
Role of Tartaric Acid in Chemical, Mechanical and Self-Healing Behaviors of a Calcium-Aluminate Cement Blend with Fly Ash F under Steam and Alkali Carbonate Environments at 270 °C

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Samples Preparation and Testing
3. Results
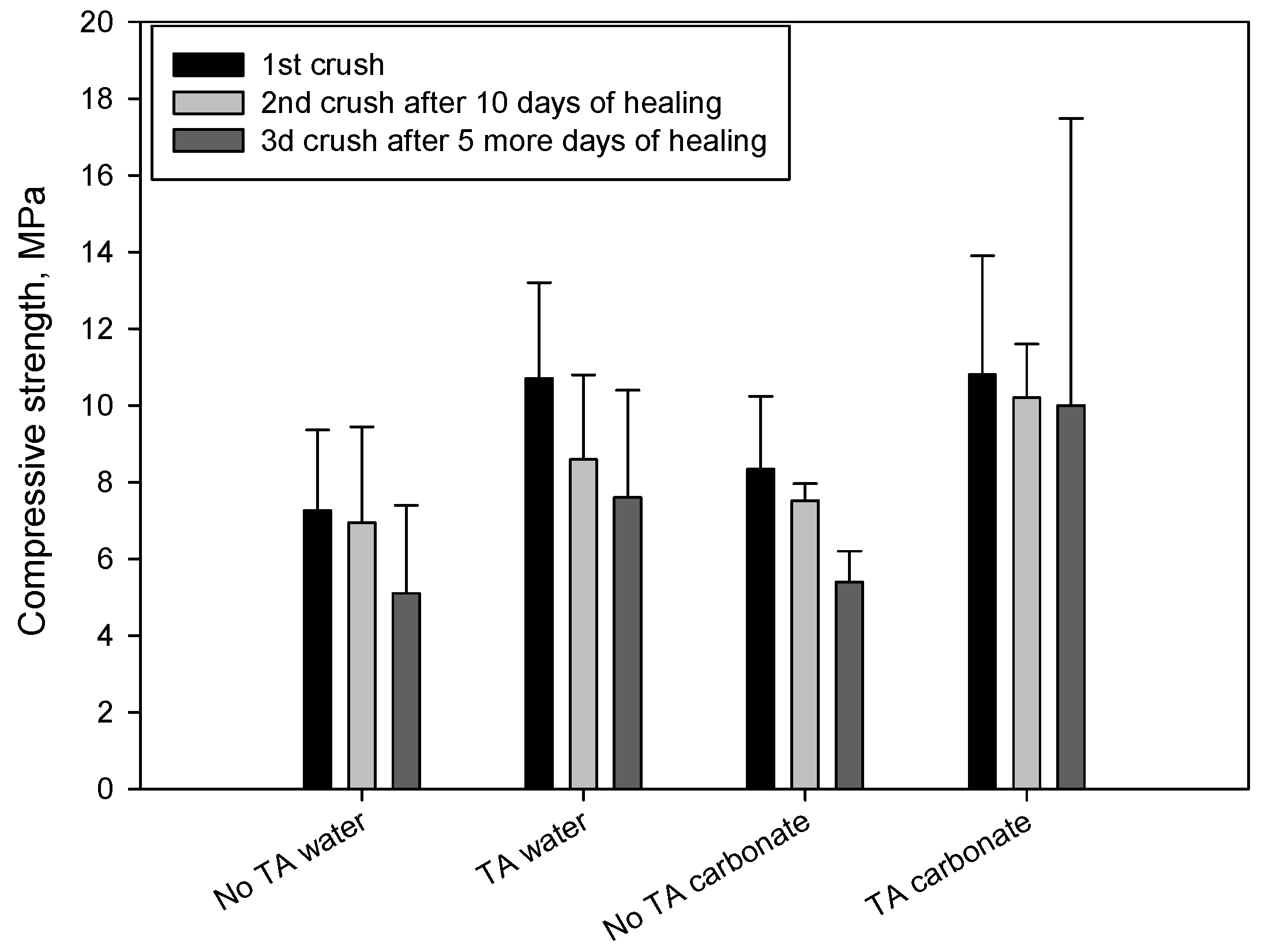
3.1. Mechanical Properties
3.2. Crack Sealing, Optical Microscope and µEDX Elemental Imaging
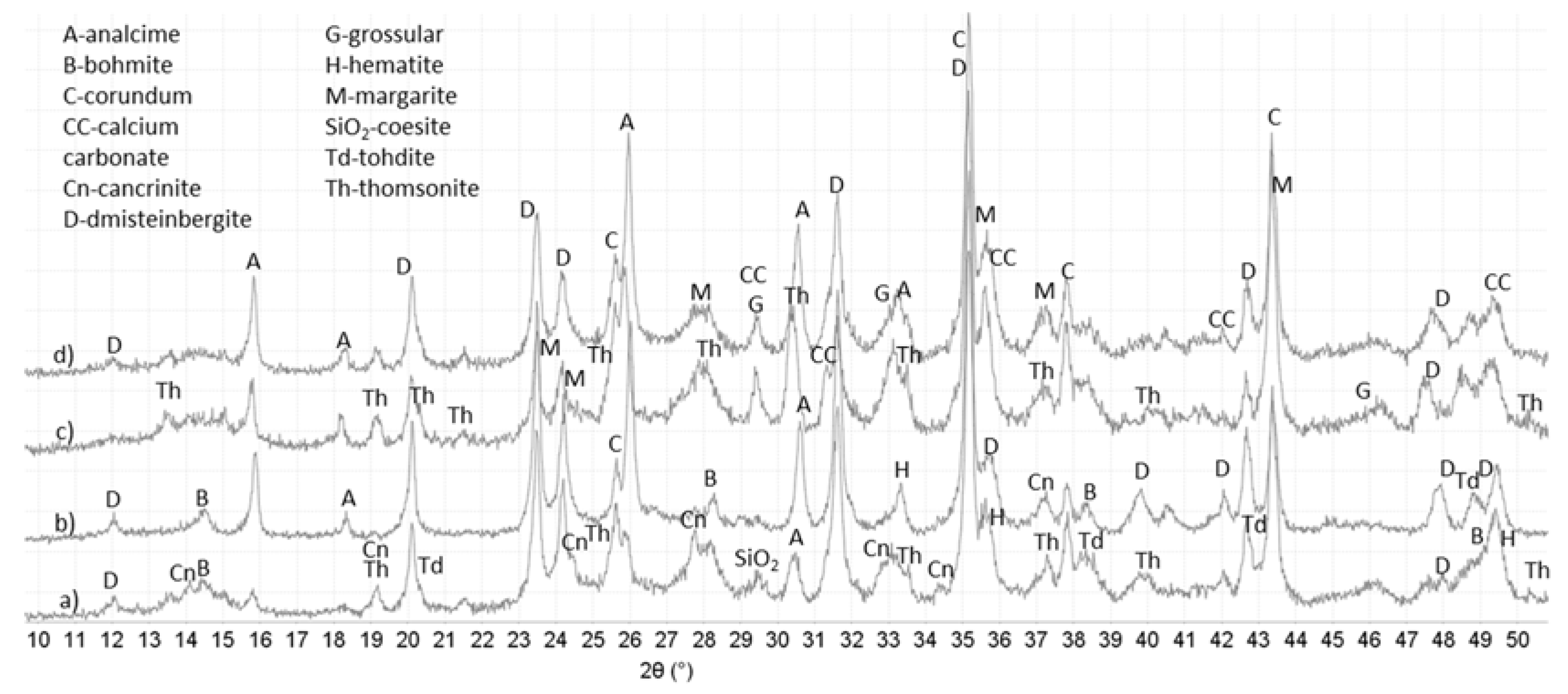
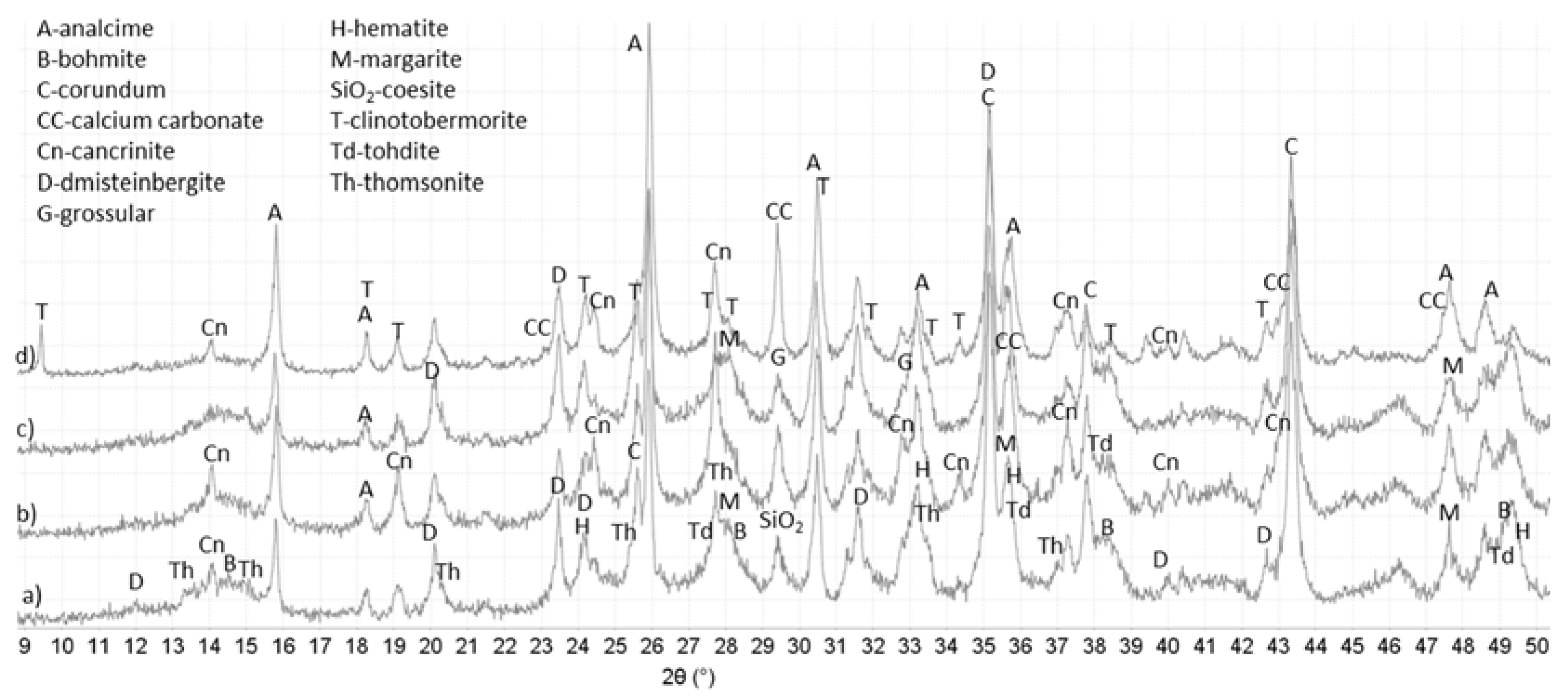
3.3. XRD Study
3.4. DTG Study
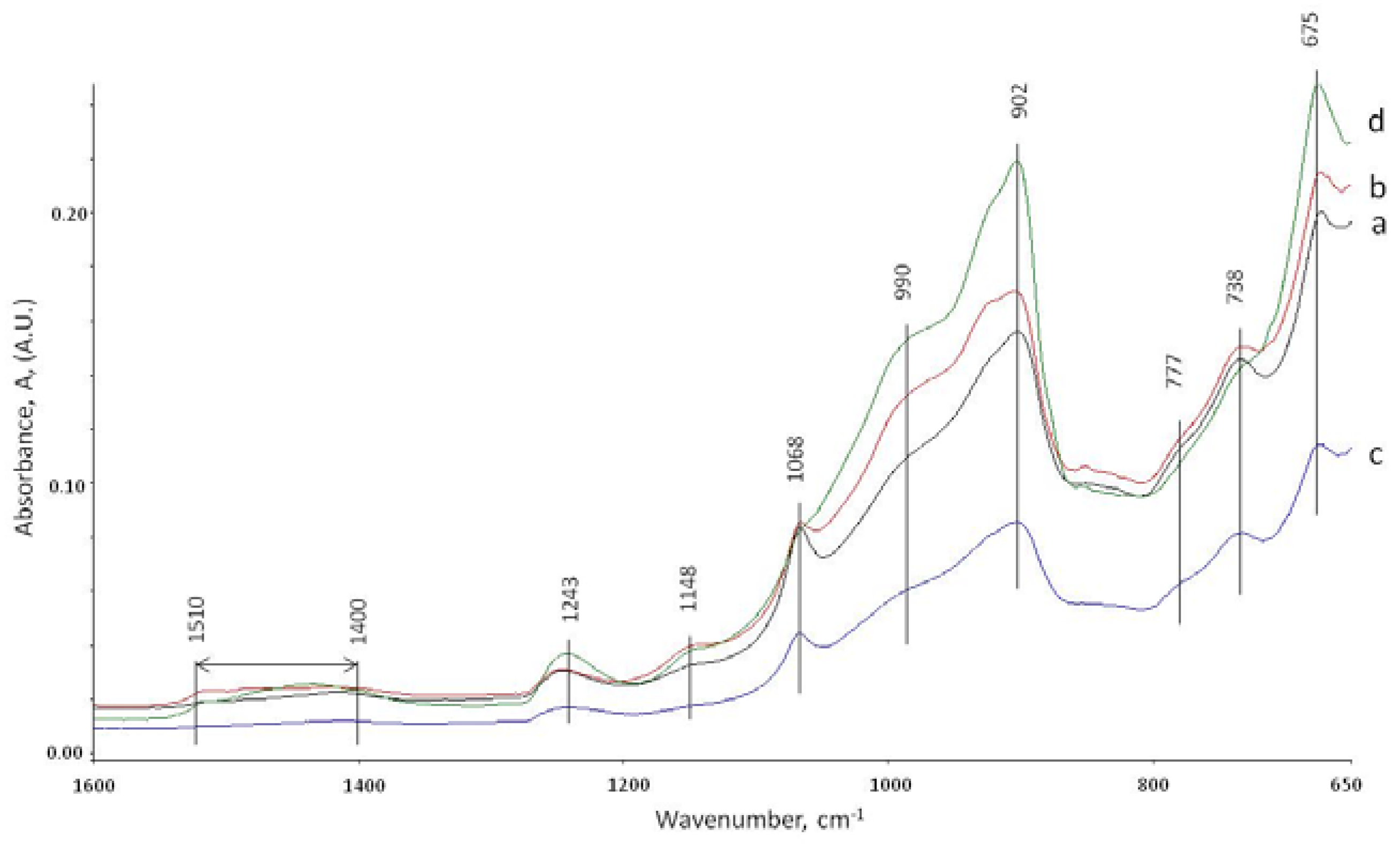
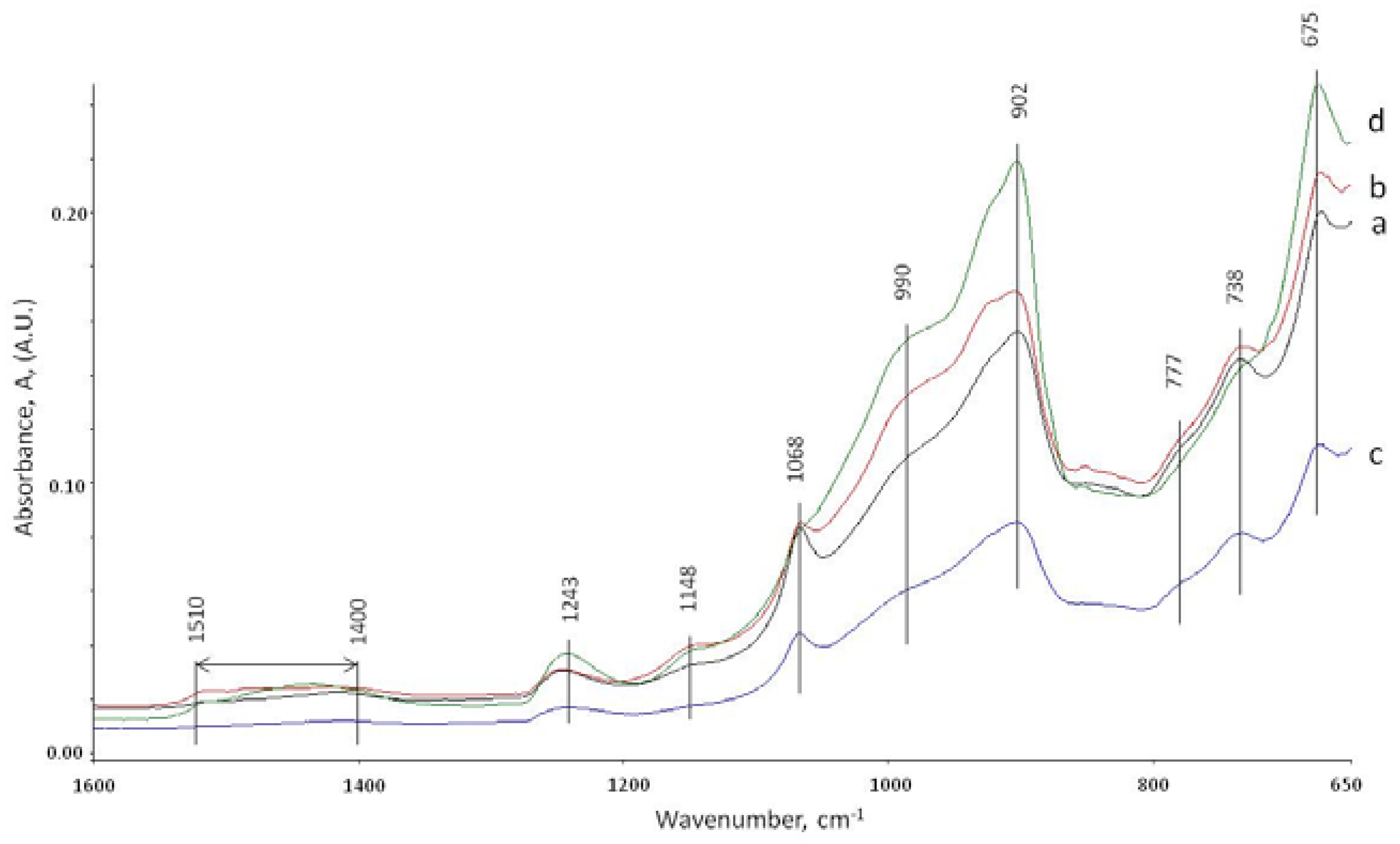
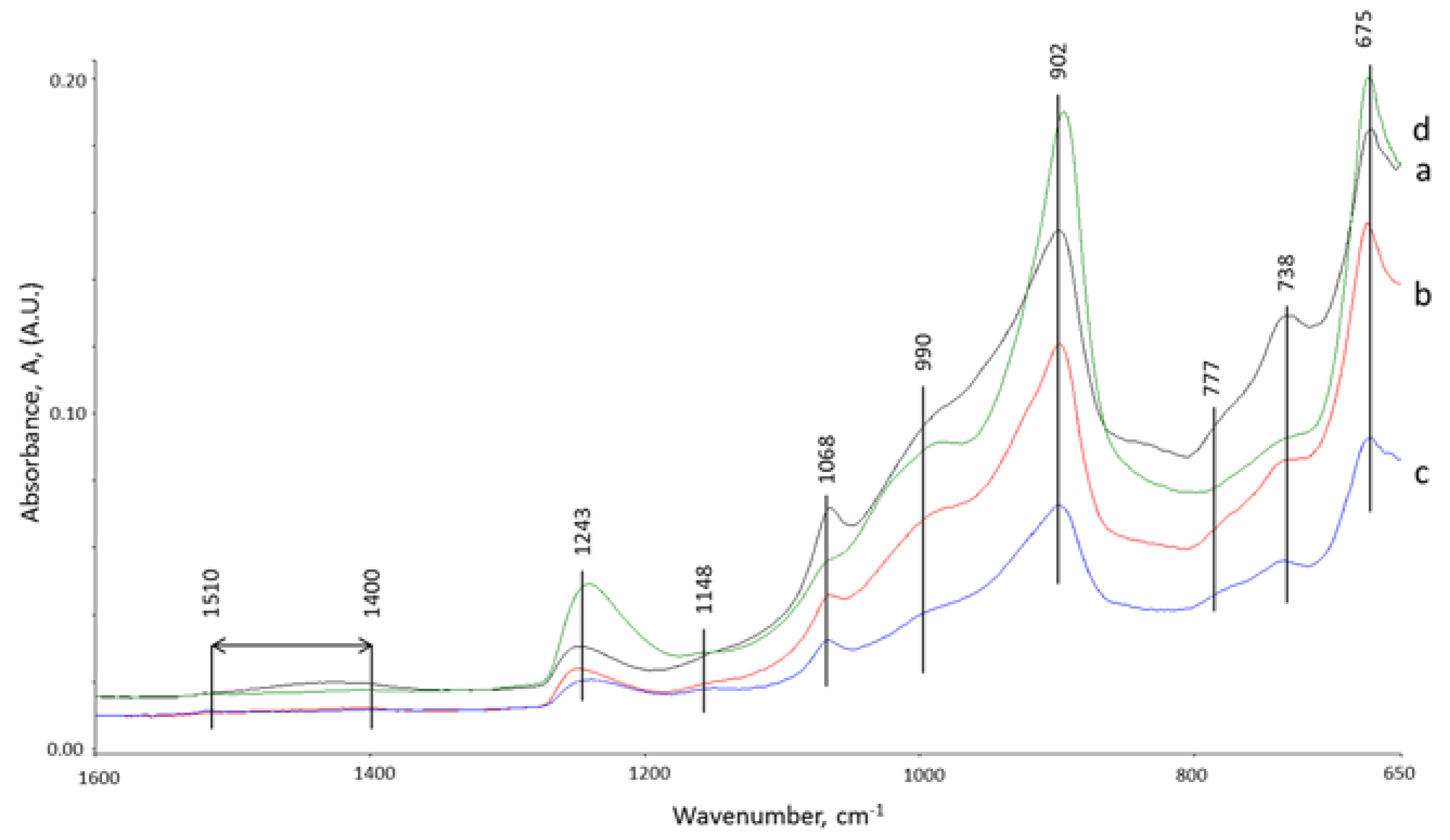
3.5. ATR-FTIR Studies
3.6. Microstructural Characterization
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TA | Tartaric Acid |
| CAC | Calcium Aluminate Cement |
| FAF | Fly Ash type F |
| SMS | Sodium Meta-Silicate |
| TSRC | Thermal Shock Resistant Cement |
References
- Yıldırım, G.; Keskin, Ö.; Keskin, S.; Sahmaran, M.; Lachemi, M. A review of intrinsic self-healing capability of engineered cementitious composites: Recovery of transport and mechanical properties. Constr. Build. Mater. 2015, 101, 10–21. [Google Scholar] [CrossRef]
- Muhammad, N.Z.; Shafaghat, A.; Keyvanfar, A.; Majid, M.Z.A.; Ghoshal, S.K.; Yasouj, S.E.M.; Ganiyu, A.A.; Kouchaksaraei, M.S.; Kamyab, H.; Taheri, M.M.; et al. Tests and methods of evaluating the self-healing efficiency of concrete: A review. Constr. Build. Mater. 2016, 112, 1123–1132. [Google Scholar] [CrossRef]
- Ahn, T.H.; Kishi, T. Crack self-healing behavior of cementitious composites incorporating various mineral admixtures. J. Adv. Concr. Technol. 2010, 8, 171–186. [Google Scholar] [CrossRef]
- Nelson, E.; Barlet-Gouedard, V. Thermal cements. In Well Cementing, 2nd ed.; Nelson, E., Guillot, D., Eds.; Schlumberger: Sugar Land, TX, USA, 2006; pp. 328–334. [Google Scholar]
- Van Tittelboom, K.; De Belie, N. Self-healing in cementitious materials—A review. Materials 2013, 6, 2182–2217. [Google Scholar] [CrossRef]
- Mostavi, E.; Asadi, S.; Hassan, M.M.; Alansari, M. Evaluation of Self-Healing Mechanisms in Concrete with Double-Walled Sodium Silicate Microcapsules. J. Mater. Civil Eng. 2015, 27. [Google Scholar] [CrossRef]
- Qureshi, T.S.; Kanellopoulos, A.; Al-Tabbaa, A. Encapsulation of expansive powder minerals within a concentric glass capsule system for self-healing concrete. Constr. Build. Mater. 2016, 121, 629–643. [Google Scholar] [CrossRef]
- Li, W.; Zhu, X.; Zhao, N.; Jiang, Z. Preparation and Properties of Melamine Urea-Formaldehyde Microcapsules for Self-Healing of Cementitious Materials. Materials 2016, 9, 152. [Google Scholar] [CrossRef]
- Li, W.; Jiang, Z.; Yang, Z.; Zhao, N.; Yuan, W. Self-Healing Efficiency of Cementitious Materials Containing Microcapsules Filled with Healing Adhesive: Mechanical Restoration and Healing Process Monitored by Water Absorption. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, V.C.; Herbert, E. Robust self-healing concrete for sustained infrastructure. J. Adv. Concr. Technol. 2012, 10, 207–218. [Google Scholar] [CrossRef]
- Lauer, K.R.; Slate, F.O. Autogeneous healing of cement paste. ACI J. 1956, 52, 1083–1097. [Google Scholar]
- Qian, S.; Zhou, J.; De Rooij, M.R.; Schlangen, E.; Ye, G.; Van Breugel, K. Self-healing behavior of strain hardening cementitious composites incorporating local waste materials. Cem. Concr. Compos. 2009, 31, 613–621. [Google Scholar] [CrossRef]
- Sahmaran, M.; Yildirim, G.; Erdem, T.K. Self-healing capabilities of cementitious composites incorporating different supplementary cementitious materials. Cem. Concr. Compos. 2013, 35, 89–101. [Google Scholar] [CrossRef]
- Yildirim, G.; Sahmaran, M.; Balcikanli, M.; Ozbay, E.; Lachemi, M. Influence of cracking and healing on the gas permeability of cementitious composites. Constr. Build. Mater. 2015, 85, 217–226. [Google Scholar] [CrossRef]
- Ozbay, M.; Sahmaran, M.; Yucel, H.E.; Erdem, T.K.; Lachemi, M.; Li, V.C. Effect of sustained flexural loading on self-healing of engineered cementitious composites. J. Adv. Concr. Technol. 2013, 11, 167–179. [Google Scholar]
- Reinhardt, H.; Jooss, M. Permeability and self-healing of cracked concrete as a function of temperature and crack width. Cem. Concr. Res. 2003, 33, 981–985. [Google Scholar] [CrossRef]
- Jacobsen, S.; Marchand, H.; Homain, S.E.M. Observation of microstructure of frost deteriorated and self-healed concrete. Cem. Concr. Res. 1995, 25, 1781–1790. [Google Scholar] [CrossRef]
- Ismail, M.; Toumi, A.; Francois, R.; Gagne, R. Effect of crack opening on local diffusion of chloride inert materials. Cem. Concr. Res. 2004, 34, 711–716. [Google Scholar] [CrossRef]
- Termkhajornkit, P.; Nawa, T.; Yamashiro, Y.; Saito, T. Self-healing ability of fly ash-cement systems. Cem. Concr. Compos. 2009, 31, 195–203. [Google Scholar] [CrossRef]
- Huang, H.; Ye, G.; Damidot, D. Effect of blast furnace slag on self-healing of microcracks in cementitious materials. Cem. Concr. Res. 2014, 60, 68–82. [Google Scholar] [CrossRef]
- Qian, S.; Zhou, J.; Schlangen, E. Influence of curing condition and precracking time on the self-healing behavior of engineered cementitious composites. Cem. Concr. Compos. 2010, 32, 686–693. [Google Scholar] [CrossRef]
- Carsana, M.; Frassoni, M.; Bertolini, L. Comparison of ground-waste-glass with other supplementary cementitious materials. Cem. Concr. Compos. 2014, 45, 39–45. [Google Scholar] [CrossRef]
- Abrams, D.A. Tests of Bond between Concrete and Steel; Bulletin 71; University of Illinois: Urbana Champaign, IL, USA, 8 December 1913. [Google Scholar]
- Loving, M.W. Autogenous Healing of Concrete; Bulletin 13; American Concrete Pipe Association: Irving, TX, USA, 1936. [Google Scholar]
- Edvardsen, C. Water permeability and autogenous healing of cracks in concrete. ACI Mater. J. 1999, 96, 448–455. [Google Scholar]
- Kishi, T.; Ahn, T.H.; Hosoda, H.; Suzuki, S.; Takaoka, H. Self-healing behavior by cementitious recrystallization of cracked concrete. In Proceedings of the first international conference on self-healing materials, Noordwijk, The Netherlands, 18–20 April 2007. [Google Scholar]
- Reinhardt, H.W.; Jonkers, H.; Van Tittelboom, K.; Snoeck, N.; De Belie, N.; De Muynck, W.; Verstraete, W.; Wang, J.; Mechtcherine, V. Recovery against environmental action. In Self-Healing Phenomena in Cement-Based Materials; De Roojik, M., Van Tittelboom, K., De Belie, N., Schlangen, E., Eds.; Springler: Victoria, Australia, 2013; pp. 65–117. [Google Scholar]
- Sahmaran, M.; Yildirim, G.; Ozbay, E.; Ahmed, K.; Lachemi, M. Self-healing ability of cementitious compositites: effect of addition of pre-soaked expanded perlite. Mag. Concr. Res. 2014, 66, 409–419. [Google Scholar] [CrossRef]
- Yildirim, G.; Sahmaran, M.; Ahmed, H. Influence of hydrated lime addition on the self-healing capability of high-volume fly ash incorporated cementitious composites. J. Mater. Civil Eng. 2014, 27. [Google Scholar] [CrossRef]
- Perez, G.; Erkizia, E.; Gaitero, J.J.; Kaltzakorta, I.; Jimenez, I.; Guerrero, A. Synthesis and characterization of epoxy encapsulating silica microcapsules and amine functionalized silica nanoparticles for development of an innovative self-healing concrete. Mater. Chem. Phys. 2015, 165, 39–48. [Google Scholar] [CrossRef]
- Qureshi, T.S.; Al-Tabbaa, A. Self-healing of drying shrinkage cracks in cement-based materials incorporating reactive MgO. Smart Mater. Struct. 2016, 25. [Google Scholar] [CrossRef]
- Kanellopoulos, A.; Qureshi, T.S.; Al-Tabbaa, A. Glass encapsulated minerals for self-healing in cement based composites. Constr. Build. Mater. 2015, 98, 780–791. [Google Scholar] [CrossRef]
- Afshinnia, K.; Rangaraju, P.R. Impact of combined use of ground glass powder and crushed glass aggregate on selected properties of Portland cement concrete. Constr. Build. Mater. 2016, 117, 263–272. [Google Scholar] [CrossRef]
- Siad, H.; Lachemi, M.; Sahmaran, M.; Hossain, K.M.A. Effect of glass powder on sulfuric acid resistance of cementitious materials. Constr. Build. Mater. 2016, 113, 163–173. [Google Scholar] [CrossRef]
- Kim, J.; Moon, J.H.; Shim, J.W.; Sim, J.; Lee, H.G.; Zi, G. Durability properties of a concrete with waste glass sludge exposed to freeze-and-thaw condition and de-icing salt. Constr. Build. Mater. 2014, 66, 398–402. [Google Scholar] [CrossRef]
- Jaroenratanapirom, D.; Sahamitmongkol, R. Effect of different mineral additives and cracking ages on self-healing performance of mortar. In Proceedings of the 6th annual concrete conference, Petchaburi, Thailand, 2010; pp. 551–556. [Google Scholar]
- ACI Committee 212, Chapter 15. In Report on Chemical Admixtures for Concrete; Report ACI 212-3R-10; American Concrete Institute (ACI): Farmington Hills, MI, USA, 2010; pp. 46–50.
- Ferrara, L.; Krelani, V.; Moretti, F. On the use of crystalline admixtures in cement based construction materials: from porosity reducers to promoters of self-healing. Smart Mater. Struct. 2016, 25. [Google Scholar] [CrossRef]
- Sisomphon, K.; Copuroglu, O.; Koenders, E.A.B. Self-healing of surface cracks in mortars with expansive additive and crystalline additive. Cem. Concr. Compos. 2012, 34, 566–574. [Google Scholar] [CrossRef]
- Roig-Flores, M.; Moscato, S.; Serna, P.; Ferrara, L. Self-healing capability of concrete with crystalline admixtures in different environments. Constr. Build. Mater. 2015, 86, 1–11. [Google Scholar] [CrossRef]
- Roig-Flores, M.; Pirritano, F.; Serna, P.; Ferrara, L. Effect of crystalline admixtures on the self-healing capability of early-age concrete studied by means of permeability and crack closing tests. Constr. Build. Mater. 2016, 114, 447–457. [Google Scholar] [CrossRef]
- Ozyurtkan, M.; Radonjic, M. An experimental study of the effect of CO2 rich brine on artificially fractured well-cement. Cem. Concr. Compos. 2014, 45, 201–208. [Google Scholar] [CrossRef]
- Pyatina, T.; Sugama, T.; Ronne, A. Self-repairing Geothermal Well Cement Composites. In GRC Transactions; Geothermal Resources Council: Sacramento, CA, USA, 2016; Volume 40. [Google Scholar]
- Pyatina, T.; Sugama, T.; Moon, J.; James, S. Effect of tartaric acid on hydration of a sodium-metasilicate-activated blend of calcium aluminate cement and fly ash F. Materials 2016, 9. [Google Scholar] [CrossRef]
- Pyatina, T.; Sugama, T. Use of Carbon Microfibers for Reinforcement of Calcium Aluminate-Class F Fly Ash cement activated with Sodium Meta-Silicate at up to 300 °C. In GRC Transactions; Geothermal Resources Council: Reno, Nevada, 2015; Volume 39. [Google Scholar]
- Winnefeld, F.; Leemann, A.; Lucuk, M.; Svoboda, P.; Neuroth, M. Assessment of phase formation in alkali activated low and high calcium fly ashes in building materials. Constr. Build. Mater. 2010, 24, 1086–1093. [Google Scholar] [CrossRef]
- Andini, S.; Cioffi, R.; Colangelo, F.; Grieco, T.; Montagnaro, F.; Santoro, L. Coal fly ash as raw material for the manufacture of geopolymer-based products. Waste Manag. 2008, 28, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lodeiro, I.; Palomo, A.; Fernandez-Jimenez, A.; Macphee, D.E. Compatibility studies between N-A-S-H and C-A-S-H gels. Study in the ternary diagram Na2O-CaO-Al2O3-SiO2-H2O. Cem. Concr. Res. 2011, 41, 923–931. [Google Scholar] [CrossRef]
- Foldvari, M. Handbook of Thermogravimetric System of Minerals and its Use in Geological Practice; Occasional papers of the Geological Institute of Hungary; Geological Institute of Hungary: Budapest, Hungary, 2011; Volume 213. [Google Scholar]
- Harada, K.; Tanaka, K.; Nagashima, K. New data on the analcime-wairakite series. Am. Mineral. 1972, 57, 924–931. [Google Scholar]
- Gosselin, C.; Scrivener, K.L. Microstructure development of calcium aluminate cements accelerated by lithium sulphate. In Calcium Aluminate Cements Proceeding of the Centenary Conference 2008; Fentiman, C.H., Mangabhai, R.J., Scrivener, K.L., Eds.; IHS BRE Press: Bracknell, UK, 2008; pp. 109–122. [Google Scholar]
- Klimesch, D.S.; Ray, A. DTA-TGA of unstirred autoclaved metakaolin-lime-quartz slurries. Formation of hydrogarnet. Thermochim. Acta. 1998, 316, 149–154. [Google Scholar] [CrossRef]
- Klimesch, D.S.; Ray, A. DTA-TGA evaluations of the CaO-Al2O3-SiO2-H2O system treated hydrothermally. Thermochim. Acta. 1999, 334, 115–122. [Google Scholar] [CrossRef]
- Breck, D.W. Zeolite molecular sieves. In Structure, Chemistry and Use; Wiley and Sons: New York, NY, USA; London, UK; Sydney, Australia; Toronto, Canada, 1974; p. 771. [Google Scholar]
- Hassan, I. The thermal behavior of cancrinite. The Canadian Mineralogist. 1996, 34, 893–900. [Google Scholar]
- Hannicutt, W.A. Characterization of Calcium-Silicate-Hydrate and Calcium-Alumino-Silicate Hydrate. Master’s Thesis, University of Illinois, Urbana-Champagne, IL, USA, 2013. [Google Scholar]
- Roy, B.N. Spectroscopic analysis of the structure of silicate glasses along the joint of xMAlO2-(1-x)SiO2 (M=Li, Na, K, Rb, Cs). J. Am. Ceramic Soc. 1987, 70, 183–192. [Google Scholar] [CrossRef]
- Husung, R.D.; Doremus, R.H. The infrared transmission spectra of four silicate glasses before and after exposure to water. J. Mater. Res. 1990, 5, 2209–2216. [Google Scholar] [CrossRef]
- Kline, A.A.; Mullins, M.E. Sol-gel kinetics followed by cylindrical attenuated total reflectance infrared spectroscopy. J. Am. Ceramic Soc. 1991, 74, 2559–25563. [Google Scholar] [CrossRef]
- Bakharev, T. Resistance of geopolymer materials to acid attack. Cem. Concr. Res. 2005, 35, 658–670. [Google Scholar] [CrossRef]
- Saikia, B.J.; Parthasarathy, G.; Sarmah, N.C. Fourier transform infrared spectroscopic estimation of crystallinity in SiO2 based rocks. Bull. Mater. Sci. 2008, 31, 775–779. [Google Scholar] [CrossRef]
- Guan, W.; Ji, F.; Fang, Z.; Fang, D.; Cheng, Y.; Yan, P.; Chen, Q. Low hydrothermal temperature synthesis of porous calcium silicate hydrate with enhanced reactivity SiO2. Ceramic Int. 2014, 40, 4415–4420. [Google Scholar] [CrossRef]
- Xyla, A.G.; Koutsoukos, P.G. Quantitative analysis of calcium carbonate polymorphs by infrared spectroscopy. J. Chem. Soc. Faraday Trans. 1989, 1, 3165–3172. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Anbalagan, G.; Pandi, S. Raman and infrared spectra of carbonate of calcite structure. J. Raman spectrosc. 2006, 37, 892–899. [Google Scholar] [CrossRef]
- Ylmen, R.; Jaglid, U. Carbonation of Portland cement studied by diffuse reflection Fourier transform infrared spectroscopy. Int. J. Concr. Struct. Mater. 2013, 7, 119–125. [Google Scholar] [CrossRef]
- Almeida, R.M.; Pantano, C.G. Structural investigation of silica gel films by infrared spectroscopy. J. Appl. Phys. 1990, 68, 4225–4232. [Google Scholar] [CrossRef]
- Fidalgo, A.; Ilharco, L.M. The defect structure of sol-gel-derived silica/polytetrahydrofuran hybrid films by FTIR. J. Non-Cryst. Solids. 2001, 283, 144–154. [Google Scholar] [CrossRef]
- Tanner, P.A.; Yan, B.; Zhang, H. Preparation and luminescence properties of sol-gel hybrid materials incorporated with europium complexes. J. Mater. Sci. 2000, 35, 4325–4328. [Google Scholar] [CrossRef]
- Kiss, A.B.; Keresztury, G.; Farkas, L. Raman and i.r. spectra and structure of bohmite (γ-AlOOH). Evidence for the recently discarded D172h space group. Spectrochim. Acta Part A: Mol. Spectrosc. 1980, 36A, 653–658. [Google Scholar] [CrossRef]
- Morterra, C.; Emanuel, C.; Cerrato, G.; Magnacca, G. Infrared study of some surface properties of bohmite (γ-AlO2H). J. Chem. Soc. Faraday Trans. 1992, 88, 339–348. [Google Scholar] [CrossRef]
- Boumaza, A.; Favaro, L.; Ledion, J.; Sattonnay, G.; Brubach, J.B.; Berthet, P.; Huntz, A.M.; Roy, P.; Tetot, R. Structural transitions in alumina nanoparticles by heat treatment. J. Solid State Chem. 2009, 182, 1171–1176. [Google Scholar] [CrossRef]
- Van Garderen, N.; Clemens, F.J.; Aneziris, C.G.; Graule, T. Improve γ-alumina support based pseudo-bohmite shaped by micro-extrusion process for oxygen carrier support application. Ceramics Int. 2012, 38, 5481–5492. [Google Scholar] [CrossRef]
- Pyatina, T.; Sugama, T. Acid resistance of calcium aluminate cement-fly ash F blends. Adv. Cem. Res. 2016, 28, 1–25. [Google Scholar] [CrossRef]
- Palomo, A.; Grutzeck, M.W.; Blanco, M.T. Alkali-activated fly ashes: A cement for the future. Cem. Concr. Res. 1999, 29, 1323–1329. [Google Scholar] [CrossRef]
- Pyatina, T.; Sugama, T. Resistance of fly ash-Portland cement blends to thermal shock. Adv. Cem. Res. 2016, 28, 121–131. [Google Scholar] [CrossRef]
- Giannopoulou, I.; Panias, D. Hydrolytic stability of sodium silicate gels in the presence of aluminum. J. Mater. Sci. 2010, 45, 5370–5377. [Google Scholar] [CrossRef]
- Pyatina, T.; Sugama, T. Set-Controlling Additive for Thermal Shock-Resistant Cement; BNL-104352-2013-IR; Brookhaven National Laboratory: Upton, NY, USA, 2013. [Google Scholar]
- Qomi, M.J.A.; Ulm, F.J.; Pellenq, R.J.M. Evidence on the dual nature of aluminum in the calcium-silicate-hydrates based on atomistic simulations. J. Am. Ceramic Soc. 2012, 95, 1128–1137. [Google Scholar]
- Myers, R.J.; Bernal, S.A.; San Nicolas, R.; Provis, J.L. Generalized structural description of calcium-sodium aluminosilicate hydrate gels: The cross-linked substituted tobermorite model. Langmuir 2013, 29, 5294–5306. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.; Clark, S.M.; Henderson, C.M.B. Hydrothermal formation of the calcium silicate hydrates, tobermorite (Ca5Si6O16(OH)2·4H2O) and xonotlite (Ca6Si6O17(OH)2): An synchrotron study. Chem. Geol. 2000, 167, 129–140. [Google Scholar] [CrossRef]




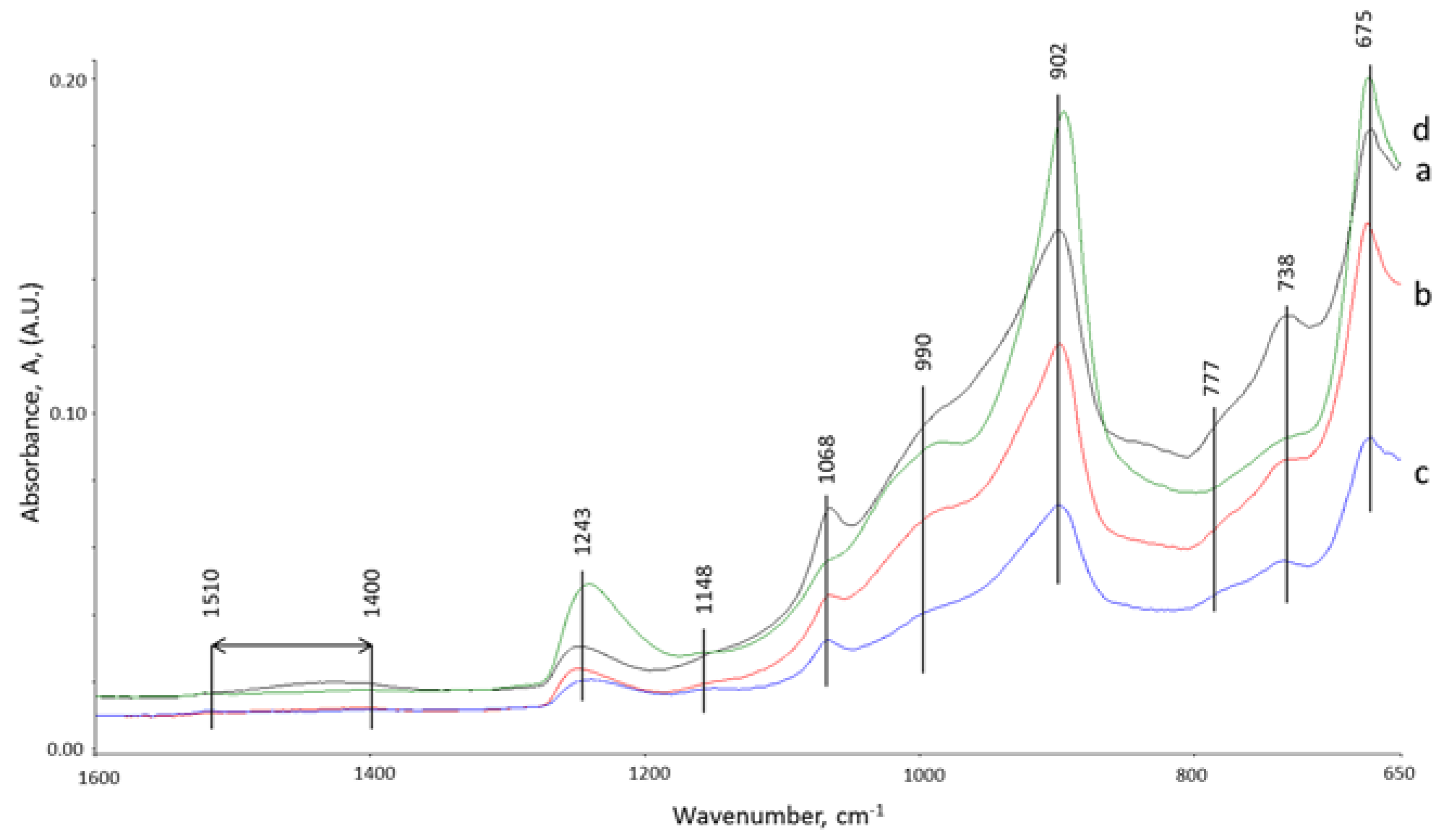



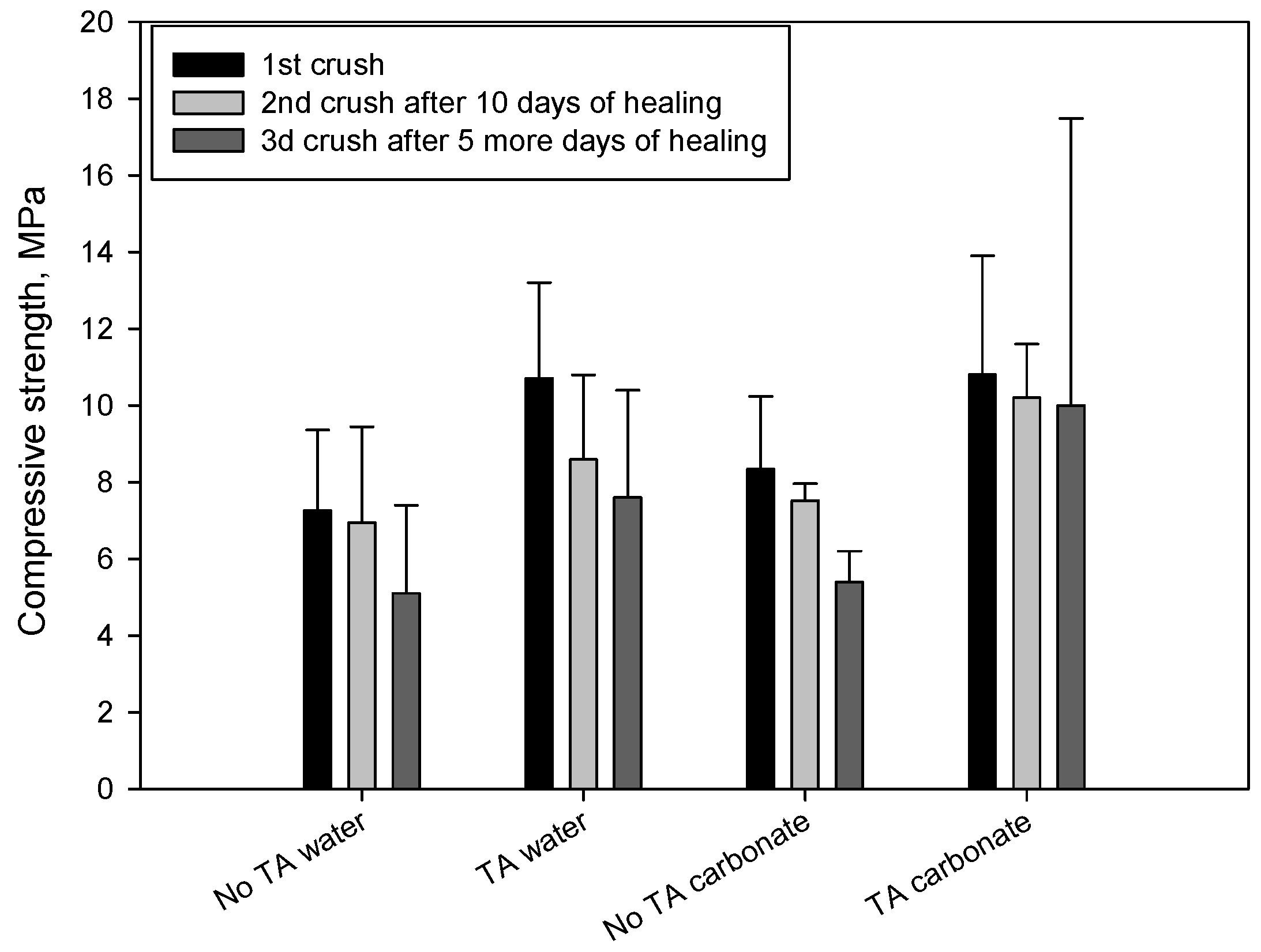{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
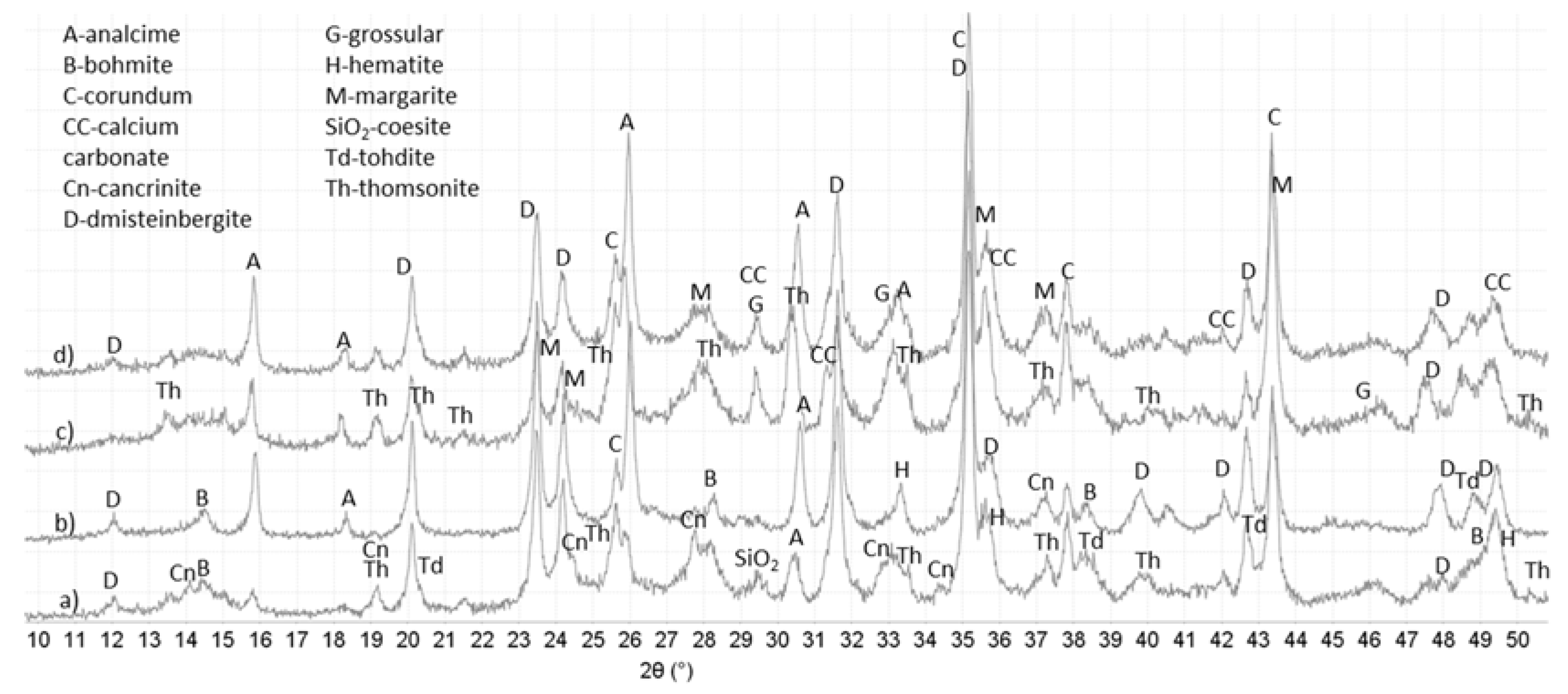{kind=link}
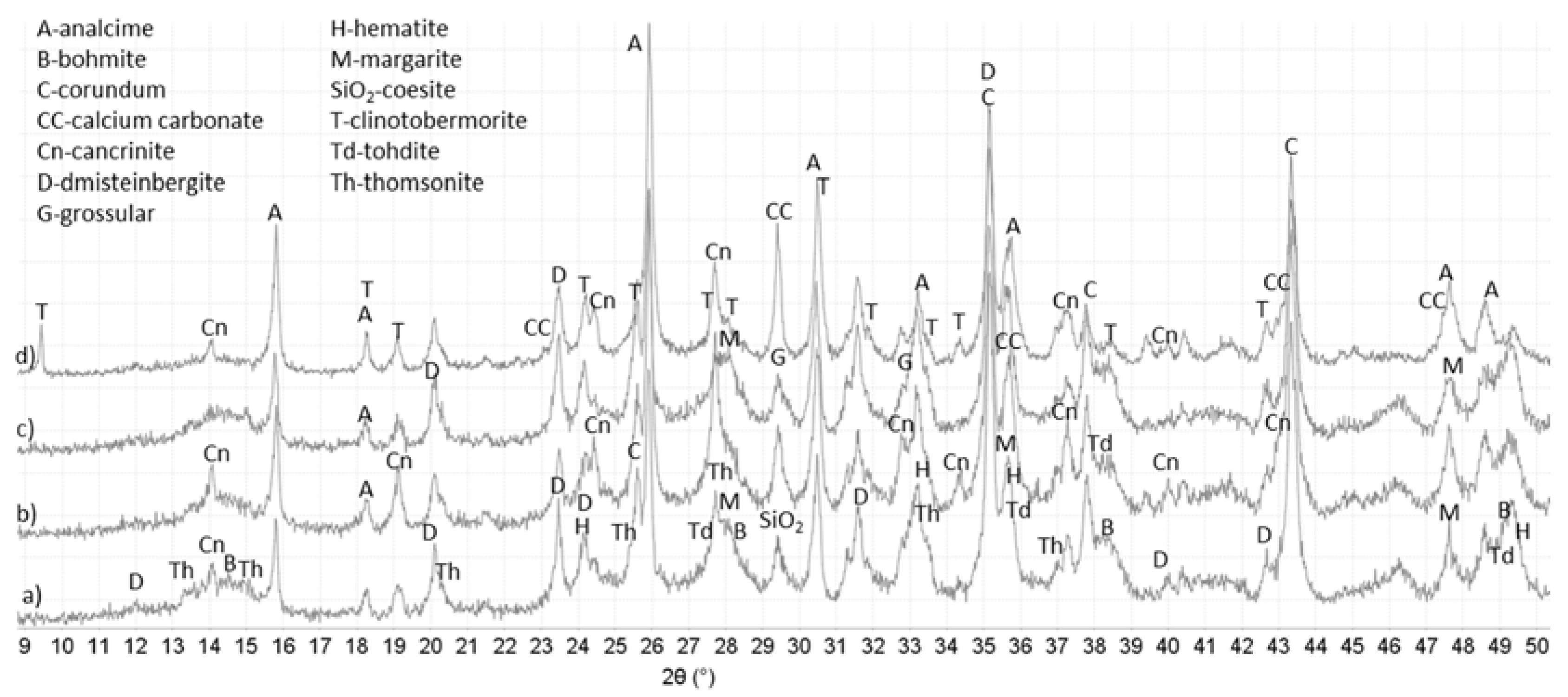{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Calcium Aluminate Cement | Flay Ash class F | Sodium Metasilicate |
|---|---|---|---|
| Al2O3 | 73.8 | 34.8 | - |
| CaO | 26.1 | 2.7 | - |
| SiO2 | - | 50.4 | 46.6 |
| Fe2O3 | 0.1 | 7.1 | - |
| Na2O | - | 0.3 | 50.5 |
| K2O | - | 3.1 | - |
| TiO2 | - | 1.6 | - |
| Sample | SiO2 | Al2O3 | CaO | CaO/Al2O3 | CaO/SiO2 | Al2O3/SiO2 |
|---|---|---|---|---|---|---|
| Samples from Healed Cracks | ||||||
| Control Water | 78 | 11 | 3.9 | 0.35 | 0.05 | 0.14 |
| Control Carbonate | 79 | 11 | 5.6 | 0.51 | 0.07 | 0.14 |
| TA Water | 69 | 19 | 3.2 | 0.17 | 0.05 | 0.27 |
| TA Carbonate | 81 | 13 | 3.2 | 0.25 | 0.04 | 0.16 |
| Core Samples | ||||||
| Control Water | 22 | 51 | 19 | 0.37 | 0.86 | 2.3 |
| Control Carbonate | 22 | 52 | 20 | 0.38 | 0.91 | 2.4 |
| TA Water | 23 | 51 | 19 | 0.37 | 0.83 | 2.2 |
| TA Carbonate | 23 | 52 | 18 | 0.35 | 0.78 | 2.3 |
| Control | TA-Modified |
|---|---|
| Core—Water | |
| 04-011-5220 Dmisteinbergite CaAl2Si2O8; 04-013-3004 Margarite-2M1 CaAl4Si2O10(OH)2; 01-078-0296 Thomsonite-Ca NaCa2Al5Si5O20(H2O)6; 04-012-1749 Analcime Na7.6Al7.6Si16.4O48(H2O)8; 04-014-1755 Tohdite Al5O7.5(H2O)0.5; 00-005-0190 Bohmite Al2(OOH)2; 00-011-0401 Hydroxysodalite Na4Al3Si3O12OH | 04-013-3004 Margarite-2M1 CaAl4Si2O10(OH)2; 01-078-0296 Thomsonite NaCa2Al5Si5O20(H2O)6; 04-011-5220 Dmisteinbergite CaAl2Si2O8; 04-013-2153 Grossular Ca3Al2(SiO4)2(OH)4; 01-072-0445 Analcime Na(AlSi2O6)(H2O) |
| Crack—Water | |
| 04-011-5220 Dmisteinbergite CaAl2Si2O8; 01-076-6569 Analcime Na0.9((Al0.9Si2)O6(H2O); 04-013-3004 Margarite-2M1 CaAl4Si2O10(OH)2; 04-014-1755 Tohdite Al5O7.5(H2O)0.5; 00-005-0190 Bohmite Al2(OOH)2 | 00-018-0276 Margarite-2M1 CaAl4Si2O10(OH)2; 01-088-2093 Thomsonite-Ca Na1.26Ca1.74(Si5.26Al4.74O20)(H2O)6; 04-011-6755 Analcime Na8Al8Si16O48(H2O)8; 04-014-1755 Tohdite Al5O7.5(H2O)0.5; 04-011-5220 Dmisteinbergite CaAl2Si2O8; 04-013-2153 Grossular Ca3Al2(SiO4)2(OH)4 |
| Core—Alkali Carbonate | |
| 00-018-0276 Margarite-2M1 CaAl2(Si2Al2)O10(OH)2; 04-011-6755 Analcime Na8Al8Si16O48(H2O)8; 01-080-6030 Cancrinite Na7.4Ca0.54(Si6.42Al5.58O24)(CO3)1.47(H2O)2; 04-011-5220 Dmisteinbergite CaSi2Al2O8; 04-013-2153 Grossular Ca3Al2(SiO4)2(OH)4; 00-005-0190 Bohmite Al2(OOH)2 | 00-018-0276 Margarite-2M1 CaAl2(Si2Al2)O10(OH)2; 04-011-7963 Analcime-R Na8Al8Si16O48(H2O)8; 01-078-0296 Thomsonite-Ca NaCa2Al5Si5O20(H2O)6; 01-075-8619 Cancrinite Na6.02Ca1.52(Si6Al6O24)(CO3)1.52(H2O)1.56; 04-011-5220 Dmisteinbergite CaSi2Al2O8; 04-013-2153 Grossular Ca3Al2(SiO4)2(OH)4; 04-014-1755 Tohdite Al5O7.5(H2O)0.5 |
| Crack—Alkali Carbonate | |
| 04-013-1895 Cancrinite Na6Ca1.5Al6(SiO4)6(CO3)1.5(H2O)1.75 04-011-7963 Analcime-R Na8Al8Si16O48(H2O)8; 04-015-7167 Coesite SiO2; 00-018-0276 Margarite-2M1 CaAl4Si2O10(OH)2; 04-011-5220 Dmisteinbergite CaSi2Al2O8; 00-005-0190 Bohmite Al2(OOH)2 | 01-074-2596 Clinotobermorite Ca5(Si6O16)(OH)2; 04-011-7963 Analcime-R Na8Al8Si16O48(H2O)8; 01-075-8620 Cancrinite Na6.02Ca1.52(Al6Si6O24)(CO3)1.52; 04-011-5220 Dmisteinbergite CaSi2Al2O8; 01-082-2720 Paragonite-2M1 Na4Al12Si12O40(OH)8 |
| Specimen | Decomposition Temperature Range, °C | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| <200 Ca(Na)-Al-Si-H Clinotobermorite (Zeolites) | 2004400 Zeolites Grossular | 400–550 Bohmite (Zeolites) | >550 Carbonates Mica group | Total | ||||||
| crack | core | crack | core | crack | core | crack | core | crack | core | |
| Carbonate-Control | 1.4 | 1.5 | 2.1 | 2.0 | 1.9 | 2.4 | 3.2 | 2.2 | 8.6 | 8.0 |
| Carbonate-TA | 1.1 | 1.5 | 2.3 | 1.9 | 1.2 | 2.3 | 9.4 | 7.1 | 14.1 | 12.8 |
| Water-Control | 0.94 | 1.3 | 1.6 | 1.5 | 1.3 | 2.2 | 7.5 | 7.1 | 11.4 | 12.0 |
| Water-TA | 1.5 | 1.7 | 2.1 | 1.9 | 1.6 | 2.1 | 2.2 | 4.2 | 7.3 | 9.9 |
| Sample | Point | Al | Ca | K | Na | Si | Possible phase |
|---|---|---|---|---|---|---|---|
| Water-Control | 1 | 5.89 | 0.33 | 1.85 | 1.28 | 26.65 | Si-rich gel |
| 2 | 9.61 | 0.53 | 0.16 | 4.68 | 22.55 | Analcime | |
| Carbonate-Control | 3 | 24.86 | 4.11 | 0.23 | 1.3 | 9.12 | Al-rich gel, dmisteinbergite precursor |
| Carbonate-TA | 4 | 6.15 | 0.14 | 1.17 | 3.71 | 25.68 | Si-rich gel |
| 5 | 10.93 | 1.62 | 0.1 | 6.0 | 20.09 | Si-rich gel | |
| 6 | 13.38 | 3.6 | 0.25 | 4.14 | 17.58 | Si-Al-rich gel | |
| 7 | 23.23 | 3.13 | 0.78 | 1.19 | 9.51 | Ca(Na)-Al-Si gel | |
| 8 | 20.75 | 5.97 | 0.21 | 1.58 | 11.16 | Ca(Na)-Al-Si gel | |
| 9 1 | 10.11 | 1.53 | 0.24 | 14.93 | 7.20 | Carbonated zeolite or cancrinite precursor |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyatina, T.; Sugama, T. Role of Tartaric Acid in Chemical, Mechanical and Self-Healing Behaviors of a Calcium-Aluminate Cement Blend with Fly Ash F under Steam and Alkali Carbonate Environments at 270 °C. Materials 2017, 10, 342. https://doi.org/10.3390/ma10040342
Pyatina T, Sugama T. Role of Tartaric Acid in Chemical, Mechanical and Self-Healing Behaviors of a Calcium-Aluminate Cement Blend with Fly Ash F under Steam and Alkali Carbonate Environments at 270 °C. Materials. 2017; 10(4):342. https://doi.org/10.3390/ma10040342
Chicago/Turabian StylePyatina, Tatiana, and Toshifumi Sugama. 2017. "Role of Tartaric Acid in Chemical, Mechanical and Self-Healing Behaviors of a Calcium-Aluminate Cement Blend with Fly Ash F under Steam and Alkali Carbonate Environments at 270 °C" Materials 10, no. 4: 342. https://doi.org/10.3390/ma10040342