
Hydrazine Borane and Hydrazinidoboranes as Chemical Hydrogen Storage Materials

Abstract
:1. Introduction
2. Brief Historical View of Hydrazine Borane
3. Hydrazine Borane
3.1. Synthesis
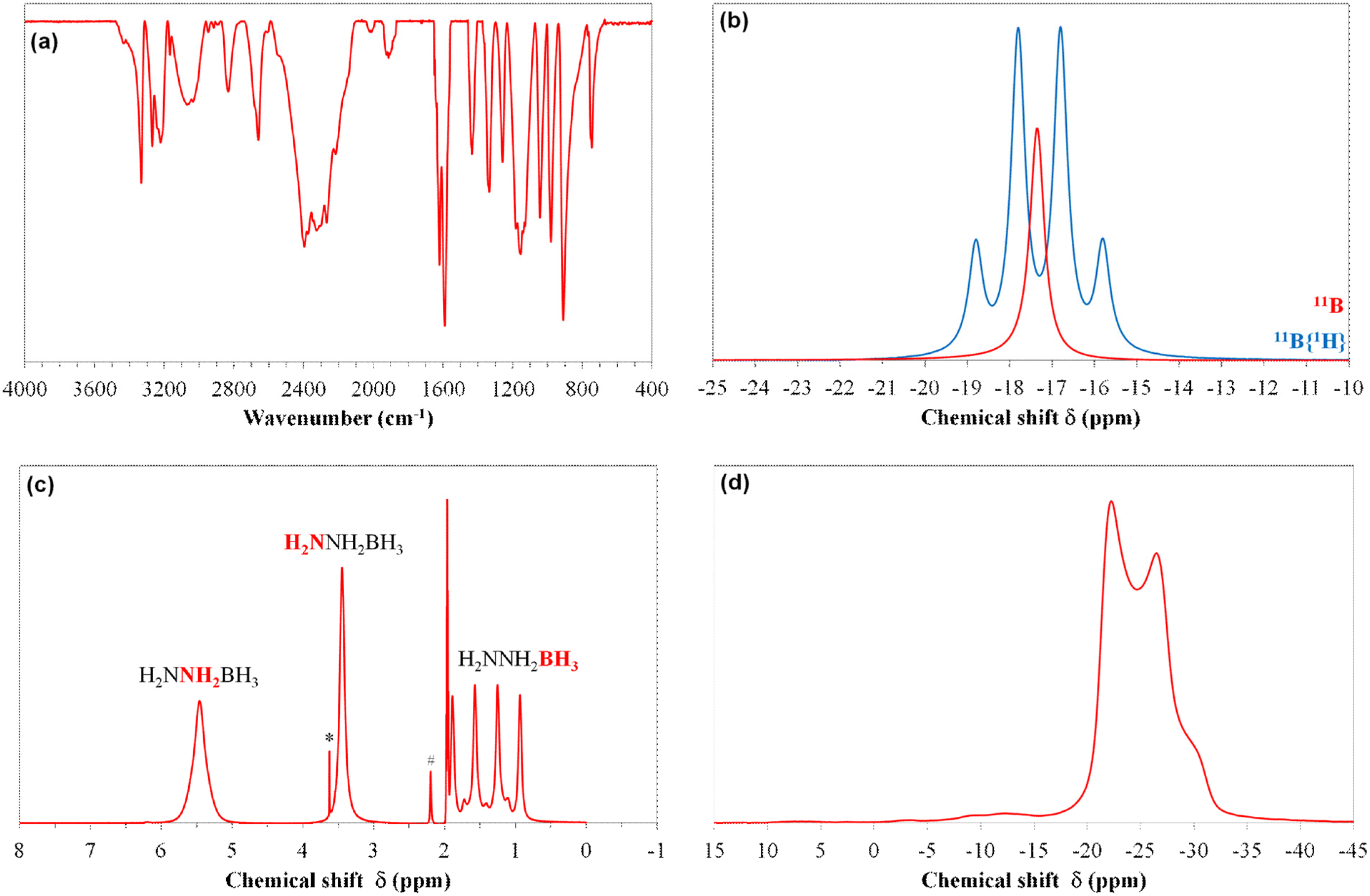
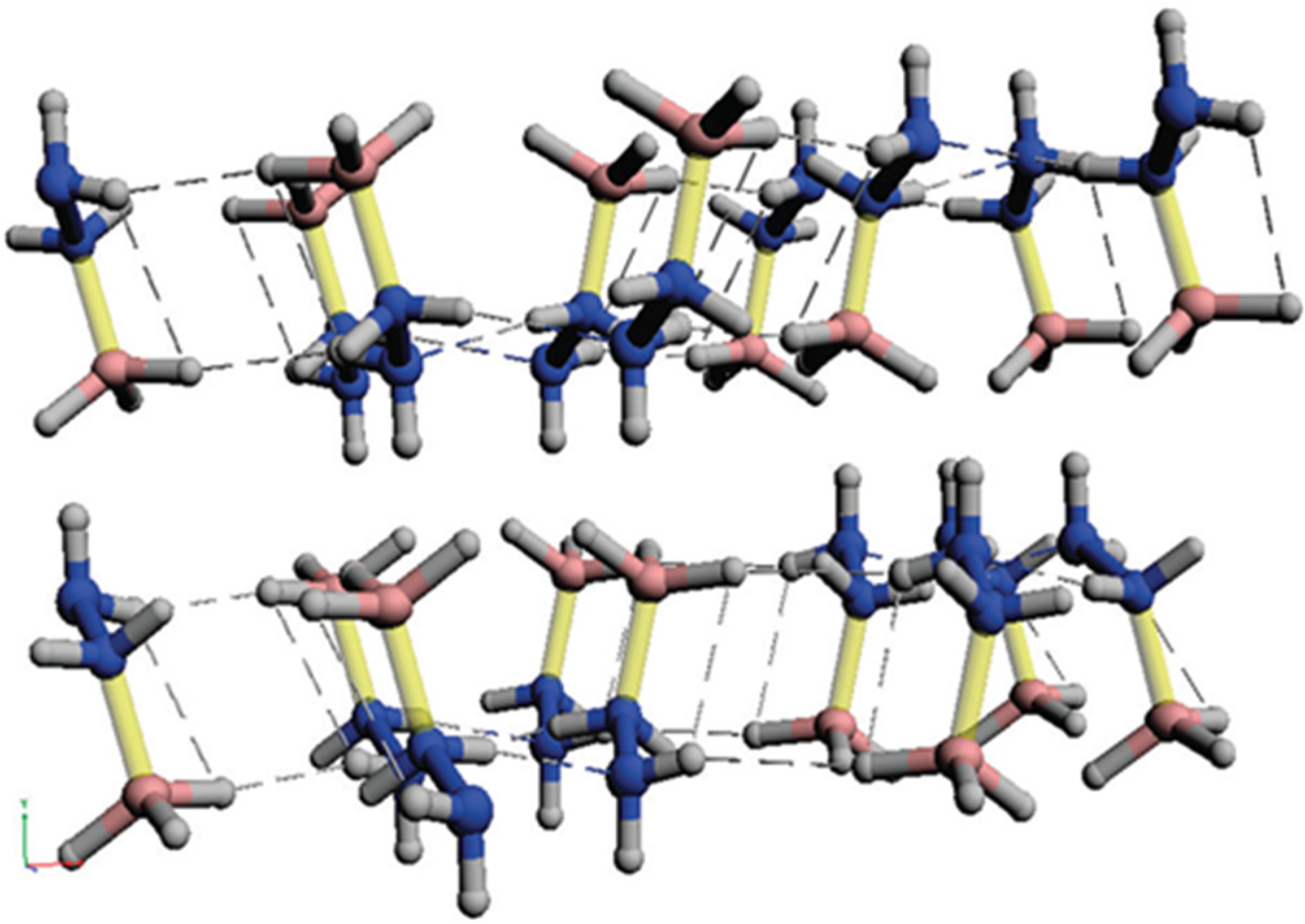
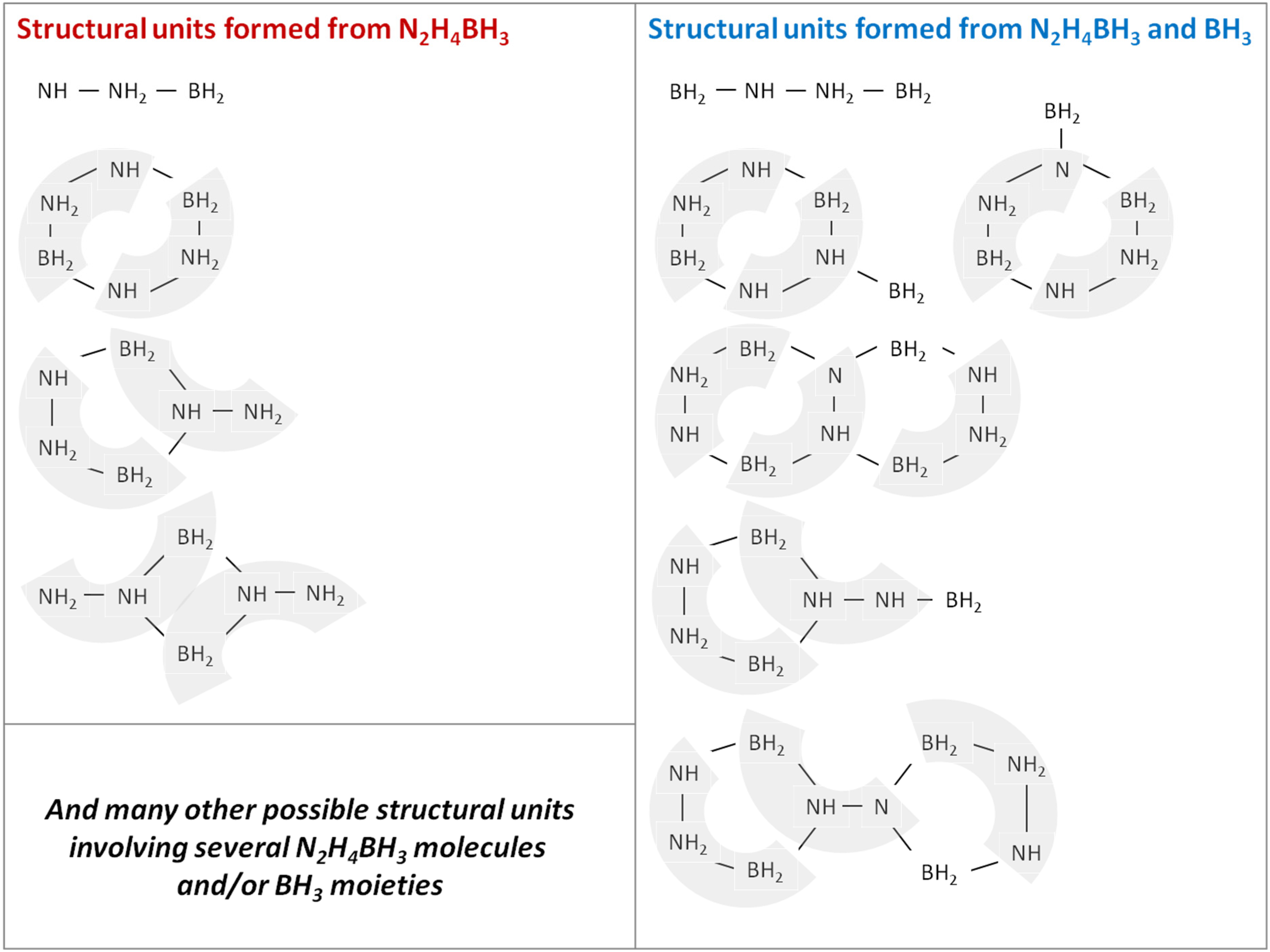
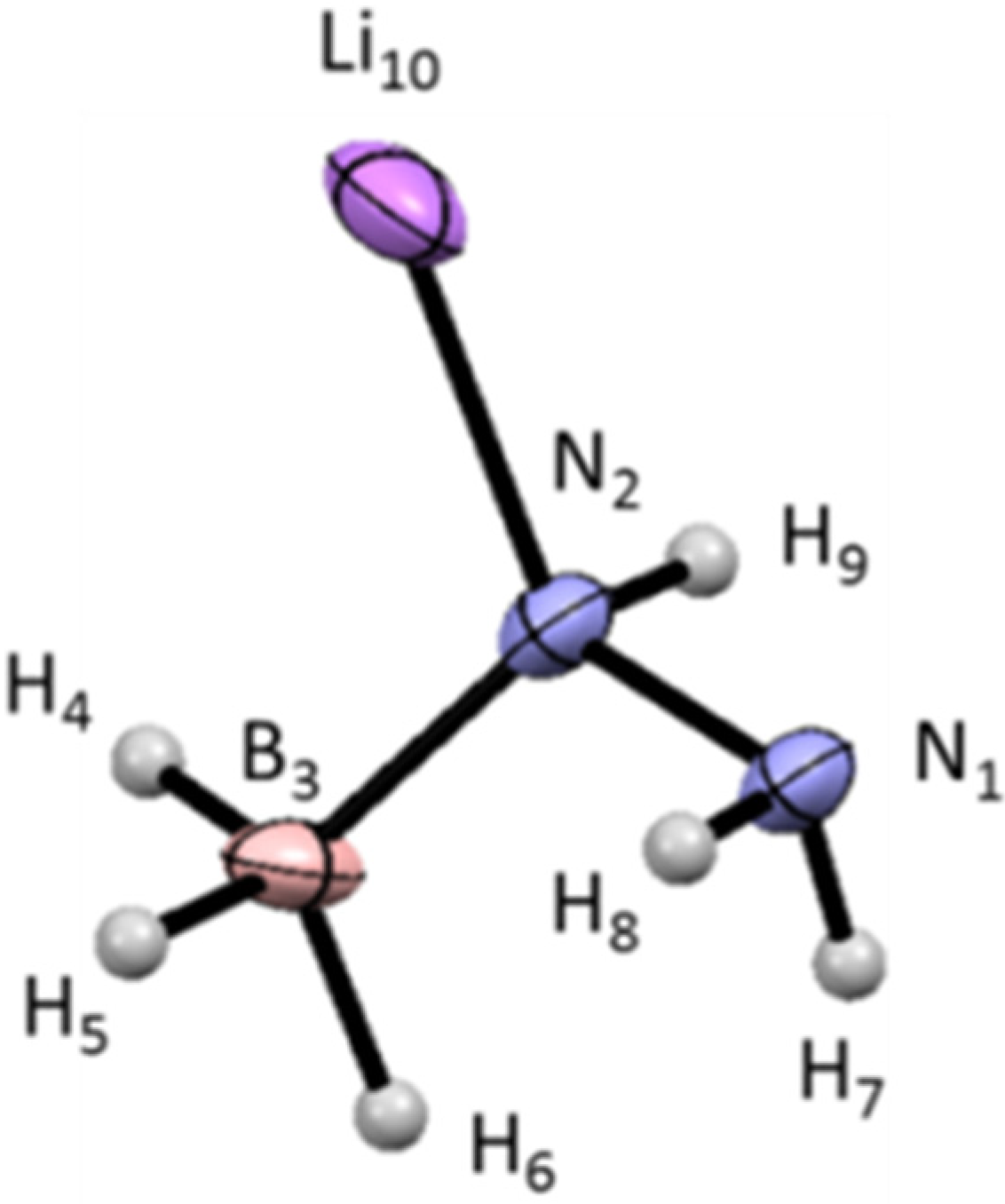
3.2. Molecular and Structural Analyses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | HB in ref. [10] | HB in ref. [31] | HB in ref. [24] | HB in ref. [32] |
|---|---|---|---|---|
| Analyzed sample | Single crystal | Single crystal | Single crystal | Powder |
| Crystal size (mm3) | 2.5 × 0.5 × 0.5 | 0.3 × 0.3 × 0.2 | 0.45 × 0.5 × 0.5 | – |
| Temperature (K) | not given | 95 | 173 | Room |
| Crystal system | Orthorhombic | Orthorhombic | Orthorhombic | Orthorhombic |
| Space group (No.) | Pccn (56) | Pbcn (60) | Pbcn (60) | Pbcn (60) |
| Z | 8 | 8 | 8 | 8 |
| a (Å) | 13.05 | 12.974(2) | 12.9788(5) | 13.1227(11) |
| b (Å) | 5.12 | 5.070(1) | 5.0616(2) | 5.1000(5) |
| c (Å) | 9.55 | 9.507(1) | 9.5087(4) | 9.5807(9) |
| B–N bond (Å) | – | 1.596 | 1.587 | 1.592 |
| N–N bond (Å) | – | 1.452 | 1.452 | 1.458 |

3.3. Stability and Solubility
4. Liquid-State Chemical Hydrogen Storage
4.1. Introductive Remark
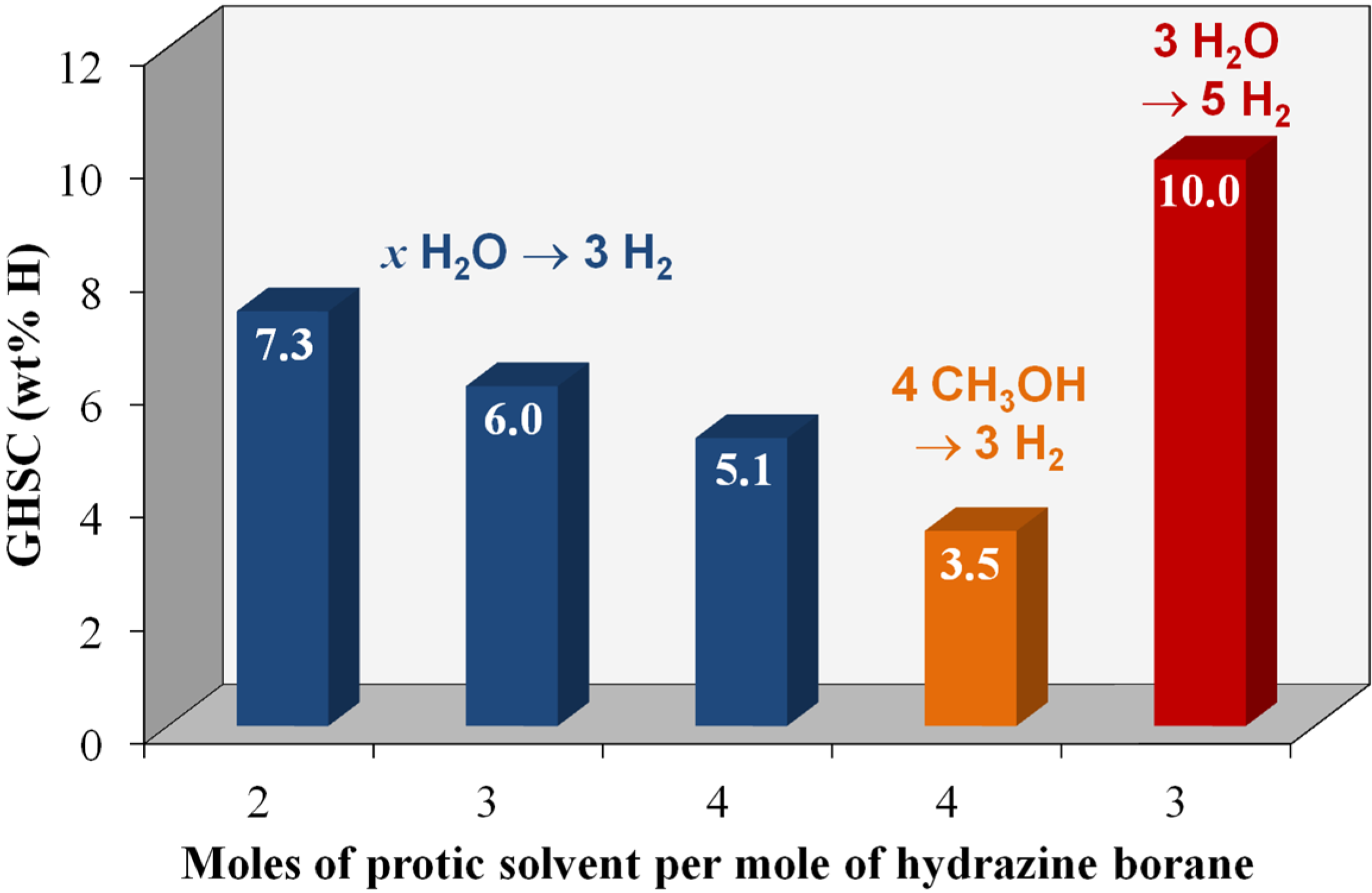
4.2. Hydrolysis of the BH3 Group of Hydrazine Borane

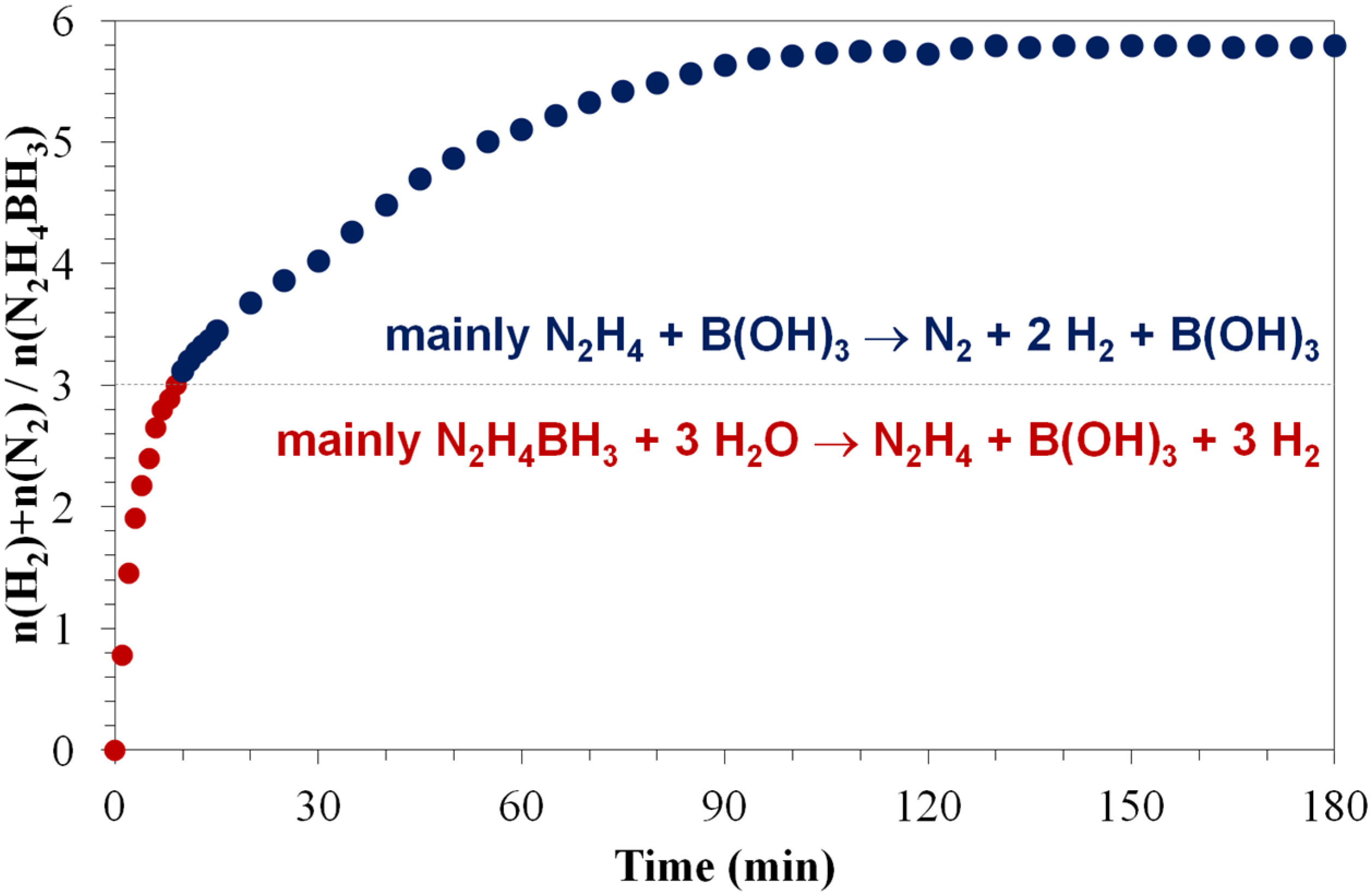
4.3. Hydrolysis of the BH3 Group and Dehydrogenation of the N2H4 Moiety of Hydrazine Borane

5. Solid-State Chemical Hydrogen Storage
5.1. Pristine Hydrazine Borane

5.2. Chemical Doping of Hydrazine Borane
5.3. Dispersion and Catalysis of Hydrazine Borane
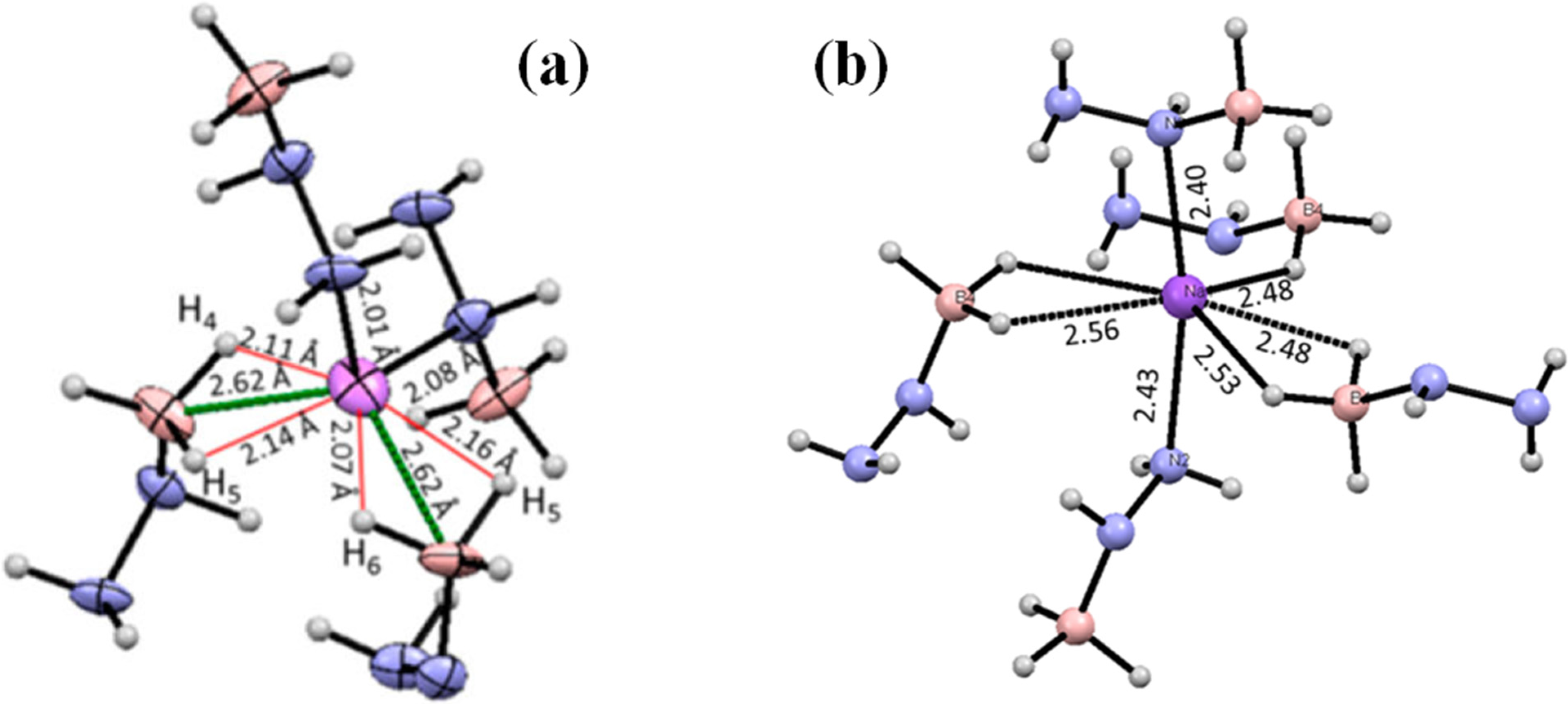
5.4. Chemical Modification of Hydrazine Borane

| MN2H3BH3 | α-LiN2H3BH3 | β-LiN2H3BH3 | NaN2H3BH3 | KN2H3BH3 |
|---|---|---|---|---|
| Reference | [32,66] | [66] | [68] | [69] |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic |
| Space group (No.) | P21/c (14) | Pbca (61) | P21/n (14) | P21 (4) |
| a (Å) | 5.8503(11) | 10.25182(11) | 4.97437(11) | 6.72102(23) |
| b (Å) | 7.4676(11) | 8.47851(10) | 7.95806(15) | 5.89299(20) |
| c (Å) | 8.8937(15) | 7.46891(8) | 9.29232(19) | 5.77795(17) |
| β (°) | 122.329(6) | – | 93.8137(11) | 108.2595(13) |
| B–N bond (Å) | 1.539 | 1.549(2) | 1.537(6) | 1.541 |
| N–N bond (Å) | 1.469 | 1.495(2) | 1.453(5) | 1.463 |


6. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Conte, M.; di Mario, F.; Iacobazzi, A.; Mattucci, A.; Moreno, A.; Ronchetti, M. Hydrogen as future energy carrier: The ENEA point of view on technology and application prospects. Energies 2009, 2, 150–179. [Google Scholar] [CrossRef]
- Armaroli, N.; Balzani, V. The hydrogen issue. ChemSusChem 2011, 4, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Mazloomi, K.; Gomes, C. Hydrogen as an energy carrier: Prospects and challenges. Renew. Sustain. Energy Rev. 2012, 16, 3024–3033. [Google Scholar] [CrossRef]
- Eberle, U.; Felderhoff, M.; Schüth, F. Chemical and physical solutions for hydrogen storage. Angew. Chem. Int. Ed. 2009, 48, 6608–6630. [Google Scholar] [CrossRef]
- Dalebrook, A.F.; Gan, W.; Grasemann, M.; Moret, S.; Laurenczy, G. Hydrogen storage: Beyond conventional methods. Chem. Commun. 2013, 49, 8735–8751. [Google Scholar] [CrossRef]
- Chamoun, R.; Demirci, U.B.; Miele, P. Cyclic dehydrogenation-(re)hydrogenation with hydrogen storage materials: An overview. Energy Technol. 2015. [Google Scholar] [CrossRef]
- Chang, F.; Zhou, J.; Chen, P.; Chen, Y.; Jia, H.; Saad, S.M.I.; Gao, Y.; Cao, X.; Zheng, T. Microporous and mesoporous materials for gas storage and separation: A review. Asia Pac. J. Chem. Eng. 2013, 8, 618–626. [Google Scholar] [CrossRef]
- Li, H.W.; Yan, Y.; Orimo, S.I.; Züttel, A.; Jensen, C.M. Recent progress in metal borohydrides for hydrogen storage. Energies 2011, 4, 185–214. [Google Scholar] [CrossRef]
- Moussa, G.; Moury, R.; Demirci, U.B.; Şener, T.; Miele, P. Boron-based hydrides for chemical hydrogen storage. Int. J. Energy Res. 2013, 37, 825–842. [Google Scholar] [CrossRef]
- Goubeau, V.J.; Ricker, E. Borinhydrazin und seine pyrolyseprodukte. Z. Anorg. Allg. Chem. 1961, 310, 123–142. [Google Scholar] [CrossRef]
- Gunderloy, F.C., Jr. Hydrazine–Mono- and—Bisborane. Inorg. Synth. 1967, 9, 13–16. [Google Scholar]
- Gunderloy, F.C., Jr. Process for Preparing Hydrazine Monoborane. U.S. Patent 3375087, 26 March 1968. [Google Scholar]
- Hough, W.V.; Hashman, J.S. Borane-Hydrazine Compounds. U.S. Patent 3298799, 1967. [Google Scholar]
- Artz, G.D.; Grant, L.R. Solid Propellant Hydrogen Generator. U.S. Patent 4468263, 1984. [Google Scholar]
- Bratton, F.H.; Reynolds, H.I. Hydrogen Generating System. U.S. Patent 3419361, 1968. [Google Scholar]
- Edwards, L.J. Hydrogen Generating Composition. U.S. Patent 3450638, 1969. [Google Scholar]
- Kirpiche, E.P.; Rubtsov, Y.I.; Manelis, G.B. Standard enthalpies for hydrazine borane and hydrazine-bis-borane. Zhurnal Neorg. Khim. 1971, 16, 2064–2064. [Google Scholar]
- Borovinskaya, I.P.; Bunin, V.A.; Merzhanov, A.G. Self-propagating high-temperature synthesis of high-porous boron nitride. Mendeleev Commun. 1997, 7, 47–48. [Google Scholar] [CrossRef]
- Rasul, G.; Prakash, G.K.S.; Olah, G.A. B–H bond protonation in mono- and diprotonated borane complexes H3BX (X = N2H4, NH2OH, and H2O2) involving hypercoordinate boron. Inorg. Chem. 1999, 38, 5876–5878. [Google Scholar] [CrossRef]
- Jepsen, L.H.; Ley, M.B.; Lee, Y.S.; Cho, Y.W.; Dornheim, M.; Jensen, J.O.; Filinchuk, Y.; Jørgensen, J.E.; Besenbacher, F.; Jensen, T.R. Boron–nitrogen based hydrides and reactive composites for hydrogen storage. Mater. Today 2014, 17, 129–135. [Google Scholar] [CrossRef]
- Chua, Y.S.; Chen, P.; Wu, G.; Xiong, Z. Development of amidoboranes for hydrogen storage. Chem. Commun. 2011, 47, 5116–5129. [Google Scholar] [CrossRef]
- Hamilton, C.W.; Baker, R.T.; Staubitz, A.; Manners, I. B–N compounds for chemical hydrogen storage. Chem. Soc. Rev. 2009, 38, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Hügle, T.; Kühnel, M.F.; Lentz, D. Hydrazine borane: A promising hydrogen storage material. J. Am. Chem. Soc. 2009, 131, 7444–7446. [Google Scholar] [CrossRef] [PubMed]
- Moury, R.; Moussa, G.; Demirci, U.B.; Hannauer, J.; Bernard, S.; Petit, E.; van der Lee, A.; Miele, P. Hydrazine borane: Synthesis, characterization, and application prospects in chemical hydrogen storage. Phys. Chem. Chem. Phys. 2012, 14, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Gunderloy, F.C., Jr. Reactions of the borohydride group with the proton donors hydroxylammonium, methoxyammonium, and hydrazinium-magnesium ions. Inorg. Chem. 1963, 2, 221–222. [Google Scholar] [CrossRef]
- Gunderloy, F.C., Jr. Preparation of Solid Boron Compounds. U.S. Patent 3159451, 1 December 1964. [Google Scholar]
- Uchida, H.S.; Hefferan, G.T. Production of Hydrazine Boranes. U.S. Patent 3119652, 28 January 1964. [Google Scholar]
- Sutton, A.; Gordon, J.C.; Ott, K.C.; Burrell, A.K. Regeneration of Ammonia Borane from Polyborazylene. U.S. Patent 20100272622, 28 October 2010. [Google Scholar]
- Sutton, A.D.; Burrell, A.K.; Dixon, D.A.; Garner, E.B., III; Gordon, J.C.; Nakagawa, T.; Ott, K.C.; Robinson, J.P.; Vasiliu, M. Regeneration of ammonia borane spent fuel by direct reaction with hydrazine and liquid ammonia. Science 2011, 331, 1426–1420. [Google Scholar] [CrossRef] [PubMed]
- Karahan, S.; Zahmakiran, M.; Özkar, S. Catalytic hydrolysis of hydrazine borane for chemical hydrogen storage: Highly efficient and fast hydrogen generation system at room temperature. Int. J. Hydrog. Energy 2011, 36, 4958–4966. [Google Scholar] [CrossRef]
- Mebs, S.; Grabowsky, S.; Förster, D.; Kickbusch, R.; Hartl, M.; Daemen, L.L.; Morgenroth, W.; Luger, P.; Paulus, B.; Lentz, D. Charge transfer via the dative N–B bond and dihydrogen contacts. Experimental and theoretical electron density studies of small Lewis acid-base adducts. J. Phys. Chem. A 2010, 114, 10185–10196. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhou, W.; Pinkerton, F.E.; Udovic, T.J.; Yildirim, T.; Rush, J.J. Metal hydrazinoborane LiN2H3BH3 and LiN2H3BH3·2N2H4BH3: Crystal structures and high-extent dehydrogenation. Energy Environ. Sci. 2012, 5, 7531–7535. [Google Scholar] [CrossRef]
- Lu, Z.H.; Xu, Q. Recent progress in boron- and nitrogen-based chemical hydrogen storage. Funct. Mater. Lett. 2012, 5, 1230001. [Google Scholar] [CrossRef]
- çelik, D.; Karahan, S.; Zahmakiran, M.; Özkar, S. Hydrogen generation from the hydrolysis of hydrazine-borane catalyzed by rhodium(0) nanoparticles supported on hydroxyapatite. Int. J. Hydrog. Energy 2011, 37, 5143–5151. [Google Scholar] [CrossRef]
- Şencanli, S.; Karahan, S.; Özkar, S. Poly(4-styrene acid-co-maleic acid) stabilized nickel(0) nanoparticles: Highly active and cost effective in hydrogen generation from the hydrolysis of hydrazine borane. Int. J. Hydrog. Energy 2013, 38, 1493–14700. [Google Scholar]
- Yadav, M.; Xu, Q. Liquid-phase chemical hydrogen storage materials. Energy Environ. Sci. 2012, 5, 9698–9725. [Google Scholar] [CrossRef]
- Li, P.Z.; Xu, Q. Metal-nanoparticles catalyzed hydrogen generation from liquid-phase chemical hydrogen storage materials. J. Chin. Chem. Soc. 2012, 59, 1181–1189. [Google Scholar] [CrossRef]
- Lu, Z.H.; Yao, Q.; Zhang, Z.; Yang, Y.; Chen, X. Nanocatalysts for hydrogen generation from ammonia borane and hydrazine borane. J. Nanomater. 2014, 2014, 729029. [Google Scholar] [CrossRef]
- Moussa, G.; Moury, R.; Demirci, U.B.; Miele, P. Borates in hydrolysis of ammonia borane. Int. J. Hydrog. Energy 2013, 38, 7888–7895. [Google Scholar] [CrossRef]
- Marrero-Alfonso, E.Y.; Beaird, A.M.; Davis, T.A.; Matthews, M.A. Hydrogen generation from chemical hydrides. Ind. Eng. Chem. Res. 2009, 48, 3703–3712. [Google Scholar] [CrossRef]
- Karahan, S.; Zahmakiran, M.; Özkar, S. Catalytic methanolysis of hydrazine borane: A new and efficient hydrogen generation system under mild conditions. Dalton Trans. 2012, 41, 4918–4918. [Google Scholar] [CrossRef]
- Özhava, D.; Kiliçaslan, N.Z.; Özkar, S. PVP-stabilized nickel(0) nanoparticles as catalyst in hydrogen generation from the methanolysis of hydrazine borane or ammonia borane. Appl. Catal. B 2014, 162, 573–582. [Google Scholar] [CrossRef]
- Demirci, U.B. The hydrogen cycle with the hydrolysis of sodium borohydride: A statistical approach for highlighting the scientific/technical issues to prioritize in the field. Int. J. Hydrog. Energy 2015. [Google Scholar] [CrossRef]
- Jiang, H.L.; Xu, Q. Catalytic hydrolysis of ammonia borane for chemical hydrogen storage. Catal. Today 2011, 170, 56–63. [Google Scholar] [CrossRef]
- Singh, S.K.; Xu, Q. Nanocatalysts for hydrogen generation from hydrazine. Catal. Sci. Technol. 2013, 3, 1889–1900. [Google Scholar] [CrossRef]
- Hannauer, J.; Akdim, O.; Demirci, U.B.; Geantet, C.; Herrmann, J.M.; Miele, P.; Xu, Q. High-extent dehydrogenation of hydrazine borane N2H4BH3 by hydrolysis of BH3 and decomposition of N2H4. Energy Environ. Sci. 2011, 4, 3355–3358. [Google Scholar] [CrossRef]
- Çakanyildirim, Ç.; Petit, E.; Demirci, U.B.; Moury, R.; Petit, J.F.; Xu, Q.; Miele, P. Gaining insight into the catalytic dehydrogenation of hydrazine borane in water. Int. J. Hydrog. Energy 2012, 37, 15983–15991. [Google Scholar]
- Zhong, D.C.; Aranishi, K.; Singh, A.K.; Demirci, U.B.; Xu, Q. The synergistic effect of Rh-Ni catalysts on the highly-efficient dehydrogenation of aqueous hydrazine borane for chemical hydrogen storage. Chem. Commun. 2012, 48, 11945–11947. [Google Scholar] [CrossRef]
- Hannauer, J.; Demirci, U.B.; Geantet, C.; Herrmann, J.M.; Miele, P. Transition metal-catalyzed dehydrogenation of hydrazine borane N2H4BH3 via the hydrolysis of BH3 and the decomposition of N2H4. Int. J. Hydrog. Energy 2012, 37, 10758–10767. [Google Scholar] [CrossRef]
- Çakanyıldırım, Ç.; Demirci, U.B.; Xu, Q.; Miele, P. Supported nickel catalysts for the decomposition of hydrazine borane N2H4BH3. Adv. Energy Res. 2013, 1, 1–12. [Google Scholar] [CrossRef]
- Clémençon, D.; Petit, J.F.; Demirci, U.B.; Xu, Q.; Miele, P. Nickel- and platinum-containing core@shell catalysts for hydrogen generation of aqueous hydrazine borane. J. Power Sourc. 2014, 260, 77–81. [Google Scholar] [CrossRef]
- Çakanyıldırım, Ç.; Demirci, U.B.; Şener, T.; Xu, Q.; Miele, P. Nickel-based bimetallic nanocatalysts in high-extent dehydrogenation of hydrazine borane. Int. J. Hydrog. Energy 2012, 37, 9722–9729. [Google Scholar] [CrossRef]
- Ben Aziza, W.; Demirci, U.B.; Xu, Q.; Miele, P. Bimetallic nickel-based nanocatalysts for hydrogen generation from aqueous hydrazine borane: Investigation of iron, cobalt and palladium as the second metal. Int. J. Hydrog. Energy 2014, 39, 16919–16926. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.T.; Kang, Y.M.; Kim, H.S.; Song, M.S.; Lee, Y.J.; Lee, P.S.; Lee, J.Y. Study on degradation of filamentary Ni catalyst on hydrolysis of sodium borohydride. J. Alloys Compd. 2004, 379, 222–227. [Google Scholar] [CrossRef]
- Demirci, U.B.; Miele, P. Cobalt in NaBH4 hydrolysis. Phys. Chem. Chem. Phys. 2010, 12, 14651–14665. [Google Scholar] [CrossRef] [PubMed]
- Demirci, U.B.; Miele, P. Cobalt-based catalysts in hydrolysis of NaBH4 and NH3BH3. Phys. Chem. Chem. Phys. 2014, 16, 6872–6885. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Dou, Y.; Liu, J.; Chen, Y.; He, S.; Wei, M.; Evans, D.G.; Duan, X. Synthesis of supported Ni@(RhNi-alloy) nanocomposites as an efficient catalyst towards hydrogen generation from N2H4BH3. Chem. Commun. 2013, 49, 9992–9994. [Google Scholar] [CrossRef]
- Zhu, Q.L.; Zhong, D.C.; Demirci, U.B.; Xu, Q. Controlled synthesis of ultrafine surfactant-free NiPt nanocatalysts towards efficient and complete hydrogen generation from hydrazine borane at room temperature. ACS Catal. 2014, 4, 4261–4268. [Google Scholar] [CrossRef]
- Vinh-Son, N.; Swinnen, S.; Matus, M.H.; Nguyen, M.T.; Dixon, D.A. The effect of the NH2 substituent on NH3: Hydrazine as an alternative for ammonia in hydrogen release in the presence of boranes and alanes. Phys. Chem. Chem. Phys. 2009, 11, 6339–6344. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Shreeve, J.M. Ionic liquid solubilized boranes as hypergolic fluids. J. Mater. Chem. 2012, 22, 11022–11024. [Google Scholar] [CrossRef]
- Zhang, Q.; Shreeve, J.M. Ionic liquid propellants: Future fuels for space propulsion. Chem. Eur. J. 2013, 19, 15446–15451. [Google Scholar] [CrossRef] [PubMed]
- Toche, F.; Chiriac, R.; Demirci, U.B.; Miele, P. Borohydride-induced destabilization of hydrazine borane. Int. J. Hydrog. Energy 2014, 39, 9321–9329. [Google Scholar] [CrossRef]
- Frueh, S.; Kellett, R.; Mallery, C.; Molter, T.; Willis, W.S.; King’ondu, C.; Suib, S.L. Pyrolytic decomposition of ammonia borane to boron nitride. Inorg. Chem. 2011, 50, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.F.; Moussa, G.; Demirci, U.B.; Toche, F.; Chiriac, R.; Miele, P. Hydrazine borane-induced destabilization of ammonia borane, and vice versa. J. Hazard. Mater. 2014, 278, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Klahn, M.; Spannenberg, A.; Beweries, T. Group 4 metallocene catalysed full dehydrogenation of hydrazine borane. Dalton Trans. 2013, 42, 14668–14672. [Google Scholar] [CrossRef] [PubMed]
- Moury, R.; Demirci, U.B.; Ban, V.; Filinchuk, Y.; Ichikawa, T.; Zeng, L.; Goshome, K.; Miele, P. Lithium hydrazinidoborane: A polymorphic material with potential for chemical hydrogen storage. Chem. Mater. 2014, 26, 3249–3255. [Google Scholar] [CrossRef]
- Qian, Z.; Pathak, B.; Ahuja, R. Energetic and structural analysis of N2H4BH3 inorganic solid and its modified material for hydrogen storage. Int. J. Hydrog. Energy 2013, 38, 6718–6725. [Google Scholar] [CrossRef]
- Moury, R.; Demirci, U.B.; Ichikawa, T.; Filinchuk, Y.; Chiriac, R.; van der Lee, A.; Miele, P. Sodium hydrazinidoborane: A chemical hydrogen-storage material. ChemSusChem 2013, 6, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Chua, Y.S.; Pei, Q.; Ju, X.; Zhou, W.; Udovic, T.J.; Wu, G.; Xiong, Z.; Chen, P.; Wu, H. Alkali metal hydride modification on hydrazine borane for improved dehydrogenation. J. Phys. Chem. C 2014, 118, 11244–11251. [Google Scholar] [CrossRef]
- Klooster, W.T.; Koetzle, T.F.; Siegbahn, P.E.M.; Richardson, T.B.; Crabtree, R.H. Study of the N–H···H–B dihydrogen bond including the crystal structure of BH3NH3 by neutron diffraction. J. Am. Chem. Soc. 1999, 121, 6337–6343. [Google Scholar] [CrossRef]
- Al-Kukhun, A.; Hwang, H.T.; Varma, A. Mechanistic studies of ammonia borane dehydrogenation. Int. J. Hydrog. Energy 2013, 38, 169–179. [Google Scholar] [CrossRef]
- Tan, Y.; Chen, X.; Chen, J.; Gu, Q.; Yu, X. The decomposition of α-LiN2H3BH3: An unexpected hydrogen release from a homopolar proton-proton pathway. J. Mater. Chem. A 2014, 2, 15627–15632. [Google Scholar] [CrossRef]
- Moury, R.; Petit, J.F.; Demirci, U.B.; Ichikawa, T.; Miele, P. Pure hydrogen-generating “doped” sodium hydrazinidoborane. Int. J. Hydrog. Energy 2015. [Google Scholar] [CrossRef]
- Unpublished results. Typically, 10 mg of hydrazine borane were weighted and transferred in an agate mortar in an argon-filled glove box where the water and oxygen concentrations were kept below 0.1 ppm. Then, KH was added onto hydrazine borane with the help of a spatula. The addition was made carefully, almost grain by grain, and at each addition the reactivity was immediate, with an explosive formation of a brown gas and dispersion of products around the mortar.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moury, R.; Demirci, U.B. Hydrazine Borane and Hydrazinidoboranes as Chemical Hydrogen Storage Materials. Energies 2015, 8, 3118-3141. https://doi.org/10.3390/en8043118
Moury R, Demirci UB. Hydrazine Borane and Hydrazinidoboranes as Chemical Hydrogen Storage Materials. Energies. 2015; 8(4):3118-3141. https://doi.org/10.3390/en8043118
Chicago/Turabian StyleMoury, Romain, and Umit B. Demirci. 2015. "Hydrazine Borane and Hydrazinidoboranes as Chemical Hydrogen Storage Materials" Energies 8, no. 4: 3118-3141. https://doi.org/10.3390/en8043118