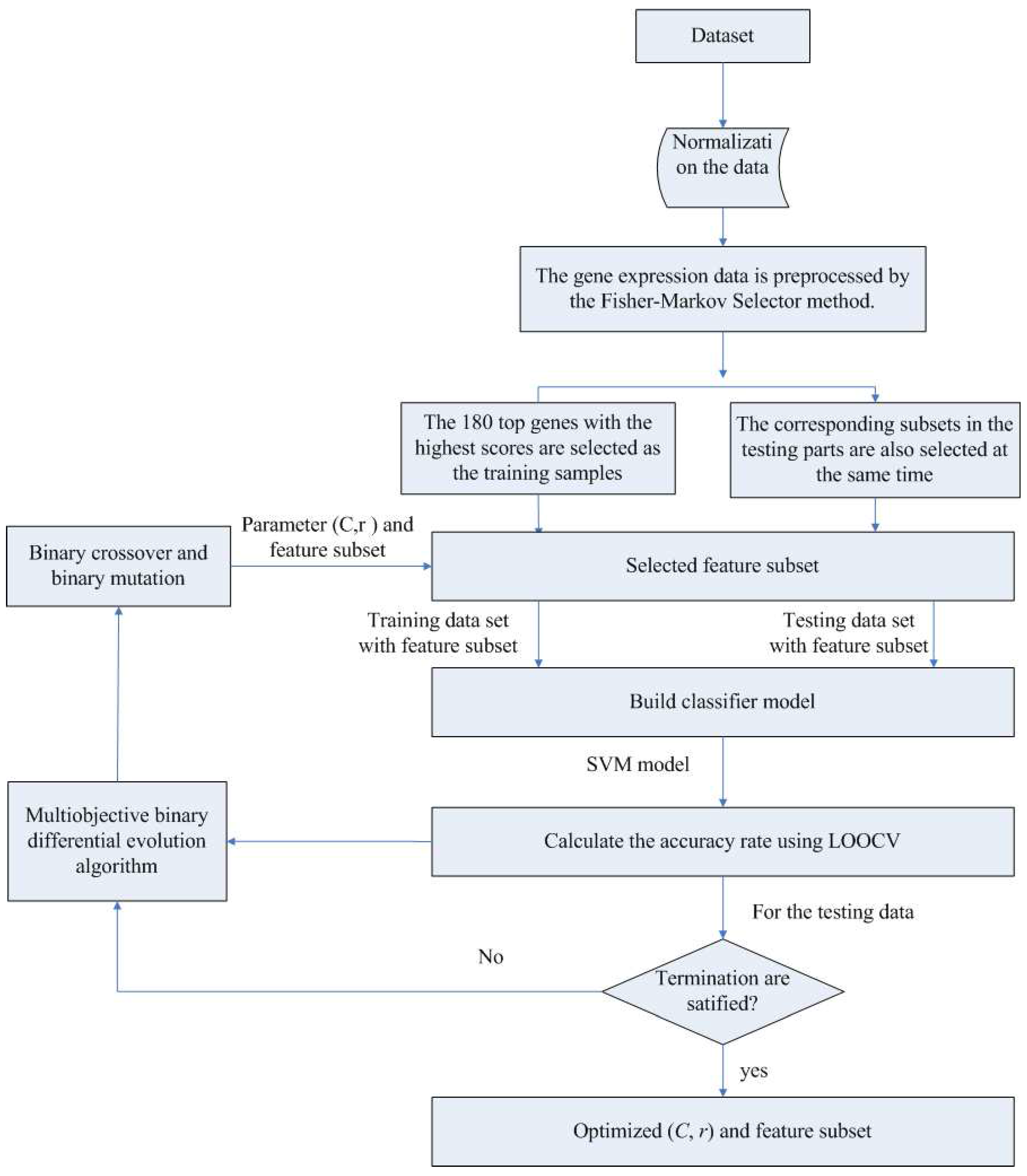
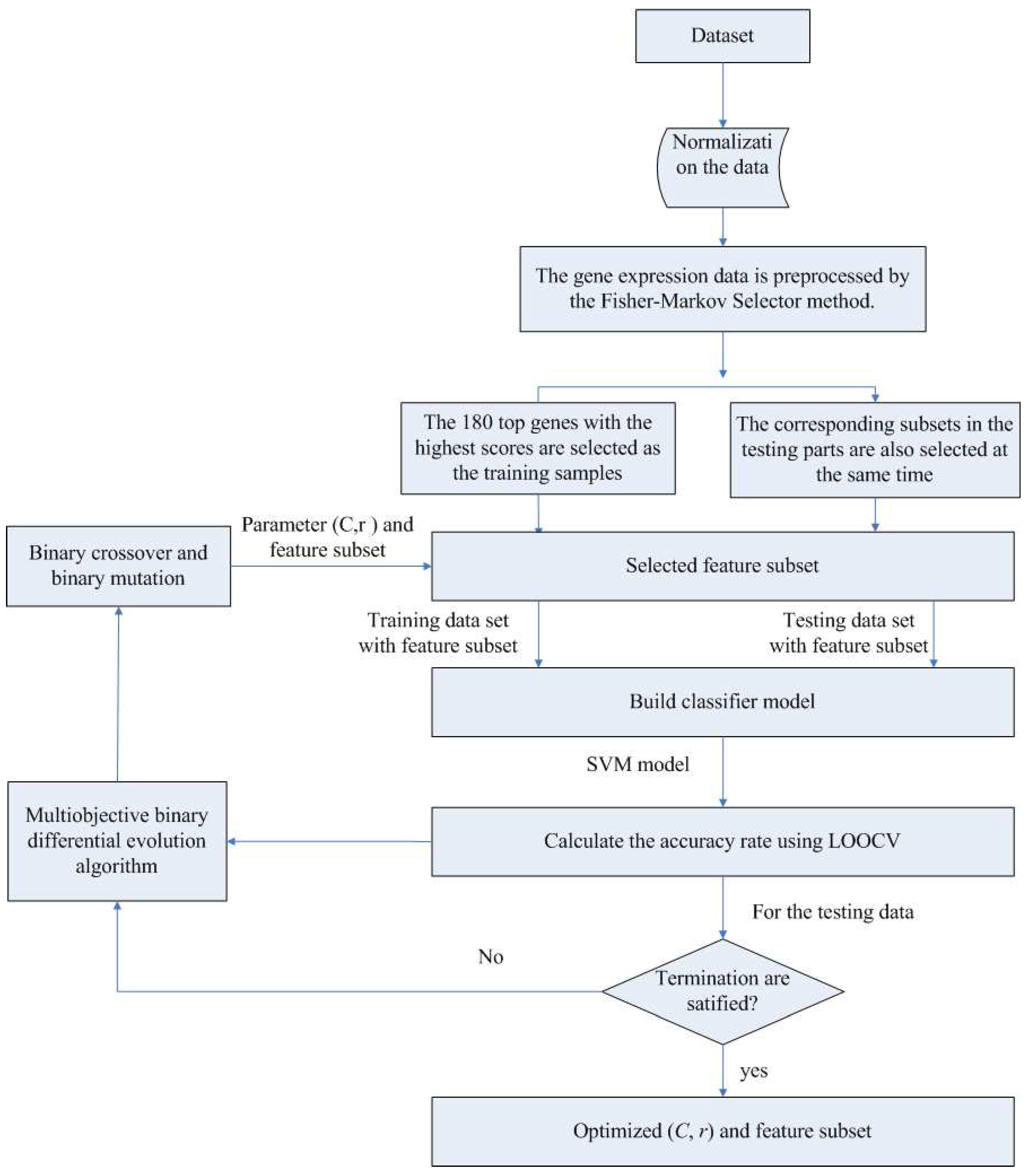
In the first component, the Fisher-Markov selector method is used to select 180 top genes with the highest scores. These selected genes will then be utilized for the second component, multiobjective binary differential evolution component. In this component, at first, a randomly-generated initial solution will be represented by a binary (0/1) string. Then, a novel binary mutation method is proposed to balance the exploration and exploitation ability during the search process. After that, the multiobjective binary differential evolution is proposed by integrating the summation of normalized objectives and diversity selection into the algorithm.
By using MOBDE, the parameters of the support vector machine (SVM) in the third component and the features subset are dynamically optimized. Specifically, for feature selection, each gene is represented as a bit of binary encoded individual, where one denotes a gene selected and zero denotes a non-selected gene. For SVM, two important parameters of RBF kernels, i.e.,
c and
γ, are taken into account. In this sense, the length of each individual is equal to
D + 2, where
D is the number of genes in the initial microarray dataset.
Table 1 shows the solution representation of the algorithm.
2.2. Multiobjective Differential Evolution Component
In this part, we shall introduce the proposed multiobjective binary differential evolution algorithm in detail.
As we know, differential evolution (DE) is a fairly novel population-based search heuristic, which is simple to implement and requires little parameter tuning compared with other search heuristics in continuous space.
The process of DE can be summarized into three major steps: mutation, cross-over and selection. In the mutation operator, the process of generating the mutation vectors
is performed by target vector
in the current population, where
D denotes the dimension of the individual,
i denotes the
i-th of individual and
G denotes the current iteration of the algorithm. In the DE algorithm, “DE/rand/1/bin” is the most common mutation strategies, as below:
where
,
and
F is the mutation factor of the differential evolution.
is the size of the population.
In the cross-over operation, a recombination of the candidate solution
and the parent
produces an offspring solution
. Usually, the binomial cross-over is accepted, which is defined as follows:
where
;
is a random number between zero and one;
is a randomly chosen index.
is the cross-over rate.
A greedy selection is used to choose the next population
between the parent population and the offspring population. The selection operation is described as follows:
As we know, the original differential evolution algorithm is a continuous optimization algorithm, but the feature selection problem is a classic binary optimization problem. Therefore, the original continuous encoding scheme of DE cannot be used directly for gene selection problems. In order to make DE suitable to solve the gene selection problem, a binary differential evolution (BDE) algorithm is proposed first. In the proposed method, the initial population is represented as a vector in which each bit is a binary value of zero or one, where one denotes this gene is selected and zero denotes a non-selected gene. The objective function values are calculated, and then, new binary populations are transported into the mutation operators. The binary cross-over operations are used to generate the trail solution. Finally, greedy selection method is used to choose the better results for the next generation.
During the reconstruction of the mutation operation, the key idea is to use some appropriate operators in place of the arithmetic operators. In [
33], He and Han used the XOR, AND and OR operations instead of the subtraction, multiplication and addition operations in the formula, which can be described as follows:
where ⊕ denotes the XOR operations, ⊗ represents the AND operation and ⊙ denotes the OR operation. Note that in Formula (
5), the use of OR operation will make the probability of a result be true. The probability of the binary “1” will be three times higher than the probability to be false (binary “0”). In other words, the binary “1” would be easily accumulated with the binary string
of the trial solution after the OR operation. This would decrease the diversity of the algorithm. Accordingly, in [
34], another novel mutation operation is proposed by considering the distance of the
and
for each dimension:
Compared with Formula (
5), the new mutation strategy can enhance the diversity of the algorithm because it does not use the OR operation. However, in this formula, the value of the previous generation will be discarded. Therefore, it cannot inherit the advantage of the original individual from the previous population.
Therefore, in this paper, we propose a new mutation strategy, which can both increase the diversity of the algorithm and take advantage of the original population, as described in the following:
As can be seen in this formula, first, it does not use the OR operation, so this operation will not harm the diversity of the algorithm. Second, the values of the previous generation, e.g., , , , will be kept with a probability. In this way, the algorithm can inherit the advantage of the original individual from the previous population. Following the binary mutation strategy, a binary cross-over operator is used to build a trial solution by combining the mutation vector and the target vector. The concept of the binary cross-over mechanism of BDE is similar to that of the original DE, though there is a difference in terms of the component data type. In BDE, the binary data is selected from the mutation vector if a random number is smaller than the cross-over rate; otherwise, the original solution is chosen to generate the trail solution. After the binary mutation and cross-over operator, the better solutions between the trial solution and the target solution will be retained to the next generation.
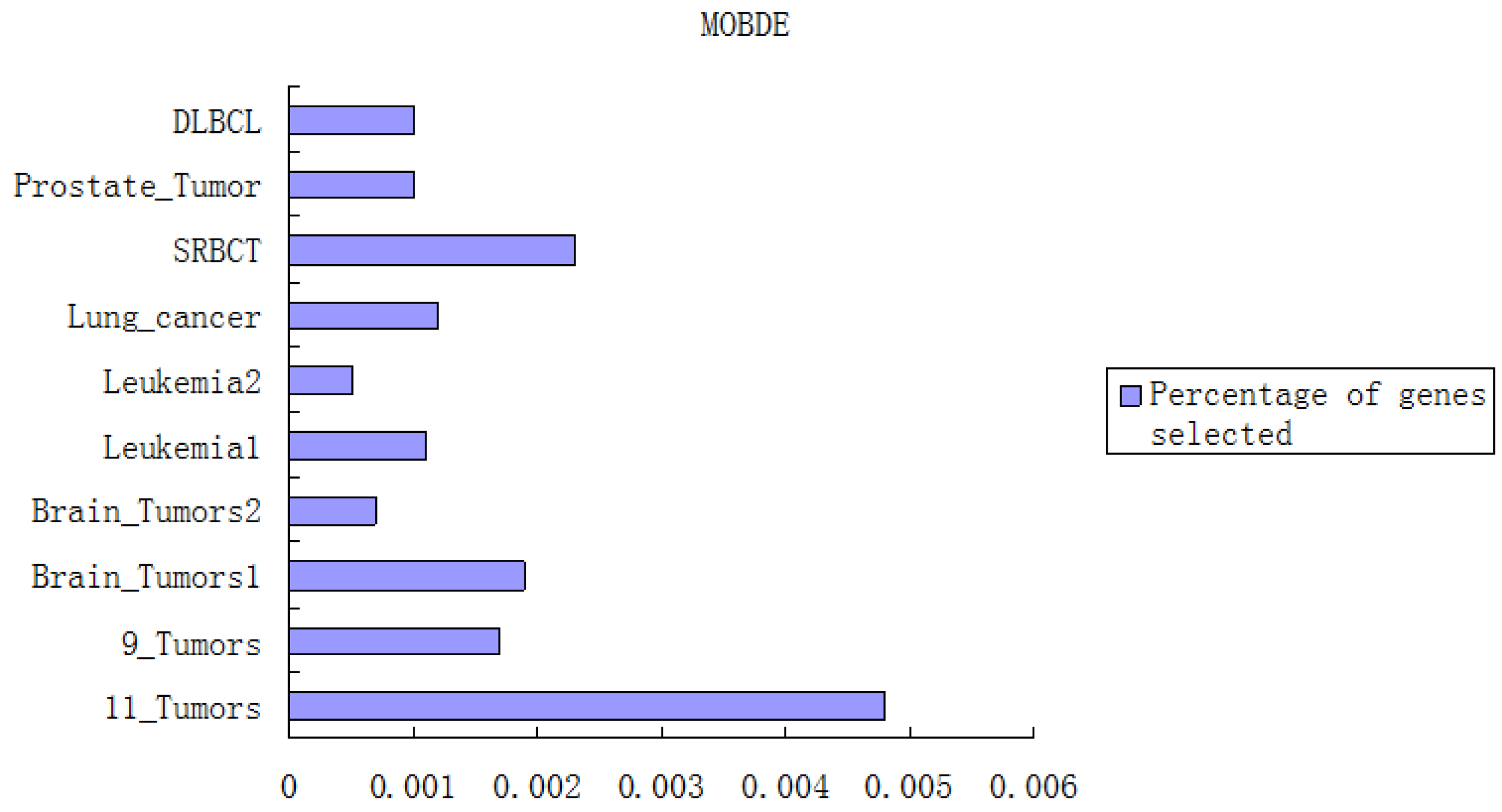
Based on the binary differential evolution algorithm, we will propose our multiobjective binary differential evolution algorithm (MOBDE). Specifically, in our method, two fitness objectives are taken into account for optimization. One is the accuracy of the classification, and the other is the number of selected genes. In order to tackle the feature selection problem, a non-dominated sorting process is often used to find the Pareto front. However, the non-dominated sorting process is always complex and time consuming. In order to solve this problem, Qu and Suganthan [
35] used the summation of the normalized objective and diversity selection, and in this paper, we use a very similar method based on the summation of the normalized objective and diversity selection for the feature selection problem. For the summation of the normalized objective, first, we need to find the maximum and minimum value for every objective and calculate the different range of every objective; then, we need to sum all normalized objective values to obtain a single value. In this way, the multiobjective problem can be regarded as a single-objective optimization problem. However, this kind of transformation may cause the problem of lacking the diversity of the population. Therefore, the diversity selection method is used to maintain the diversity of the algorithm.
The preferential set and backup set are generated from the current population, and three rules are used to select the sets in the next process:
The preferential set can be selected in the next process firstly.
The backup set will be chosen based on the summation of the normalized objective and diversity selection if the preferential set is not sufficient for the solution.
While the individuals in the store exceed the maximum size, the required number of solutions will be randomly chosen from the preferential set.
Based on the above discussion, we can show the framework of our multiobjective binary differential evolution algorithm as follows in Algorithm 1.
| Algorithm 1 Algorithm description of the MOBDE algorithm |
Set the generation counter ; and randomly initialize a population of individuals . Initialize the parameters F, . Evaluate the fitness for each individual in P. Return the non-dominated solutions from the individual P. while stopping criteria is not satisfied do for to do select randomly for to D do if then if & then else if & then else end if else end if end for end for Calculate the objective function for the new population Select the better individual based on the summation of normalized objectives and diversified selection Update the archive based on the new individual end while
|
2.3. Support Vector Machines
In our system, the support vector machine with the leave-one-out cross-validation serves as the evaluator of the multiobjective binary differential evolution algorithm. Let
,
and
,
be a set of training samples and the corresponding labels, respectively. Vapnik and Cortes [
36] defined the SVM method as follows:
where
ω is a normal vector to the hyperplane and
b is a constant, such that
represents the Euclidean distance between the hyperplane and the original feature space. The
is the slack variables to control the training errors, and
C is a penalty parameter of SVM. In this paper, the radial basis function (RBF) is used in SVM to obtain the optimal solution for classification. Considering two samples
,
,
and
are the different samples, the RBF function is calculated by using
, where
is the width of the Gaussian.
is the kernel function.
For the RBF kernel function,
C and
γ are the very important parameters, and the performance of SVM depends on the choice of kernel function in terms of the parameters
C and
γ. If the value of
C is large, the accuracy value of the training will perform better, but the test rate will perform worse. Meanwhile, if the value of
C is small, the accuracy rate will be unsatisfactory, though the test accuracy rate may be high. Sometimes, the parameter
γ has a more effective effect on the test phase than the parameter
C. In order to optimize the feature selection and parameter simultaneously, in the modified MOBDE, each individual is encoded to a string of binary bits associated with the number of genes, and the parameters
C and
γ of the SVM will be dynamically optimized by a real code differential evolution in Equations (
3) and (
4). Specifically, the constrained ranges of the value of
C and
γ are [
, 15] and [
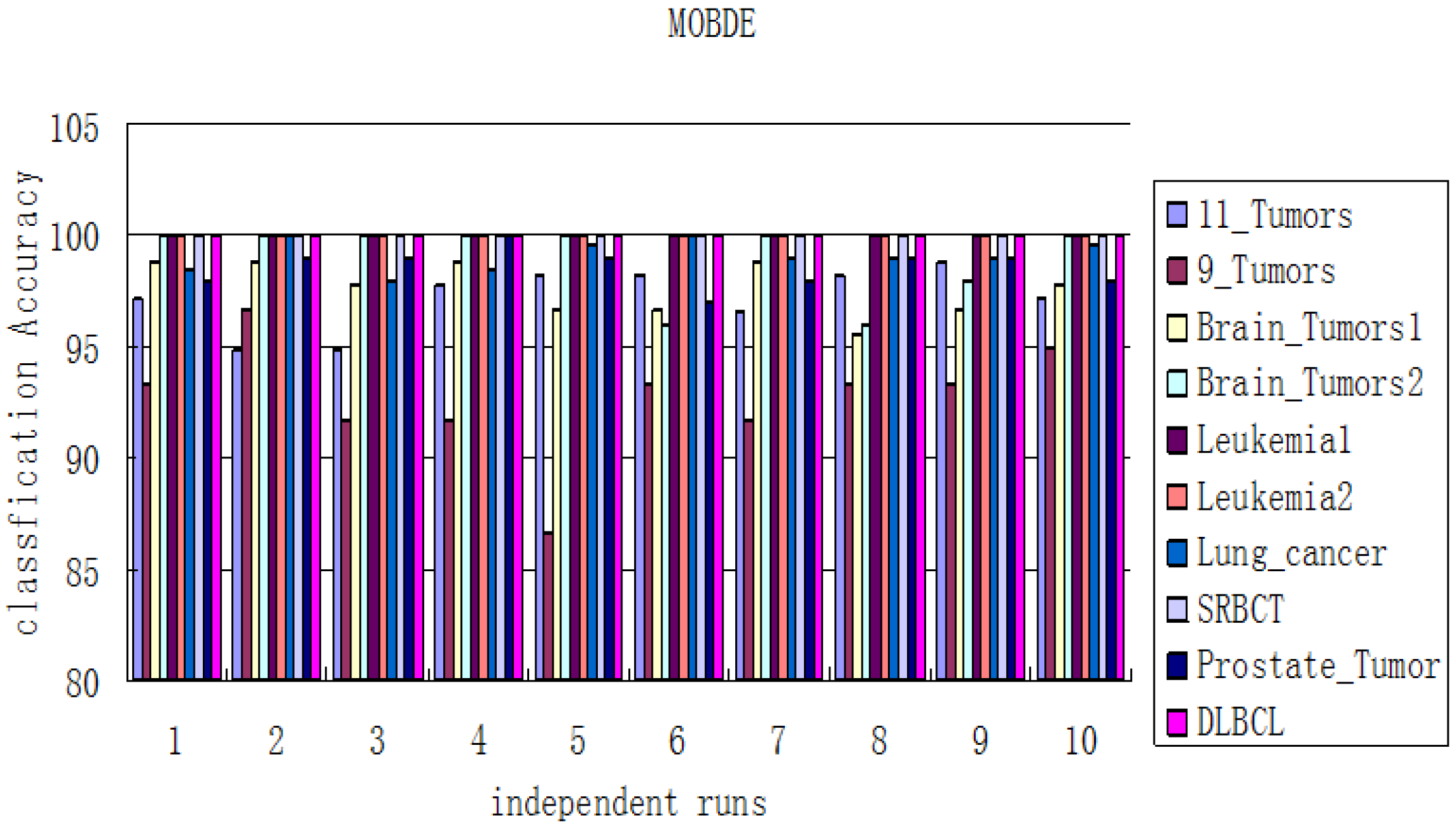
, 5] respectively. In our method, the classification accuracy of the prediction models and the number of selected genes derived from all datasets will be measured by the LOOCV procedure discussed in
Section 2.






{kind=link}
{kind=link}
{kind=link}
{kind=link}