Spectroscopic, Electrochemical and DFT Studies of Phosphorescent Homoleptic Cyclometalated Iridium(III) Complexes Based on Substituted 4-Fluorophenylvinyl- and 4-Methoxyphenylvinylquinolines

 , , and
, , and
Abstract
:
1. Introduction
2. Results
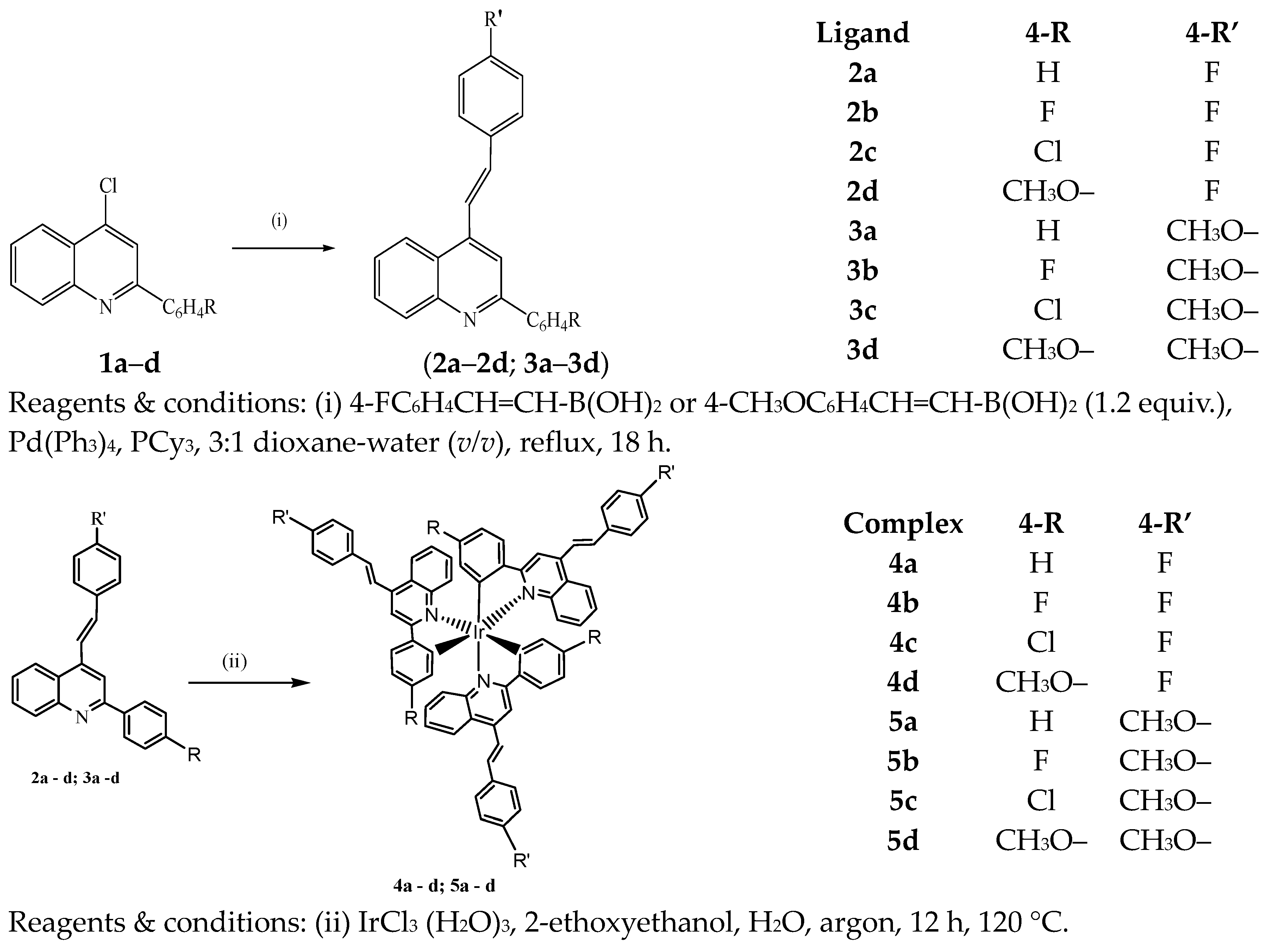
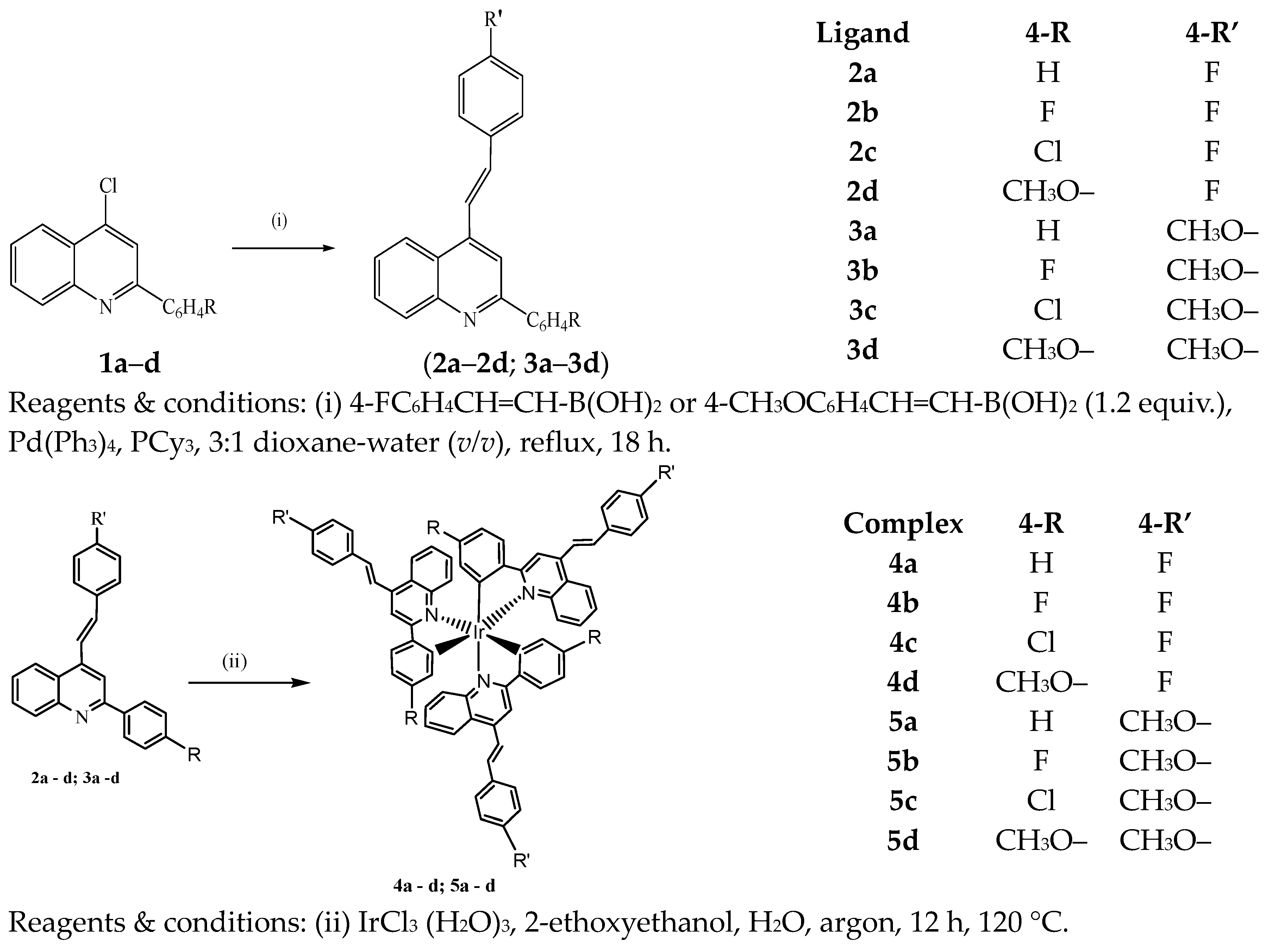
2.1. Synthesis
2.2. Electronic Absorption and Luminescence Spectra of Ligands
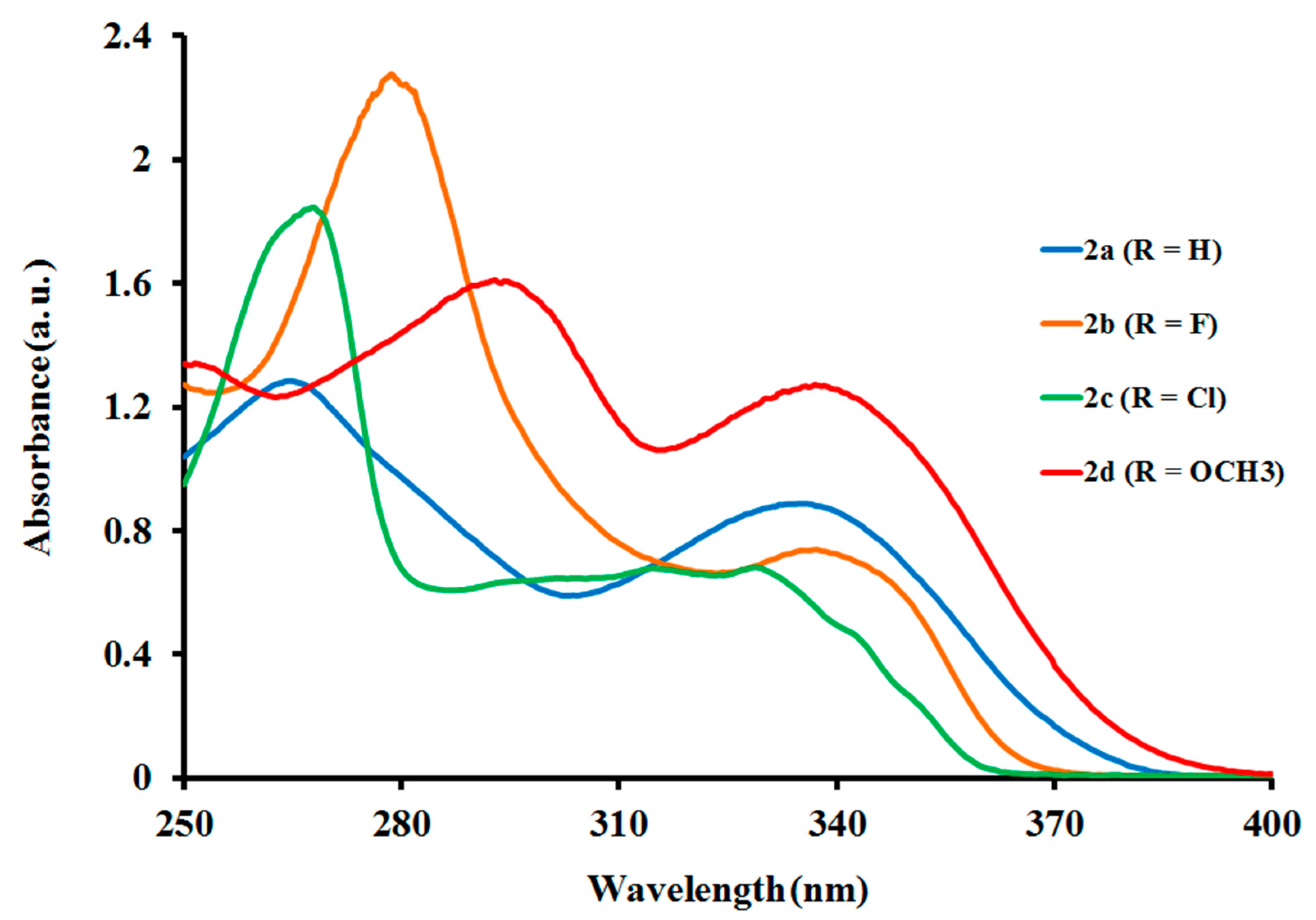
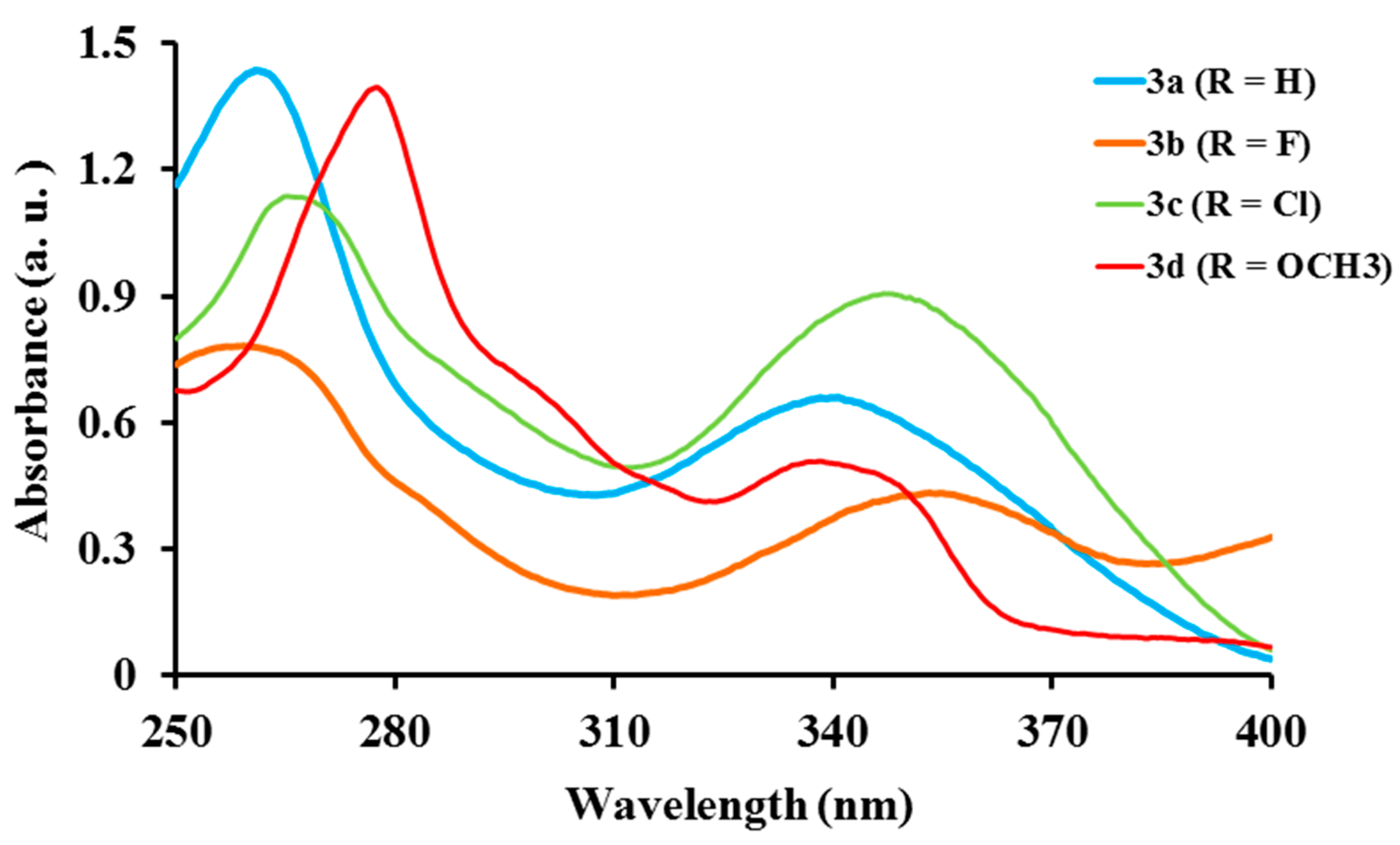
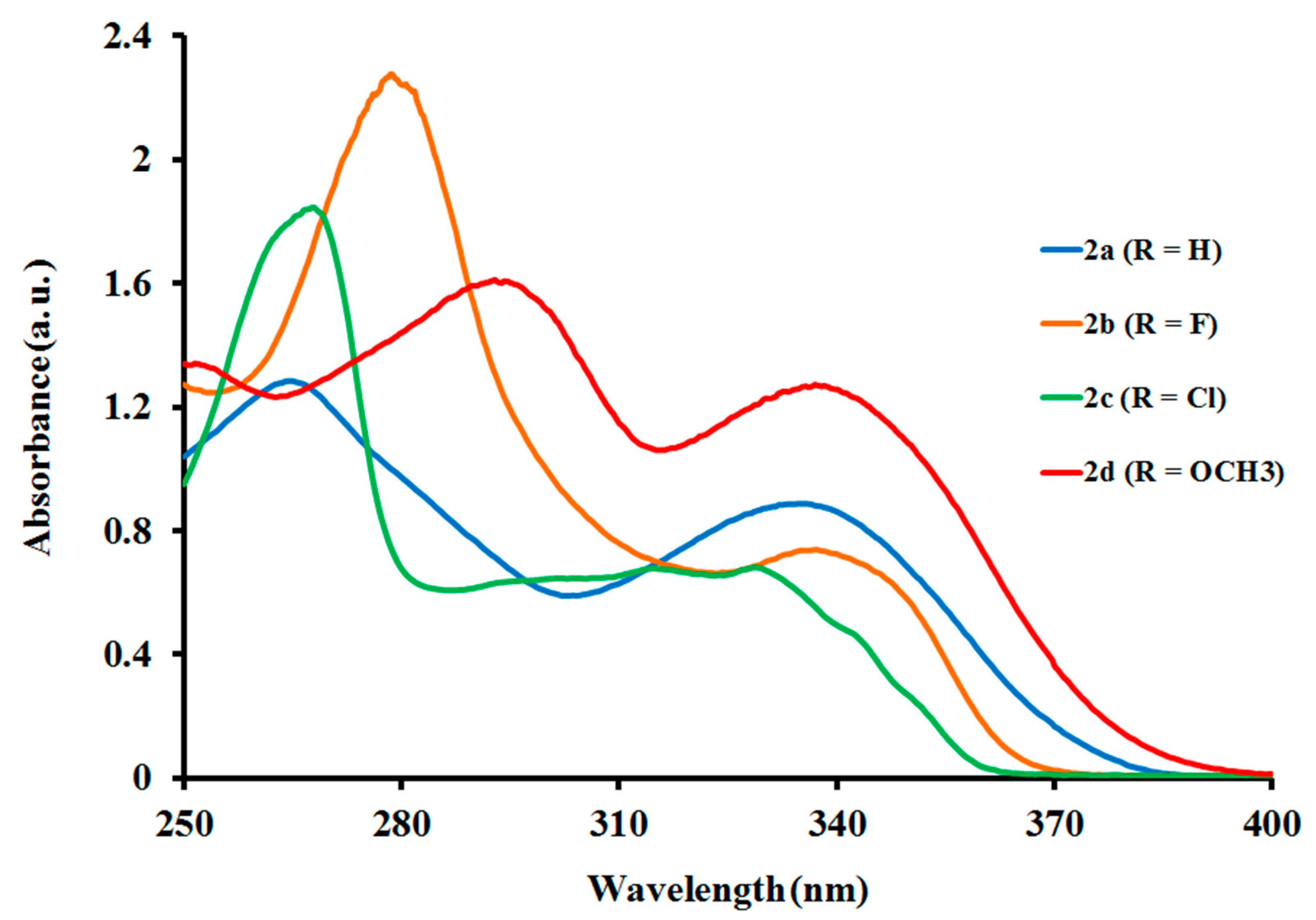
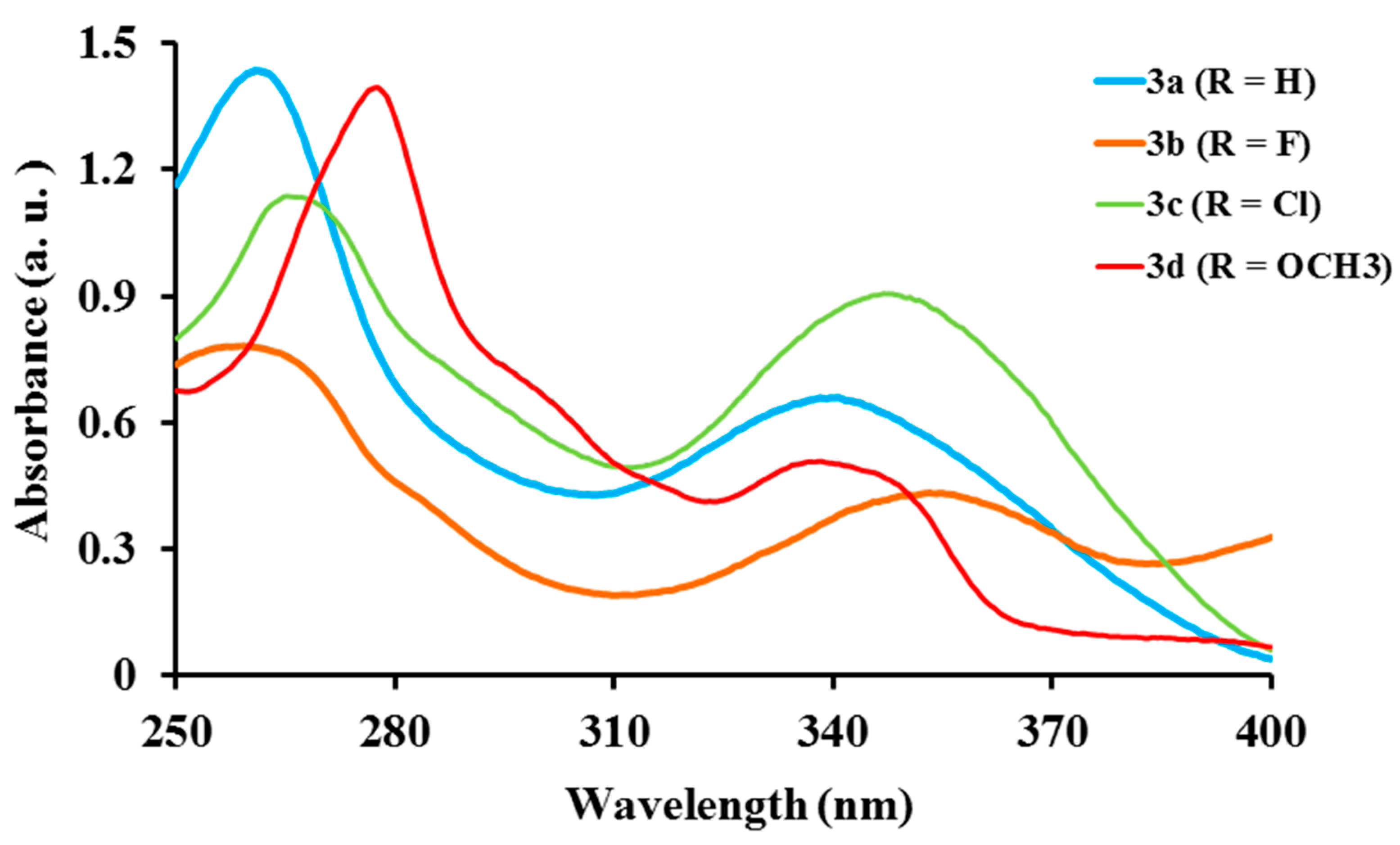
2.2.1. Electronic Absorption of Ligands
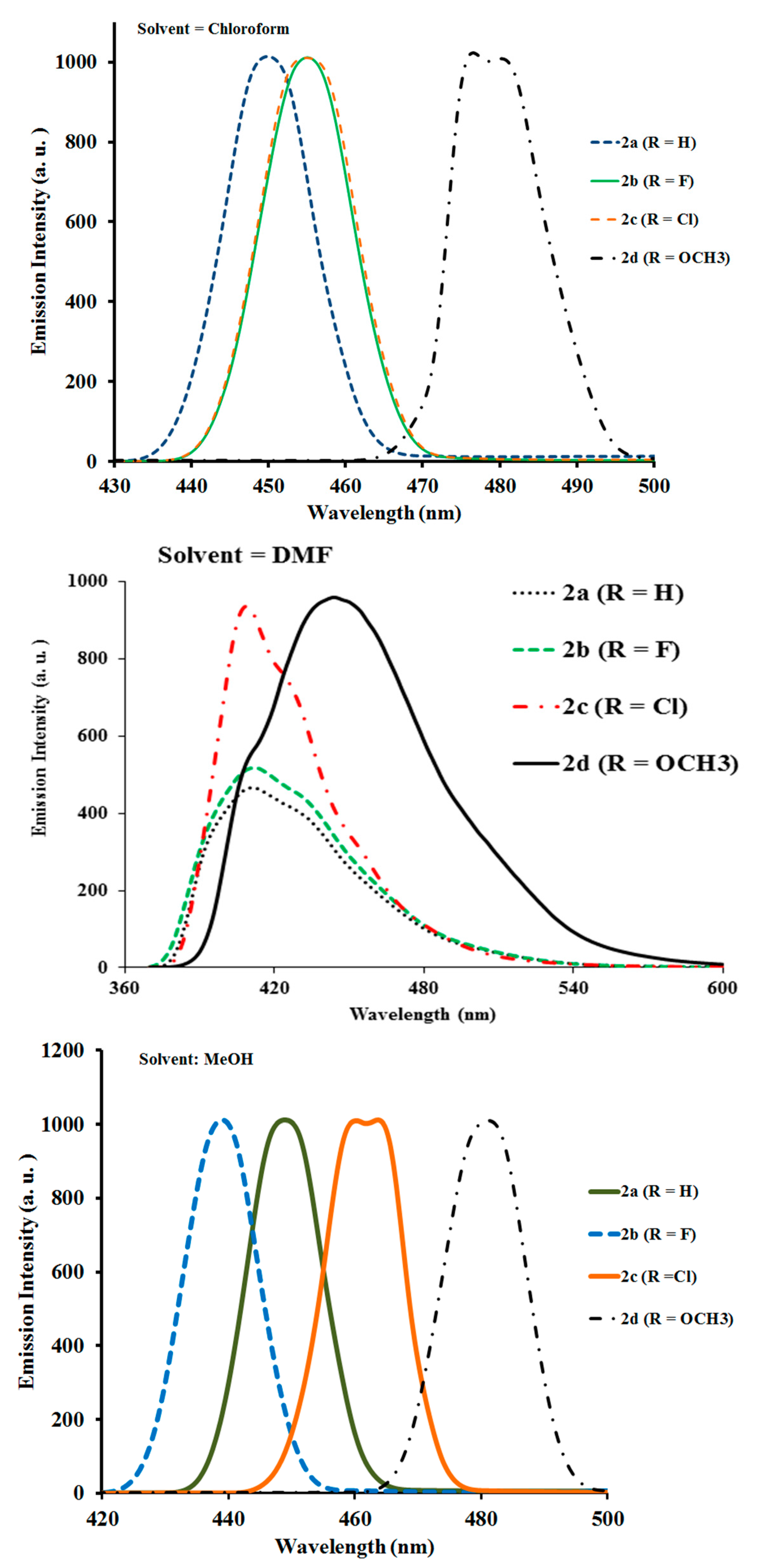
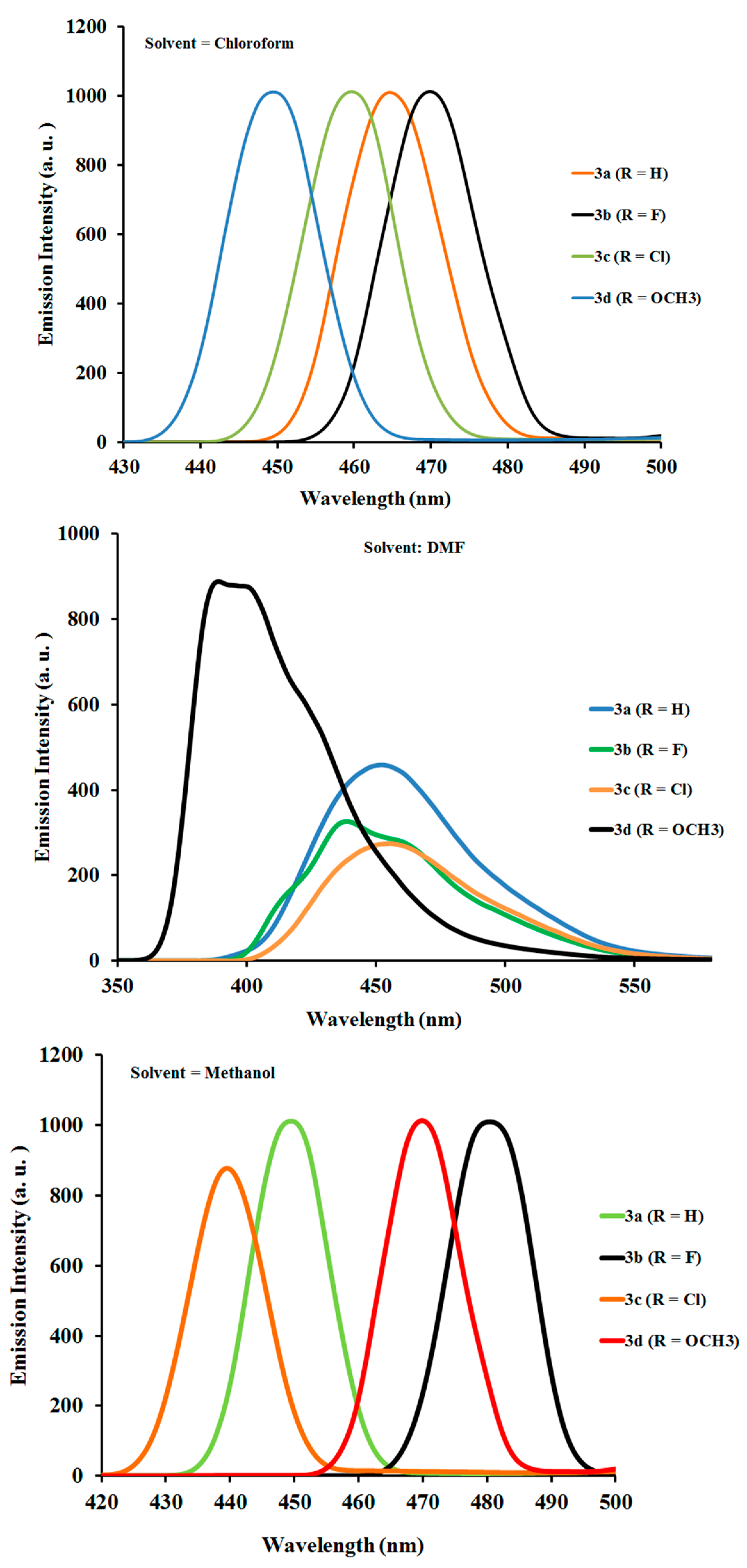
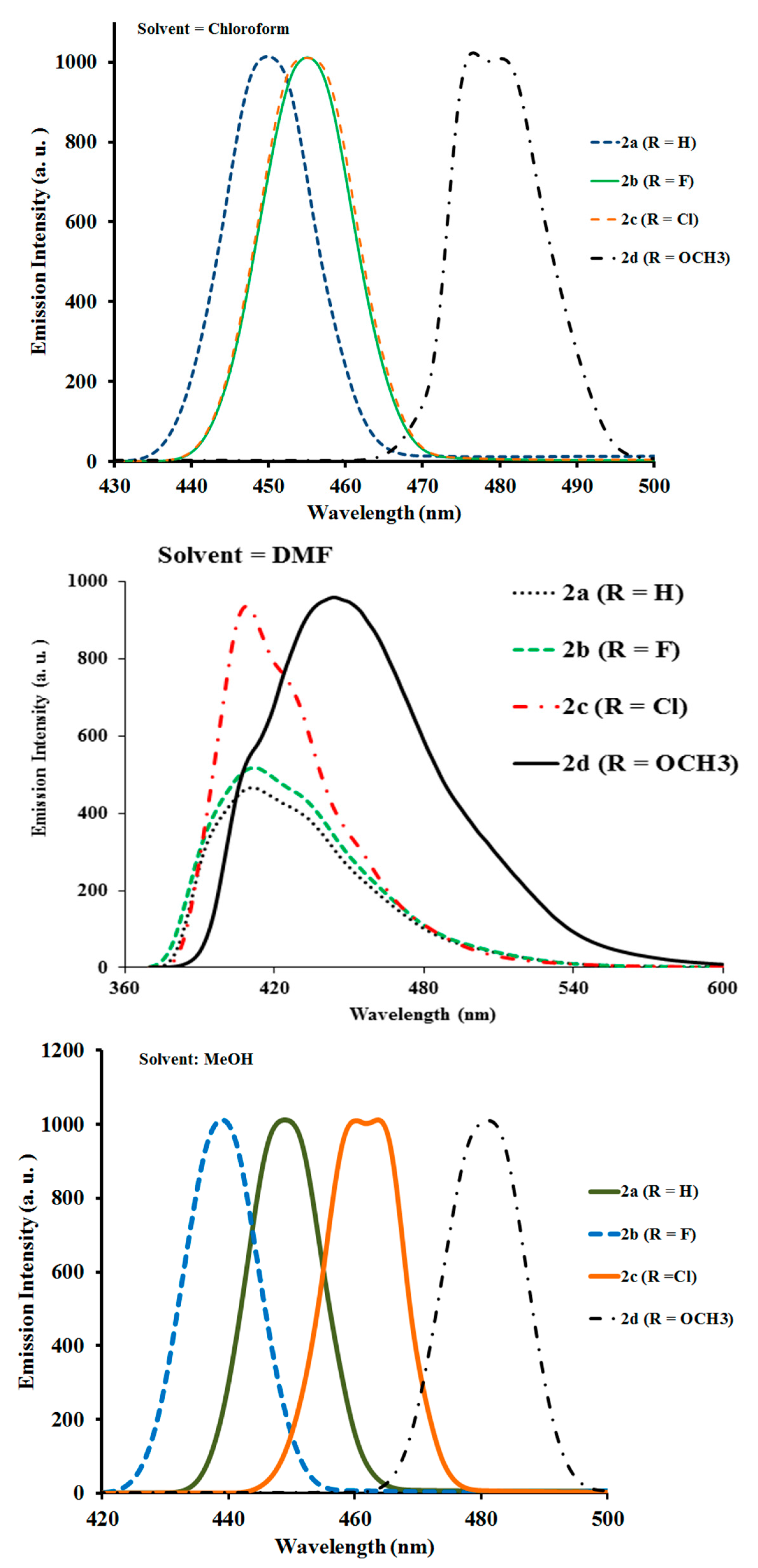
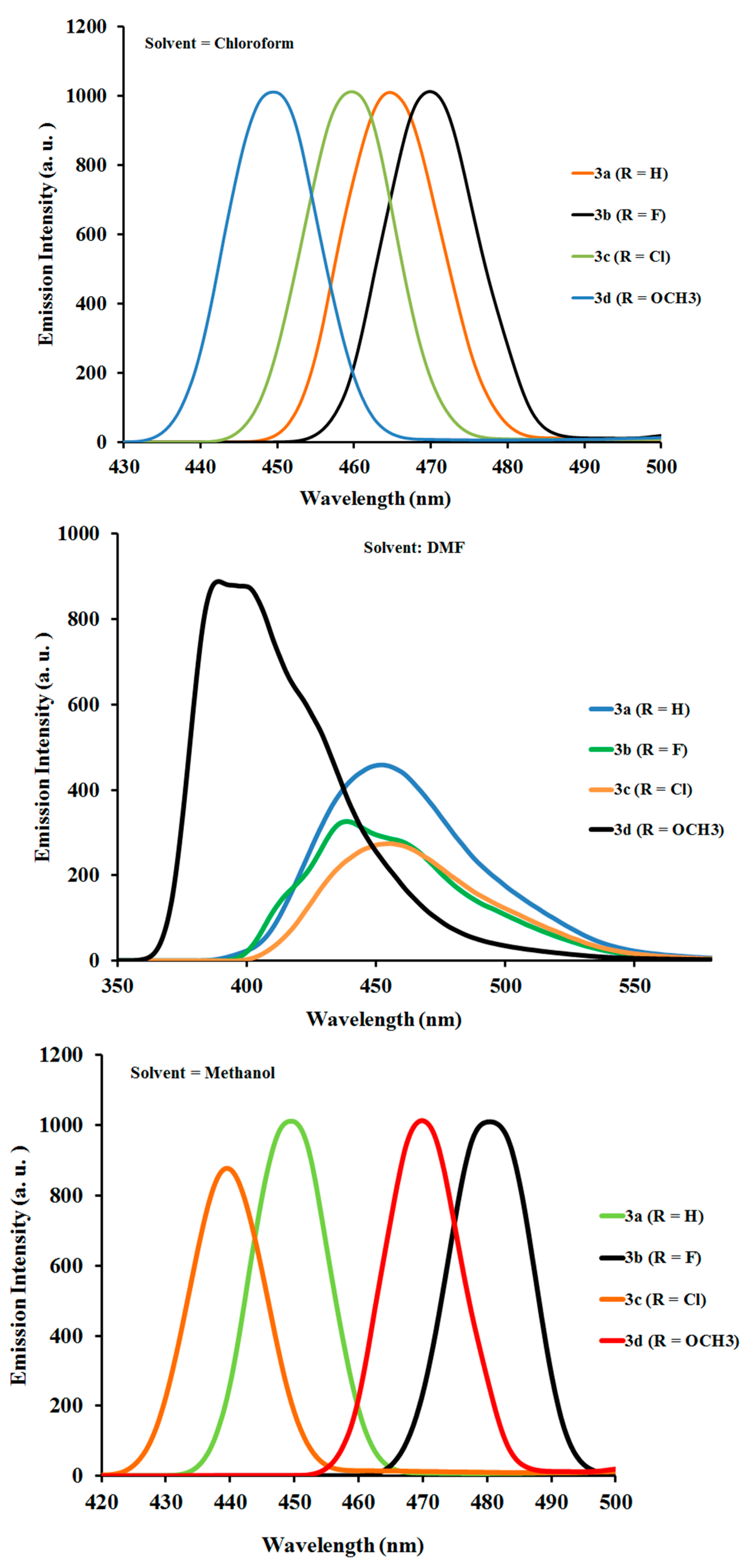
2.2.2. Photoluminescence Properties of Ligands
2.3. Electronic Absorption and Luminescence Spectra of Complexes
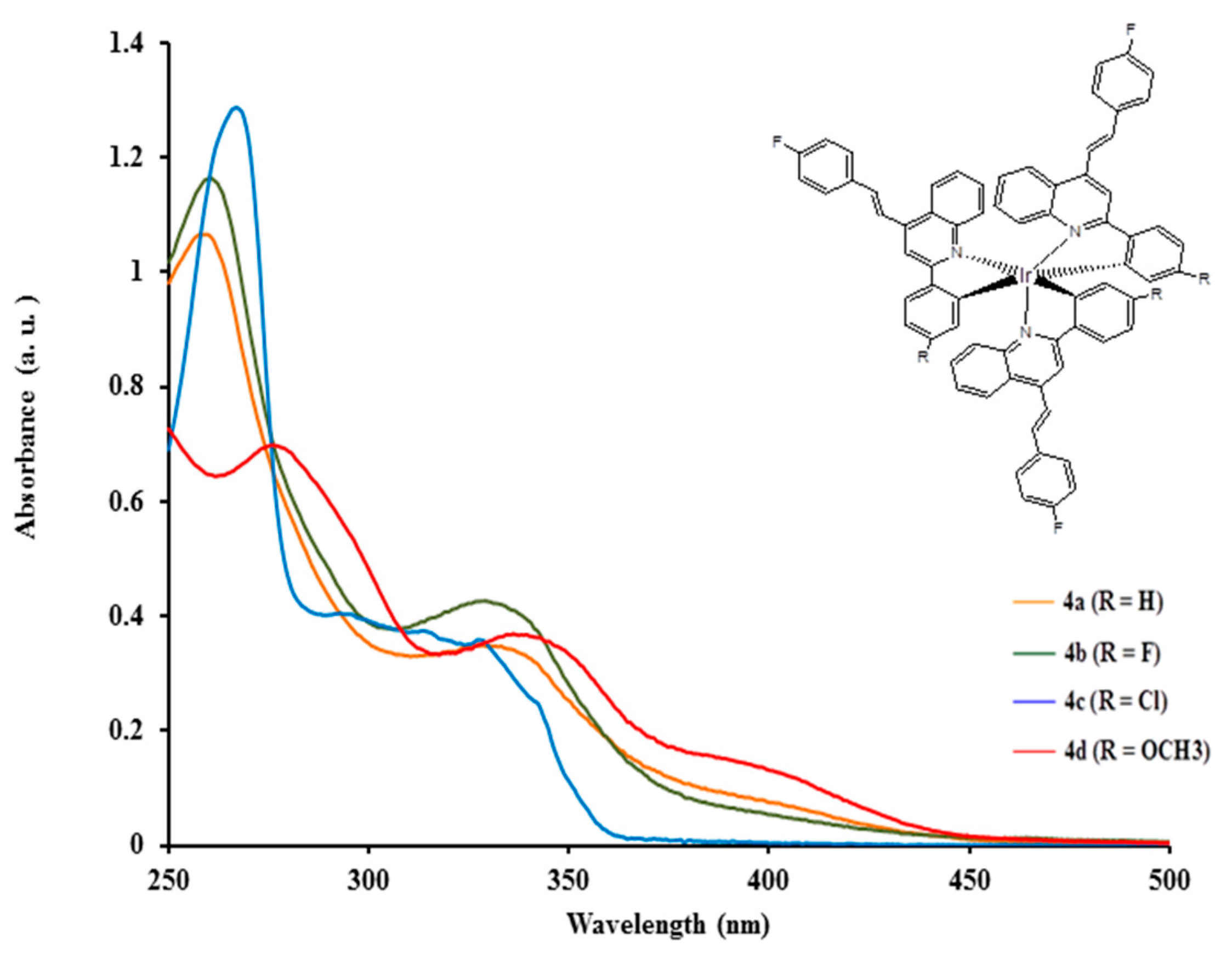
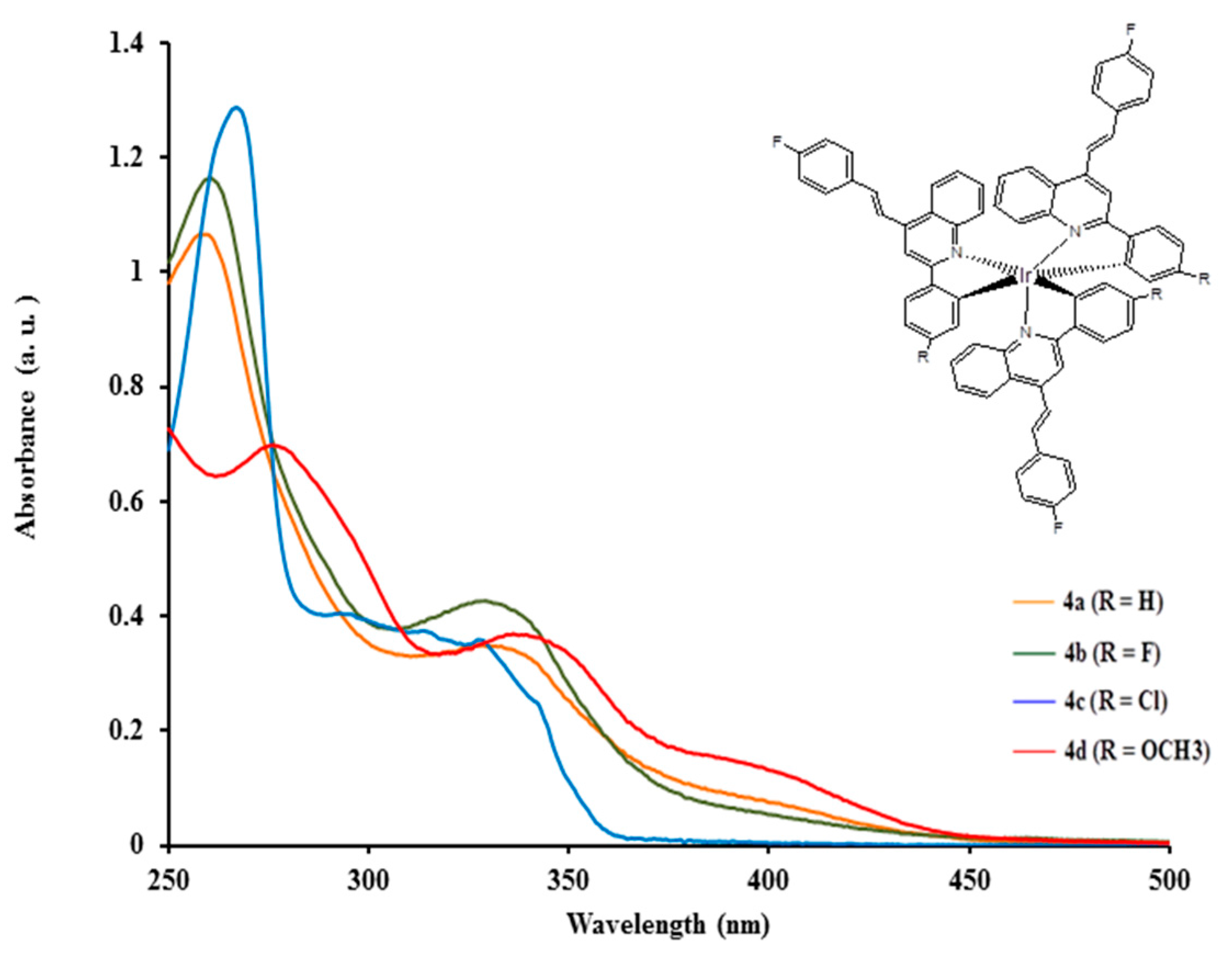
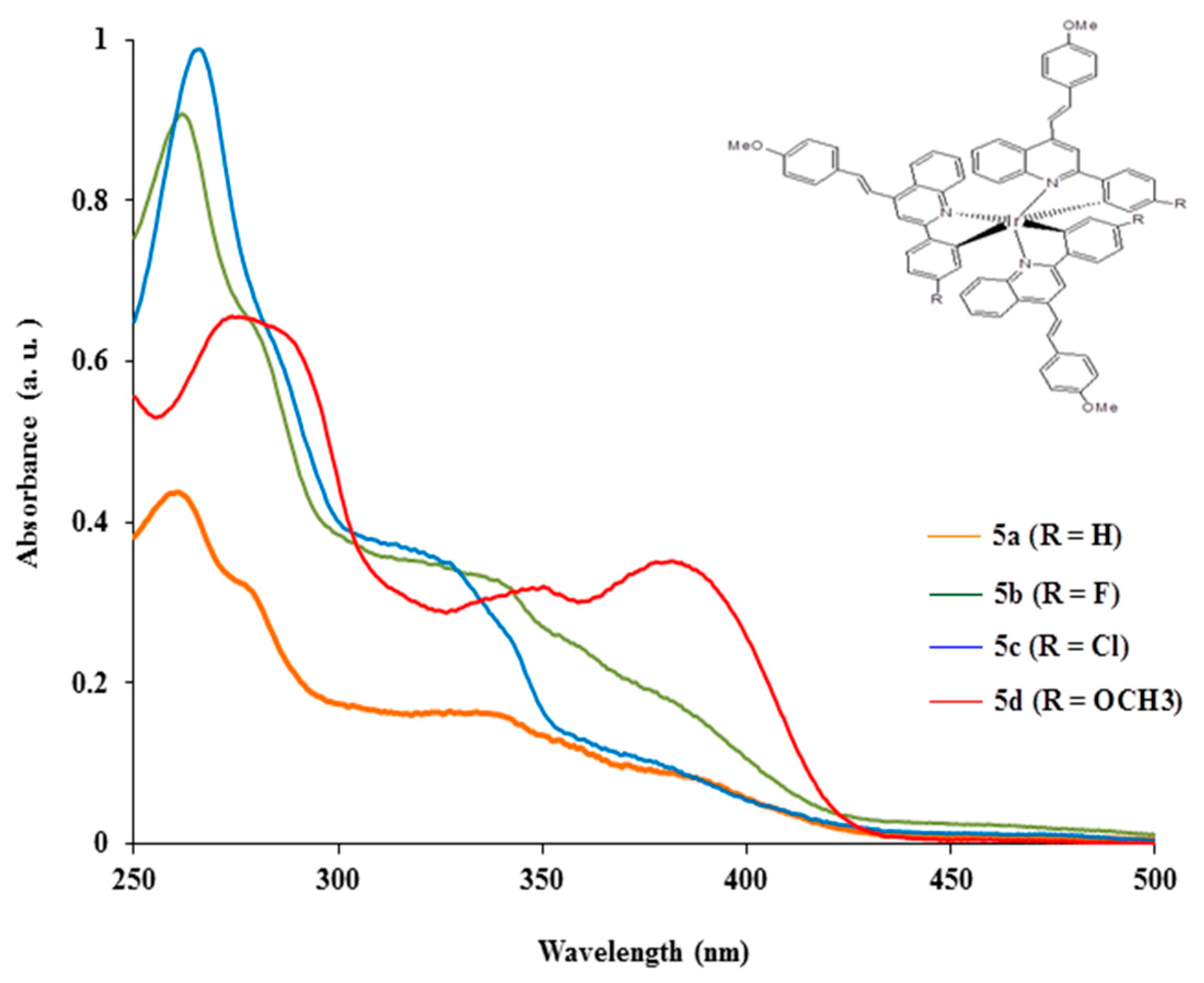
2.3.1. Electronic Absorption of Complexes
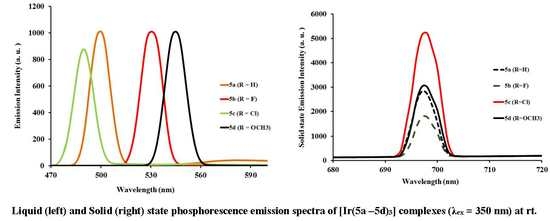
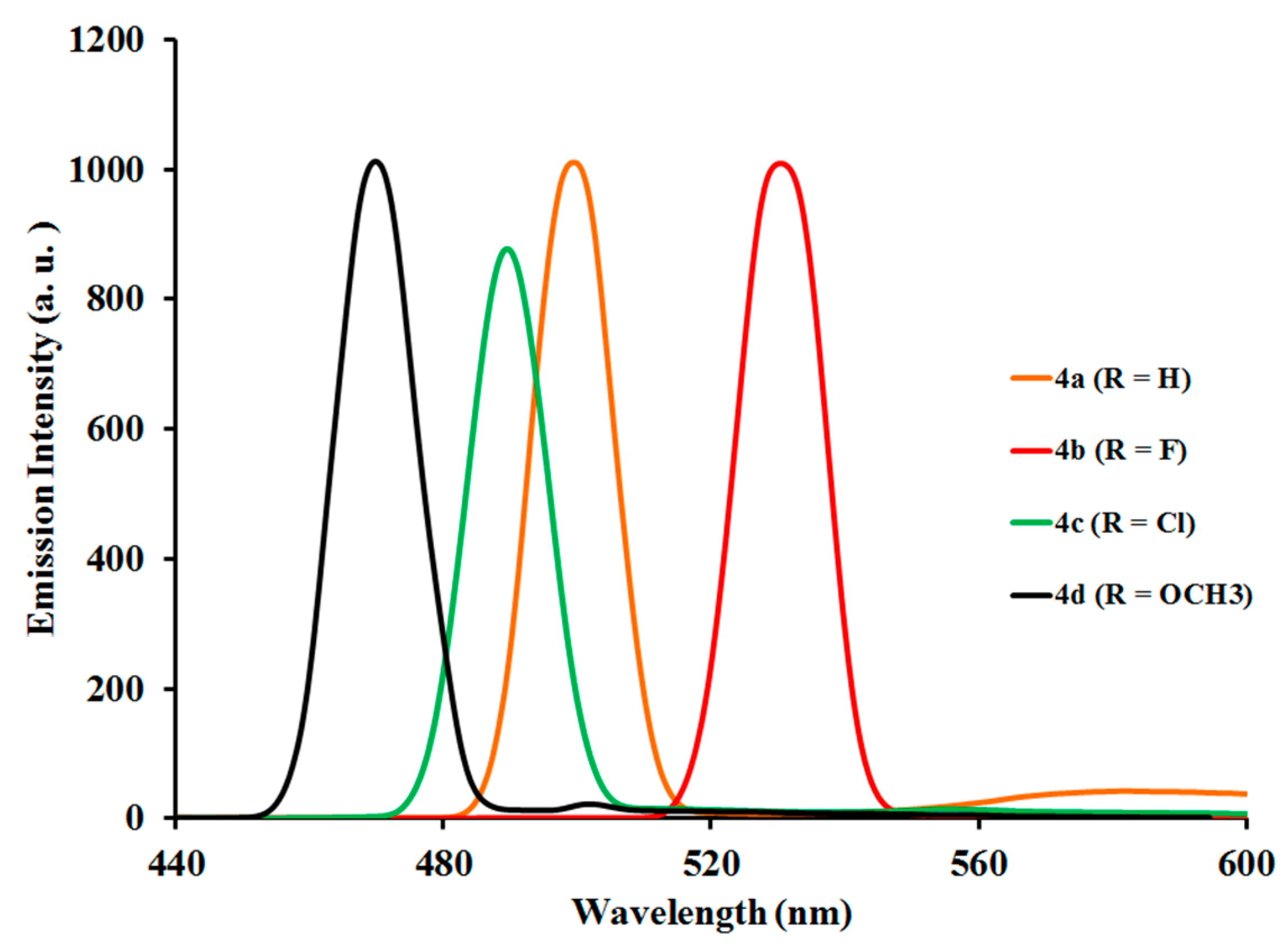
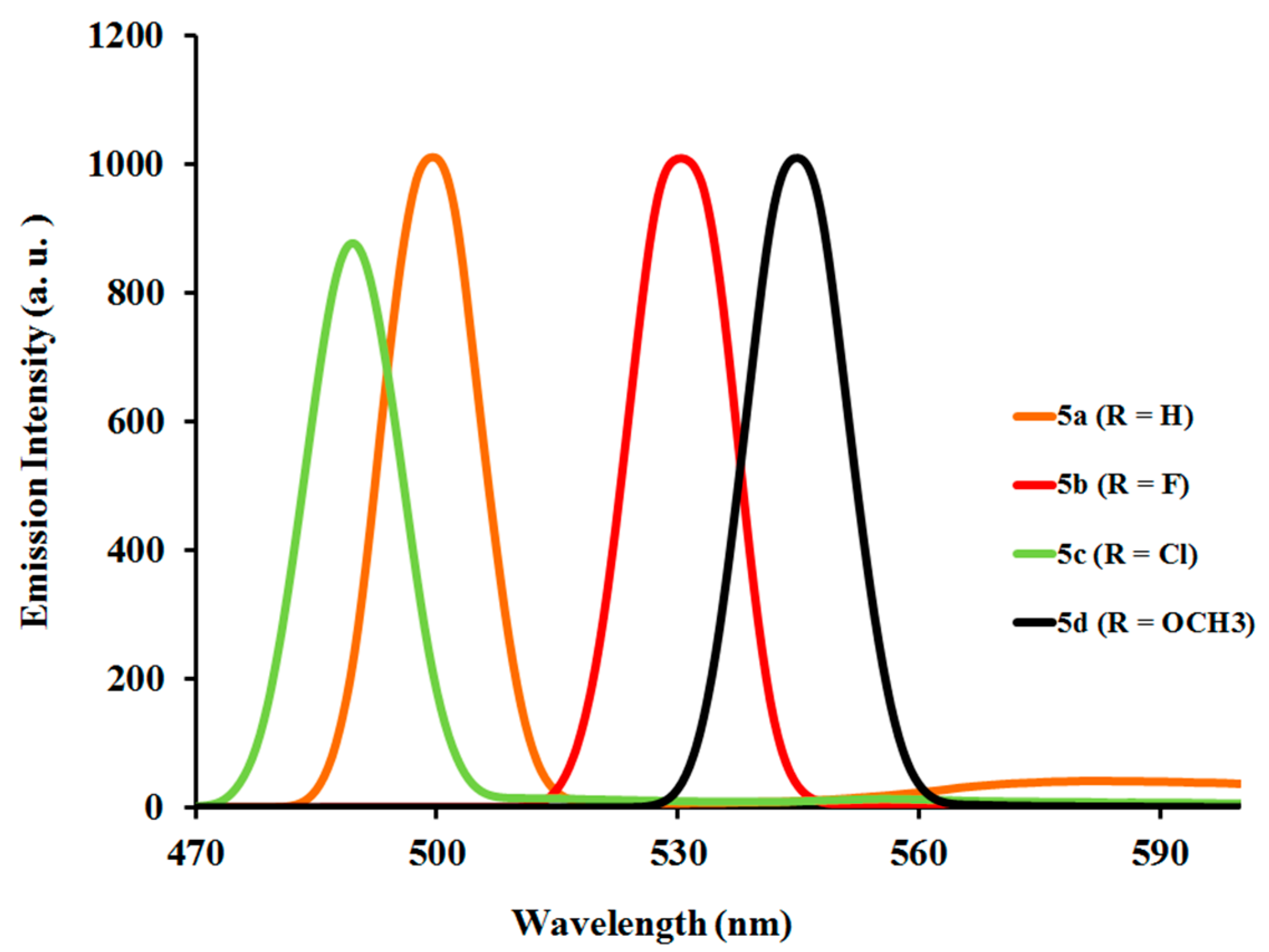
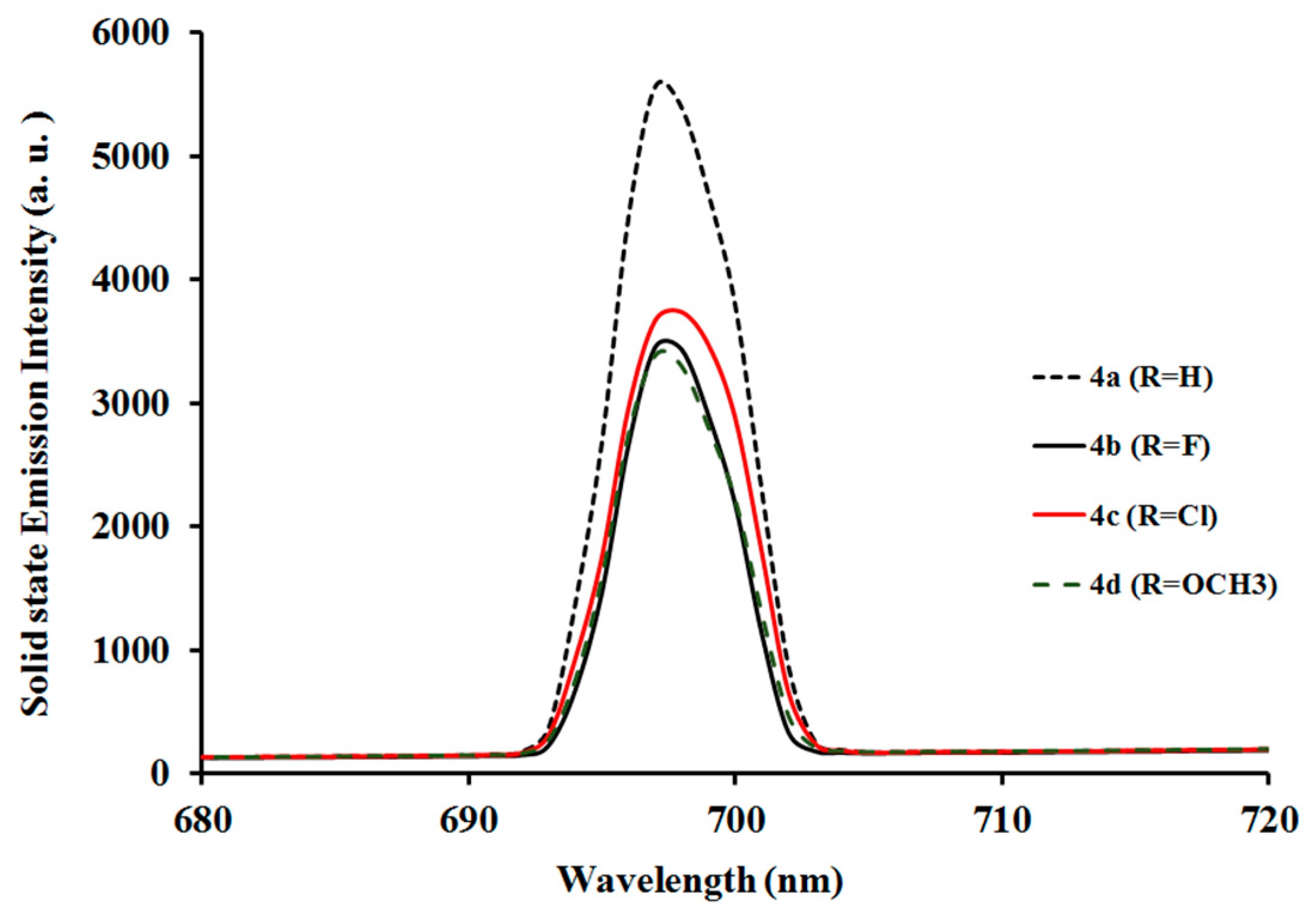
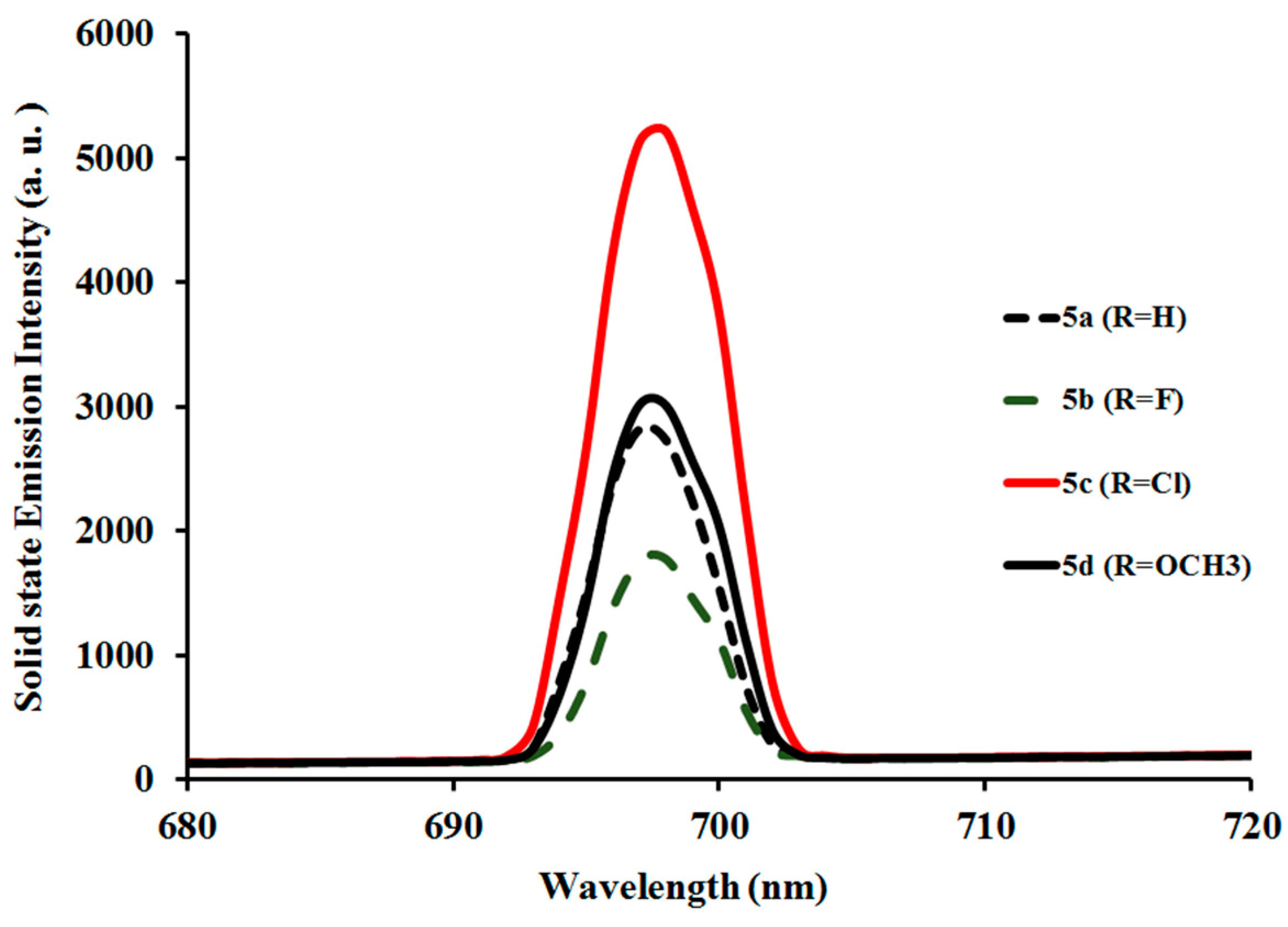
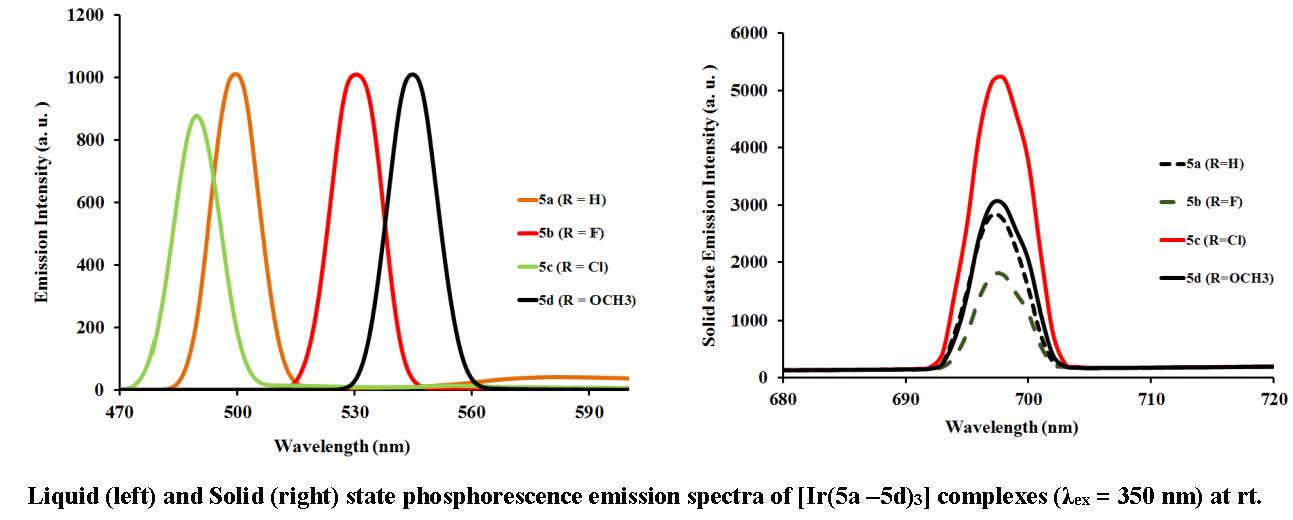
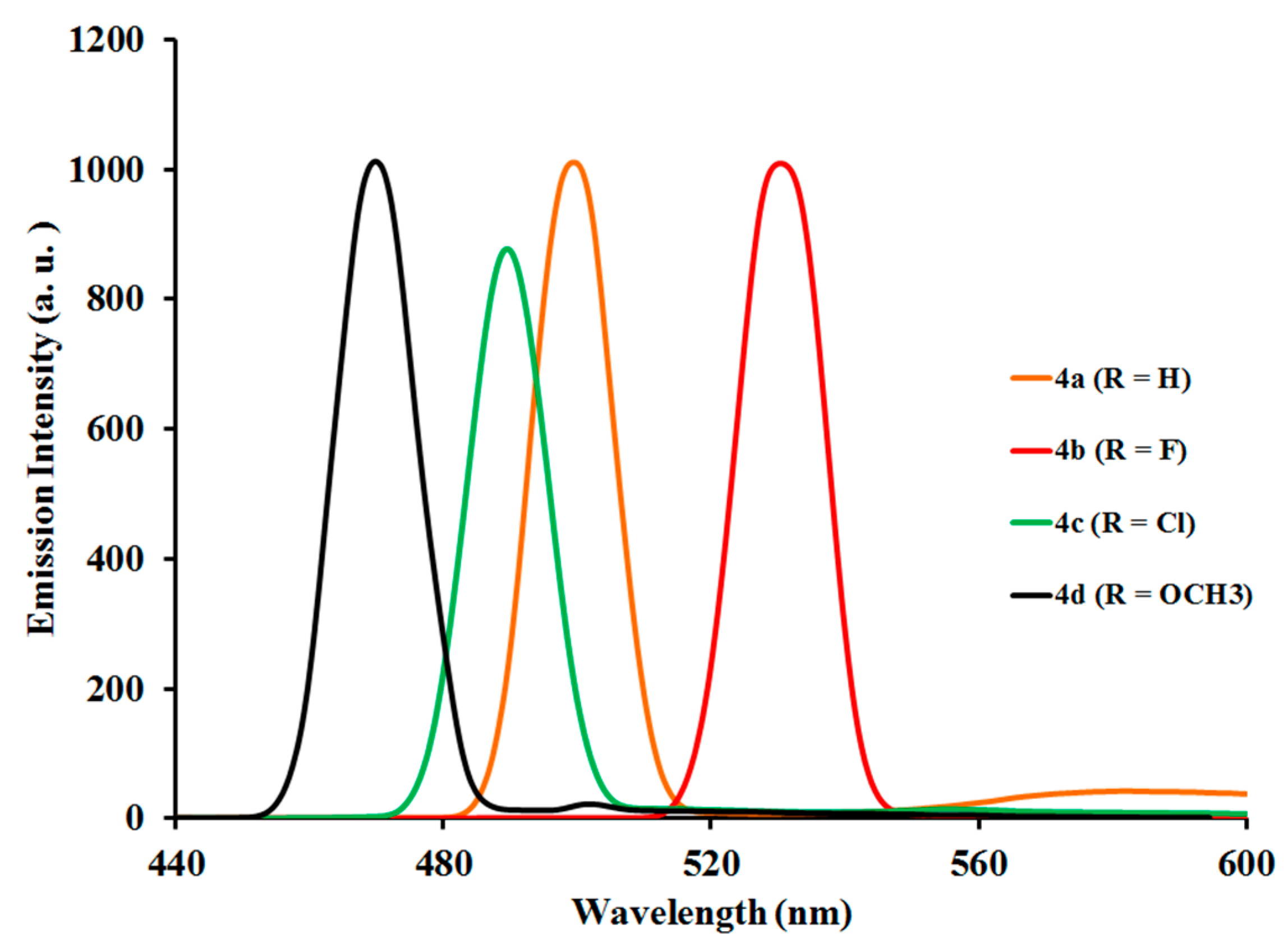
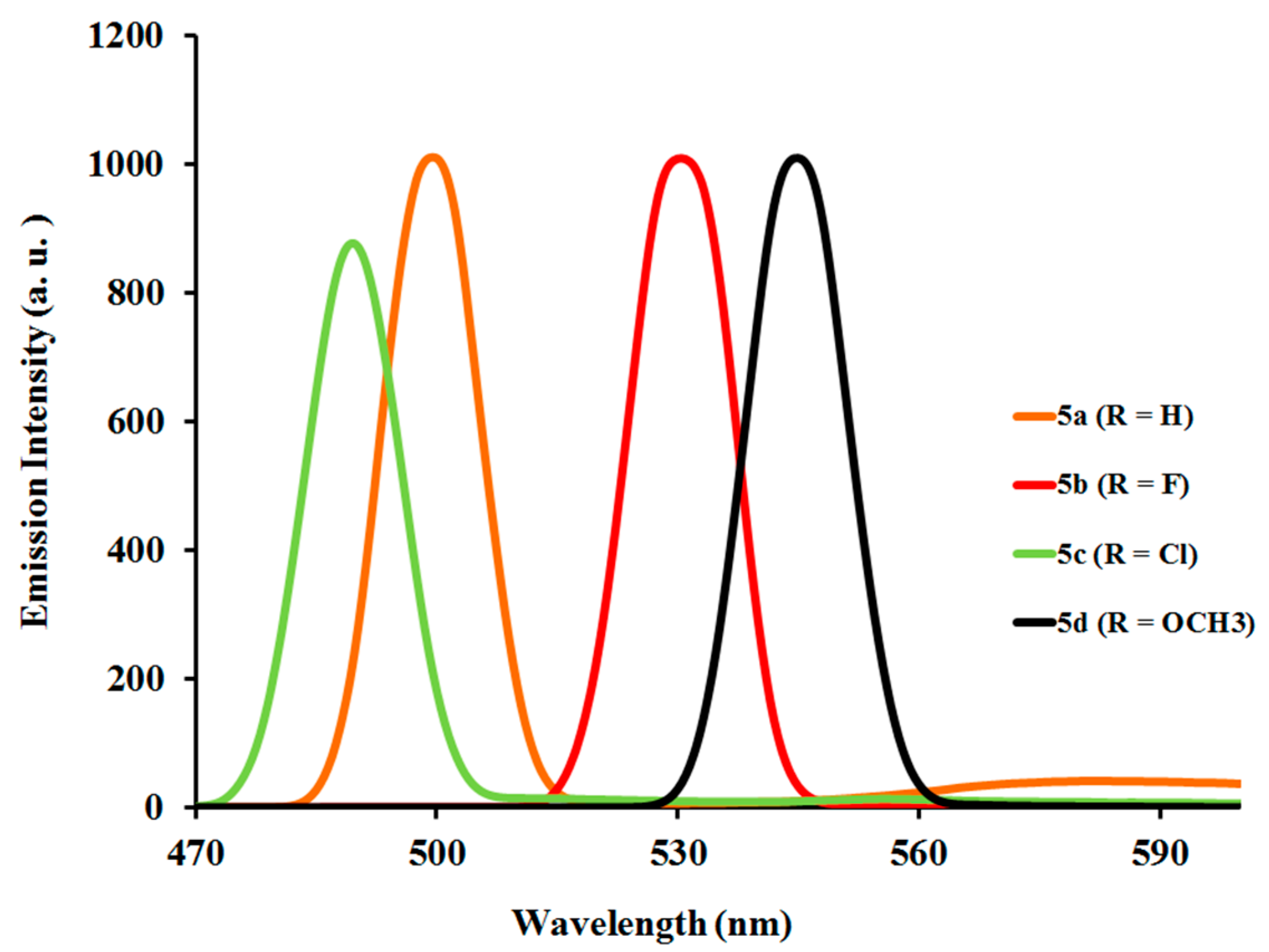
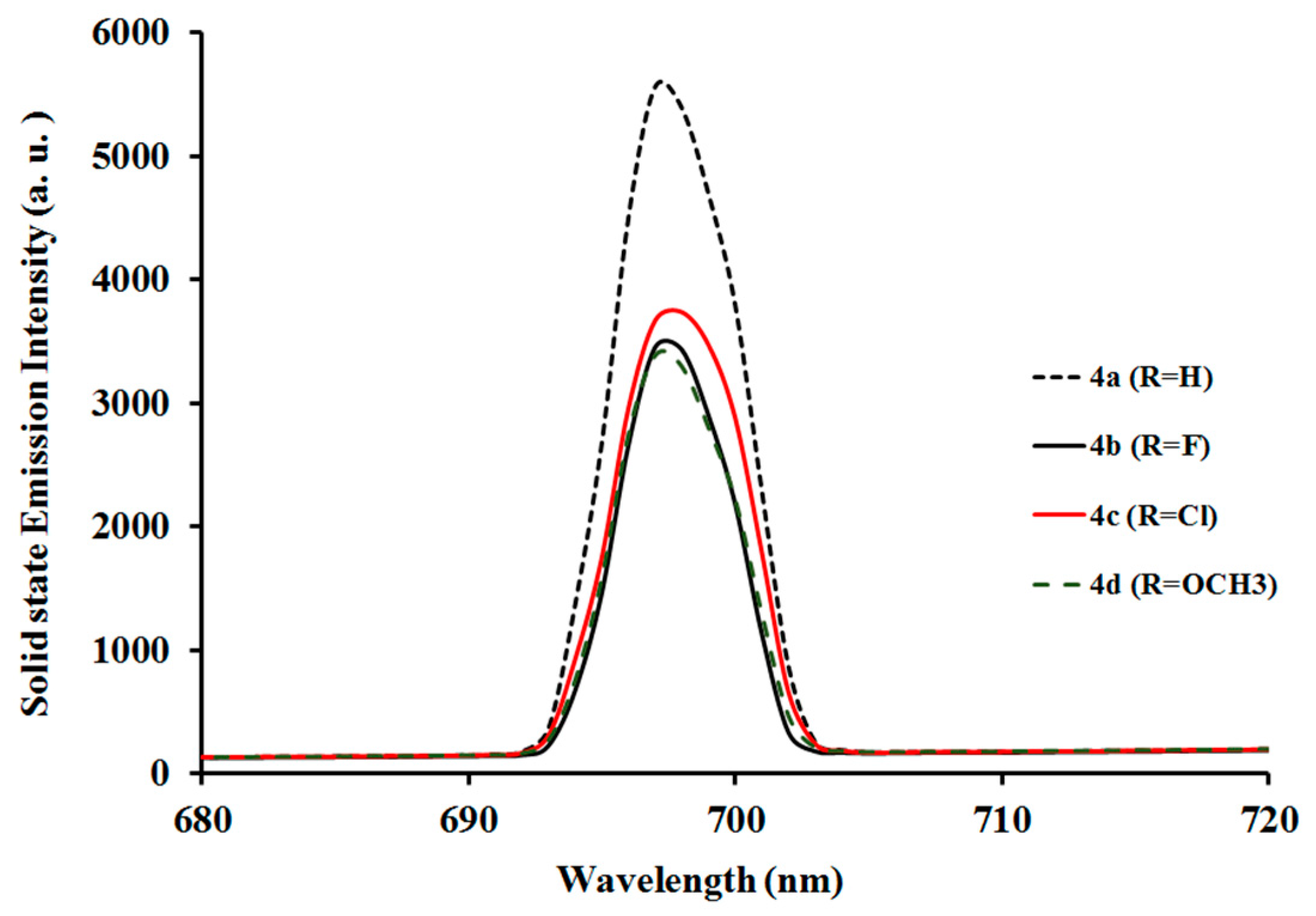
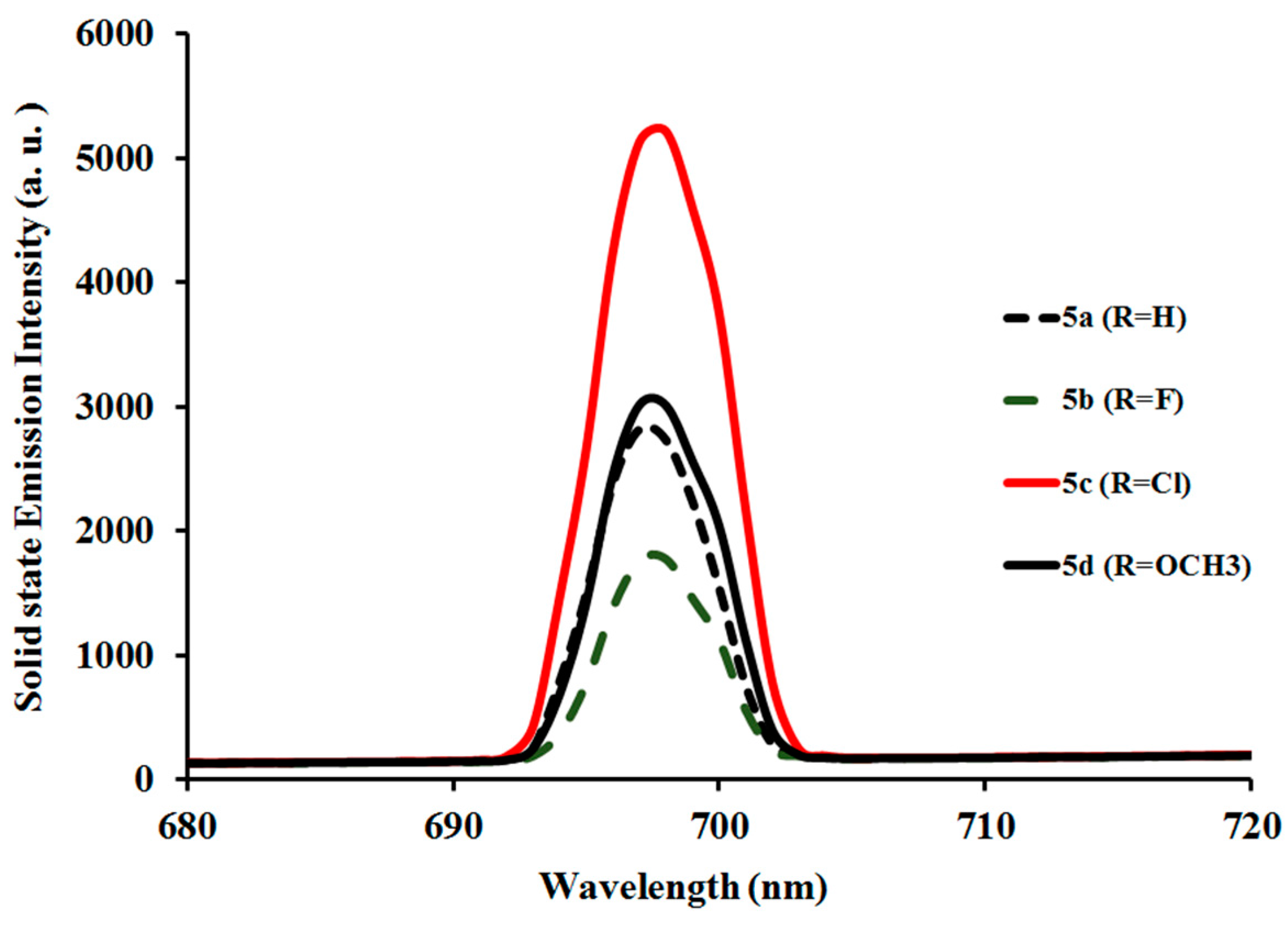
2.3.2. Photoluminescent Properties of Complexes
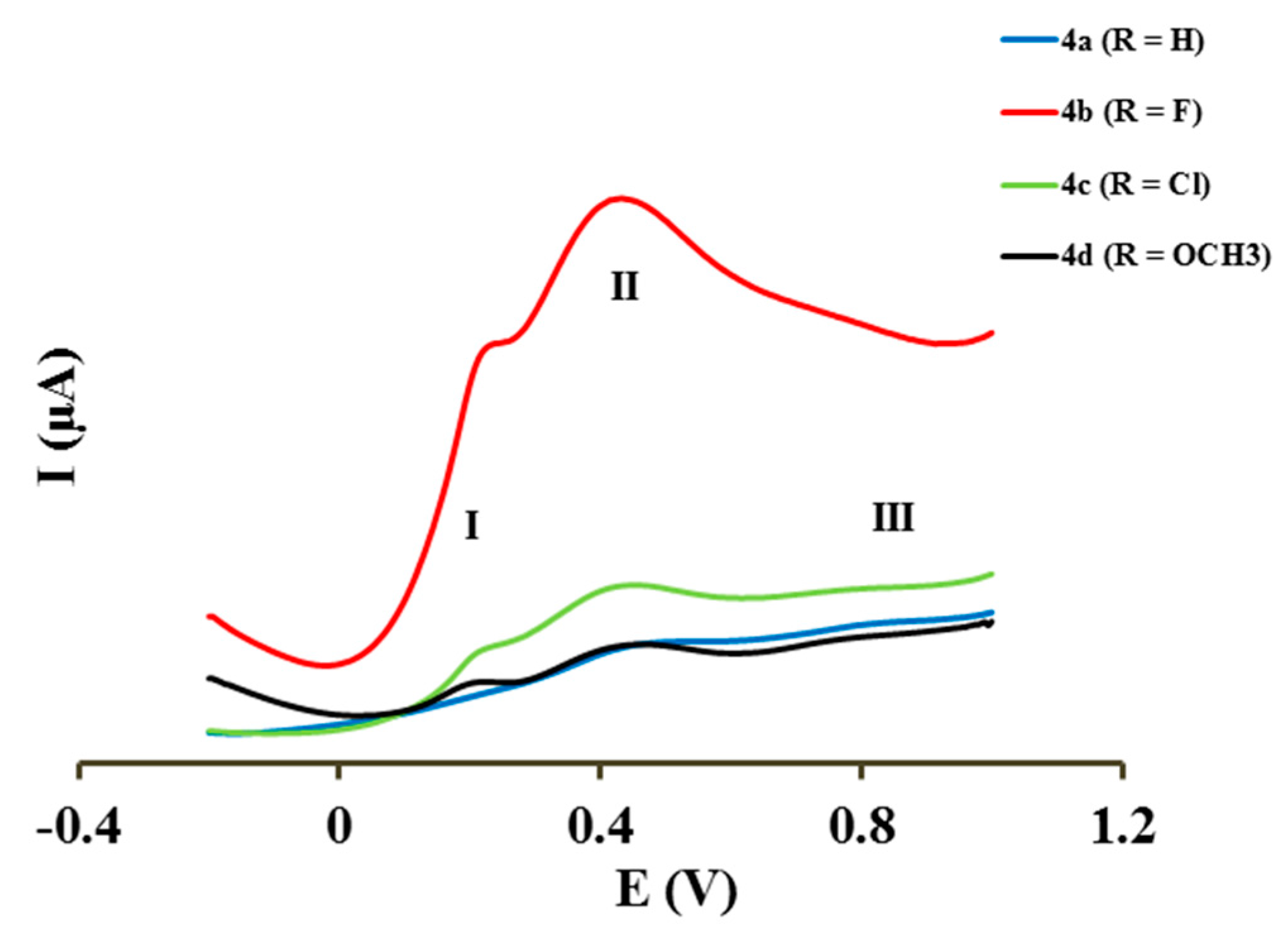
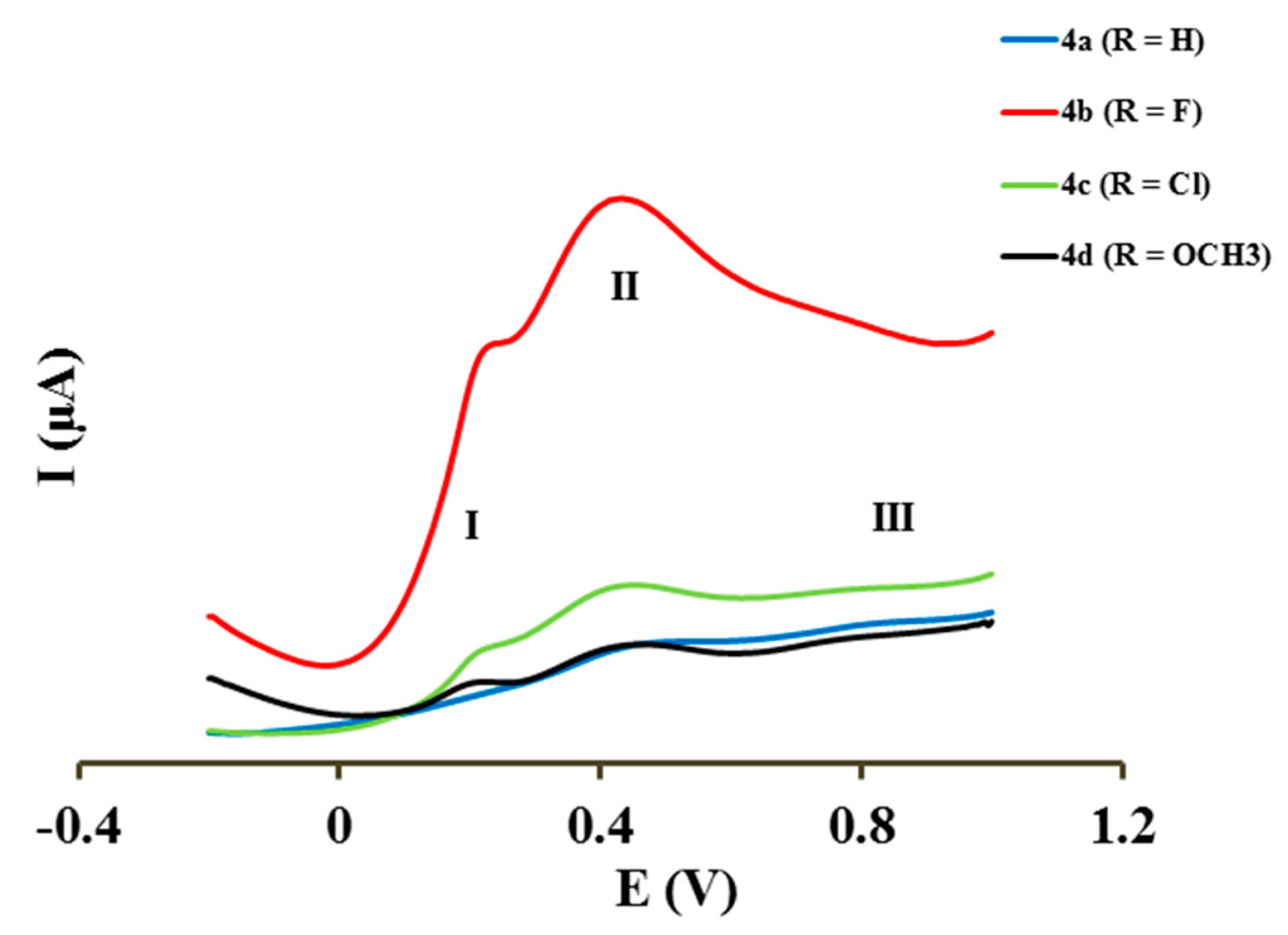
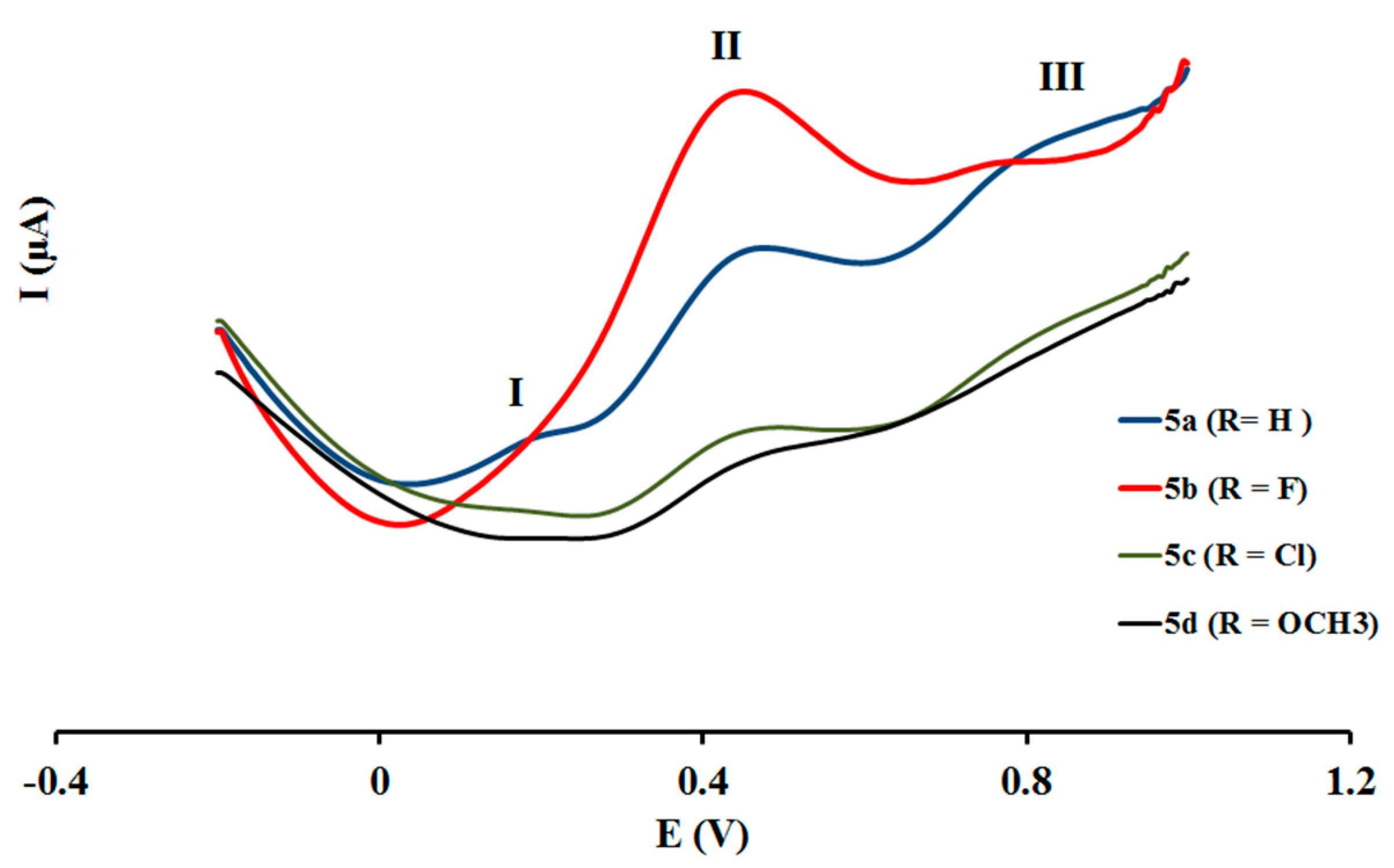
2.4. Electrochemical Studies
3. Materials and Methods
3.1. Typical Procedure for the Suzuki–Miyaura Cross-Coupling of 1 with Arylboronic Acids
3.2. Synthetic Method and Characterization of Iridium(III) Complexes 4a–d and 5a–d
3.3. DFT Computation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Welter, S.; Brunner, K.; Hofstraat, J.W.; De Cola, L. Electroluminescent device with reversible switching between red and green emission. Nature 2003, 421, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Beltran, J.; Lemaur, V.; Cornil, J.; Hartmann, D.; Sarfert, W.; Frohlich, R.; Bizzarri, C.; De Cola, L. Iridium metal complexes containing N-heterocyclic carbene ligands for blue-light-emitting electrochemical cells. Inorg. Chem. 2010, 49, 9891–9901. [Google Scholar] [CrossRef] [PubMed]
- Slinker, J.D.; Gorodetsky, A.A.; Lowry, M.S.; Wang, J.J.; Parker, S.; Rohl, R.; Bernhard, S.; Malliarasa, G.G. Efficient yellow electroluminescence from a single layer of a cyclometalated iridium complex. J. Am. Chem. Soc. 2004, 126, 2763–2767. [Google Scholar] [CrossRef] [PubMed]
- Bolink, H.J.; Coronado, E.; Costa, R.D.; Lardies, N.; Orti, E. Near-quantitative internal quantum efficiency in a light-emitting electrochemical cell. Inorg. Chem. 2008, 47, 9149–9151. [Google Scholar] [CrossRef] [PubMed]
- Bolink, H.J.; Cappelli, L.; Coronado, E.; Gratzel, M.; Orti, E.; Costa, R.D.; Viruela, P.M.; Nazeeruddin, M.K. Stable single-layer light-emitting electrochemical cell using 4,7-Diphenyl-1,10-phenanthroline-bis(2-phenylpyridine)iridium(III) hexafluorophosphate. J. Am. Chem. Soc. 2006, 128, 14786–14787. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-C.; Fang, F.-C.; Hwu, T.-Y.; Hsieh, H.-H.; Chen, H.-F.; Lee, G.-H.; Peng, S.-M.; Wong, K.-T.; Wu, C.-C. Highly efficient orange and green solid-state light-emitting electrochemical cells based on cationic IrIII complexes with enhanced steric hindrance. Adv. Funct. Mater. 2007, 17, 1019–1027. [Google Scholar] [CrossRef]
- Colman, E.Z.; Slinker, J.D.; Parker, J.B.; Malliaras, G.G.; Bernhard, S. Improved turn-on times of light-emitting electrochemical cells. Chem. Mater. 2008, 20, 388–396. [Google Scholar] [CrossRef]
- Dixon, I.M.; Collin, J.P.; Sauvage, J.P.; Flamigni, L.; Encinas, S.; Barigelletti, F. A family of luminescent coordination compounds: Iridium(III) polyimine complexes. Chem. Soc. Rev. 2000, 29, 385–391. [Google Scholar] [CrossRef]
- Ulbricht, C.; Beyer, B.; Friebe, C.; Winter, A.; Schubert, U.S. Recent developments in the application of phosphorescent iridium(III) complex systems. Adv. Mater. 2009, 21, 4418–4441. [Google Scholar] [CrossRef]
- Chi, Y.; Chou, R.-T. Transition-metal phosphors with cyclometalating ligands: Fundamentals and applications. Chem. Soc. Rev. 2010, 39, 638–655. [Google Scholar] [CrossRef] [PubMed]
- Haugland, R.P. The Handbook—A Guide to Fluorescent Probes and Labelling Technologies, 10th ed.; Molecular Probes Inc.: Eugene, OR, USA, 2005. [Google Scholar]
- Lo, K.K.-W.; Li, S.P.-Y.; Zhang, K.Y. Development of luminescent iridium(III) polypyridine complexes as chemical and biological probes. New J. Chem. 2011, 35, 265–287. [Google Scholar] [CrossRef]
- Fernandez-Moreira, V.; Thorp-Greenwood, F.L.; Coogan, M.P. Application of d6 transition metal complexes in fluorescence cell imaging. Chem. Commun. 2010, 46, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Margapoti, E.; Shukla, V.; Valore, A.; Sharma, A.; Dragonetti, C.; Kitts, C.C.; Roberto, D.; Murgia, M.; Ugo, R.; Muccini, M. Excimer emission in single layer electroluminescent devices based on [Ir(4,5-diphenyl-2-methylthiazolo)2(5-methyl-1,10-phenanthroline)]+[PF6]−. J. Phys. Chem. C 2009, 113, 12517–12522. [Google Scholar] [CrossRef]
- Margapoti, E.; Muccini, M.; Sharma, A.; Colombo, A.; Dragonetti, C.; Robertob, D.; Valore, A. Optoelectronic properties of OLEC devices based on phenylquinoline and phenylpyridine ionic iridium complexes. Dalton Trans. 2012, 41, 9227–9231. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Kuddes, D.D.; Naquin, C.A.; Hesterberg, T.W.; Kusmierz, C.; Holliday, B.J.; Slinker, J.D. Improving light-emitting electrochemical cells with ionic additives. Appl. Phys. Lett. 2013, 102, 203305–203309. [Google Scholar] [CrossRef]
- Meier, S.B.; Hartmann, D.; Tordera, D.; Bolink, H.J.; Winnackera, A.; Sarfert, W. Dynamic doping and degradation in sandwich-type light-emitting electrochemical cells. Phys. Chem. Chem. Phys. 2012, 14, 10886–10890. [Google Scholar] [CrossRef] [PubMed]
- Zaarour, M.; Guerchais, V.; Le Bozec, H.; Dragonetti, C.; Righetto, S.; Roberto, D.; De Angelis, F.; Fantacci, S.; Lobello, M.G. An investigation on the second order nonlinear optical response of tris-cyclometallated Ir(III) complexes with various substituted 2-phenylpyridines. Dalton Trans. 2013, 42, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Zaarour, M.; Singh, A.; Latouche, C.; William, J.G.; Ledoux-Rak, I.; Zyss, J.; Boucekkine, A.; Le Bozec, H.; Guerchais, V.; Dragonetti, C.; et al. Linear and nonlinear optical properties of tris cyclometalated phenylpyridine Ir(III) complexes incorporating π-conjugated substituents. Inorg. Chem. 2013, 52, 7987–7994. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Fasani, E. A specialist periodical report. Photochemistry 2016, 43, 148–172. [Google Scholar]
- Tsuboyama, A.; Iwawaki, H.; Furugori, M.; Mukaide, T.; Kamatani, J.; Igawa, S.; Moriyama, T.; Miura, S.; Takiguchi, T.; Okada, S.; et al. Homoleptic cyclometalated iridium complexes with highly efficient red phosphorescence and application to organic light-emitting diode. J. Am. Chem. Soc. 2003, 125, 12971–12979. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.-I.; Su, H.-J.; Shu, C.-F.; Luo, L.; Diau, W.-G.; Cheng, C.-H.; Duan, J.-P.; Lee, G.-H. Tuning the emission and morphology of cyclometalated iridium complexes and their applications to organic light-emitting diodes. J. Mater. Chem. 2005, 15, 1035–1042. [Google Scholar] [CrossRef]
- Park, Y.H.; Park, G.Y.; Kim, Y.S. Heteroleptic iridium(III) complexes with phenylpridine and diphenylquinoline derivative ligands. Mol. Cryst. Liq. Cryst. 2007, 462, 197–207. [Google Scholar] [CrossRef]
- Dahule, H.K.; Dhoble, S.J. Effect of substituents on the photoluminescence performance of Ir(III) complexes: Synthesis and photo physical properties. J. Chem. Biol. Phys. Sci. 2012, 2, 1539–1550. [Google Scholar]
- Lupton, J.M.; Samuel, I.D.W.; Frampton, M.J.; Beavington, R.; Burn, P.L. Control of electrophosphorescence in conjugated dendrimer light-emitting diodes. Adv. Funct. Mater. 2001, 11, 287–294. [Google Scholar] [CrossRef]
- Lo, S.-C.; Richards, G.J.; Markham, J.P.J.; Namdas, E.B.; Sharma, S.; Burn, P.L. A light-blue phosphorescent dendrimer for efficient solution-processed light-emitting diodes. Adv. Funct. Mater. 2005, 15, 1451–1458. [Google Scholar] [CrossRef]
- Li, X.; Chen, Z.; Zhao, Q.; Shen, L.; Li, F.; Yi, T.; Cao, Y.; Huang, C. Nonconjugated dendritic iridium(III) complexes with tunable pyridine-based ligands: Synthesis, photophysical, electrochemical, and electroluminescent properties. Inorg. Chem. 2007, 46, 5518–5527. [Google Scholar] [CrossRef] [PubMed]
- Lowry, M.S.; Bernhard, S. Synthetically tailored excited states: Phosphorescent, cyclometalated iridium(III) complexes and their applications. Chem. Eur. J. 2006, 12, 7970–7977. [Google Scholar] [CrossRef] [PubMed]
- Adeloye, A.O.; Mphahlele, M.J. 2,4-Diarylquinolines: Synthesis, absorption and emission properties. J. Chem. Res. 2014, 38, 254–259. [Google Scholar] [CrossRef]
- Aranda, B.S.; Aguirre, P.; Moya, S.A.; Bonneau, M.; Williams, J.G.; Toupet, L.; Escadeillas, M.; Le Bozec, H.; Guerhais, V. Luminescent bis-cyclometallated iridium(III) complexes containing phosphine-based ligands: Influence of the P^N bridge. Polyhedron 2015, 86, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Grushin, V.V.; Herron, N.; LeCloux, D.D.; Marshall, W.J.; Petrov, V.A.; Wang, Y. New, efficient electroluminescent materials based on organometallic Ir complexes. Chem. Commun. 2001, 16, 1494–1495. [Google Scholar] [CrossRef]
- Jung, S.O.; Kang, Y.; Kim, H.S.; Kim, Y.H.; Lee, C.L.; Kim, J.J.; Lee, S.K.; Kwon, S.K. Effect of substitution of methyl groups on the luminescence performance of IrIII complexes: Preparation, structures, electrochemistry, photophysical properties and their applications in organic light-emitting diodes (OLEDs). Eur. J. Inorg. Chem. 2004, 16, 3415–3423. [Google Scholar] [CrossRef]
- Lee, S.J.; Park, K.M.; Yang, K.; Kang, Y. Blue phosphorescent Ir(III) complex with high color purity: fac-Tris(2’,6’-difluoro-2,3’-bipyridinato-N,C4’)iridium(III). Inorg. Chem. 2009, 48, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Lowry, M.S.; Hudson, W.R.; Pascal, R.A., Jr.; Bernhard, S. Accelerated luminophore discovery through combinatorial synthesis. J. Am. Chem. Soc. 2004, 126, 14129–14135. [Google Scholar] [CrossRef] [PubMed]
- Lowry, M.S.; Goldsmith, J.I.; Slinker, J.D.; Rohl, R.; Pascal, R.A., Jr.; Malliaras, G.G.; Bernhard, S. Single-layer electroluminescent devices and photoinduced hydrogen production from an ionic iridium(III) complex. Chem. Mater. 2005, 17, 5712–5719. [Google Scholar] [CrossRef]
- Laskar, I.R.; Chen, T.M. Tuning of wavelengths: Synthesis and photophysical studies of iridium complexes and their applications in organic light emitting devices. Chem. Mater. 2004, 16, 111–117. [Google Scholar] [CrossRef]
- Coppo, P.; Plummer, E.A.; De Cola, L. Tuning iridium(III) phenylpyridine complexes in the “almost blue” region. Chem. Commun. 2004, 15, 1774–1775. [Google Scholar] [CrossRef] [PubMed]
- Sengottuvelan, N.; Seo, H.-J.; Kang, S.K.; Kim, Y.-I. Tuning photophysical and electrochemical properties of heteroleptic cationic iridium(III) complexes containing substituted 2-phenylquinoxaline and biimidazole. Bull. Korean Chem. Soc. 2010, 31, 2309–2314. [Google Scholar] [CrossRef]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Kwong, R.; Tsyba, I.; Bortz, M.; Mui, B.; Bau, R.; Thompson, M.E. Synthesis and characterization of phosphorescent cyclometalated iridium complexes. Inorg. Chem. 2001, 40, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.G.; Brunold, T.C.; Riedener, T.; Gudel, H.U.; Fortsch, M.; Burgi, H. Facial tris cyclometalated rhodium(3+) and iridium(3+) complexes: Their synthesis, structure, and optical spectroscopic properties. Inorg. Chem. 1994, 33, 545–550. [Google Scholar] [CrossRef]
- Park, J.; Park, J.S.; Park, Y.G.; Lee, J.Y.; Kang, J.W.; Liu, J.; Dai, L.; Jin, S.H. Synthesis, characterization of the phenylquinoline-based on iridium(III) complexes for solution processable phosphorescent organic light-emitting diodes. Org. Electron. 2013, 14, 2114–2123. [Google Scholar] [CrossRef]
- Joshi, N.B.; Gangola, P.; Pant, D.D. Internal heavy atom effect on the radiative and non-radiative rate constants in xanthene dyes. J. Lumin. 1979, 21, 111–118. [Google Scholar] [CrossRef]
- Duprez, V.; Biancardo, M.; Spanggaard, H.; Krebs, F.C. Synthesis of conjugated polymers containing terpyridine–ruthenium complexes: Photovoltaic applications. Macromolecules 2005, 38, 10436–10448. [Google Scholar] [CrossRef]
- Schmid, B.; Graces, F.O.; Watts, R.J. Synthesis and characterizations of cyclometalated iridium(III) solvento complexes. Inorg. Chem. 1994, 33, 9–14. [Google Scholar] [CrossRef]
- Colombo, M.G.; Gudel, H.U. Synthesis and high-resolution optical spectroscopy of bis[2-(2-thienyl)pyridinato-C3,N’](2,2’-bipyridine)iridium(III). Inorg. Chem. 1993, 32, 3081–3087. [Google Scholar] [CrossRef]
- Ichimura, K.; Kobayashi, T.; King, K.A.; Watts, R.J. Excited-state absorption spectroscopy of ortho-metalated iridium(III) complexes. J. Phys. Chem. 1987, 91, 6104–6106. [Google Scholar] [CrossRef]
- King, K.A.; Spellane, P.J.; Watts, R.J. Excited-state properties of a triply ortho-metalated iridium(III) complex. J. Am. Chem. Soc. 1985, 107, 1431–1432. [Google Scholar] [CrossRef]
- Balton, C.B.; Murtaza, Z.; Shaver, R.J.; Rillema, D.P. Excited-state properties of platinum(II) complexes containing biphenyl as a ligand: Complexes of the type [(bph)PtL2], where L = monodentate or saturated bidentate ligands. Inorg. Chem. 1992, 31, 3230–3235. [Google Scholar]
- Wong, W.Y.; Zhou, G.J.; Yu, X.M.; Kwok, H.S.; Tang, B.Z. Amorphous diphenylaminofluorene-functionalized iridium complexes for high-efficiency electrophosphorescent light-emitting diodes. Adv. Funct. Mater. 2006, 16, 838–846. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Berlin, Germany, 2006; 954p, ISBN 978-0-387-31278-1. [Google Scholar]
- Eisinger, J. A variable temperature, UV luminescence spectrograph for small samples. Photochem. Photobiol. 1969, 94, 15–21. [Google Scholar]
- Kasha, M. Paths of molecular excitation. Radiat. Res. 1960, 2, 243–275. [Google Scholar] [CrossRef] [PubMed]
- Ge, G.; Zhang, G.; Guo, H.; Chuai, Y.; Zou, D. Yellow organic light-emitting diodes based on phosphorescent iridium(III) pyrazine complexes: Fine tuning of emission. Inorg. Chim. Acta 2009, 362, 2231–2236. [Google Scholar] [CrossRef]
- Dragonetti, C.; Falciola, L.; Mussini, P.; Righetto, S.; Roberto, D.; Ugo, R.; Valore, A.; Angelis, D.F.; Fantacci, S.; Sgamellotti, A.; et al. The role of substituents on functionalized 1,10-phenanthroline in controlling the emission properties of cationic iridium(III) complexes of interest for electroluminescent devices. Inorg. Chem. 2007, 46, 8533–8547. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F.; Fantacci, S.; Evans, N.; Klein, C.; Zakeeruddin, S.M.; Moser, J.E.; Kalyanasundaram, K.; Bolink, H.J.; Gratzel, M.; Nazeeruddin, M.K. Controlling phosphorescence color and quantum yields in cationic iridium complexes: A combined experimental and theoretical study. Inorg. Chem. 2007, 46, 5989–6001. [Google Scholar] [CrossRef] [PubMed]
- Hasan, K.; Pal, A.K.; Auvray, T.; Zysman-Colman, E.; Hanan, G.S. Blue-green emissive cationic iridium(III) complexes using partially saturated strongly-donating guanidyl-pyridine/-pyrazine ancillary ligands. Chem. Commun. 2015, 51, 14060–14063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, I.; Dreyse, P.; Cortes-Arriagada, D.; Sundararajan, M.; Morgado, C.; Brito, I.; Roldan-Carmona, C.; Bolink, H.J.; Loeb, B. A comparative study of Ir(III) complexes with pyrazino[2,3-f][1,10]phenanthroline and pyrazino[2,3-f][4,7]phenanthroline ligands in light-emitting electrochemical cells (LECs). Dalton Trans. 2015, 44, 14771–14781. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Qiao, J.; Duan, I.; Dong, G.; Zhang, D.; Wang, L.; Qiu, Y. Toward highly efficient solid-state white light-emitting electrochemical cells: Blue-green to red emitting cationic iridium complexes with imidazole-type ancillary ligands. Adv. Funct. Mater. 2009, 19, 2950–2960. [Google Scholar] [CrossRef]
- Sunahara, H.; Urano, Y.; Kijima, H.; Nagano, T. Design and synthesis of a library of BODIPY-based environmental polarity sensors utilizing photoinduced electron-transfer-controlled fluorescence ON/OFF switching. J. Am. Chem. Soc. 2007, 129, 5597–5604. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Sheikhet, I. Quantum Chemical and Statistical Theory of Solutions—A Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Frisch, M.J.E.A.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D. 01, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Dooley, R.; Milfeld, K.; Guiang, C.; Pamidighantam, S.; Allen, G. From proposal to production: Lessons learned developing the computational chemistry grid cyberinfrastructure. J. Grid Comput. 2006, 4, 195–208. [Google Scholar] [CrossRef]
- Milfeld, K.; Guiang, C.; Pamidighantam, S.; Giuliani, J. Cluster computing through an application-oriented computational chemistry grid. In Proceedings of the 2005 Linux Clusters: The HPC Revolution, NCSA University of Illinois, CHPC University of New Mexico and IBM, Chapel Hill, NC, USA, 25–28 April 2005. [Google Scholar]
- Dooley, R.; Allen, G.; Pamidighantam, S. Computational chemistry grid: Production cyberinfrastructure for computational chemistry. In Proceedings of the 13th Annual Mardi Gras Conference, CCT, Louisiana State University, Baton Rouge, LA, USA, 3–5 February 2005; p. 83. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 4a | Literature [30] | |
|---|---|---|
| Ir–N | 2.299 | 2.173 |
| Ir–C | 2.018 | 2.017 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adeloye, A.O.; Mphahlele, M.J.; Adekunle, A.S.; Rhyman, L.; Ramasami, P. Spectroscopic, Electrochemical and DFT Studies of Phosphorescent Homoleptic Cyclometalated Iridium(III) Complexes Based on Substituted 4-Fluorophenylvinyl- and 4-Methoxyphenylvinylquinolines. Materials 2017, 10, 1061. https://doi.org/10.3390/ma10101061
Adeloye AO, Mphahlele MJ, Adekunle AS, Rhyman L, Ramasami P. Spectroscopic, Electrochemical and DFT Studies of Phosphorescent Homoleptic Cyclometalated Iridium(III) Complexes Based on Substituted 4-Fluorophenylvinyl- and 4-Methoxyphenylvinylquinolines. Materials. 2017; 10(10):1061. https://doi.org/10.3390/ma10101061
Chicago/Turabian StyleAdeloye, Adewale O., Malose J. Mphahlele, Abolanle S. Adekunle, Lydia Rhyman, and Ponnadurai Ramasami. 2017. "Spectroscopic, Electrochemical and DFT Studies of Phosphorescent Homoleptic Cyclometalated Iridium(III) Complexes Based on Substituted 4-Fluorophenylvinyl- and 4-Methoxyphenylvinylquinolines" Materials 10, no. 10: 1061. https://doi.org/10.3390/ma10101061