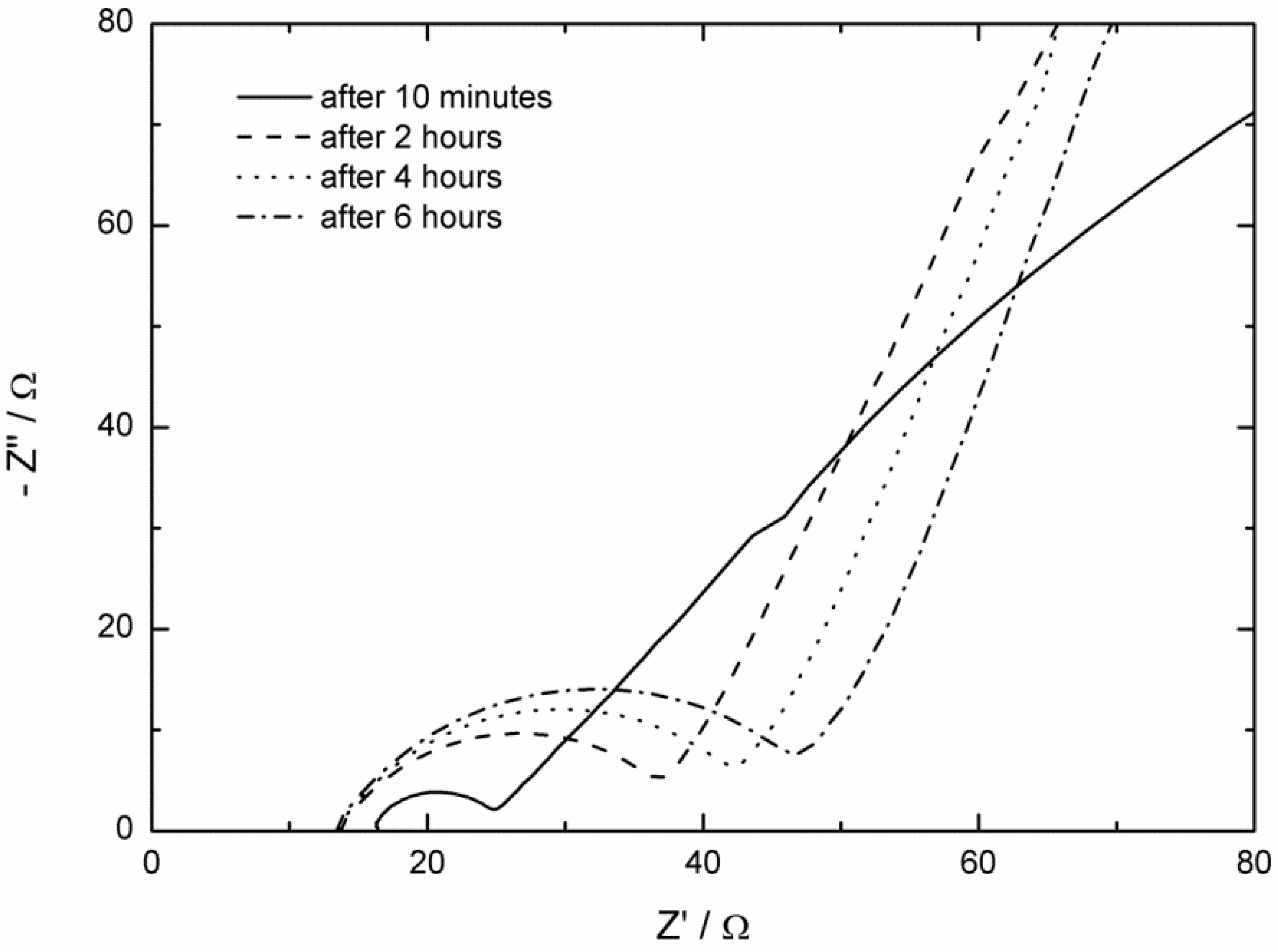
2.2. AC Impedance Measurements
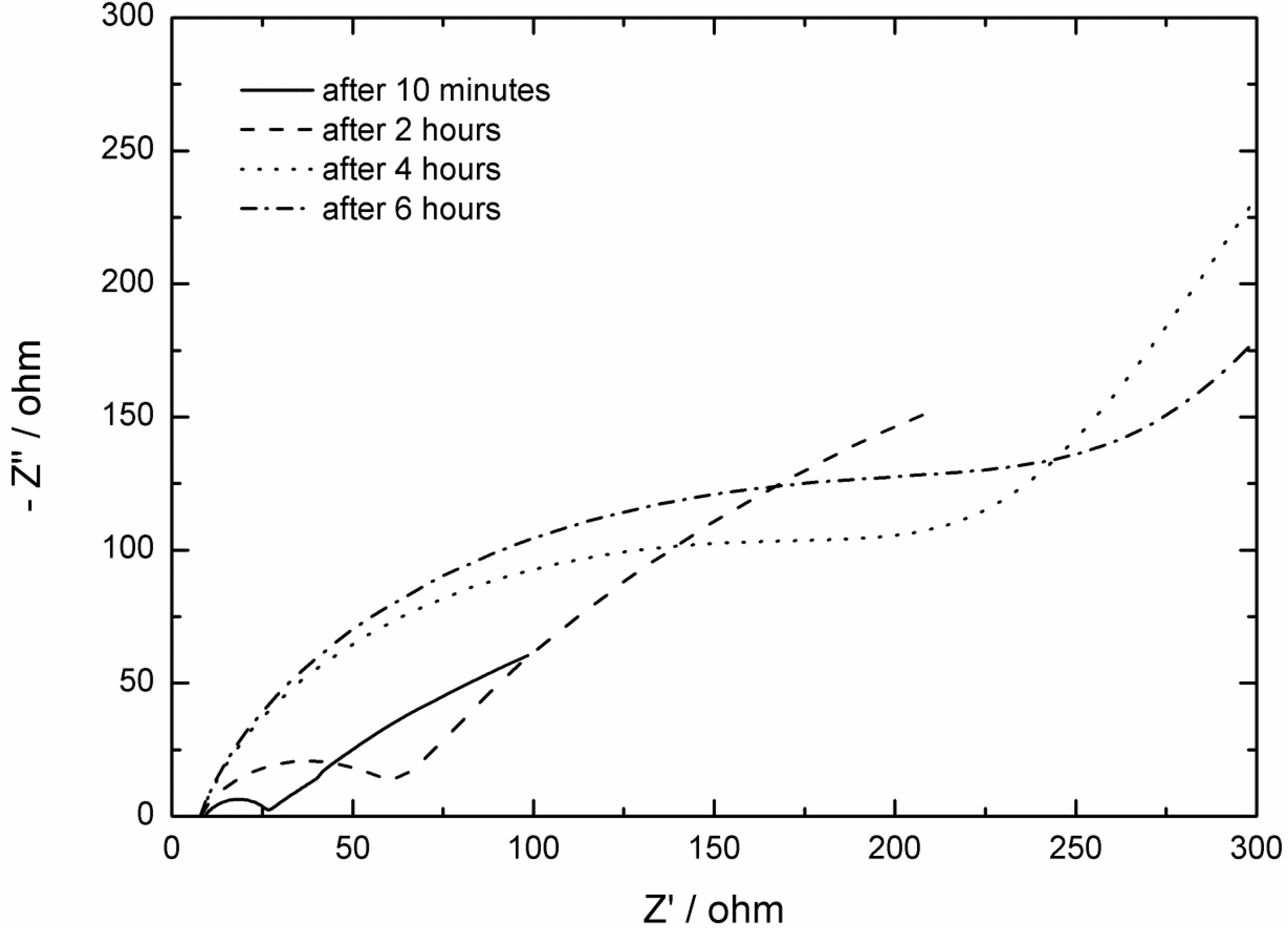
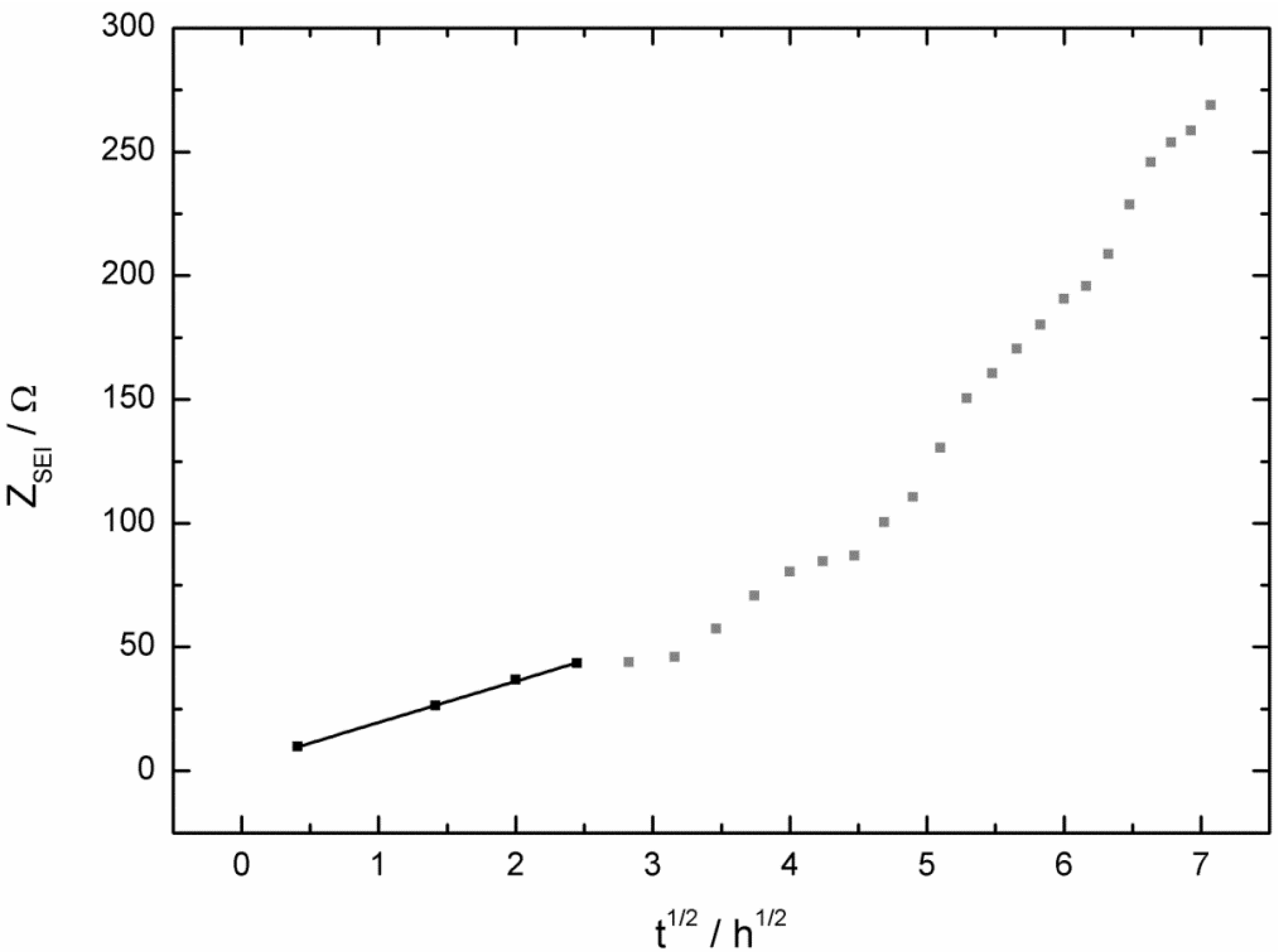
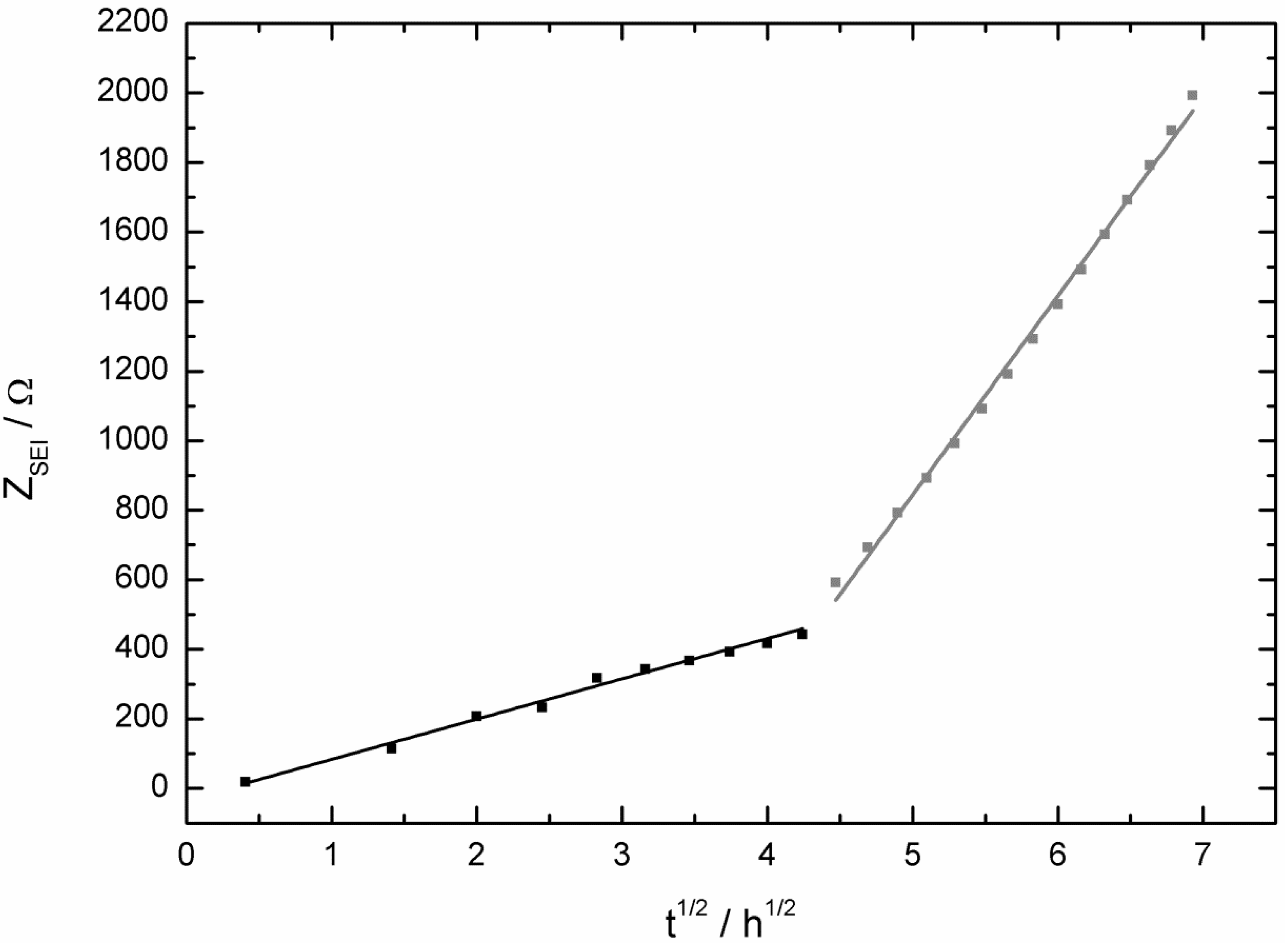
3. Experimental Section
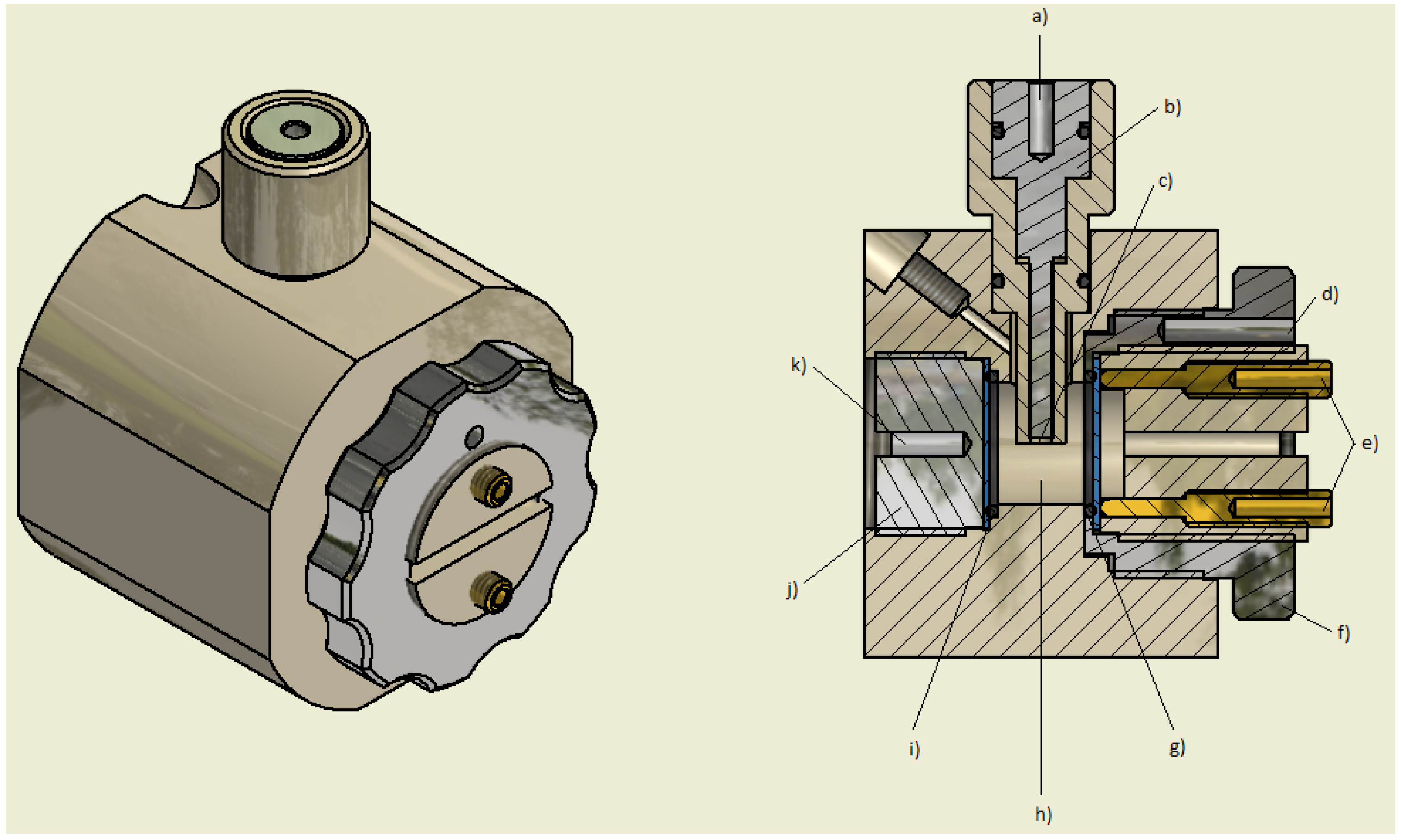
4. Conclusions
- -
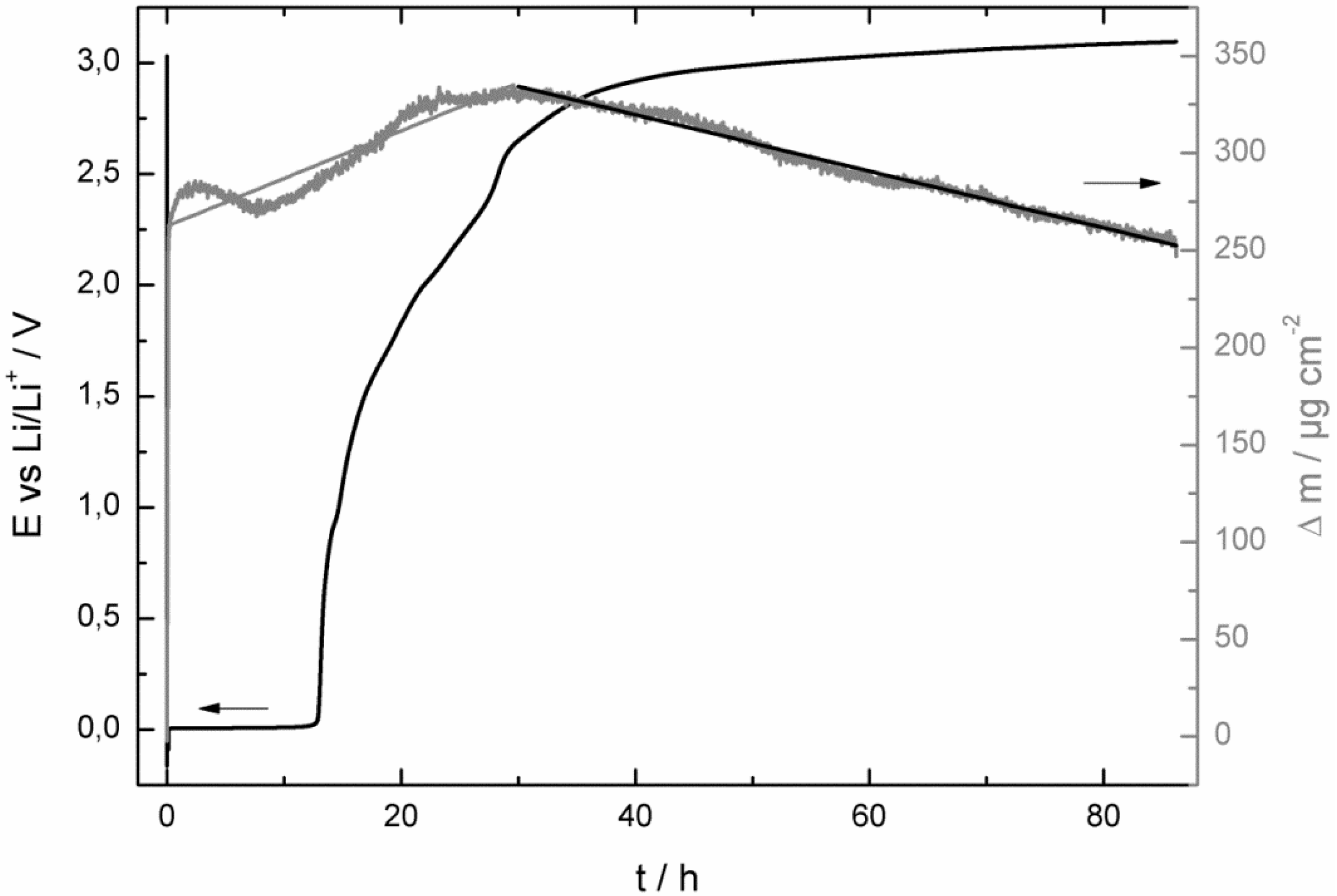
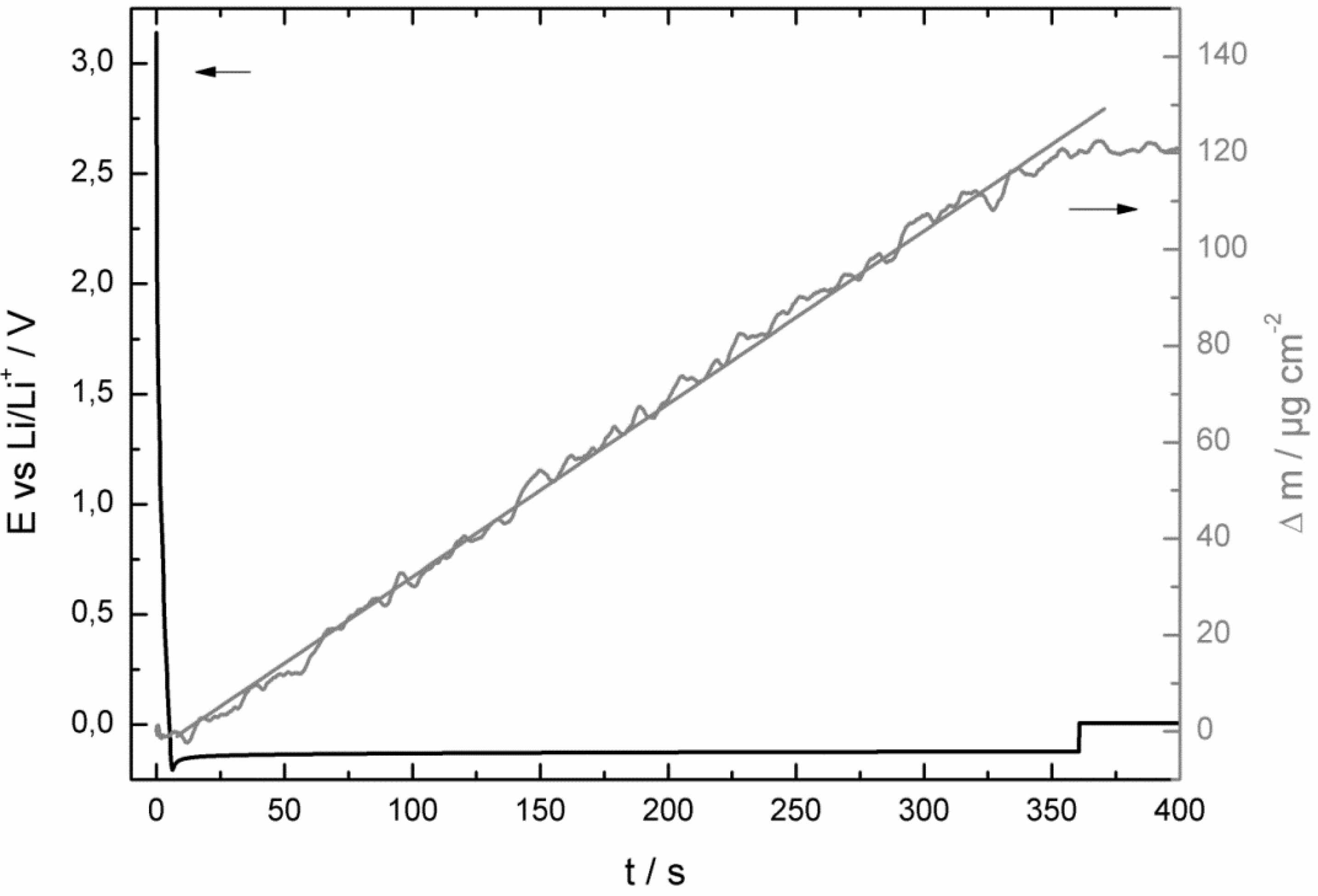
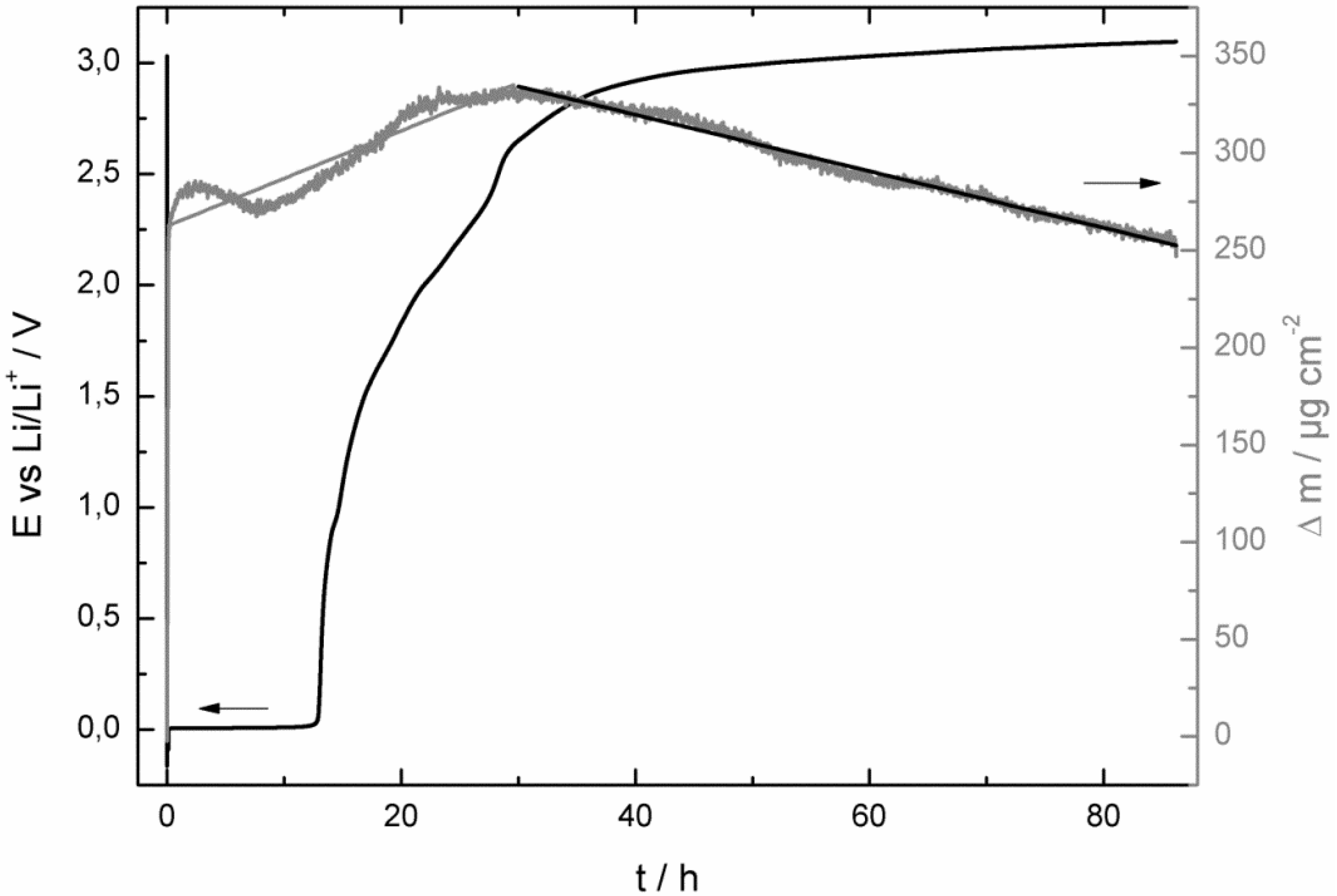
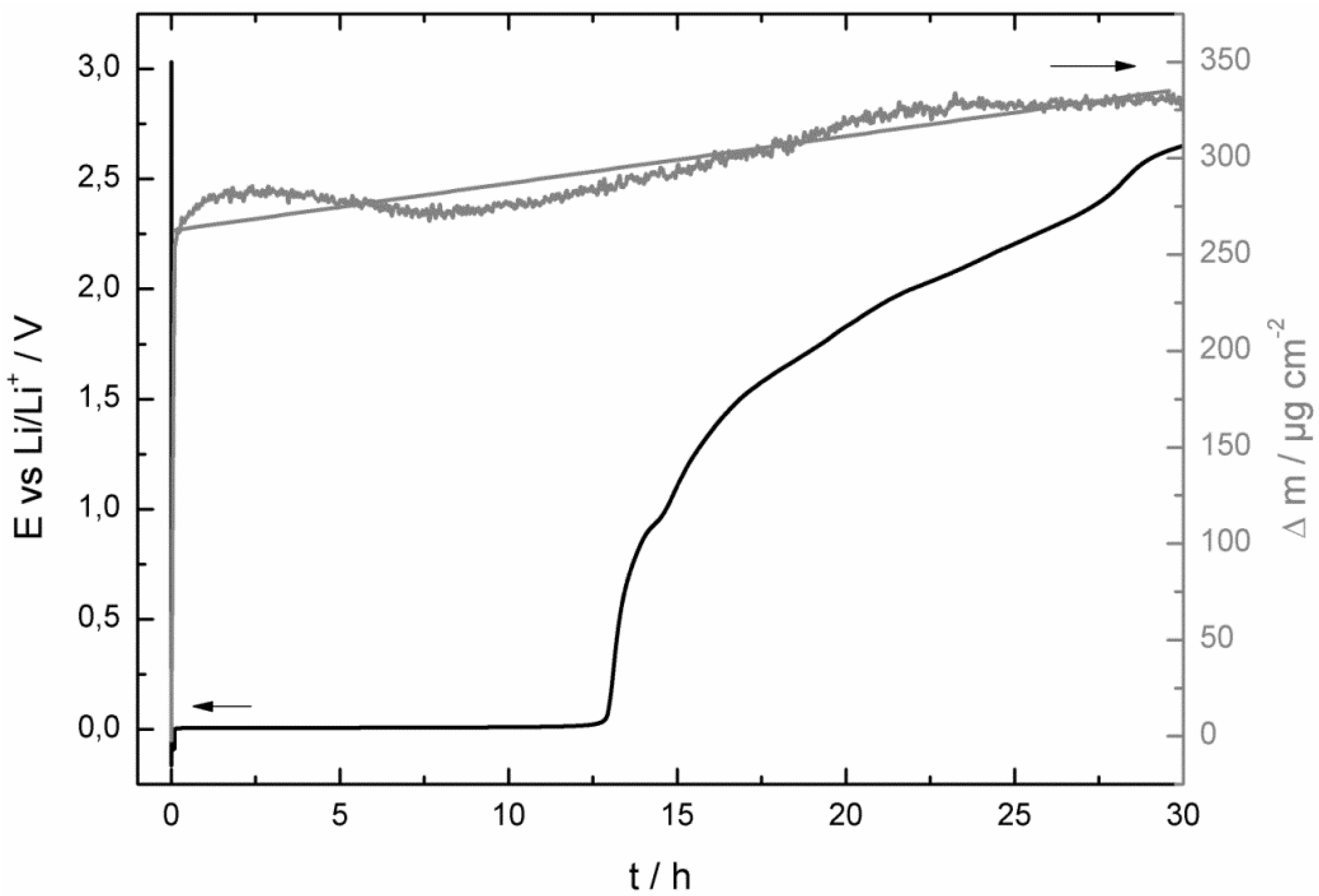
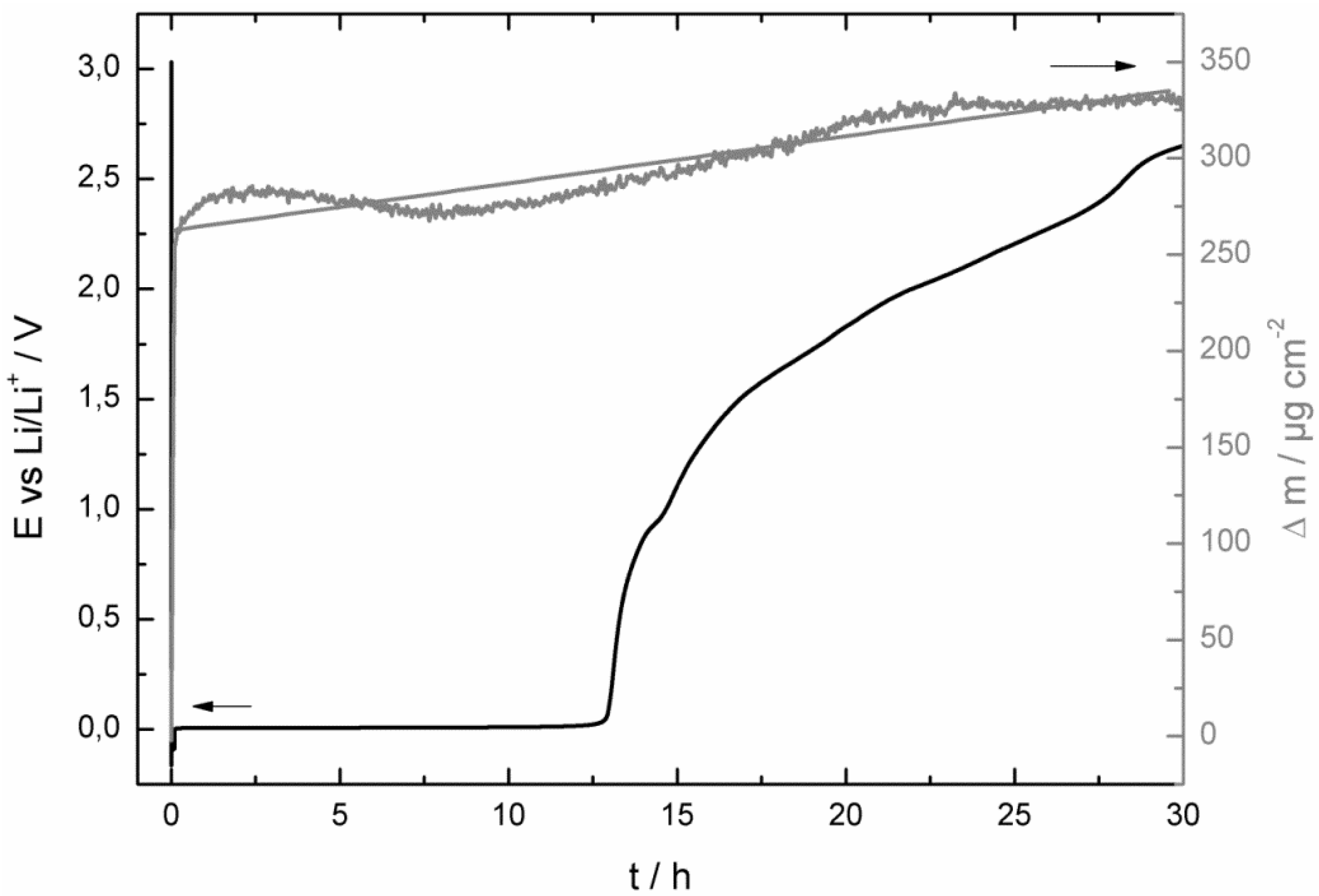
- at the beginning of the corrosion process the potential stayed for some hours at 0.0 V vs. Li/Li+ and increased then consistently until it reached the potential of the bare copper foil;
- -
- at least as long as the potential stayed at 0.0 V vs. Li/Li+ the mass of the quartz sensor increased and after reaching a maximum it started to decrease;
- -
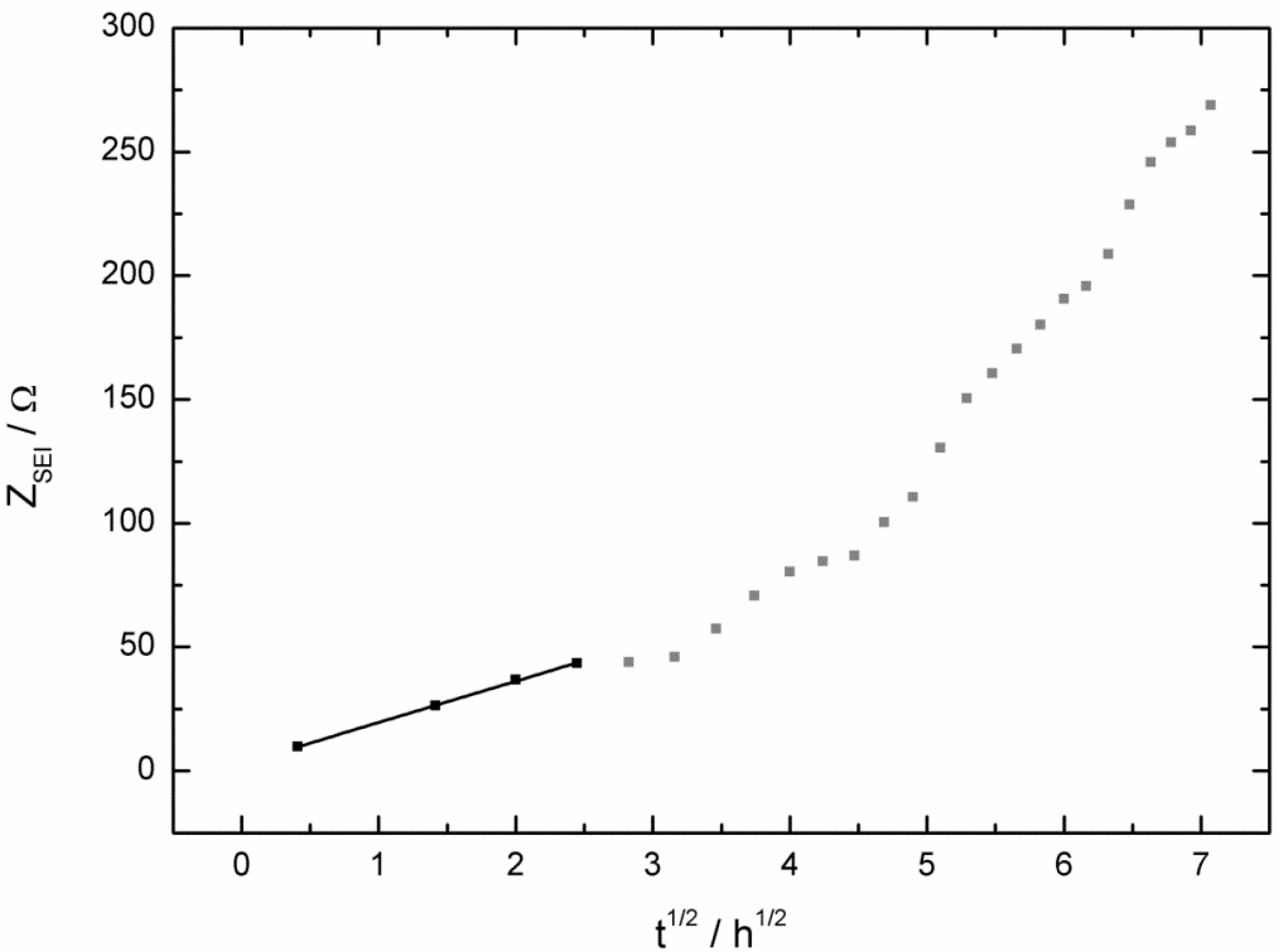
- the SEI growth is only diffusion-limited at the beginning of the corrosion experiment, probably as long the potential stayed at 0.0 V vs. Li/Li+.
- -
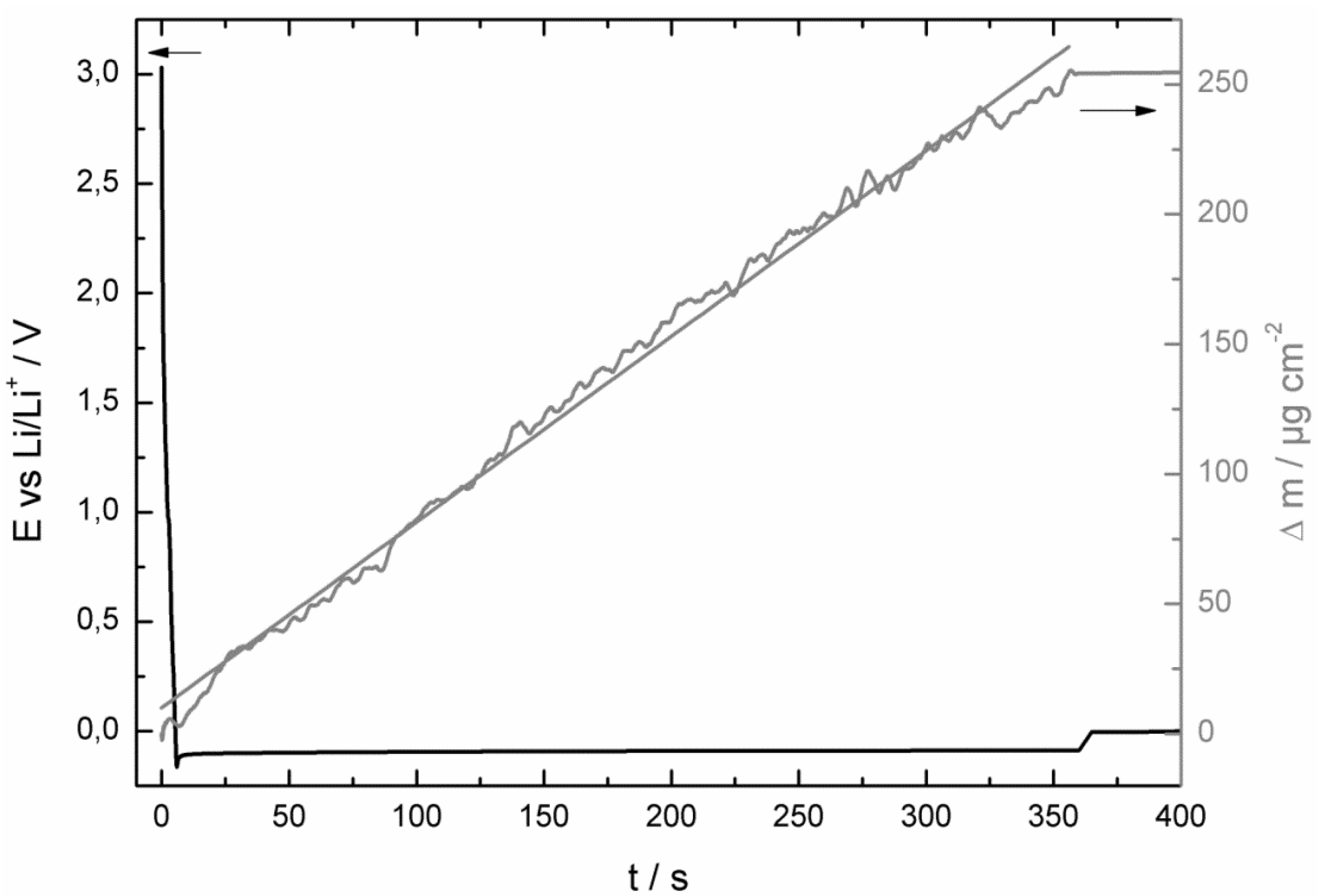
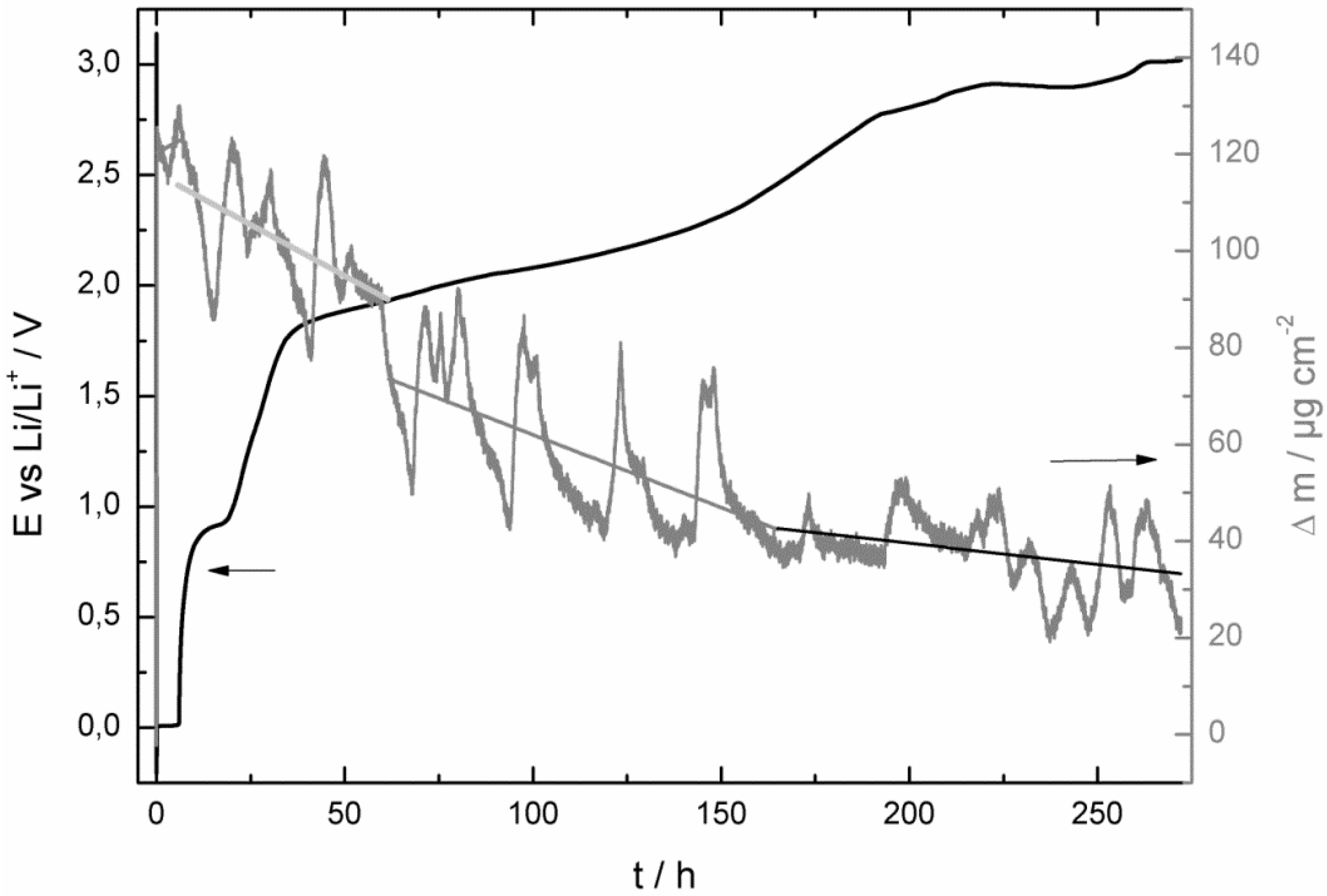
- at the beginning of the corrosion measurement the potential stayed longer at 0.0 V vs. Li/Li+ for the LiPF6-based electrolyte;
- -
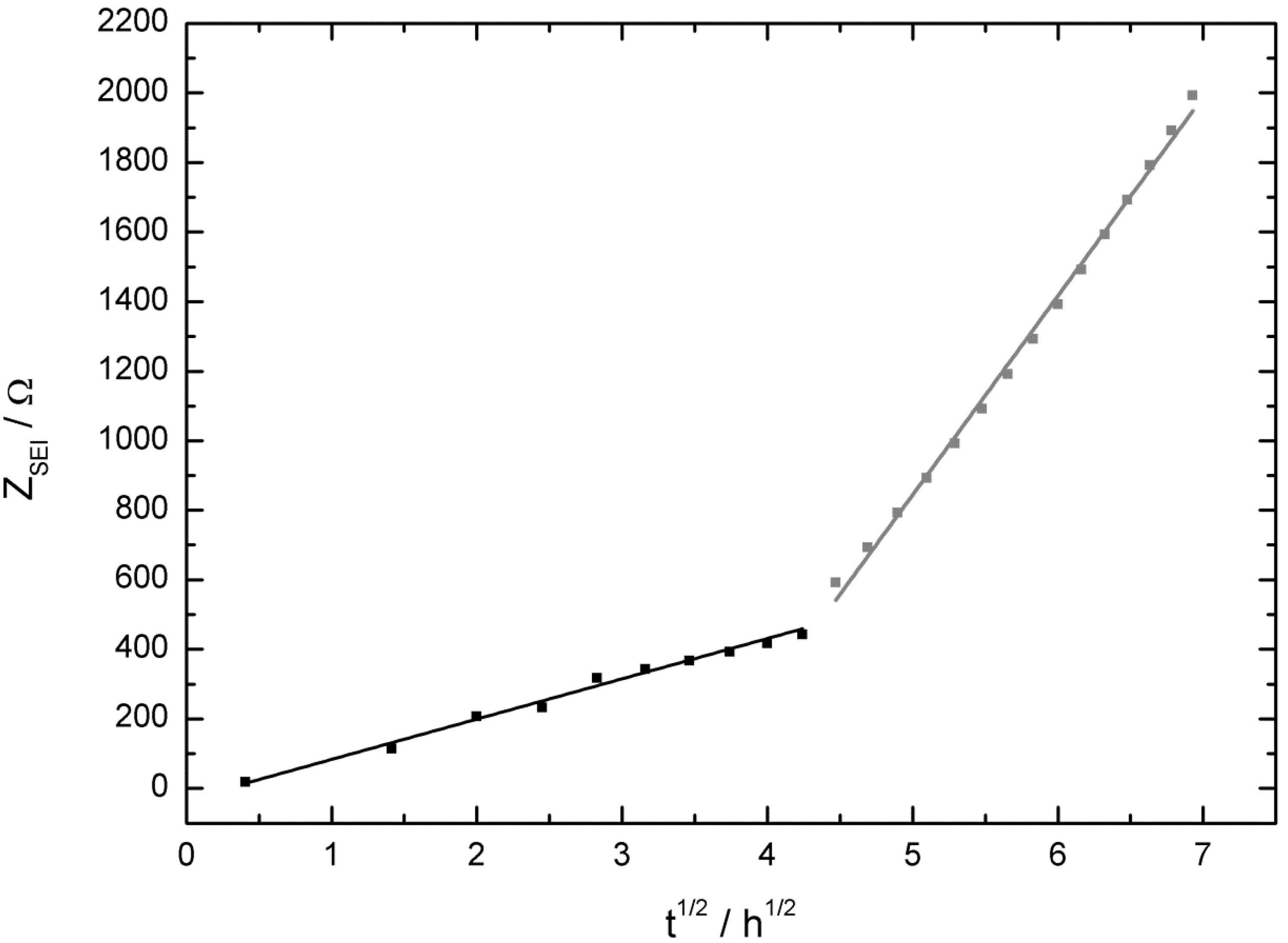
- then the potential increased also much faster to the initial potential of the bare copper foil for the LiPF6-based (after 86 h) electrolyte than for the LiDFOB-based electrolyte (after 272 h);
- -
- the mass increase at the beginning of the corrosion measurement of the LiDFOB-based electrolyte took only place when the potential stayed at 0.0 V vs. Li/Li+, whereas the mass increase of the LiPF6-based electrolyte continued until the potential had already reached a value of 2.6 V vs. Li/Li+;
- -
- the mass decrease of the quartz sensor in the LiPF6-based electrolyte showed a linear decrease until the end of the experiment, whereas the mass decrease of the quartz sensor of the LiDFOB-based electrolyte consisted of a series of huge mass fluctuations.
Acknowledgments
Conflict of Interest
References
- Vetter, J.; Novak, P.; Wagner, M.; Veit, C.; Möller, K.; Besenhard, J.; Winter, M.; Wohlfahrt-Mehrens, M.; Vogler, C.; Hammouche, A. Ageing mechanisms in lithium-ion batteries. J. Power Sources. 2005, 147, 269–281. [Google Scholar] [CrossRef]
- Winter, M.; Imhof, R.; Joho, F.; Novak, P. FTIR and DEMS investigations on the electroreduction of chloroethylene carbonate-based electrolyte solutions for lithium-ion cells. J. Power Sources. 1999, 82, 818–823. [Google Scholar] [CrossRef]
- Wrodnigg, G.H.; Besenhard, J.O.; Winter, M. Cyclic and acyclic sulfites: new solvents and electrolyte additives for lithium ion batteries with graphitic anodes? J. Power Sources. 2001, 97, 592–594. [Google Scholar] [CrossRef]
- Tasaki, K.; Goldberg, A.; Winter, M. On the difference in cycling behaviors of lithium-ion battery cell between the ethylene carbonate- and propylene carbonate-based electrolytes. Electrochim. Acta. 2011, 56, 10424–10435. [Google Scholar] [CrossRef]
- Olivier, J.P.; Winter, M. Determination of the absolute and relative extents of basal plane surface area and “non-basal plane surface” area of graphites and their impact on anode performance in lithium ion batteries. J. Power Sources. 2001, 97, 151–155. [Google Scholar] [CrossRef]
- Wagner, M.R.; Raimann, P.R.; Möller, K.C.; Besenhard, J.O.; Winter, M. Electrolyte decomposition reactions on tin- and graphite-based anodes are different. Electrochem. Solid St. 2004, 7, A201–A205. [Google Scholar] [CrossRef]
- Schranzhofer, H.; Bugajski, J.; Santner, H.; Korepp, C.; Moller, K.; Besenhard, J.; Winter, M.; Sitte, W. Electrochemical impedance spectroscopy study of the SEI formation on graphite and metal electrodes. J. Power Sources. 2006, 153, 391–395. [Google Scholar] [CrossRef]
- Wudy, F.; Schedlbauer, T.; Stock, C.; Gores, H.J. Impedance scanning QCM studies of aniline electropolymerization in aqueous phosphoric acid solutions to determine the most effective acid concentration. Acta Chim. Slov. 2009, 56, 65–69. [Google Scholar]
- Wudy, F.; Multerer, M.; Stock, C.; Schmeer, G.; Gores, H.J. Rapid impedance scanning QCM for electrochemical applications based on miniaturized hardware and high-performance curve Fitting. Electrochim. Acta. 2008, 53, 6568–6574. [Google Scholar] [CrossRef]
- Kramer, E.; Schedlbauer, T.; Hoffmann, B.; Terborg, L.; Nowak, S.; Gores, H.J.; Passerini, S.; Winter, M. Mechanism of anodic dissolution of the aluminum current collector in 1 M LiTFSI EC:DEC 3:7 in rechargeable lithium batteries. J. Electrochem. Soc. 2013, 160, A356–A360. [Google Scholar] [CrossRef]
- Song, S.W.; Richardson, T.J.; Zhuang, G.V.; Devine, T.M.; Evans, J.W. Effect on aluminum corrosion of LiBF4 addition into lithium imide electrolyte: A study using the EQCM. Electrochim. Acta. 2004, 49, 1483–1490. [Google Scholar] [CrossRef]
- Simbeck, T.; Thomaier, S.; Stock, C.; Riedl, E.; Gores, H.J. Measurement of adsorption kinetics of benzotriazole on copper surfaces via impedance scanning quartz crystal microbalance studies. Electrochem. Commun. 2011, 13, 803–805. [Google Scholar] [CrossRef]
- Simbeck, T.; Hammer, M.M.; Thomaier, S.; Stock, C.; Riedl, E.; Gores, H.J. Kinetics of adsorption of poly(vinylimidazole) (PVI) onto copper surfaces investigated by quartz crystal microbalance studies. J. Solid State Electr. 2012, 16, 3467–3472. [Google Scholar] [CrossRef]
- Aurbach, D.; Moshkovich, M. A study of lithium deposition-dissolution processes in a few selected electrolyte solutions by electrochemical quartz crystal microbalance. J. Electrochem. Soc. 1998, 145, 2629–2639. [Google Scholar] [CrossRef]
- Sauerbrey, G. Verwendung Von Schwingquarzen zur Wägung dunner Schichten und zur Mikrowägung. Z. Phys. 1959, 155, 206–222. [Google Scholar] [CrossRef]
- Aurbach, D.; Zaban, A. The application of EQCM to the study of the electrochemical-behavior of propylene carbonate solutions. J. Electroanal. Chem. 1995, 393, 43–53. [Google Scholar] [CrossRef]
- Bund, A.; Schneider, O.; Dehnke, A. Combining AFM and EQCM for the in situ investigation of surface roughness effects during electrochemical metal depositions. Phys. Chem. Chem. Phys. 2002, 4, 3552–3554. [Google Scholar] [CrossRef]
- Kanazawa, K.K.; Gordon, J.G. Frequency of a quartz microbalance in contact with liquid. Anal. Chem. 1985, 57, 1770–1771. [Google Scholar] [CrossRef]
- Stock, C.; Moosbauer, D.; Zugmann, S.; Simbeck, T.; Amereller, M.; Gores, H.J. A novel method for in situ measurement of solubility via impedance scanning quartz crystal microbalance studies. Chem. Commun. 2011, 47, 6984–6986. [Google Scholar] [CrossRef]
- Larush-Asraf, L.; Biton, A.; Teller, H.; Zinigrad, E.; Aurbach, D. On the electrochemical and thermal behavior of lithium bis(oxalato)borate (LiBOB) solutions. J. Power Sources. 2007, 174, 400–407. [Google Scholar] [CrossRef]
- Moller, K.; Santner, H.; Kern, W.; Yamaguchi, S.; Besenhard, J.; Winter, M. In situ characterization of the SEI formation on graphite in the presence of a vinylene group containing film-forming electrolyte additives. J. Power Sources. 2003, 119, 561–566. [Google Scholar] [CrossRef]
- Tavassol, H.; Buthker, J.W.; Ferguson, G.A.; Curtiss, L.A.; Gewirth, A.A. Solvent oligomerization during SEI formation on model systems for Li-ion battery anodes. J. Electrochem. Soc. 2012, 159, A730–A738. [Google Scholar] [CrossRef]
- Wang, F.M.; Shieh, D.T.; Cheng, J.H.; Yang, C.R. An investigation of the salt dissociation effects on solid electrolyte interface (SEI) formation using linear carbonate-based electrolytes in lithium ion batteries. Solid State Ionics. 2010, 180, 1660–1666. [Google Scholar] [CrossRef]
- Bucur, R.V.; Carlsson, J.O.; Mecea, V.M. Quartz-crystal mass sensors with glued foil electrodes. Sensor Actuat. B Chem. 1996, 37, 91–95. [Google Scholar] [CrossRef]
- Zugmann, S.; Moosbauer, D.; Amereller, M.; Schreiner, C.; Wudy, F.; Schmitz, R.; Schmitz, R.; Isken, P.; Dippel, C.; Muller, R.; et al. Electrochemical characterizaiton of electrolytes for lithium-ion batteries based on lithium difluoromono(oxalato)borate. J. Power Sources 2011, 196, 1417–1424. [Google Scholar] [CrossRef]
- Moosbauer, D.; Zugmann, S.; Amereller, M.; Gores, H.J. Effect of ionic liquids as additives on lithium electrolytes: Conductivity, electrochemical stability, and aluminum corrosion. J. Chem. Eng. Data. 2010, 55, 1794–1798. [Google Scholar] [CrossRef]
- Schedlbauer, T.; Krüger, S.; Schmitz, R.; Schmitz, R.W.; Schreiner, C.; Gores, H.J.; Passerini, S.; Winter, M. Lithium difluoro(oxalato)borate: A promising salt for lithium metal based secondary batteries? Electrochim. Acta. 2013, 92, 102–107. [Google Scholar] [CrossRef]
- Zhang, S.S. An unique lithium salt for the improved electrolyte of Li-ion battery. Electrochem. Commun. 2006, 8, 1423–1428. [Google Scholar] [CrossRef]
- Zhang, S.S. Electrochemical study of the formation of a solid electrolyte interface on graphite in a LiBC(2)O(4)F(2)-based electrolyte. J. Power Sources. 2007, 163, 713–718. [Google Scholar] [CrossRef]
- Chen, Z.H.; Liu, J.; Amine, K. Lithium difluoro(oxalato)borate as salt for lithium-ion batteries. Electrochem. Solid St. 2007, 10, A45–A47. [Google Scholar] [CrossRef]
- Xu, M.Q.; Zhou, L.; Hao, L.S.; Xing, L.D.; Li, W.S.; Lucht, B.L. Investigation and application of lithium difluoro(oxalate)borate (LiDFOB) as additive to improve the thermal stability of electrolyte for lithium-ion batteries. J. Power Sources 2011, 196, 6794–6801. [Google Scholar] [CrossRef]
- Lex-Balducci, A.; Schmitz, R.; Schmitz, R.W.; Müller, R.A.; Amereller, M.; Moosbauer, D.; Gores, H.J.; Winter, M. Lithium borates for lithium-ion battery electrolytes. ECS Trans. 2010, 25, 13–17. [Google Scholar]
- Zugmann, S.; Fleischmann, M.; Amereller, M.; Gschwind, R.M.; Winter, M.; Gores, H.J. Salt diffusion coefficients, concentration dependence of cell potentials, and transference numbers of lithium difluoromono(oxalato)borate-based solutions. J. Chem. Eng. Data. 2011, 56, 4786–4789. [Google Scholar] [CrossRef]
- Zugmann, S.; Fleischmann, M.; Amereller, M.; Gschwind, R.M.; Wiemhofer, H.D.; Gores, H.J. Measurement of transference numbers for lithium ion electrolytes via four different methods, a comparative study. Electrochim. Acta. 2011, 56, 3926–3933. [Google Scholar] [CrossRef]
- Besenhard, J.O.; Winter, M. Insertion reactions in advanced electrochemical energy storage. Pure Appl. Chem. 1998, 70, 603–608. [Google Scholar] [CrossRef]
- Winter, M.; Appel, W.; Evers, B.; Hodal, T.; Moller, K.; Schneider, I.; Wachtler, M.; Wagner, M.; Wrodnigg, G.; Besenhard, J. Studies on the anode/electrolyte interface in lithium ion batteries. Monatsh. Chem. 2001, 132, 473–486. [Google Scholar] [CrossRef]
- Wu, Q.L.; Lu, W.Q.; Miranda, M.; Honaker-Schroeder, T.K.; Lakhsassi, K.Y.; Dees, D. Effects of lithium difluoro(oxalate)borate on the performance of Li-rich composite cathode in Li-ion battery. Electrochem. Commun. 2012, 24, 78–81. [Google Scholar] [CrossRef]
- Herzig, T.; Schreiner, C.; Bruglachner, H.; Jordan, S.; Sclunidt, M.; Gores, H.J. Temperature and concentration dependence of conductivities of some new semichelatoborates in acetonitrile and comparison with other borates. J. Chem. Eng. Data. 2008, 53, 434–438. [Google Scholar] [CrossRef]
- Kraemer, E.P.; Winter, M. Dependency of aluminum collector corrosion in lithium ion batteries on the electrolyte solvent. ECS Electrochem. Letters. 2012, 1, C9–C11. [Google Scholar] [CrossRef]
- Amereller, M.; Multerer, M.; Schreiner, C.; Lodermeyer, J.; Schmid, A.; Barthel, J.; Gores, H.J. Investigation of the hydrolysis of lithium bis[1,2-oxalato(2-)O,O'] borate (LiBOB) in water and acetonitrile by conductivity and NMR measurements in comparison to some other borates. J. Chem. Eng. Data. 2009, 54, 468–471. [Google Scholar] [CrossRef]
- Terborg, L.; Nowak, S.; Passerini, S.; Winter, M.; Karst, U.; Haddad, P.R.; Nesterenko, P.N. Ion chromatographic determination of hydrolysis products of hexafluorophosphate salts in aqueous solution. Anal. Chim. Acta. 2012, 714, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Lux, S.F.; Lucas, I.T.; Pollak, E.; Passerini, S.; Winter, M.; Kostecki, R. The mechanism of HF formation in LiPF6 based organic carbonate electrolytes. Electrochem. Commun. 2012, 14, 47–50. [Google Scholar] [CrossRef]
- Winter, M.; Besenhard, J.O. Rechargeable batteries. Chem. Unserer Zeit. 1999, 33, 320–332. [Google Scholar] [CrossRef]
- Wagner, R.; Preschitschek, N.; Passerini, S.; Leker, J.; Winter, M. Current research trends and prospects among the various materials and designs used in lithium-based batteries. J. Appl. Electrochem. 2013, 1, 1–16. [Google Scholar]
- Zhu, Y.; Li, Y.; Bettge, M.; Abraham, D.P. Positive electrode passivation by LiDFOB electrolyte additive in high-capacity lithium-ion cells. J. Electrochem. Soc. 2012, 159, A2109–A2117. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; John Wiley & Sons: New York, NY, USA, 2001; pp. 60–62. [Google Scholar]
- Ohue, K.; Utsunomiya, T.; Hatozaki, O.; Yoshimoto, N.; Egashira, M.; Morita, M. Self-discharge behavior of polyacenic semiconductor and graphite negative electrodes for lithium-ion batteries. J. Power Sources. 2011, 196, 3604–3610. [Google Scholar] [CrossRef]
- Winter, M. The solid electrolyte interphase—The most important and the least understood solid electrolyte in rechargeable Li batteries. Z. Phys. Chem. 2009, 223, 1395–1406. [Google Scholar] [CrossRef]
- Morita, M.; Aoki, S.; Matsuda, Y. AC-impedance behavior of lithium electrode in organic electrolyte-solutions containing additives. Electrochimica Acta. 1992, 37, 119–123. [Google Scholar] [CrossRef]
- Xu, K. Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 2004, 104, 4303–4417. [Google Scholar] [CrossRef] [PubMed]
- Aurbach, D.; Ein Eli, Y.; Markovsky, B.; Zaban, A.; Luski, S.; Carmeli, Y.; Yamin, H. The study of electrolyte-solutions based on ethylene and diethyl carbonates for rechargeable Li batteries. 2. Graphite-Electrodes. J. Electrochem. Soc. 1995, 142, 2882–2890. [Google Scholar] [CrossRef]
- Aurbach, D.; Zaban, A.; Schechter, A.; Ein Eli, Y.; Zinigrad, E.; Markovsky, B. The Study of Electrolyte-Solutions Based on Ethylene and Diethyl Carbonates for Rechargeable Li Batteries.1. Li Metal Anodes. J. Electrochem. Soc. 1995, 142, 2873–2882. [Google Scholar] [CrossRef]
- Basile, A.; Hollenkamp, A.F.; Bhatt, A. I.; O'Mullane, A.P. Extensive charge–discharge cycling of lithium metal electrodes achieved using ionic liquid electrolytes. Electrochem. Commun. 2013, 27, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Rauh, R.D.; Brummer, S.B. Effect of additives on lithium cycling in propylene carbonate. Electrochim. Acta. 1977, 22, 75–83. [Google Scholar] [CrossRef]
- Atkins, P.W. Physikalische Chemie, 3rd ed.; WILEY-VCH Verlag: Weinheim, Germany, 2001; pp. 938–942. [Google Scholar]
- Mark, B.T.; Orazem, E. Electrochemical Impedance Spectroscopy, 1st ed.; John Willey & Sons: Hoboken, NJ, USA, 2008; pp. 60–97. [Google Scholar]
- Aurbach, D.; Zaban, A. Impedance spectroscopy of lithium electrodes. 1. General behavior in propylene carbonate solutions and the correlation to surface-chemistry and cycling efficiency. J. Electroanal. Chem. 1993, 348, 155–179. [Google Scholar] [CrossRef]
- Schreiner, C.; Amereller, M.; Gores, H.J. Chloride-free method to synthesise new ionic liquids with mixed borate anions. Chemistry A Eur. J. 2009, 15, 2270–2272. [Google Scholar] [CrossRef]
- Herzig, T.; Schreiner, C.; Gerhard, D.; Wasserscheid, P.; Gores, H.J. Characterisation and properties of new ionic liquids with the difluoromono[1,2-oxalato(2-)-O,O']borate anion. J. Fluor. Chem. 2007, 128, 612–618. [Google Scholar] [CrossRef]
- Kologo, S.; Eyraud, M.; Bonou, L.; Vacandio, F.; Massiani, Y. Voltametry and EQCM study of copper oxidation in acidic solution in presence of chloride ions. Electrochim. Acta. 2007, 52, 3105–3113. [Google Scholar] [CrossRef]
- Moosbauer, D. Electrochemical Characterization of Electrolytes and Electrodes for Lithium-Ion-Batteries: Development of a New Measurement Method for Electrochemical Investigations on Electrodes with the EQCM. Ph.D. Dissertation, University of Regensburg, Regensburg, Germany, 2010. [Google Scholar]
- Vatankhah, G.; Lessard, J.; Jerkiewicz, G.; Zolfaghari, A.; Conway, B.E. Dependence of the reliability of electrochemical quartz-crystal nanobalance mass responses on the calibration constant, C-f: Analysis of three procedures for its determination. Electrochim. Acta. 2003, 48, 1613–1622. [Google Scholar] [CrossRef]
© 2013 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schedlbauer, T.; Hoffmann, B.; Krüger, S.; Gores, H.J.; Winter, M. Results from a Novel Method for Corrosion Studies of Electroplated Lithium Metal Based on Measurements with an Impedance Scanning Electrochemical Quartz Crystal Microbalance. Energies 2013, 6, 3481-3505. https://doi.org/10.3390/en6073481
Schedlbauer T, Hoffmann B, Krüger S, Gores HJ, Winter M. Results from a Novel Method for Corrosion Studies of Electroplated Lithium Metal Based on Measurements with an Impedance Scanning Electrochemical Quartz Crystal Microbalance. Energies. 2013; 6(7):3481-3505. https://doi.org/10.3390/en6073481
Chicago/Turabian StyleSchedlbauer, Tanja, Björn Hoffmann, Steffen Krüger, Heiner Jakob Gores, and Martin Winter. 2013. "Results from a Novel Method for Corrosion Studies of Electroplated Lithium Metal Based on Measurements with an Impedance Scanning Electrochemical Quartz Crystal Microbalance" Energies 6, no. 7: 3481-3505. https://doi.org/10.3390/en6073481