Customized Peptide Biomaterial Synthesis via an Environment-Reliant Auto-Programmer Stigmergic Approach

 , , , ,
, , , ,  and
and
Abstract
:
1. Introduction
Stigmergy
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. The “In Solution” Digestion Protocol for Peptide Biomaterial Synthesis by the Stigmergic Process
2.2.2. Characterization of Peptide Biomaterial Synthesized by the Stigmergic Process
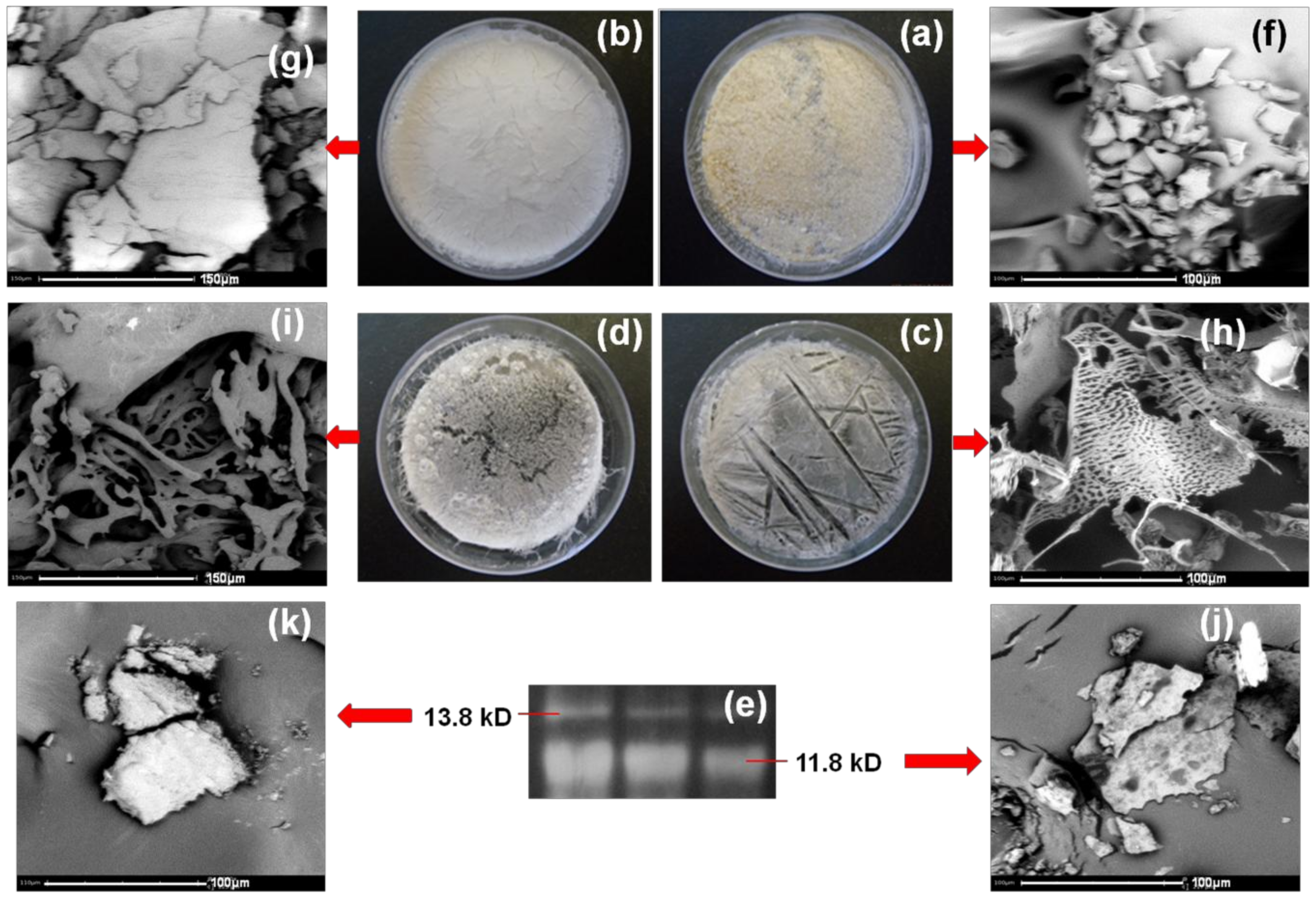
Molecular Weight Determination of Stigmergized Peptides using SDS-PAGE
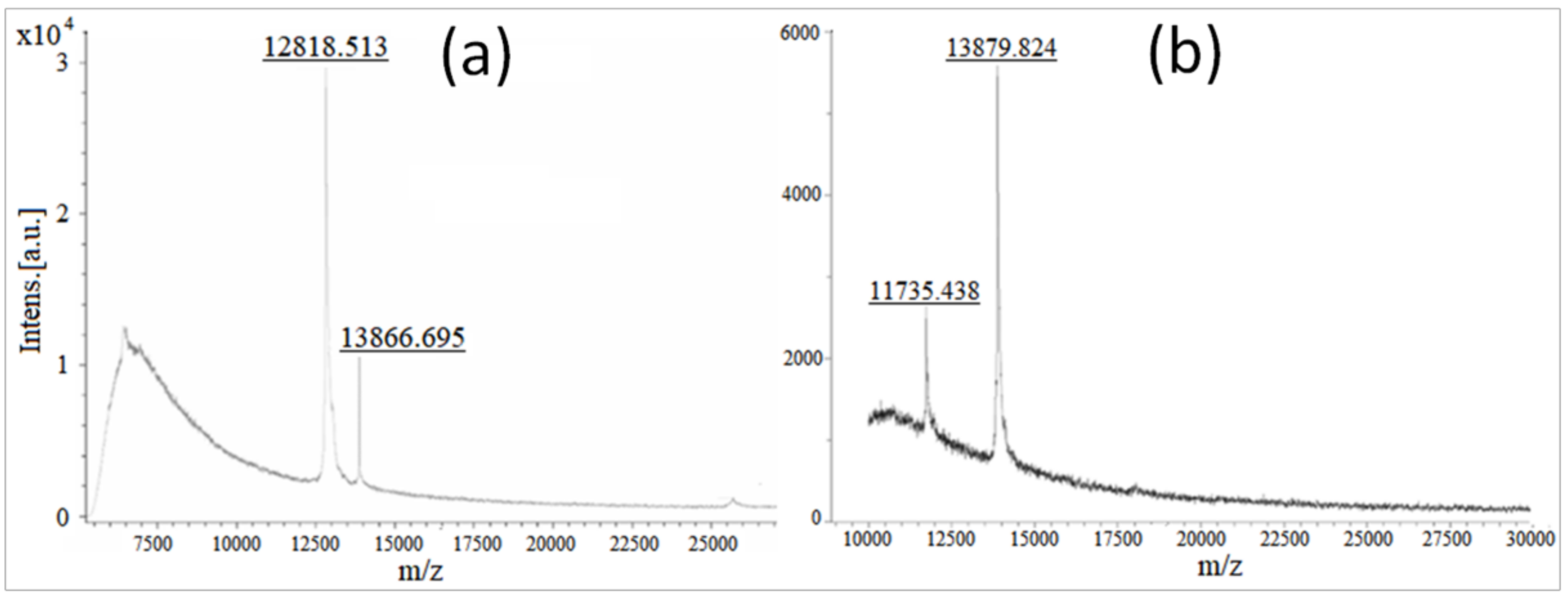
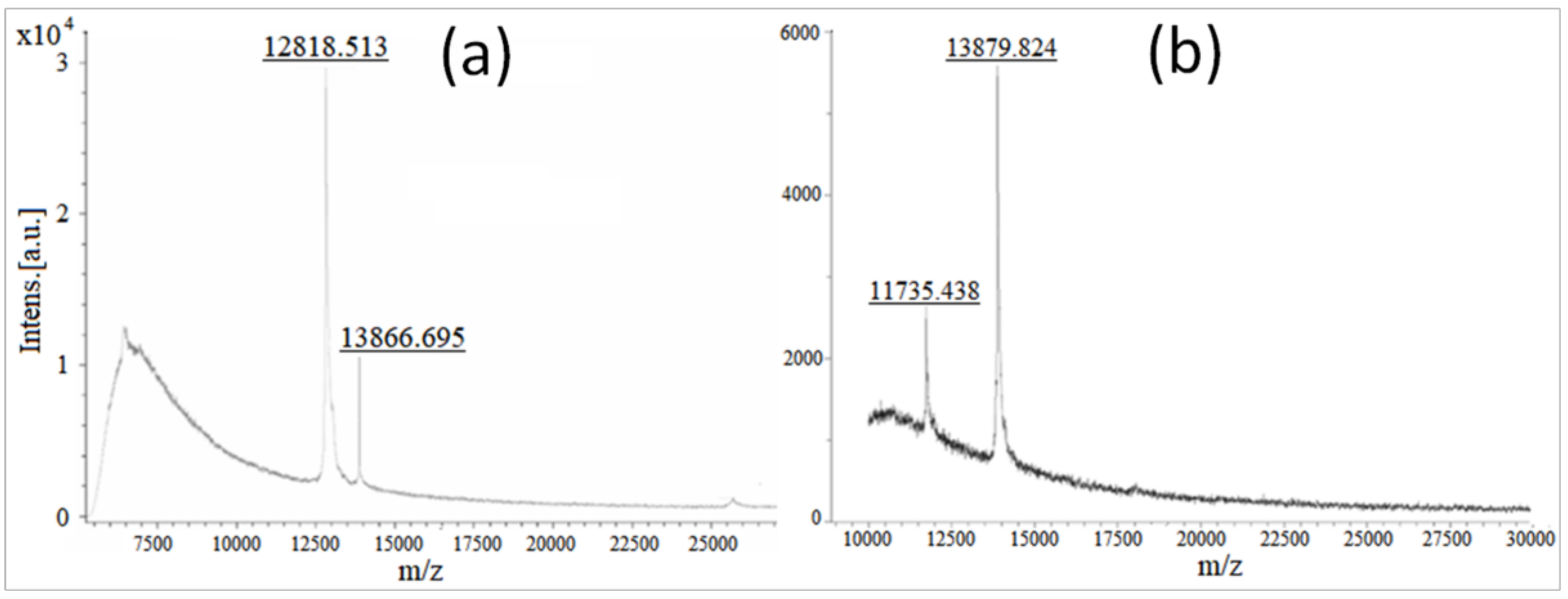
Molecular Weight Confirmation of Stigmergized Peptides using MALDI-TOF Analysis
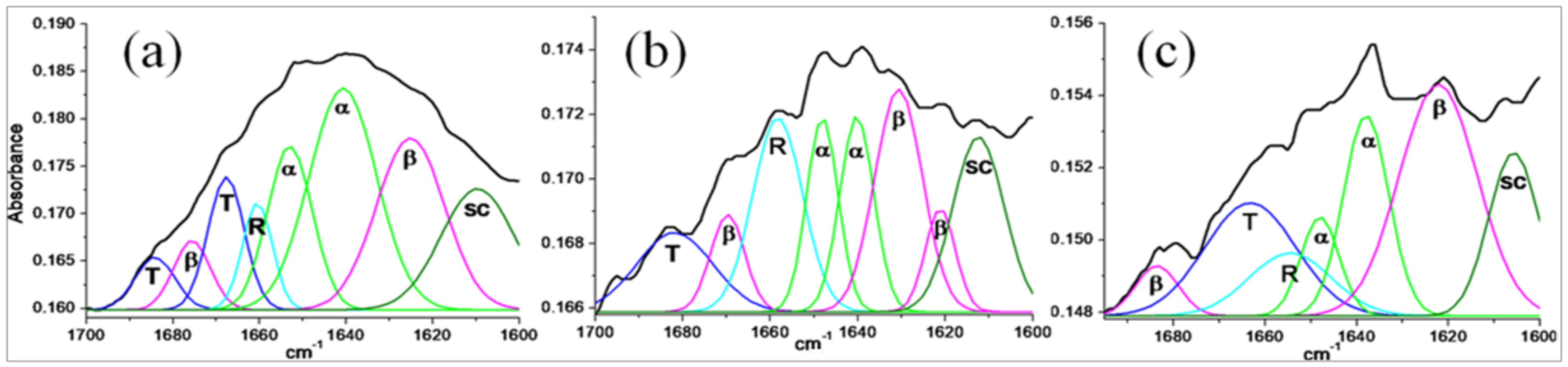
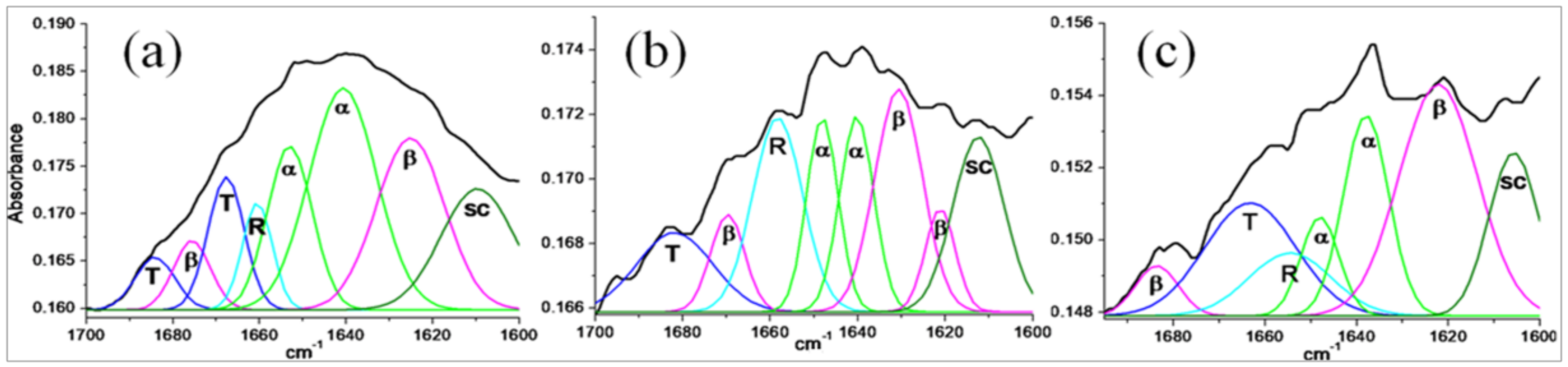
Secondary Structure Analysis of Stigmergized Peptides
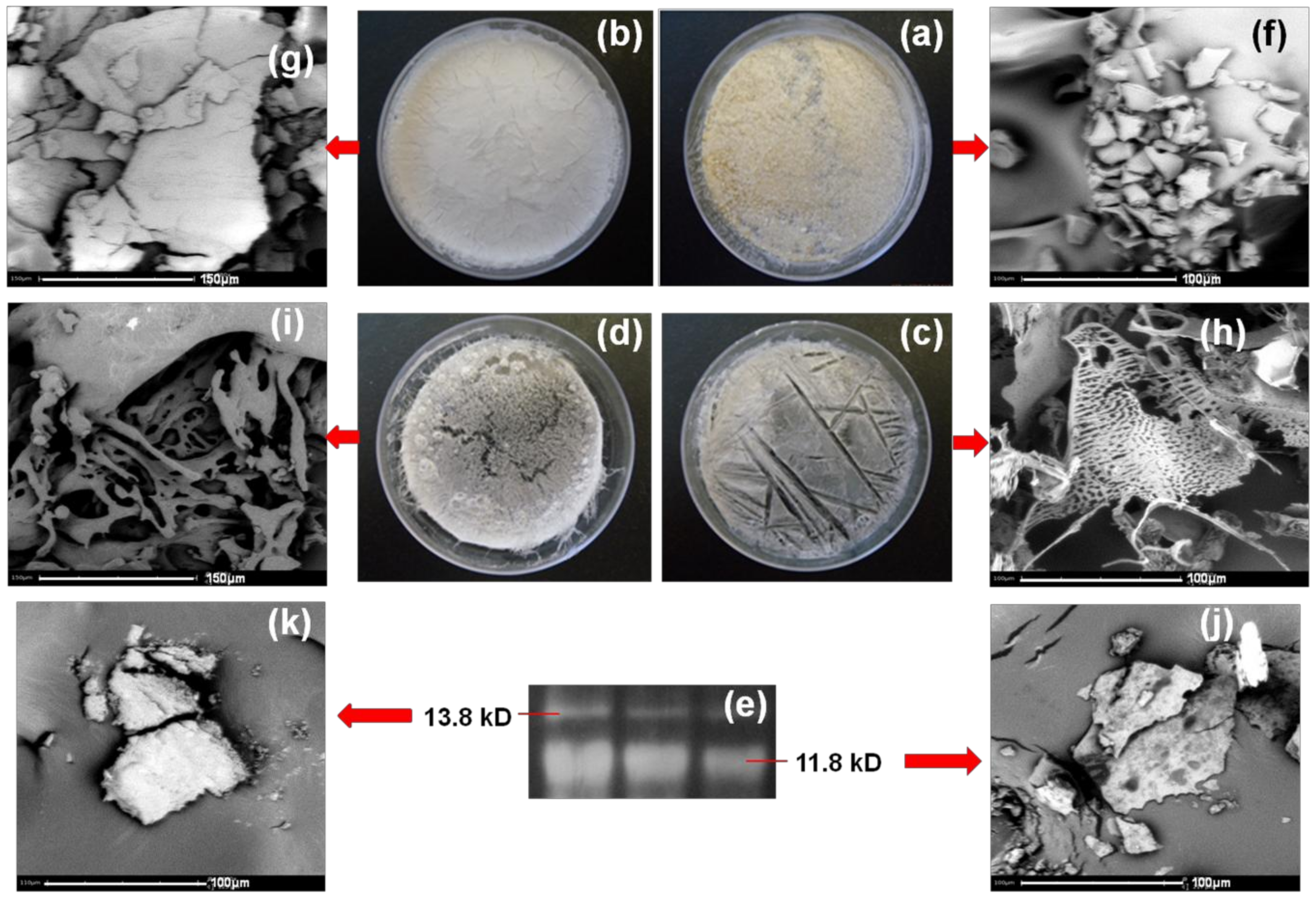
Morphology of Stigmergized Peptides
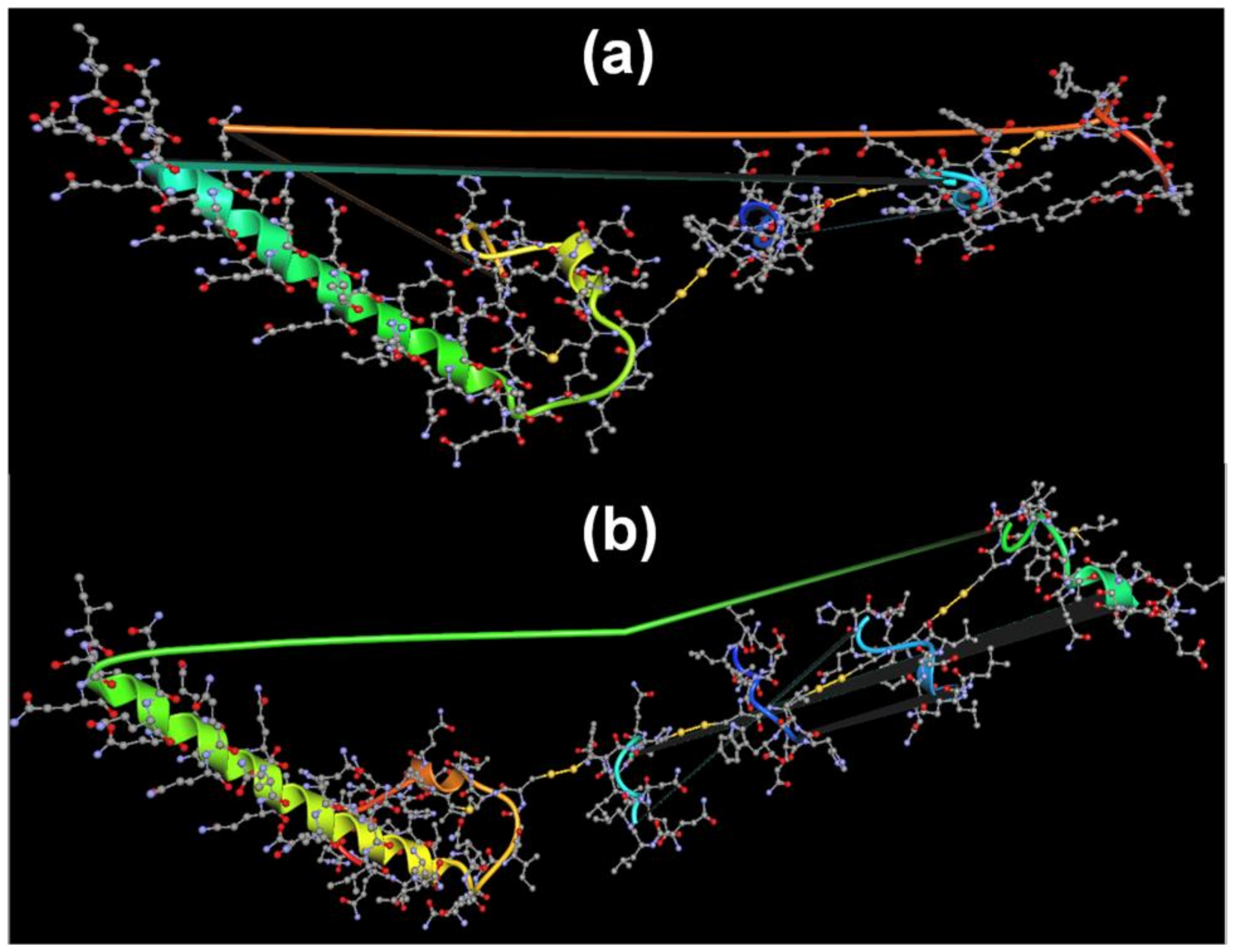
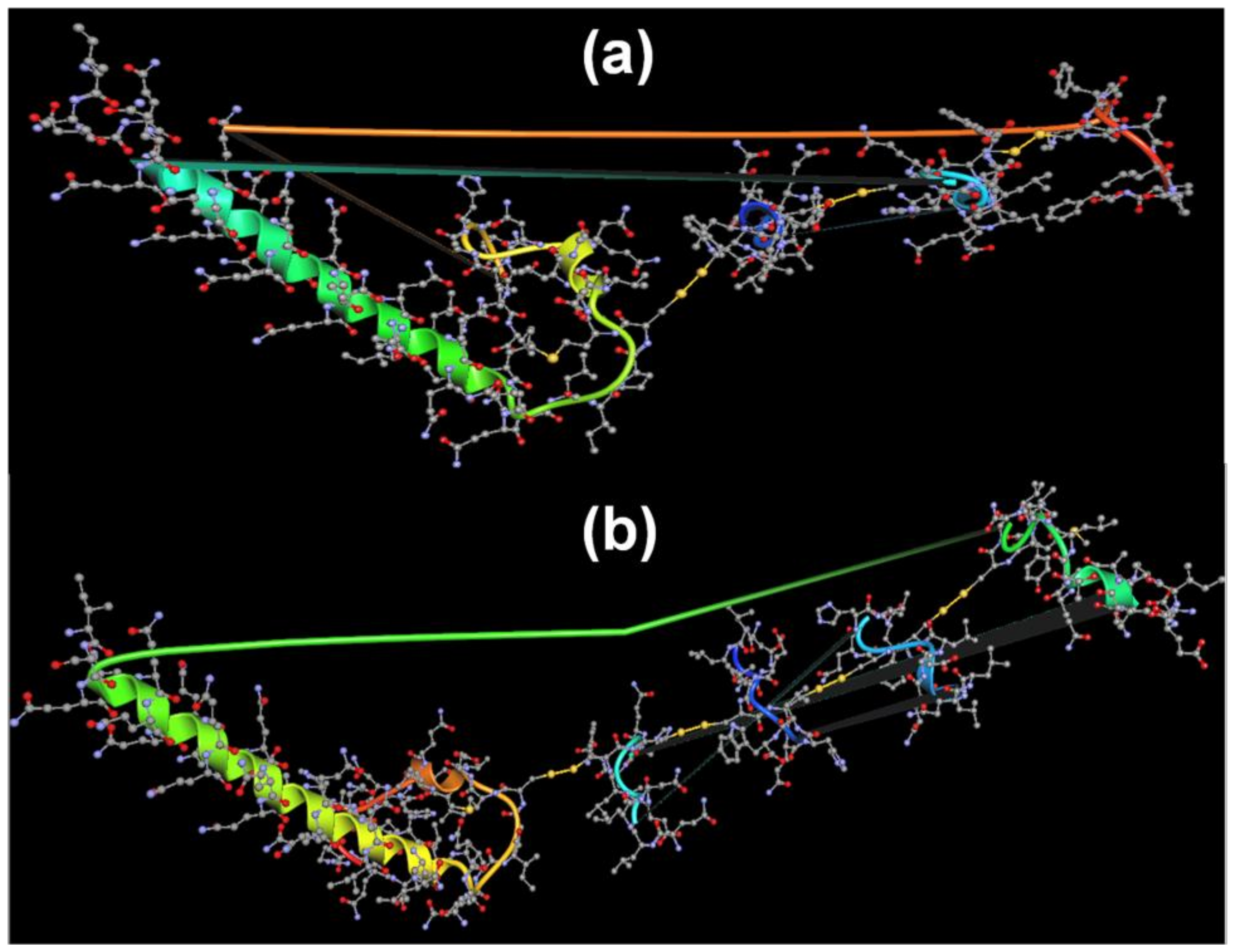
Predicted Self-Organization of Stigmergized Peptides

3. Results
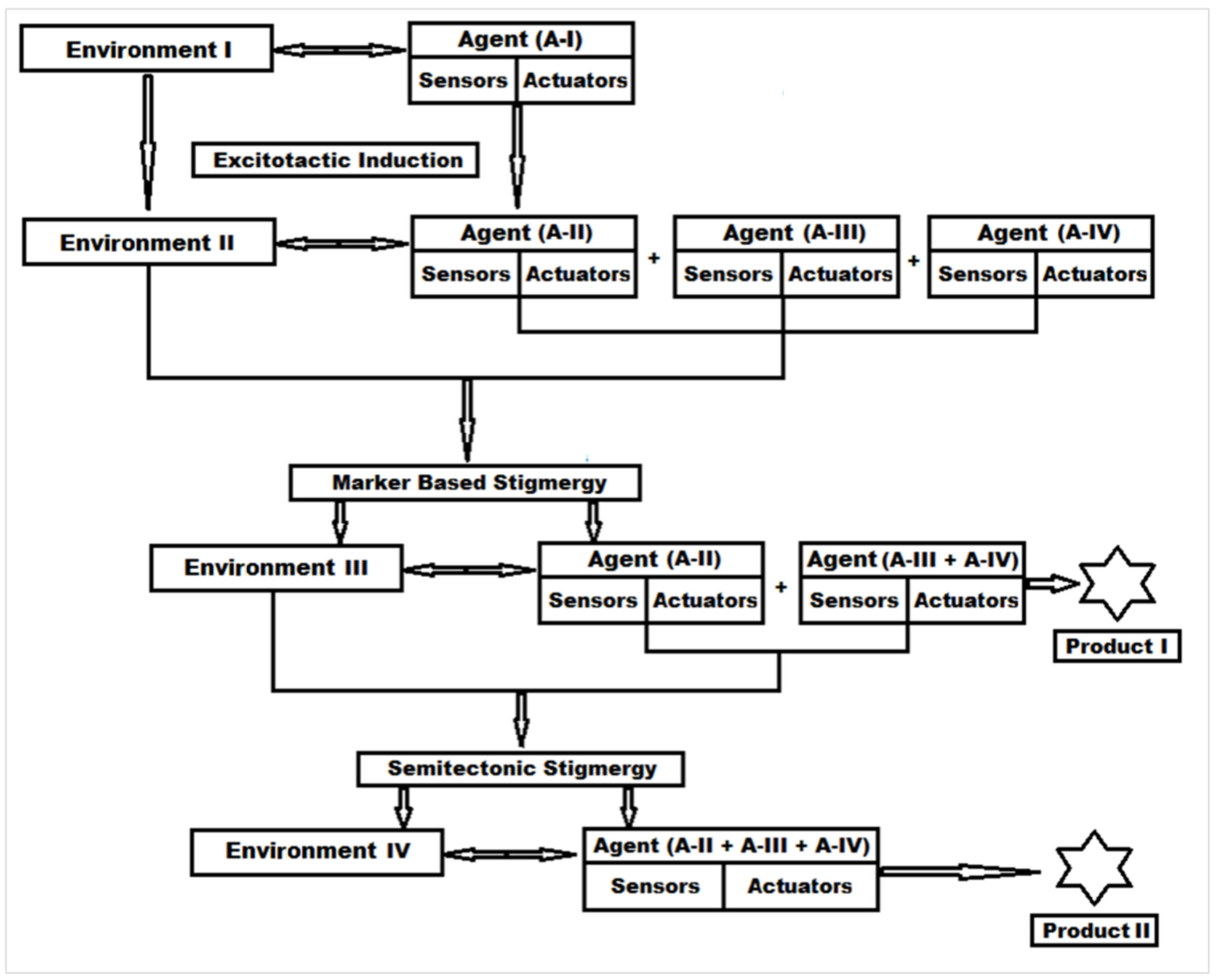
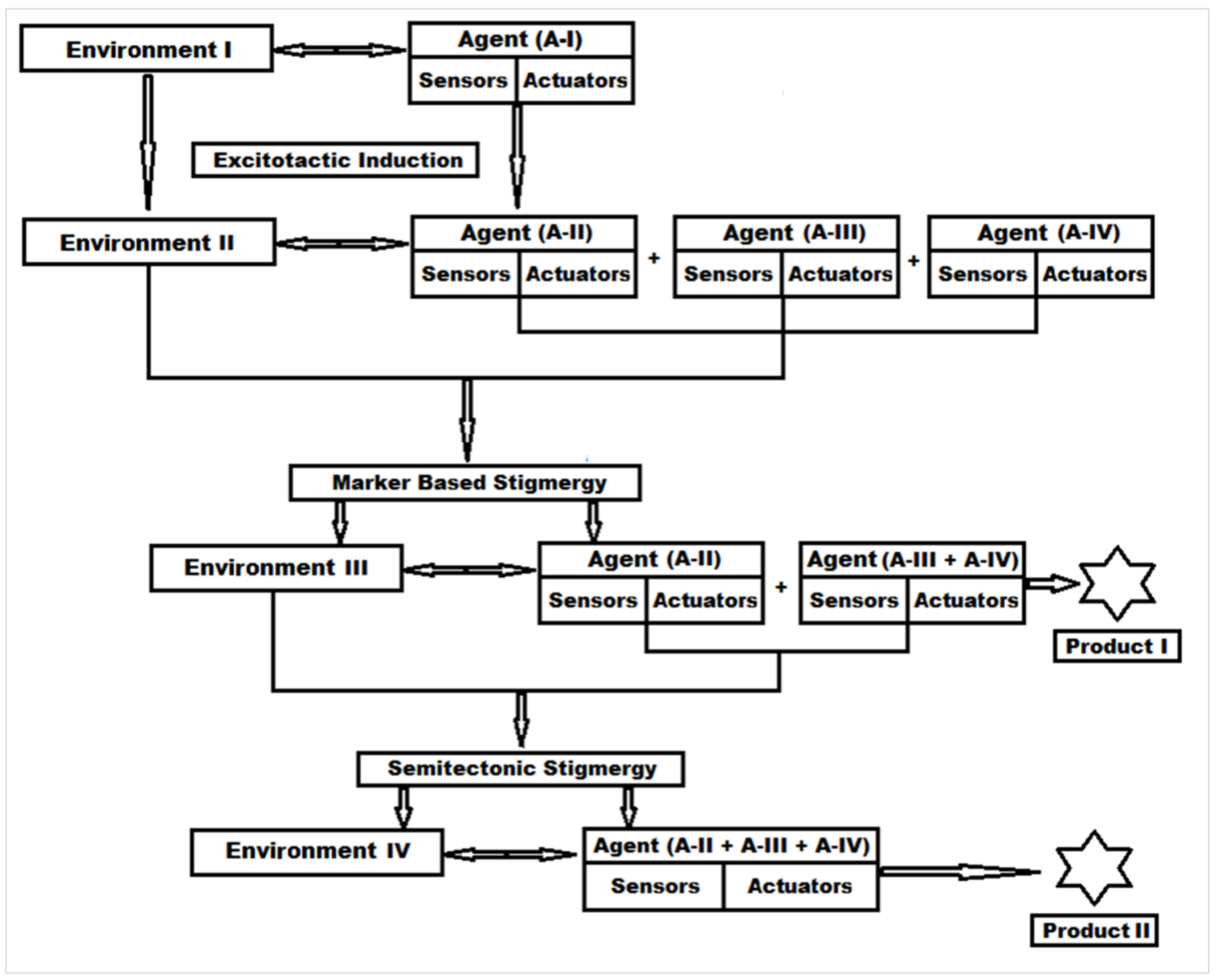
3.1. Stigmergy-Based Mechanism of Protein Digestion and New Peptide Biomaterial Synthesis
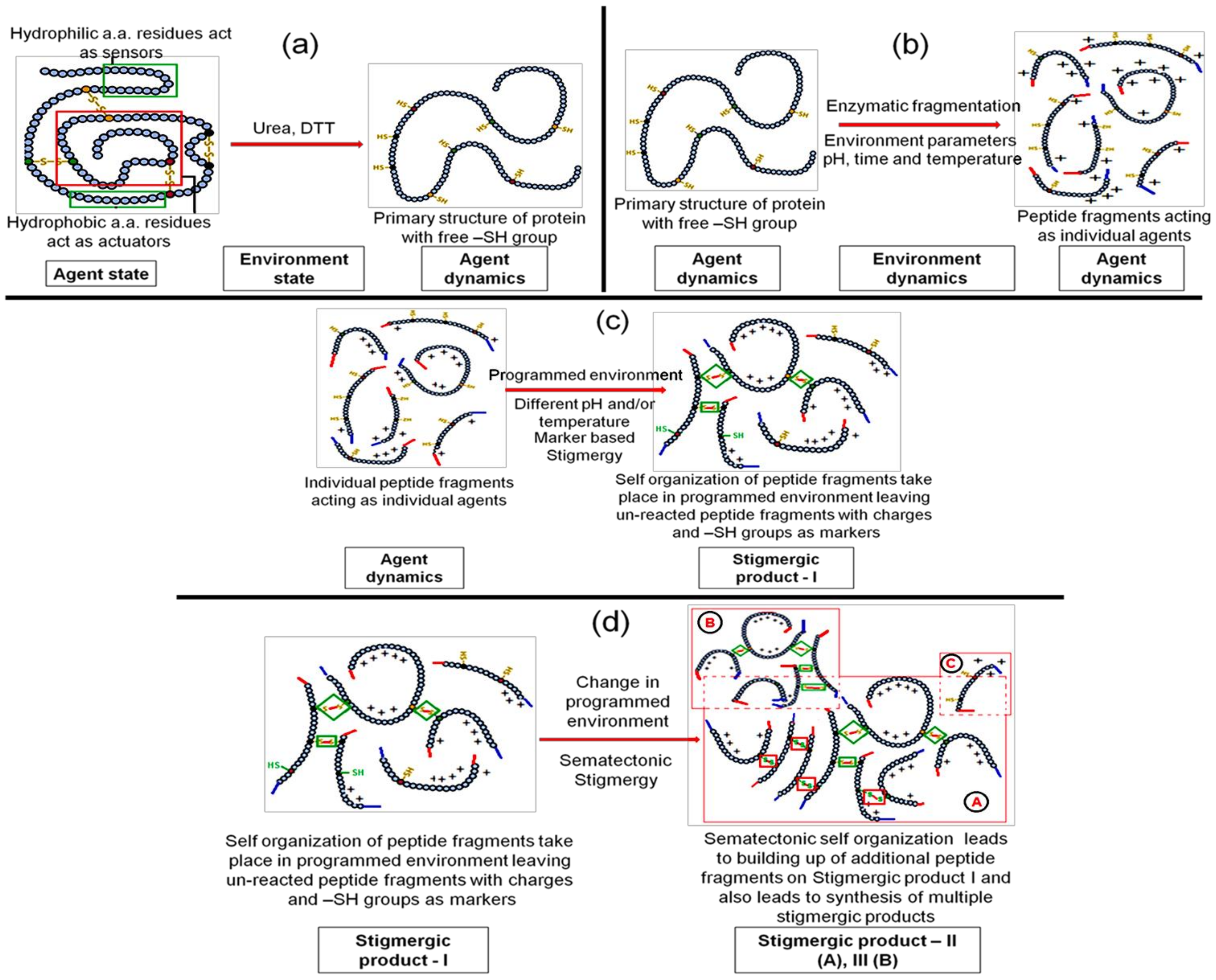
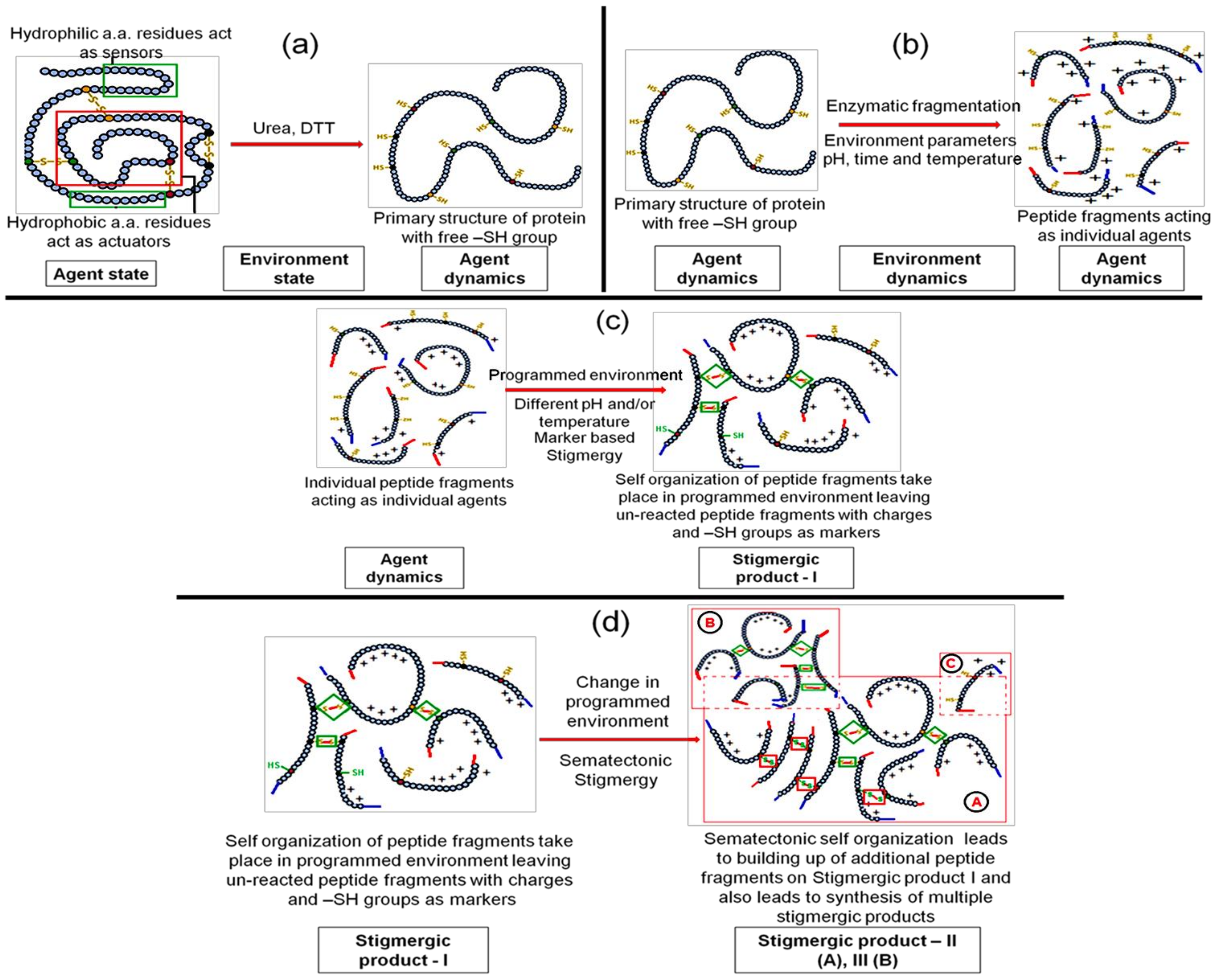
3.1.1. Step 1—Engineering “Agent-State” to “Agent-Dynamic”
3.1.2. Step 2—Engineering the Environment’s Dynamic and Multi-Agent Systems
3.1.3. Step 3—Marker Based Stimulus to Generate Stigmergic Product
3.1.4. Step 4—Sematectonic Emergence of Stigmergic Products
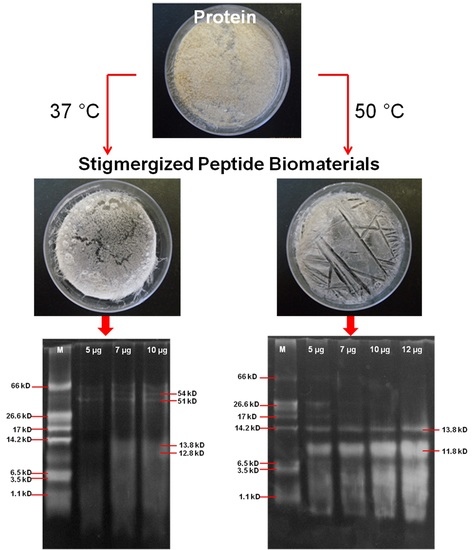
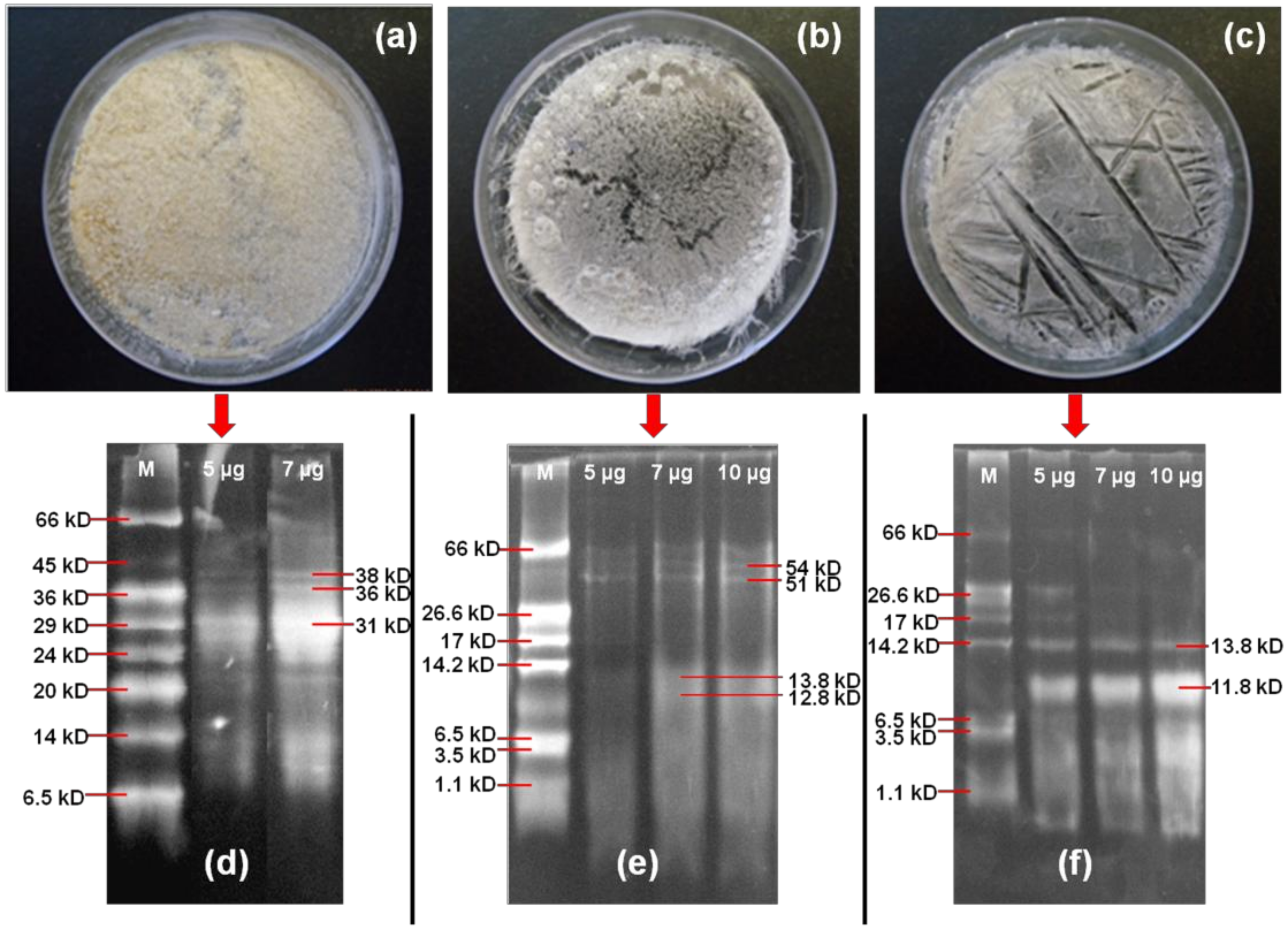
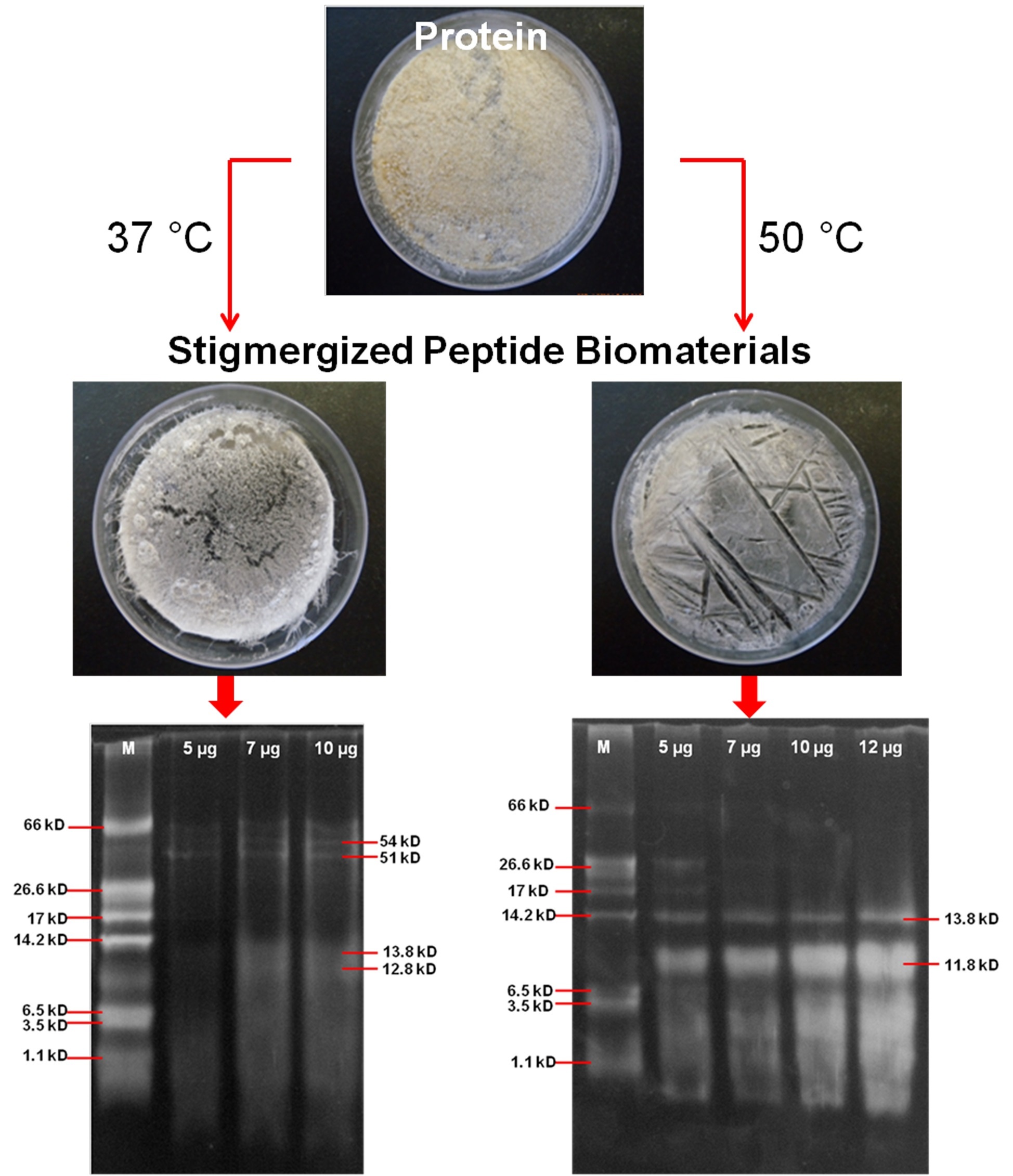
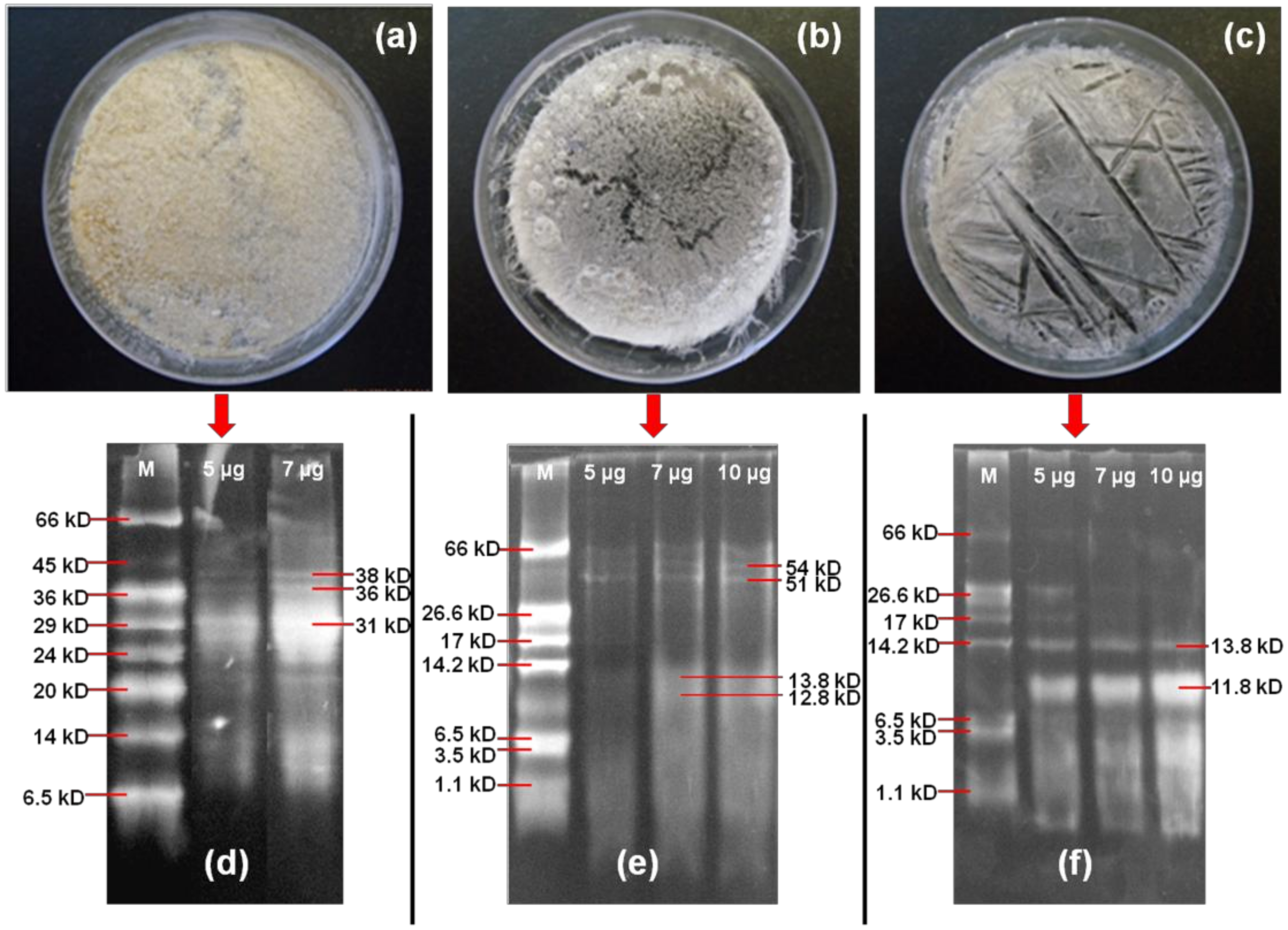
3.2. Characterization of Synthesized Peptide Biomaterial
3.2.1. Molecular Weight Analysis of Stigmergized Peptides
3.2.2. Molecular Weight Confirmation of Stigmergized Peptides
3.2.3. Secondary Structure Analysis of Stigmergized Peptides
3.2.4. Morphology of Stigmergize Peptides
3.2.5. Predicted Self-Organization of Stigmergize Peptides
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhao, X.; Pan, F.; Lu, J.R. Recent development of peptide self-assembly. Prog. Nat. Sci. 2008, 18, 653–660. [Google Scholar] [CrossRef]
- Rad-Malekshahi, M.; Lempsink, L.; Amidi, M.; Hennink, W.E.; Mastrobattista, E. Biomedical applications of self-assembling peptides. Bioconj. Chem. 2016, 27, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, A.; Zhang, S.; Hauser, C.A.E. Short self-assembling peptides as building blocks for modern nanodevices. Trends Biotechnol. 2012, 30, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.G.; Webber, M.J.; Stupp, S.I. Self-assembly of peptide amphiphiles: From molecules to nanostructures to biomaterials. Biopolymers 2010, 94, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Muraoka, T.; Cheetham, A.G.; Stupp, S.I. Self-assembly of giant peptide nanobelts. Nano Lett. 2009, 9, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Bonabeau, E. Editor’s Introduction: Stigmergy. Artif. Life 1999, 5, 95–96. [Google Scholar] [CrossRef]
- Marsh, L.; Onof, C. Stigmergic epistemology, stigmergic cognition. Cognit. Syst. Res. 2008, 9, 136–149. [Google Scholar] [CrossRef] [Green Version]
- Heylighen, F. Stigmergy as a universal coordination mechanism I: Definition and components. Cognit. Syst. Res. 2016, 38, 4–13. [Google Scholar] [CrossRef]
- Rocher, A.; Soriano, F.; Molina, E.; González-Limas, G.; Méndez, E. Characterization of distinct alpha- and gamma-type gliadins and low molecular weight components from wheat endosperm as coeliac immunoreactive proteins. Biochim. Biophys. Acta 1995, 1247, 143–148. [Google Scholar] [CrossRef]
- Moiler, S.; Wieser, H. The location of disulphide bonds in α-type gliadins. J. Cereal Sci. 1995, 22, 21–27. [Google Scholar] [CrossRef]
- Vermachova, M.; Purkrtova, Z.; Santrucek, J.; Jolivet, P.; Chardot, T.; Kodicek, M. Combining chymotrypsin/trypsin digestion to identify hydrophobic proteins from oil bodies. Methods Mol. Biol. 2014, 1072, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.; Zhou, R.; Thirumalai, D.; Berne, B.J. Urea denaturation by stronger dispersion interactions with proteins than water implies a 2-stage unfolding. Proc. Natl. Acad. Sci. USA 2008, 105, 16928–16933. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Gundry, R.L.; White, M.Y.; Murray, C.I.; Kane, L.A.; Fu, Q.; Stanley, B.A.; Van Eyk, J.E. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr. Protoc. Mol. Biol. 2009, 90, 10–25. [Google Scholar] [CrossRef]
- Purcell, J.M.; Kasarda, D.D.; Wu, C.S.C. Secondary structures of wheat alpha and omega gliadin proteins: Fourier transform infrared spectroscopy. J. Cereal Sci. 1988, 7, 21–32. [Google Scholar] [CrossRef]
- Pannequin, R.; Morel, G.; Thomas, A. The performance of product-driven manufacturing control: An emulation-based benchmarking study. Comput. Ind. 2009, 60, 195–203. [Google Scholar] [CrossRef]
- Wallqvist, A.; Covell, D.G.; Thirumalai, D. Hydrophobic interactions in aqueous urea solutions with implications for the mechanism of protein denaturation. J. Am. Chem. Soc. 1998, 120, 427–428. [Google Scholar] [CrossRef]
- Zangi, R.; Zhou, R.; Berne, B.J. Urea’s action on hydrophobic interactions. J. Am. Chem. Soc. 2009, 131, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Giuggiolia, L.; Pottsa, J.R.; Rubensteind, D.I.; Levin, S.A. Stigmergy, collective actions, and animal social spacing. Proc. Natl. Acad. Sci. USA 2013, 110, 16904–16909. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke Parunak, H. Expert Assessment of Human–Human Stigmergy. Analysis for the Canadian Defence Organization; Altarum Institute: Ann Arbor, MI, USA, 2005; Available online: www.dtic.mil/get-tr-doc/pdf?AD=ADA440006 (accessed on 25 May 2015).
- Van Dyke Parunak, H. A survey of environments and mechanisms for human-human stigmergy. In Lecture Notes in Computer Science, Proceedings of the Environments for Multi-Agent Systems II (E4MAS 2005), Utrecht, The Netherlands, 25 July 2005; Weyns, D., Van Dyke Parunak, H., Michel, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 3830, ISBN 978-3-540-32615-1. [Google Scholar] [CrossRef]
- Asgeirsson, B.; Bjarnason, J.B. Structural and kinetic properties of chymotrypsin from Atlantic cod (Gadus morhua). Comparison with bovine trypsin. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1991, 9, 327–335. [Google Scholar] [CrossRef]
- Perera, E.; Moyano, F.J.; Díaz, M.; Perdomo-Morales, R.; Montero-Alejo, V.; Alonso, E.; Carrillo, O.; Galich, G.S. Polymorphism and partial characterization of digestive enzymes in the spiny lobster Panulirus argus. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 150, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.T.; Zhai, J.; Hoffmann, S.V.; Aguilar, M.I.; Augustin, M.A.; Wooster, T.J.; Day, L. Conformational changes to deamidated wheat gliadins and β-casein upon adsorption to oil–water emulsion interfaces. Food Hydrocoll. 2012, 27, 91–101. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragment | Mass | Position | Peptide Sequence |
|---|---|---|---|
| 1 | 7345.1270 | 88–149 | SQPQQPISQQQQQQQQQQQQQQQQQQILQQILQQQLIPCM DVVLQQHNIAHGRSQVLQQS TY |
| 2 | 5158.8066 | 31–74 | LGQQQPFPPQQPYPQPQPFPSQQPYLQLQPFPQPQLPYSQ PQPF |
| 3 | 4136.6144 | 162–197 | QIPEQSQCQAIHNVVHAIIL HQQQKQQQQPSSQVSF |
| 4 | 3493.9273 | 1–30 | VRVPVPQLQPQNPSQQQPQE QVPLVQQQQF |
| 5 | 2363.5747 | 212–232 | RPSQQNPQAQGSVQPQQLPQ F |
| 6 | 2078.4291 | 233–250 | EEIRNLALQTLPAMCNVY |
| 7 | 1626.7918 | 75–87 | RPQQPYPQPQPQY |
| 8 | 1590.7556 | 198–211 | QQPLQQYPLGQGSF |
| 9 | 1513.7924 | 150–161 | QLLQELCCQHLW |
| 10 | 1121.3595 | 251–260 | IAPYCTIPPF |
| Protein/Stigmergized Peptides | Secondary Structure (%) | |||
|---|---|---|---|---|
| α-Helix (α) | β-Sheet (β) | β-Turns (T) | Disordered (R and SC) | |
| α-Gliadin | 39 | 22 | 17 | 22 |
| 11.8 kD | 62 | 10 | 17 | 11 |
| 13.8 kD | 64 | 06 | 20 | 10 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badhe, R.V.; Kumar, P.; Choonara, Y.E.; Marimuthu, T.; Du Toit, L.C.; Bijukumar, D.; Chejara, D.R.; Mabrouk, M.; Pillay, V. Customized Peptide Biomaterial Synthesis via an Environment-Reliant Auto-Programmer Stigmergic Approach. Materials 2018, 11, 609. https://doi.org/10.3390/ma11040609
Badhe RV, Kumar P, Choonara YE, Marimuthu T, Du Toit LC, Bijukumar D, Chejara DR, Mabrouk M, Pillay V. Customized Peptide Biomaterial Synthesis via an Environment-Reliant Auto-Programmer Stigmergic Approach. Materials. 2018; 11(4):609. https://doi.org/10.3390/ma11040609
Chicago/Turabian StyleBadhe, Ravindra V., Pradeep Kumar, Yahya E. Choonara, Thashree Marimuthu, Lisa C. Du Toit, Divya Bijukumar, Dharmesh R. Chejara, Mostafa Mabrouk, and Viness Pillay. 2018. "Customized Peptide Biomaterial Synthesis via an Environment-Reliant Auto-Programmer Stigmergic Approach" Materials 11, no. 4: 609. https://doi.org/10.3390/ma11040609