Mechanical, Anisotropic, and Electronic Properties of XN (X = C, Si, Ge): Theoretical Investigations

Tianjin Key Laboratory for Civil Aircraft Airworthiness and Maintenance, Civil Aviation University of China, Tianjin 300300, China
*
Author to whom correspondence should be addressed.
Materials 2017, 10(8), 912; https://doi.org/10.3390/ma10080912
Submission received: 12 June 2017
/
Revised: 25 July 2017
/
Accepted: 3 August 2017
/
Published: 8 August 2017
Abstract
:The structural, mechanical, elastic anisotropic, and electronic properties of Pbca-XN (X = C, Si, Ge) are investigated in this work using the Perdew–Burke–Ernzerhof (PBE) functional, Perdew–Burke–Ernzerhof for solids (PBEsol) functional, and Ceperly and Alder, parameterized by Perdew and Zunger (CA–PZ) functional in the framework of density functional theory. The achieved results for the lattice parameters and band gap of Pbca-CN with the PBE functional in this research are in good accordance with other theoretical results. The band structures of Pbca-XN (X = C, Si, Ge) show that Pbca-SiN and Pbca-GeN are both direct band gap semiconductor materials with a band gap of 3.39 eV and 2.22 eV, respectively. Pbca-XN (X = C, Si, Ge) exhibits varying degrees of mechanical anisotropic properties with respect to the Poisson’s ratio, bulk modulus, shear modulus, Young’s modulus, and universal anisotropic index. The (001) plane and (010) plane of Pbca-CN/SiN/GeN both exhibit greater elastic anisotropy in the bulk modulus and Young’s modulus than the (100) plane.
1. Introduction
In the last few decades, nitride-based ceramics such as silicon nitride (Si3N4) have attracted increasing attention from researchers in the ceramics, mechanical, and aerospace industries, as well as in fields such as solar cells, as they have a wide range of applications [1,2,3,4,5,6,7,8]. This is due to their significant chemical stability, good compression resistance, corrosion resistance, high hardness, good mechanical properties, and good optical performance characteristics. Other stoichiometries like Si3N4, SiN2, and Si2N2(NH) have also been proposed to exist [9,10,11,12,13,14]. Silicon and germanium-based compounds and alloys such as the Si/Ge-group-III and Si/Ge-group-V compounds have been widely investigated [15,16,17].
CxNy with different stoichiometries is often used as a potential superhard material [18,19,20,21]. Li et al. [18] have reported a novel body-centered tetragonal CN2 named bct-CN2, using the newly-developed particle swarm optimization algorithm for crystal structure prediction. They found that the hardness of bct-CN2 is 77.4 GPa, and it is an indirect wide gap semiconductor material with a band gap of 3.6 eV. Wang et al. [19] suggested a new carbon nitride phase consisting of sp3 hybridized bonds, with cubic symmetry and a P213 space group (i.e., cg-CN). Unlike most of the other superhard materials that are insulators or semiconductors, it is a metallic compound, and its Vickers hardness is 82.56 GPa. They found that cg-CN is the most favorable stable crystal structure, with carbon nitride with 1:1 stoichiometry. Using the particle swarm optimization technique, Wei et al. [20] proposed a cubic superhard phase of C3N (c-C3N) with a Vickers hardness of 65 GPa, which is more energetically favorable than the recently proposed o-C3N [21]. o-C3N was proposed by Hao et al. [21]. It has a C2221 phase, and its Vickers hardness is 76 GPa.
CN2, SiN2, and GeN2 were proposed by Manyali et al. [22] using first-principles calculations; they found that SiN2 and GeN2 both have mechanical stability, SiN2 and GeN2 are characterized by an indirect band gap, and the optical spectra of GeN2 is within the solar spectrum for CN2 and SiN2. The structural, elastic, electronic, and optical properties of Si3N2 [23] have been calculated using density functional theory. First, Si3N2 has both mechanical and dynamical stability at ambient pressure, and it is still stable at 10–20 GPa. The first-principles plane-wave pseudo-potential (PW-PP) method was applied to investigate the mechanical properties, thermal properties, and phase transition characters of Ge3N4 by Luo et al. [24]. The β→wII→γ phase transitions of Ge3N4 were also successfully predicted by them; at 300 K, the calculated Pt of the β→wII transition is 10.7 GPa, and the calculated Pt of the β→γ transition is 14.26 GPa at 1200 K. The bulk moduli of β-Ge3N4, wII-Ge3N4, and γ-Ge3N4 are 179 GPa, 187 GPa, and 220 GPa, respectively. Pseudocubic-Si3P4 and Ge3P4 [25] were proposed by first-principles calculations for investigating the electronic, mechanical, and optical properties of pseudocubic-Si3P4 and Ge3P4. The bulk modulus and shear modulus of pseudocubic-Si3P4 and Ge3P4 are 76 GPa and 58 GPa, and 60 GPa and 47 GPa, respectively. In addition, pseudocubic-Si3P4 and Ge3P4 are both indirect and narrow band gap semiconductor materials, with band gaps of 0.24 eV and 0.13 eV, respectively.
Recently, Wei et al. [26] investigated the stability and electronic and mechanical properties of Pbca-CN using first-principles calculations. The electronic properties and elastic anisotropy in bulk modulus, shear modulus, and Poisson’s ratio of Pbca-CN are not fully represented. We proposed Pbca-SiN and Pbca-GeN (space group: Pbca), which have a structure based on Pbca-CN, with silicon atoms or germanium atoms substituting carbon atoms. In this work, the stability as well as structural, mechanical, electronic, and elastic anisotropy properties of Pbca-XN (X = C, Si, Ge) were systematically investigated.
2. Materials and Methods
The theoretical calculations were carried out using first-principles density functional theory (DFT) [27,28]. The calculations were performed using the Cambridge Serial Total Energy Package (CASTEP) code [29]. The generalized gradient approximation (GGA) parameterized by Perdew–Burke–Ernzerhof (PBE) [30] functional, Perdew–Burke–Ernzerhof for solids (PBEsol) [31] functional, and the local density approximation (LDA) parameterized by Ceperly and Alder, parameterized by Perdew and Zunger (CA-PZ) [32,33] exchange-correlation functional were employed for the self-consistent total energy calculations and geometry optimization. The C/Si/Ge: 2s22p2/3s23p2/4s24p2 and N: 2s22p3 electrons were explicitly treated as valence electrons. The energy cutoff for the plane wave basis set was chosen to be 520/500/440 eV for CN/SiN/GeN in the Pbca phase. The conjugate gradient method was used for the relaxation of structural parameters. The k-point samplings with 2π × 0.025 Å−1 (7 × 9 × 10/5 × 7 × 8/5 × 7 × 8) in the Brillouin zone were performed using the Monkhorst–Pack scheme [34] for CN/SiN/GeN in Pbca phase. The structural parameters optimizations were determined using the Broyden–Fletcher–Goldfarb–Shenno (BFGS) algorithm [35], with the flowing thresholds for converged structures: energy change less than 5 × 10−6 eV per atom, residual force below 0.01 eV/Å, stress less than 0.02 GPa, and displacement of atoms during the geometry optimization less than 0.0005 Å. The phonon frequencies were calculated using linear response theory [36]. The electronic band structures of the CN/SiN/GeN in Pbca phase were calculated utilizing the Heyd–Scuseria–Ernzerhof (HSE06) [37,38] hybrid functional.
3. Discussion
3.1. Structural Properties
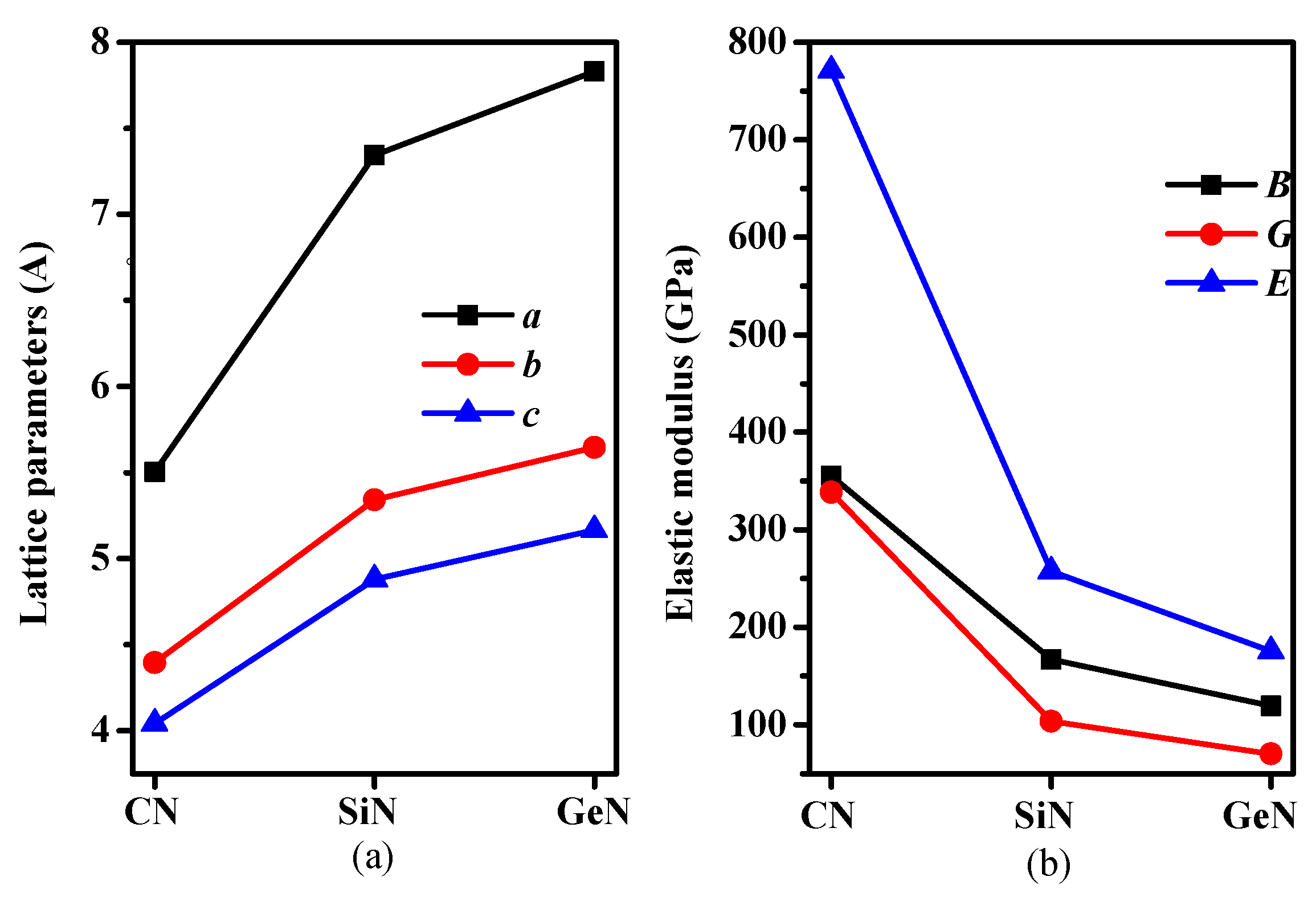
In the newly-formed (Pbca phase) solid, all the nitrogen atoms have sp2 hybridizations and all the carbon/silicon/germanium atoms have sp3 hybridization with their nearest neighboring N and C/Si/Ge atoms. The crystal structures of CN/SiN/GeN in Pbca phase are shown in Figure 1a. The C/Si/Ge atoms and N atoms consist of zigzag six-membered rings and eight-membered rings. The C/Si/Ge and N atoms are located at Wyckoff 8c (0.1396, 0.0722, 0.0205)/(0.1507, 0.0794, 0.0291)/(0.1476, 0.0769, 0.0310) and 8c (0.8154, 0.8661, 0.6318)/(0.8038, 0.8690, 0.6241)/(0.8093, 0.8725, 0.6197) sites in Pbca-CN/SiN/GeN, respectively. The crystal structures of CN/SiN/GeN in the Pbca phase along the (001) direction and (010) direction are shown in Figure 1b,c, respectively. The eight-membered rings are normal to the (001) direction in the structure of Pbca-CN/SiN/GeN, and the six-membered rings are normal to the (010) direction. The optimal lattice parameters of Pbca-CN/SiN/GeN, together with the previous results [27,39] of Pbca-CN are listed in Table 1. The optimized lattice parameters are a = 5.504 Å, b = 4.395 Å, and c = 4.041Å, which are in excellent agreement with [27,39]. In addition, taking into account the van der Waals forces, we also calculated the lattice parameters of Pbca-CN/SiN/GeN and diamond, c-BN using the dispersion-corrected Perdew–Burke–Ernzerhof (PBE + D) [40]. For diamond and c-BN, the theoretical results obtained by the GGA-PBE level (diamond: 3.566 Å for PBE level, 3.526 Å for CA-PZ [41], experimental value 3.567 Å [42]; c-BN: 3.626 Å for PBE level, 3.569 Å for CA-PZ [43], experimental value 3.620 Å [44]) are closer to the experimental values; the obtained results of c-BN and diamond using PBE + D are not much different from those obtained by PBE functional compared to corresponding experimental values, so the results obtained by the GGA-PBE level are all used in our paper. The lattice parameters of Pbca-XN with X changing from C to Ge are illustrated in Figure 2a. It is clear that the lattice parameters of Pbca-XN increase with X changing from C to Ge. From CN to SiN, the lattice parameters increase 31.4%, 21.52%, and 20.7% for a, b, and c of SiN compared to CN, while the lattice parameters increase 8.25%, 5.69%, and 5.84% for a, b, and c of GeN compared to SiN, respectively. This is because the average bond length of Si-N (1.751 Å) is much greater than that of the C–N bond (1.452 Å), and the average bond length of Ge-N (1.871 Å) is slightly longer than that of the Si–N bond.
3.2. Mechanical Properties
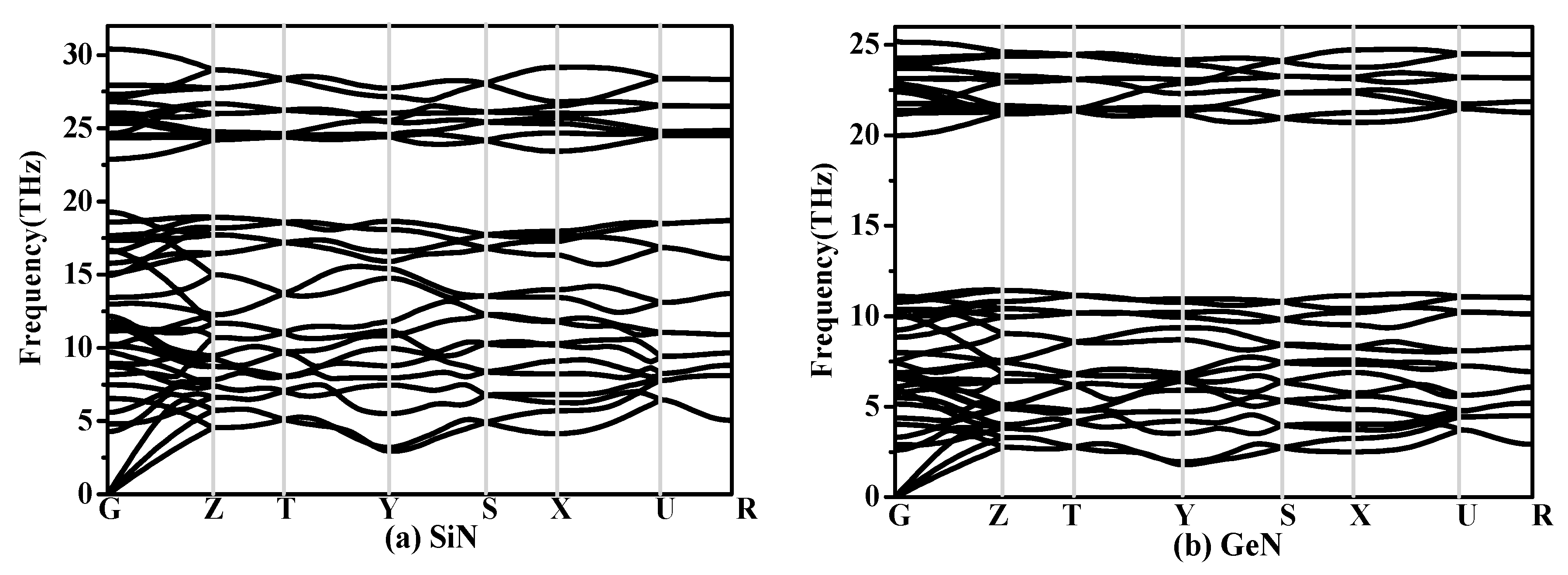
The calculated elastic constants and elastic moduli of CN/SiN/GeN in the Pbca phase are listed in Table 2. The calculated elastic constants and elastic modulus of Pbca-CN are excellent agreement with the previous report [26]. For an orthorhombic phase, the criteria of mechanical stability are [45]: Cii > 0, i = 1–6; C11C22 − C > 0; C11C22C33 + 2C12C13C23 − C11C − C22C − C33C > 0, where the Cij is elastic constant of the material. The mechanical stability of a phase can be confirmed by using the elastic constants. The SiN/GeN in the Pbca phase both satisfy the above mechanical stability criteria. The SiN/GeN in the Pbca phase show mechanical stability under ambient pressure. The phonon dispersion curve can show dynamic stability; the phonon dispersion curves of SiN/GeN in the Pbca phase are illustrated in Figure 3. There is no imaginary frequency in the Brillouin zone, which means SiN/GeN in the Pbca phase can be dynamically stable under ambient pressure. The elastic moduli of Pbca-XN with X changing from C to Ge are illustrated in Figure 2b. It is clear that the elastic moduli of Pbca-XN decrease with X changing from C to Ge. The elastic constants and elastic moduli of other SixNy compounds [22,46,47] are also listed in Table 2. The bulk modulus B of Pbca-SiN is slightly smaller than that of SiN2, o-Si3N4, and t-Si3N4, while it is slightly larger than Si3N2 and t-Si3N4. The shear modulus G and Young’s modulus E of Pbca-SiN are similar to the bulk modulus of Pbca-SiN. For Pbca-GeN, its bulk modulus is as large as that of GeN2. However, its shear modulus and Young’s modulus are slightly smaller than that of GeN2.
Brittleness and ductility of materials are important properties in crystal physics and engineering sciences. Pugh [48] proposed the ratio of bulk to shear modulus (B/G) as an indication of ductile verses brittle characters. If B/G > 1.75, the material is characterized by a ductile manner; otherwise, the material has a brittle character. The Poisson’s ratio v is consistent with B/G, but refers to brittle compounds, usually with a small v (less than 0.26) [49]. The B/G ratio of Pbca-CN/SiN/GeN is 1.12 (1.11 [26]), 1.63, and 1.70; it is revealed that Pbca-CN/SiN/GeN are all brittle materials, and Pbca-CN has the most brittleness. For Poisson’s ratio v, we obtained the same conclusion.
The Debye temperature (ΘD) is a fundamental physical property, and correlates with many physical properties of solids (e.g., specific heat and the thermal coefficient) [50]. Debye temperature ΘD can be estimated by elastic moduli. The Debye temperature can be estimated from the average sound velocity by the following equation based on elastic constant evaluations [51]: ΘD = (h/kB) (3nρNA/4πM)1/3vm, where h is the Planck constant, kB is Boltzmann’s constant, n is the number of atoms in the molecule, NA is the Avogadro number, M is the molecular weight, and ρ is the density. The average sound velocity vm can be calculated as follows: vm = [(2/v + 1/v)/3]−1/3, where vl = [(B + 4G/3)/ρ]1/2, and vt = (G/ρ)1/2, where B and G are bulk modulus and shear modulus, vl is the longitudinal sound velocity, and vt is the transverse sound velocity. In addition, we can obtain the sound velocity in the main directions of a material according to the elastic constants. For the (001) propagation direction in orthorhombic symmetry, polarization direction (001)vl = (C33/ρ)1/2, (100)vt1 = (C55/ρ)1/2, and (010)vt2 = (C44/ρ)1/2. For the (010) propagation direction, polarization direction (010)vl = (C22/ρ)1/2, (100)vt1 = (C66/ρ)1/2, and (001)vt2 = (C44/ρ)1/2. For the (100) propagation direction, polarization direction (100)vl = (C11/ρ)1/2, (010)vt1 = (C66/ρ)1/2, and (100)vt2 = (C55/ρ)1/2 [49,52].
The calculated results of Debye temperature, longitudinal sound velocity, and transverse sound velocity of Pbca-XN (X = C, Si, Ge) are all listed in Table 3. The densities of Pbca-XN (X = C, Si, Ge) are also listed in Table 3. For Pbca-XN (X = C, Si, Ge), in the (001) propagation direction, the (001) polarization direction has the largest sound velocity. The longitudinal sound velocity in the (010) propagation direction aligns with the (001) polarization direction, and the longitudinal sound velocity in the (100) propagation direction aligns with the (010) polarization direction. The longitudinal sound velocity is generally larger than the transverse sound velocity, mainly because the elastic constants that determine the longitudinal sound velocity are greater than those of the transverse sound velocity. In addition, for the same the propagation direction and polarization direction, the sound velocity decreases with X changing from C to Ge. Furthermore, the Debye temperature of Pbca-XN (X = C, Si, Ge) decreases with X changing from C to Ge. For Pbca-SiN, the Debye temperature is 863 K; it is slightly smaller than that of m-Si3N4 (892 K), o-Si3N4 (1107 K), and t-Si3N4 (949 K) [53]. The longitudinal sound velocity and transverse sound velocity of Pbca-XN (X = C, Si, Ge) are different along different directions; this shows that the sound velocity of Pbca-XN (X = C, Si, Ge) is also anisotropic.
3.3. Electronic Properties
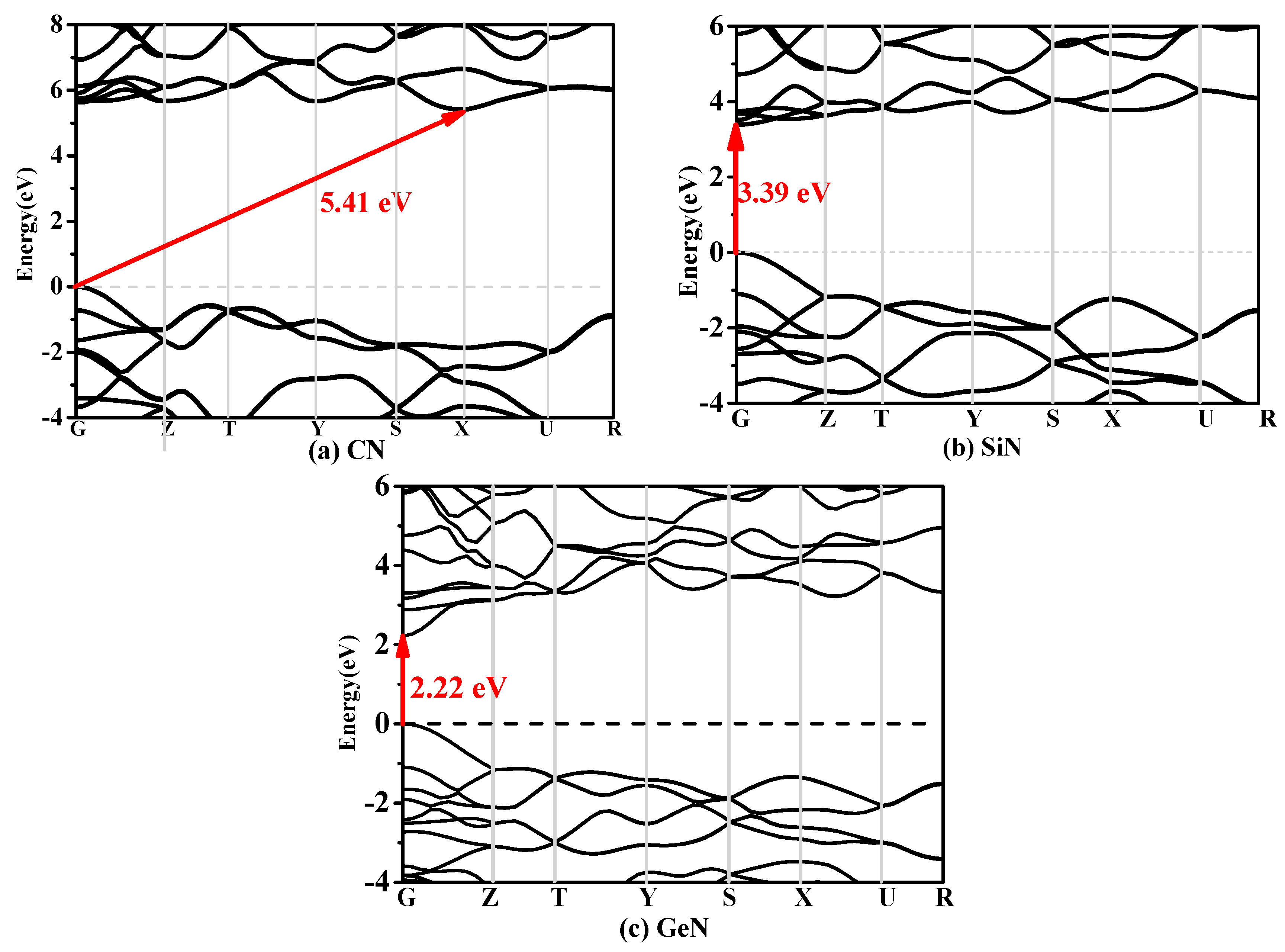
In solid-state physics and semiconductor physics, the band structure of a solid or a material describes the energy that is forbidden or permitted by electrons. The band structure of a material determines a variety of properties—especially its electronic and optical properties. It is known that since the calculated band gap with DFT is usually underestimated by 30–50%, the band gap should be greater than the calculated results with the PBE functional. Hence, the band structures of Pbca-CN/SiN/GeN calculated utilizing the Heyd–Scuseria–Ernzerhof (HSE06) [37,38] hybrid functional are shown in Figure 4a–c, respectively. The band gap of Pbca-CN is 5.41 eV within the HSE06 hybrid functional and 3.96 eV within the PBE functional; the results of the PBE functional of Pbca-CN are in excellent agreement with previous report [26]. The valence band maximum is located at the G point in the Brillouin zone, whereas the conduction band minimum is located at the X point. That is to say, Pbca-CN is an indirect semiconductor with a band gap of 3.94 eV. In contrast, the valence band maximums of Pbca-SiN and Pbca-GeN are all located at the G point in the Brillouin zone; it is shown that Pbca-SiN and Pbca-GeN are both direct semiconductors with band gaps of 3.39 eV and 2.22 eV, respectively. In addition, the Fermi level decreases as the carbon atoms change into silicon atoms; the silicon atoms then change into germanium atoms, and the Fermi level also decreases. Similar to the lattice constants and the elastic moduli, the Fermi level changes rapidly when the carbon atom is replaced by silicon atoms. The Fermi levels of Pbca-CN/SiN/GeN are 11.02 eV, 2.26 eV, and 0.01 eV, respectively.
3.4. Elastic Anisotropy Properties
The elastic anisotropy properties are an important characteristic of materials. Along the different crystallographic directions, various elastic moduli exhibit different values. In this work, we mainly investigated the anisotropy of Poisson’s ratio v, shear modulus, bulk modulus, and Young’s modulus in different planes and different directions. The Poisson's ratio v and shear modulus G have two unit vectors (a, b) and three angles [43,54], so they have a maximum value and a minimum value in the same direction, while the Young's modulus has only two unit vectors (a, b) and a two-angle description [43,54], so it is in the same direction with only one value. The Poisson’s ratio v of Pbca-CN/SiN/GeN in the (001) plane (namely the xy or ab plane), the (010) plane (namely the xz or ac plane), and the (100) plane (namely the yz or bc plane) are displayed in Figure 5a–c, respectively. The dashed line and solid line represent the maximum value and minimum value of Poisson’s ratio in different directions in the (001) plane, (010) plane, and (100) plane; the cyan line, red line, and blue line represent the Poisson’s ratio v of Pbca-CN/SiN/GeN in (001) plane, (010) plane, and (100) plane in Figure 5, respectively. From Figure 5a–c, it is obvious that the Poisson’s ratio v of Pbca-CN/SiN/GeN exhibits a larger anisotropy. In the (001), (010), and (100) plane, along almost all directions, the Pbca-GeN exhibits the largest Poisson ratio. The positions of the maximum values are all located at θ = 1.57, φ = 4.73 (more details see [43,54]) for Pbca-CN/SiN/GeN; the angles θ and φ are measured in radians. The minimum values of Pbca-CN/SiN/GeN occupy the position θ = 2.33, φ = 1.87; θ = 1.46, φ = 1.06; and θ = 0.83, φ = 4.73, respectively.
The shear moduli G of Pbca-CN/SiN/GeN in the (001), (010), and (100) planes are displayed in Figure 6a–c, respectively. The cyan line, red line, and blue line represent the Poisson’s ratio v of Pbca-CN/SiN/GeN in Figure 6, and the dashed line and solid line represent the maximum value and minimum value of the shear modulus, respectively. The maximum shear moduli G of Pbca-CN/SiN/GeN are 469 GPa, 150 GPa, and 107 GPa, and the minimum shear moduli of Pbca-CN/SiN/GeN are 379 GPa, 106 GPa and 74 GPa, respectively. From Figure 6a,c, with X change from C to Ge, the shape of the minimum value for shear modulus is increasingly rounded in the (001) plane and (100) plane, while in the (010) plane the shape of the minimum is closer to a square. The ratios Gmax/Gmin of Pbca-CN/SiN/GeN are 1.24, 1.42, and 1.53; in other words, the elastic anisotropy in shear modulus becomes larger and larger with X changing from C to Ge.
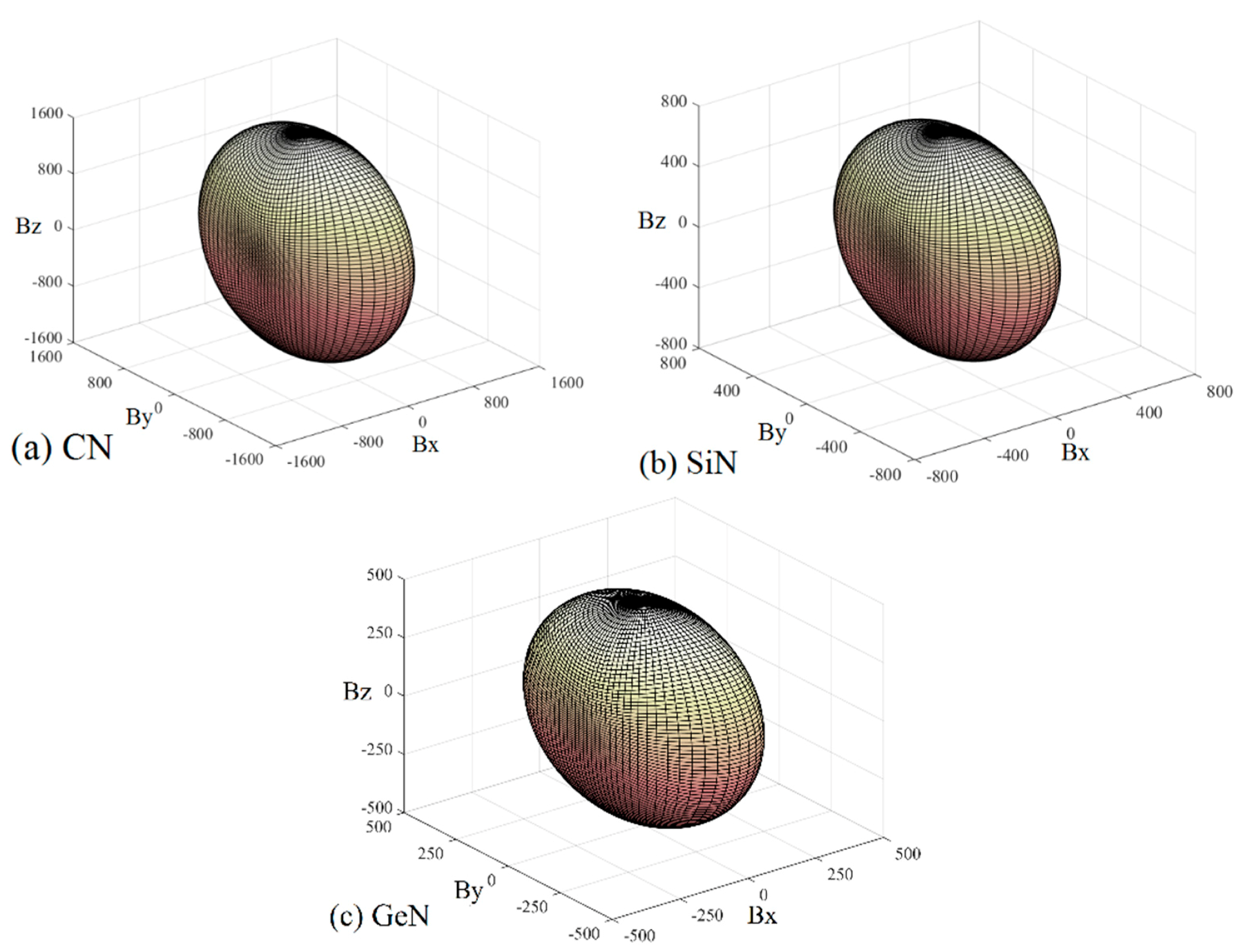
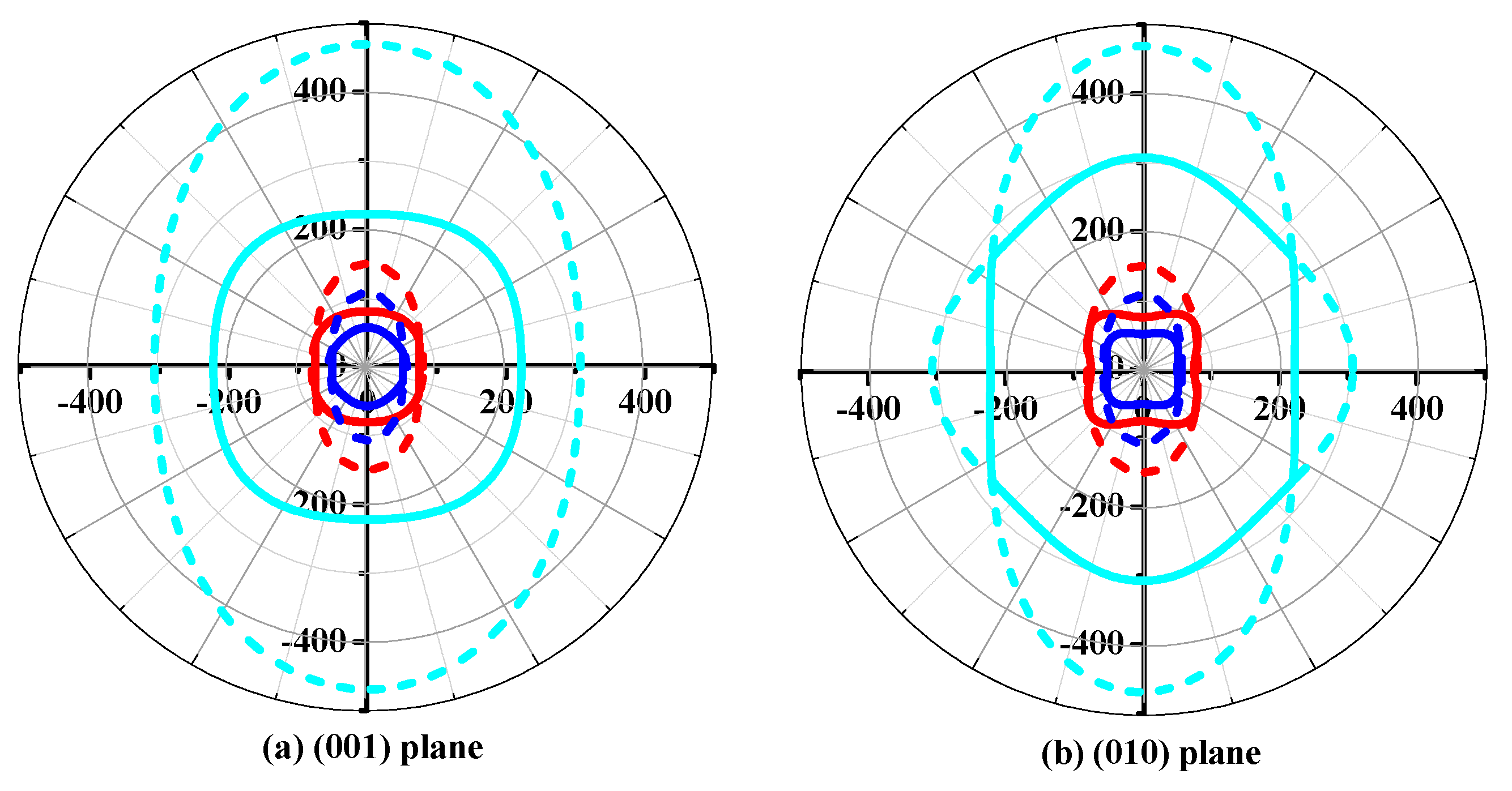
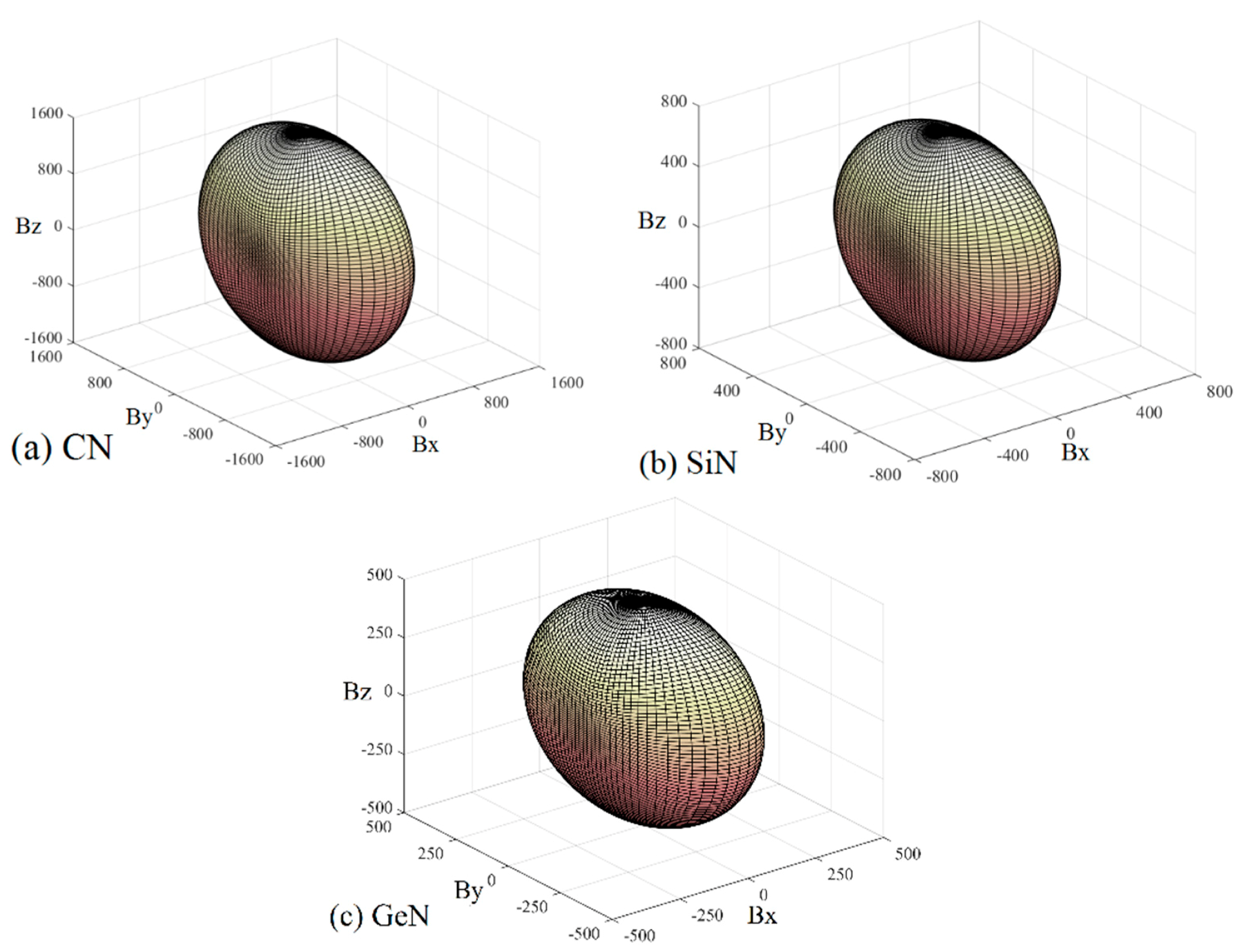
Young’s modulus is a measure of the stiffness of a solid material. It is a mechanical property of linear elastic solid materials. It defines the relationship between stress (force per unit area) and strain (proportional deformation) in a material. To study the elastic anisotropy in more detail, a variation of Young’s modulus with crystallographic direction is displayed in a three-dimensional manner. The directional dependence of Young’s modulus E for orthorhombic crystal is [55]: E−1 = lS11 + lS22 + lS33 + 2llS12 + 2llS13 + 2llS23 + llS66 + llS55 + llS44, where l1, l2, and l3 are the direct cosines of the [uvw] direction, and Sij refers to the elastic compliance constants. The three-dimensional surface representations of Young’s modulus E for Pbca-CN/SiN/GeN are illustrated in Figure 7a–c. For an isotropic system, the three-dimensional directional dependence exhibits a spherical shape. If there is a deviation of degrees from the spherical shape, it reflects the material exhibiting elastic anisotropy [56]. From Figure 7a–c, it is obvious that the shape of the three-dimensional directional dependence does not exhibit a spherical shape, and the shapes of the three-dimensional directional dependence for Pbca-CN/SiN/GeN all exhibit mechanical anisotropy in Young’s modulus.
To further understand the elastic anisotropy of the Young’s modulus along different directions, the dependence of the Young’s modulus on orientation is investigated when we take the tensile axis within a given plane. Let α be the angle of between (100) and (uv0) for the (001) plane; the Young’s modulus between (100) and (uv0) for the (001) plane can be expressed as: E−1 = S11cos4α + S22sin4α + 2S12sin2αcos2α + S66sin2αcos2α. Let β be the angle of between (001) and (u0w) for the (010) plane; the Young’s modulus between (001) and (u0w) for the (010) plane can be calculated as: E−1 = S11sin4β + S33cos4β + (2S13sin22β + S55sin22β)/4. Let γ be the angle of between (001) and (0vw) for the (001) plane, the Young’s modulus between (001) and (0vw) for the (001) plane can be estimated as: E−1 = S22sin4γ + S33cos4γ + (2S23sin22γ + S44sin22γ)/4. The two-dimensional representations of Young’s modulus in the (001) plane, (010) plane, and (100) plane for Pbca-CN/SiN/GeN are illustrated in Figure 7d–f, respectively. The cyan line, red line, and blue line represent the Poisson’s ratio v of Pbca-CN/SiN/GeN, respectively. From Figure 7d–f, the (001) plane and (010) plane of Pbca-CN/SiN/GeN exhibit a larger elastic anisotropy in Young’s modulus than the (100) plane. Pbca-CN has a maximum of Emax = 1034 GPa and a minimum of Emin = 447 GPa. The calculated results of elastic anisotropy in Young’s modulus for Pbca-CN are in excellent agreement with [26]. Pbca-CN/SiN/GeN has a maximum of Emax = 380/241 GPa and a minimum of Emin = 179/120 GPa. In order to quantify the elastic anisotropy, we introduce a ratio; that is, the ratio of the maximum and minimum Young’s modulus (ratio Emax/Emin). The greater the ratio Emax/Emin, the greater the maximum and minimum differences, and the greater the anisotropy of the material. Through the values of the ratio Emax/Emin = 2.31, 2.12, and 2.01, it is shown that the elastic anisotropy in Young’s modulus for Pbca-XN (X = C, Si, Ge) decreases with X changing from C to Ge. In addition, the maximum values of Pbca-CN/SiN/GeN all occupy the position θ = 0, φ = 0; that is, the maximum values of Pbca-XN (X = C, Si, Ge) all occupy the z (c) axis, while the minimum values of Pbca-CN/SiN/GeN do not occupy the same position (x (a) axis). The minimum value of Pbca-SiN is located at θ = 1.32, φ = 0, but the minimum value of Pbca-CN/GeN occupies the position of θ = π/2, φ = 0. For the orthorhombic phase, the dependence of the bulk modulus B along the crystallographic direction is expressed by: B−1 = (S11 + S12 + S13)l1 + (S12 + S22 + S23)l2 + (S13 + S23 + S33)l3. The three-dimensional surface representations of bulk modulus B for Pbca-CN/SiN/GeN are illustrated in Figure 8a–c. The anisotropy of the bulk modulus of Pbca-XN (X = C, Si, Ge) is similar to that of Young’s modulus; the (001) plane and (010) plane of Pbca-CN/SiN/GeN exhibit a larger elastic anisotropy in bulk modulus than the (100) plane.
In addition, apart from the Poisson’s ratio, shear modulus, and Young’s modulus, there is another significant physical quantity which describes the elastic anisotropy of a material: the universal anisotropic index AU [57], which is defined as AU = 5GV/GR + BV/BR − 6, where G and B are the shear modulus and bulk modulus, and the subscripts V and R denote the Voigt and Reuss approximations, respectively. The calculated universal anisotropic indices of Pbca-XN (X = C, Si, Ge) are 0.717, 0.671, and 0.662, respectively. The elastic anisotropy in the universal anisotropic index AU of Pbca-XN (X = C, Si, Ge) is similar to the bulk modulus, Young’s modulus, and shear modulus; it also decreases with X changing from C to Ge. Furthermore, for Pbca-CN, the universal anisotropic index is slightly smaller than that of m-C3N4 (0.798 [58]), while it is much higher than that of t-C3N4 (0.305 [58]). The universal anisotropic index of Pbca-SiN is slightly larger than that of o-Si3N4 (0.582 [49]), but it is smaller than that of m-Si3N4 (0.968) and t-Si3N4 (1.231) [49].
4. Conclusions
The structural, mechanical, electronic, and elastic anisotropy properties of CN, SiN, and GeN in orthorhombic phase were performed using DFT calculations in this work. SiN and GeN are mechanically and dynamically stable, fulfilling the Born stability criteria for an orthorhombic phase and phonon spectra, respectively. PBE function predicts lattice parameters that agree well with the previous report. From band gap calculations with the HSE06 function, SiN and GeN are direct band gap semiconductor materials with band gap of 3.39 eV and 2.22 eV, while CN has an indirect band gap with band gap of 5.41 eV. The elastic moduli of Pbca-XN (X = C, Si, Ge) such as Young’s moduli, bulk moduli, shear moduli, Poisson’s ratio, and sound velocities have also been reported in this work. The Debye temperature, longitudinal sound velocities, and transverse sound velocities are also estimated using the elastic constants. The elastic anisotropy calculations showed that Pbca-XN (X = C, Si, Ge) exhibited anisotropy in bulk modulus, shear modulus, Poisson’s ratio, Young’s modulus, and AU. Besides, the elastic anisotropy in bulk modulus, shear modulus, Poisson’s ratio, Young’s modulus, and AU for Pbca-XN (X = C, Si, Ge) decreases with X changing from C to Ge.
Acknowledgments
This work was supported by the Natural Science Foundation of China (No. 61601468), the Fundamental Research Funds for the Central Universities (No. 3122014C024), and the Fund for Scholars of Civil Aviation University of China (No. 2013QD06X). Q.Y. FAN (School of Microelectronics, Xidian University) is thanked for allowing to use the CASTEP code in Materials Studio.
Author Contributions
Zhenyang Ma and Xuhong Liu designed the project; Zhenyang Ma, Xuhong Liu, Qingynag Fan, and Dayun Wang performed the calculations; Zhenyang Ma, Xuhong Liu, Xinhai Yu, and Chunlei Shi determined the results; Zhenyang Ma and Xuhong Liu wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fang, C.M.; de Wijs, G.; Hintzen, H.T.; de With, G. Phonon spectrum and thermal properties of cubic Si3N4 from first-principles calculations. J. Appl. Phys. 2003, 93, 5175–5180. [Google Scholar] [CrossRef]
- Riley, F.L. Silicon Nitride and Related Materials. J. Am. Ceram. Soc. 2000, 83, 245–265. [Google Scholar] [CrossRef]
- Horvath-Bordon, E.; Riedel, R.; Zerr, A.; McMillan, P.F.; Auffermann, G.; Prots, Y.; Bronger, W.; Kniep, R.; Kroll, P. High-pressure chemistry of nitride-based materials. Chem. Soc. Rev. 2006, 35, 987–1014. [Google Scholar] [CrossRef] [PubMed]
- Zerr, A.; Riedel, R.; Sekine, T.; Lowther, J.E.; Ching, W.Y.; Tanaka, I. Recent advances in new hard high-pressure nitrides. Adv. Mater. 2006, 18, 2933–2948. [Google Scholar] [CrossRef]
- Xu, B.; Dong, J.; McMillan, P.; Shebanova, O.; Salamat, A. Equilibrium and metastable phase transitions in silicon nitride at high pressure: A first-principles and experimental study. Phys. Rev. B 2011, 84, 014113. [Google Scholar] [CrossRef]
- Kuwabara, A.; Matsunaga, K.; Tanaka, I. Lattice dynamics and thermodynamical properties of silicon nitride polymorphs. Phys. Rev. B 2008, 78, 064104. [Google Scholar] [CrossRef]
- Zerr, A.; Kempf, M.; Schwarz, M.; Kroke, E.; Goken, M.; Riedel, R. Elastic Moduli and Hardness of Cubic Silicon Nitride. J. Am. Ceram. Soc. 2002, 85, 86–90. [Google Scholar] [CrossRef]
- Ching, W.; Mo, S.D.; Ouyang, L.Z.; Rulis, P. Theoretical prediction of the structure and properties of cubic spinel nitrides. J. Am. Ceram. Soc. 2002, 85, 75–80. [Google Scholar] [CrossRef]
- Weihrich, R.; Eyert, V.; Matar, S.F. Structure and electronic properties of new model dinitride systems: A density-functional study of CN2, SiN2, and GeN2. Chem. Phys. Lett. 2003, 373, 636–641. [Google Scholar] [CrossRef]
- Johnson, W.C. Nitrogen compounds of germanium I. the preparation and properties of germanic nitride. J. Am. Chem. Soc. 1930, 52, 5160–5165. [Google Scholar] [CrossRef]
- Salamat, A.; Woodhead, K.; McMillan, P.F.; Cabrera, R.Q.; Rahman, A.; Adriaens, D.; Corà, F.; Perrillat, J.P. Tetrahedrally bonded dense C2N3H with a defective wurtzite structure: X-ray diffraction and Raman scattering results at high pressure and ambient conditions. Phys. Rev. B 2009, 80, 397–403. [Google Scholar] [CrossRef]
- Manyali, G.S.; Warmbier, R.; Quandt, A. Computational study of the structural, electronic and optical properties of M2N2(NH): M = C, Si, Ge, Sn. Comput. Mater. Sci. 2013, 79, 710–714. [Google Scholar] [CrossRef]
- Yu, B.H.; Chen, D. First-principles study on the electronic structure and phase transition of α-, β- and γ-Si3N4. Acta Phys. Sin. 2012, 19, 197102. [Google Scholar]
- Manyali, G.S.; Warmbier, R.; Quandt, A.; Lowther, J.E. Ab initio study of elastic properties of super hard and graphitic structures of C3N4. Comput. Mater. Sci. 2014, 69, 229–303. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T. The mechanical and electronic properties of carbon-rich silicon carbide. Materials 2016, 9, 333. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Y.; Chai, C.C.; Fan, Q.Y.; Yang, Y.T. Theoretical prediction of new C–Si alloys in C2/m-20 structure. Chin. Phys. B 2017, 26, 046101. [Google Scholar] [CrossRef]
- Wang, Q.K.; Chai, C.C.; Fan, Q.Y.; Yang, Y.T. Physical properties of C-Si alloys in C2/m structure. Commun. Theor. Phys. 2017, 68, 259–268. [Google Scholar] [CrossRef]
- Li, Q.; Liu, H.Y.; Zhou, D.; Zheng, W.T.; Wu, Z.J.; Ma, Y.M. A novel low compressible and superhard carbon nitride: Body-centered tetragonal CN2. Phys. Chem. Chem. Phys. 2012, 14, 13081–13087. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Bao, K.; Tian, F.B.; Meng, X.; Chen, C.B.; Dong, B.W.; Li, D.; Liu, B.B.; Cui, T. Cubic gauche-CN: A superhard metallic compound predicted via first-principles calculations. J. Chem. Phys. 2010, 133, 044512. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhang, Q.; Yan, H.Y.; Zhang, M.G. Cubic C3N: A new superhard phase of carbon-rich nitride. Materials 2016, 9, 840. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Liu, H.Y.; Lei, W.W.; Tang, X.; Lu, J.; Liu, D.; Li, Y.W. Prediction of a Superhard Carbon-rich C-N Compound Comparable to Diamond. J. Phys. Chem. C 2015, 119, 28614–28619. [Google Scholar] [CrossRef]
- George, S.M.; Robert, W.; Alexander, Q. First-principles studies of the structural, electronic and optical properties of dinitrides CN2, SiN2 and GeN2. Comput. Mater. Sci. 2014, 95, 706–711. [Google Scholar]
- George, S.M.; Robert, W.; Alexander, Q. First-principles study of Si3N2. Comput. Mater. Sci. 2015, 96, 140–145. [Google Scholar]
- Luo, Y.S.; Cang, Y.P.; Chen, D. Determination of the finite-temperature anisotropic elastic and thermal properties of Ge3N4: A first-principles study. Comput. Condens. Matter 2014, 1, 1–7. [Google Scholar] [CrossRef]
- Khan, G.G.; Clark, S.J.; Bandyopadhyay, N.R. Electronic, mechanical and optical properties of Si3P4 and Ge3P4: An ab initio study. Int. J. Mod. Phys. B 2010, 28, 5487–5494. [Google Scholar] [CrossRef]
- Wei, Q.; Zhang, M.G.; Yan, H.Y. First-principles investigation of high pressure Pbca phase of carbon mononitride. Phys. Lett. A 2016, 380, 3217–3221. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principlesmethods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Ceperley, D.M.; Alder, B.J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 1980, 45, 566–568. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2006, 118, 8207–8215, Erratum in J. Chem. Phys. 2006, 124, 219906. [Google Scholar] [CrossRef]
- Khazaei, M.; Tripathi, M.N.; Kawazoe, Y. First-principles simulation of cyanogen under high pressure: Formation of paracyanogen and an insulating carbon nitride solid. Phys. Rev. B 2011, 83, 134111. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.Y.; Wei, Q.; Chai, C.C.; Yan, H.Y.; Zhang, M.G.; Lin, Z.Z.; Zhang, Z.X.; Zhang, J.Q.; Zhang, D.Y. Structural, mechanical, and electronic properties of P3m1-BCN. J. Phys. Chem. Solids 2015, 79, 89–96. [Google Scholar] [CrossRef]
- Xing, M.J.; Li, B.H.; Yu, Z.T.; Chen, Q. Monoclinic C2/m-20 carbon: A novel superhard sp3 carbon allotrope. RSC Adv. 2016, 6, 32740–32745. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Han, Z.; Liu, X.H.; Yu, X.H.; Wang, D.Y.; Tian, Y. Pnma-BN: Another Boron Nitride polymorph with interesting physical properties. Nanomaterials 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Petrescu, M.L. Boron nitride theoretical hardness compared to carbon polymorphs. Diamond Relat. Mater. 2004, 13, 1848–1853. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B. 2014, 90, 224104. [Google Scholar] [CrossRef]
- Cang, Y.P.; Lian, S.B.; Yang, H.M.; Chen, D. Predicting physical properties of tetragonal, monoclinic and orthorhombic M3N4 (M = C, Si, Sn) polymorphs via first-principles calculations. Chin. Phys. Lett. 2016, 33, 066301. [Google Scholar] [CrossRef]
- Chen, D.; Cheng, K.; Qi, B.Y. The electronic, optical, and thermodynamical properties of tetragonal, monoclinic, and orthorhombicM3N4 (M = Si, Ge, Sn): A first-principles study. Chin. Phys. B. 2017, 26, 046303. [Google Scholar] [CrossRef]
- Pugh, S.F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Duan, Y.H.; Sun, Y.; Peng, M.J.; Zhou, S.G. Anisotropic elastic properties of the Ca–Pb compounds. J. Alloy. Compd. 2014, 595, 14–21. [Google Scholar] [CrossRef]
- Hao, Y.J.; Chen, X.R.; Cui, H.L.; Bai, Y.L. First-principles calculations of elastic constants of c-BN. Physica B 2006, 382, 118–122. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Wei, Q.; Yan, H.Y.; Zhang, M.G.; Zhang, Z.X.; Zhang, J.Q.; Zhang, D.Y. Elastic and electronic properties of Pbca-BN: First-principles calculations. Comput. Mater. Sci. 2014, 85, 80–87. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Zhou, P.K.; Yang, Y.T. Elastic anisotropy and electronic properties of Si3N4 under pressures. AIP Adv. 2016, 6, 085207. [Google Scholar] [CrossRef]
- Marmier, A.; Lethbridge, Z.A.D.; Walton, R.I.; Smith, C.W.; Parker, S.C.; Evans, K.E. ElAM: A computer program for the analysis and representation of anisotropic elastic properties. Comput. Phys. Commun. 2010, 181, 2102–2115. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.R.; Zhang, H.R.; Zhang, G.T.; Wei, Q.; Yuan, Y.Q. First-principles investigation on elastic and thermodynamic properties of Pnnm-CN under high pressure. AIP Adv. 2016, 6, 125040. [Google Scholar] [CrossRef]
- Hu, W.C.; Liu, Y.; Li, D.J.; Zeng, X.Q.; Xu, C.S. First-principles study of structural and electronic properties of C14-type Laves phase Al2Zr and Al2Hf. Comput. Mater. Sci. 2014, 83, 27–34. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T. Two novel C3N4 phases: Structural, mechanical and electronic properties. Materials 2016, 9, 427. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The crystal structures of (a) CN/SiN/GeN in the Pbca phase; and CN/SiN/GeN in the Pbca phase along the (b) (001) direction and (c) (010) direction. The blue and red spheres represent the N atoms and C/Si/Ge atoms.
Figure 1.
The crystal structures of (a) CN/SiN/GeN in the Pbca phase; and CN/SiN/GeN in the Pbca phase along the (b) (001) direction and (c) (010) direction. The blue and red spheres represent the N atoms and C/Si/Ge atoms.
Figure 2.
(a) Lattice parameters and (b) elastic moduli for Pbca-CN/SiN/GeN with PBE level.
Figure 3.
The phonon spectra of (a) Pbca-SiN and (b) Pbca-GeN with PBE level.
Figure 4.
The band structures of (a) Pbca-CN; (b) Pbca-SiN; and (c) Pbca-CN GeN with the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional with PBE level.
Figure 4.
The band structures of (a) Pbca-CN; (b) Pbca-SiN; and (c) Pbca-CN GeN with the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional with PBE level.
Figure 5.
The two-dimensional representation of Poisson’s ratio in the (a) (001) plane; (b) (010) plane; and (c) (100) plane for Pbca-CN/SiN/GeN with PBE level. The cyan line, red line, and blue line represent the Poisson’s ratio v of Pbca-CN/SiN/GeN, respectively.
Figure 5.
The two-dimensional representation of Poisson’s ratio in the (a) (001) plane; (b) (010) plane; and (c) (100) plane for Pbca-CN/SiN/GeN with PBE level. The cyan line, red line, and blue line represent the Poisson’s ratio v of Pbca-CN/SiN/GeN, respectively.

Figure 6.
The two-dimensional representation of Young’s modulus in the (a) (001) plane; (b) (010) plane; and (c) (100) plane for Pbca-CN/SiN/GeN with PBE level. The cyan line, red line, and blue line represent the Poisson’s ratio v of Pbca-CN/SiN/GeN, respectively.
Figure 6.
The two-dimensional representation of Young’s modulus in the (a) (001) plane; (b) (010) plane; and (c) (100) plane for Pbca-CN/SiN/GeN with PBE level. The cyan line, red line, and blue line represent the Poisson’s ratio v of Pbca-CN/SiN/GeN, respectively.

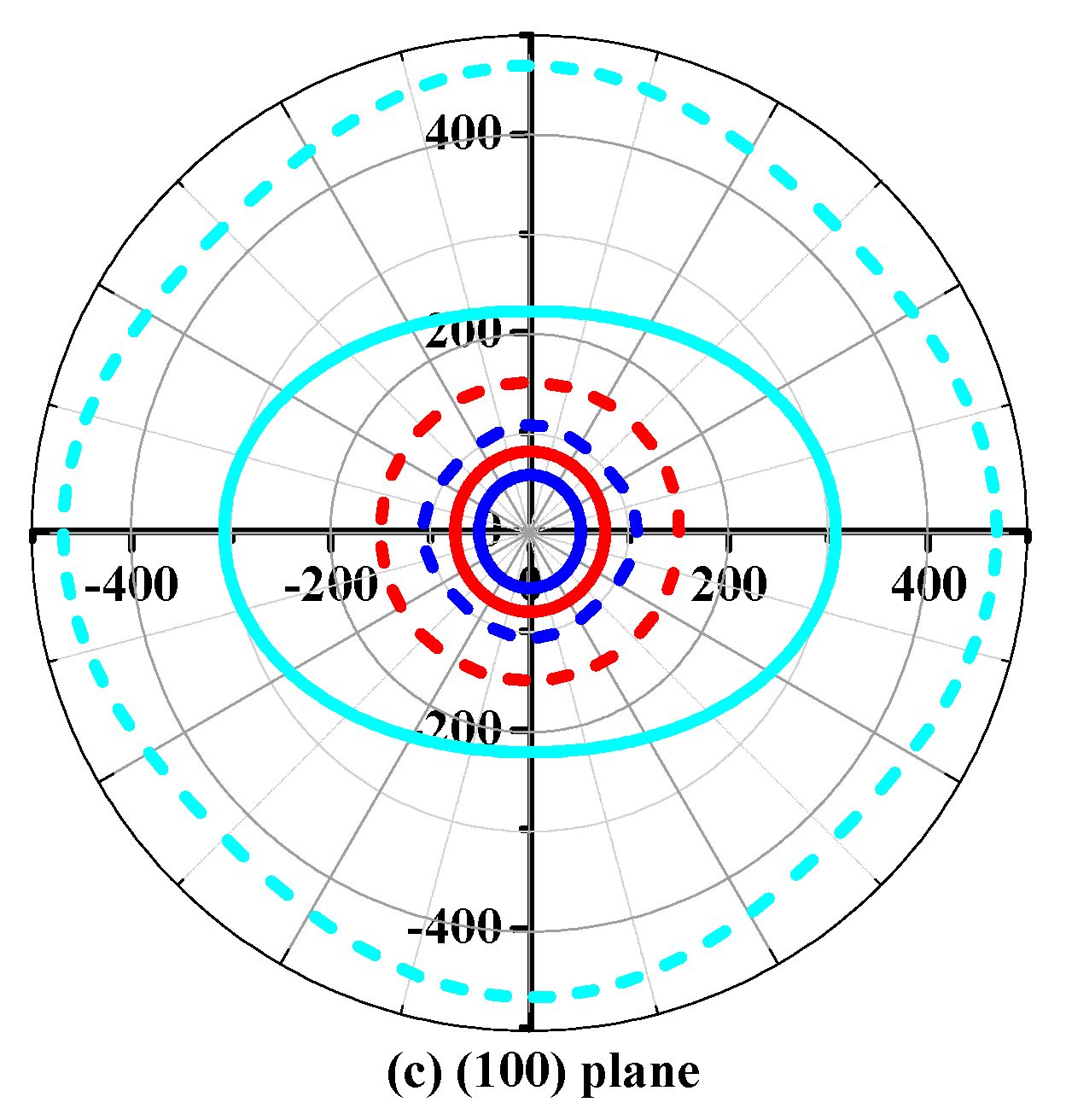
Figure 7.
The surface constructions of Young’s modulus for (a) Pbca-CN; (b) Pbca-SiN; and (c) Pbca-GeN with PBE level. Two-dimensional representation of Young’s modulus in the (d) (001) plane; (e) (010) plane; and (f) (100) plane for Pbca-CN/SiN/GeN with PBE level. The cyan line, red line, and blue line represent the Poisson’s ratio v of Pbca-CN/SiN/GeN, respectively.
Figure 7.
The surface constructions of Young’s modulus for (a) Pbca-CN; (b) Pbca-SiN; and (c) Pbca-GeN with PBE level. Two-dimensional representation of Young’s modulus in the (d) (001) plane; (e) (010) plane; and (f) (100) plane for Pbca-CN/SiN/GeN with PBE level. The cyan line, red line, and blue line represent the Poisson’s ratio v of Pbca-CN/SiN/GeN, respectively.
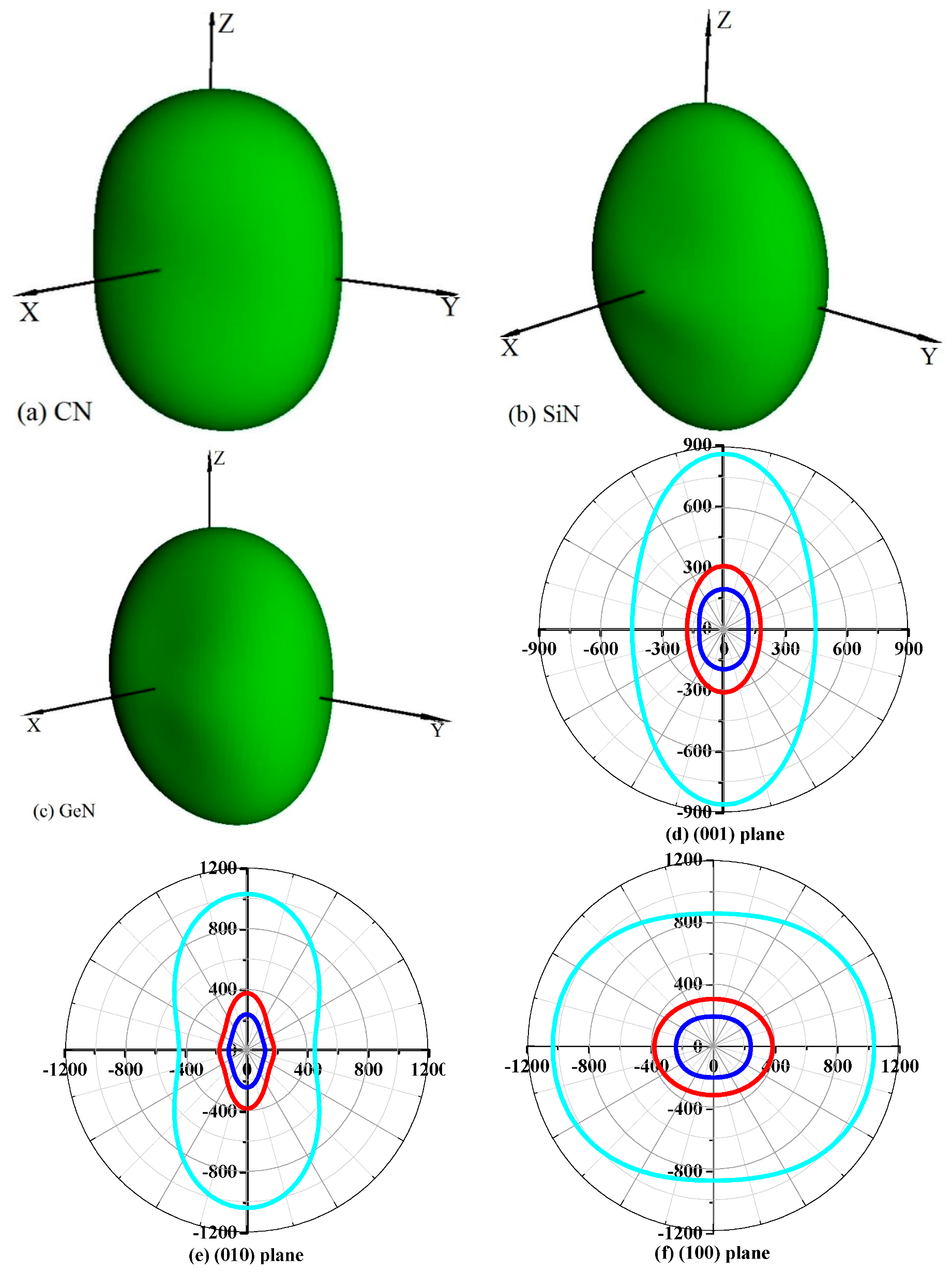
Figure 8.
The surface constructions of bulk modulus in the (a) (001) plane; (b) (010) plane; and (c) (100) plane for Pbca-CN/SiN/GeN with PBE level.
Figure 8.
The surface constructions of bulk modulus in the (a) (001) plane; (b) (010) plane; and (c) (100) plane for Pbca-CN/SiN/GeN with PBE level.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The lattice parameters (in Å) of Pbca-CN/SiN/GeN using different functionals.
| Materials | PBE | PBEsol | CA-PZ | PBE + D | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a | b | c | a | b | c | a | b | c | a | b | c | |
| CN | 5.504 | 4.395 | 4.041 | 5.461 | 4.384 | 4.029 | 5.402 | 4.352 | 3.998 | 5.484 | 4.385 | 4.029 |
| 5.514 1 | 4.396 | 4.041 | ||||||||||
| 5.514 2 | 4.396 | 4.041 | ||||||||||
| SiN | 7.234 | 5.341 | 7.226 | 7.339 | 5.333 | 4.867 | 7.226 | 5.257 | 4.798 | 7.281 | 5.322 | 4.856 |
| GeN | 7.831 | 5.645 | 7.578 | 7.823 | 5.625 | 5.136 | 7.578 | 5.488 | 5.011 | 7.744 | 5.610 | 5.129 |
| c-BN | 3.626 | 3.612 | 3.569 | 3.600 | ||||||||
| Diamond | 3.566 | 3.558 | 3.526 | 3.566 | ||||||||
Table 2.
The calculated elastic constants Cij (in GPa) and bulk moduli B (in GPa), shear moduli G (in GPa), Young’s moduli E (in GPa), and Poisson’s ratio v of Pbca-CN/SiN/GeN and other CxNy, SixNy, and GexNy compounds with PBE level.
Table 2.
The calculated elastic constants Cij (in GPa) and bulk moduli B (in GPa), shear moduli G (in GPa), Young’s moduli E (in GPa), and Poisson’s ratio v of Pbca-CN/SiN/GeN and other CxNy, SixNy, and GexNy compounds with PBE level.
| Materials | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 | B | G | E | v |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CN | 491 | 169 | 139 | 922 | 122 | 1080 | 469 | 307 | 222 | 356 | 319 | 771 | 0.139 |
| CN 1 | 495 | 174 | 145 | 934 | 124 | 1112 | 465 | 313 | 243 | 363 | 326 | 754 | 0.154 |
| SiN | 221 | 107 | 88 | 367 | 90 | 422 | 150 | 75 | 81 | 170 | 104 | 257 | 0.243 |
| SiN2 2 | 836 | 1269 | 397 | 313 | 407 | 386 | 879 | 0.140 | |||||
| SiN2 3 | 442 | 75 | 58 | 610 | 133 | 133 | 237 | 76 | 71 | 191 | 138 | 333 | 0.200 |
| Si3N2 4 | 261 | 97 | 68 | 152 | 73 | 190 | 0.290 | ||||||
| o-Si3N4 5 | 581 | 181 | 55 | 587 | 132 | 483 | 244 | 88 | 197 | 262 | 179 | 436 | 0.221 |
| t-Si3N4 5 | 277 | 152 | 145 | 312 | 178 | 207 | 194 | 126 | 311 | 0.233 | |||
| m-Si3N4 5 | 241 | 39 | 139 | 457 | 55 | 358 | 88 | 128 | 86 | 165 | 104 | 259 | 0.239 |
| GeN | 159 | 85 | 69 | 243 | 55 | 272 | 107 | 51 | 57 | 119 | 70 | 176 | 0.255 |
| GeN2 3 | 260 | 40 | 22 | 350 | 94 | 145 | 138 | 45 | 44 | 119 | 85 | 205 | 0.210 |
| o-Ge3N4 6 | 203 | 122 | 305 | 0.250 | |||||||||
| t-Ge3N4 6 | 147 | 87 | 218 | 0.253 | |||||||||
| m-Ge3N4 6 | 124 | 73 | 183 | 0.254 |
Table 3.
The density (in g/cm3), sound velocities (in m/s), average sound velocity (in m/s), and the Debye temperature (in K) for Pbca-CN/SiN/GeN with PBE level.
Table 3.
The density (in g/cm3), sound velocities (in m/s), average sound velocity (in m/s), and the Debye temperature (in K) for Pbca-CN/SiN/GeN with PBE level.
| Materials | CN | SiN | GeN | |
|---|---|---|---|---|
| ρ | 3.536 | 2.922 | 5.039 | |
| (100) | (100)vl | 11,784 | 8697 | 5617 |
| (010)vtl | 7927 | 5265 | 3363 | |
| (001)vt2 | 9318 | 5066 | 3181 | |
| (010) | (010)vl | 16,148 | 11,207 | 6944 |
| (100)vt1 | 7927 | 5265 | 3363 | |
| (001)vt2 | 11,517 | 7165 | 4608 | |
| (001) | (001)vl | 17,477 | 12,018 | 7347 |
| (100)vt1 | 9318 | 5066 | 3181 | |
| (010)vt2 | 11,517 | 7165 | 4608 | |
| vl | 14,865 | 10,278 | 6491 | |
| vt | 9498 | 5966 | 3727 | |
| vm | 10,437 | 6620 | 4140 | |
| ΘD | 1702 | 863 | 508 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ma, Z.; Liu, X.; Yu, X.; Shi, C.; Wang, D. Mechanical, Anisotropic, and Electronic Properties of XN (X = C, Si, Ge): Theoretical Investigations. Materials 2017, 10, 912. https://doi.org/10.3390/ma10080912
AMA Style
Ma Z, Liu X, Yu X, Shi C, Wang D. Mechanical, Anisotropic, and Electronic Properties of XN (X = C, Si, Ge): Theoretical Investigations. Materials. 2017; 10(8):912. https://doi.org/10.3390/ma10080912
Chicago/Turabian StyleMa, Zhenyang, Xuhong Liu, Xinhai Yu, Chunlei Shi, and Dayun Wang. 2017. "Mechanical, Anisotropic, and Electronic Properties of XN (X = C, Si, Ge): Theoretical Investigations" Materials 10, no. 8: 912. https://doi.org/10.3390/ma10080912
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.