Syntheses and Functional Properties of Phthalocyanines

1
Department of Sustainable Engineering, College of Industrial Technology, Nihon University, 1-2-1 Izumi-cho, Narashino-shi, Chiba-ken 275-8575 Japan
2
Department of Applied Molecular Chemistry, College of Industrial Technology, Nihon University, 1- 2-1 Izumi-cho, Narashino-shi, Chiba-ken 275-8575 Japan
3
Research Institute of Chemical Science, Technology and Education, 8-37-1-104 Narashinodai, Funabashi-shi Chiba-ken 274-0063 Japan
*
Author to whom correspondence should be addressed.
#
Current address: Tokyo Chemical Industry Co. Ltd., 4-10-2 Nihombashi-honcho, Chuo-ku, Tokyo 103-0023 Japan
Materials 2009, 2(3), 1127-1179; https://doi.org/10.3390/ma2031127
Submission received: 27 June 2009
/
Revised: 19 August 2009
/
Accepted: 24 August 2009
/
Published: 28 August 2009
(This article belongs to the Special Issue Functional Colorants)
Abstract
:Metal phthalocyanine tetrasulfonic acids, metal phthalocyanine octacarboxylic acids, metal octakis(hexyloxymethyl)phthalocyanines, and metal anthraquinocyanines have been synthesized. Then, zinc bis(1,4-didecylbenzo)-bis(3,4-pyrido)porphyrazines, the cyclotetramerization products of a 1:1 mixture of 3,6-didecylphthalonitrile and 3,4-dicyanopyridine, were synthesized. Futher, subphthalocyanine and its derivatives, with substituents such as thiobutyl and thiophenyl moieties were synthesized. Electrochemical measurements were performed on the abovementioned phthalocyanine derivatives and analogues in order to examine their electron transfer abilities and electrochemical reaction mechanisms in an organic solvent. Moreover, 1,4,8,11,15,18,22,25-octakis(thio-phenylmethyl)phthalocyanes were synthesized. The Q-bands of the latter compounds appeared in the near-infrared region. Furthermore, non-colored transparent films in the visible region can be produced.
1. Introduction
Phthalocyanine derivatives, which have a similar structure to porphyrin, have been utilized in important functional materials in many fields. Their useful properties are attributed to their efficient electron transfer abilities. The central cavity of phthalocyanines is known to be capable of accomodating 63 different elemental ions, including hydrogens (metal-free phthalocyanine, H2-PC). A phthalocyanine containing one or two metal ions is called a metal phthalocyanine (M-PC). In the last decade, as a result of their high electron transfer abilities, M-PCs have been utilized in many fields such as molecular electronics, optelectronics, photonics, etc [1,2,3,4,5,6] (Figure 1). The functions of M-PCs are almost universally based on electron transfer reactions because of the 18 π electron conjugated ring system found in their molecular structure.
Figure 1.
Typical function of phthalocyanine derivatives.
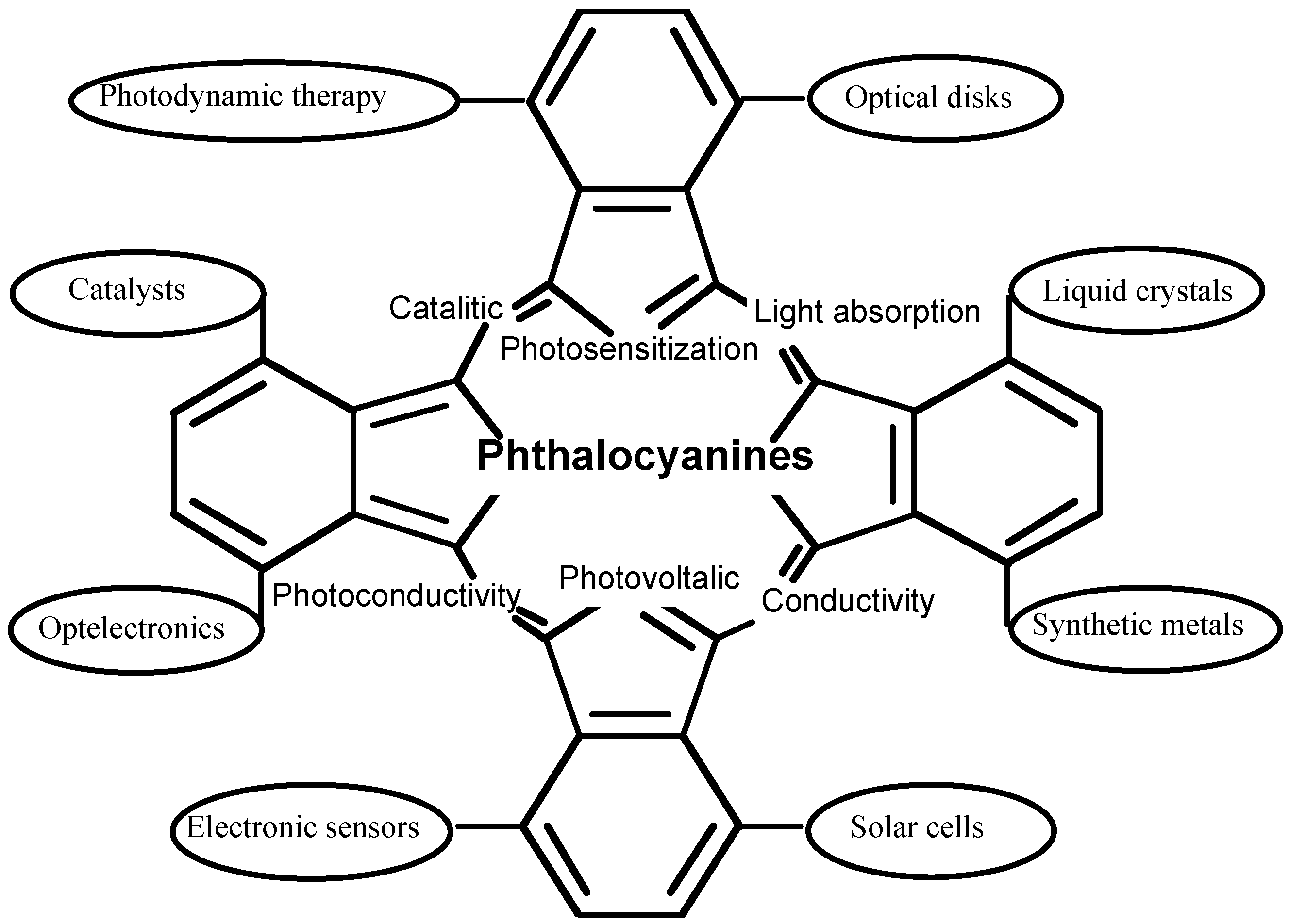
Further, particular derivatives are known to have potential as second-generation photosensitisers for photodynamic therapy (PDT) of cancer [7] because they show strong absorption of the far-red light between the wavelengths of 600 and 850 nm, which has greater tissue penetration properties [8], and satisfactory photosensitization of singlet oxygen [9].
For some applications, the lower solubility of unsubstituted M-PCs can present problems, but low solubility in common organic solvents can be overcome by the introduction of appropriate substituents onto the ring system. In this context M-PC analogues containing a pyridine (Py) ring in place of one or more of the benzenoid rings are particularly interesting compounds because quaternization of the pyridine nitrogen is expected to confer solubility in aqueous media.
Tetrapyridoporphyrazine M-PC analogues in which all four benzenoid rings are replaced by pyridinoid rings were first synthesized by Linstead and his co-workers in 1937 [10]. They obtained an insoluble product from the self-condensation of 3,4-dicyanopyridine which was presumably a mixture of ‘positional isomers’ or regioisomers. Subsequently, Yokote and Shibamiya reported the synthesis and dying properties of some unsubstituted benzopyridopyridoporphyrazines [11,12] and the ring system attracted the attention of other groups, resulting in a substantial increase in the number of known derivatives [13,14]. Yokote and Shibamiya also reported the synthesis of unsubstituted benzopyridoporphyrazines containing a mixture of benzenoid and pyridinoid rings by cross cyclotetramerization of phthalic anhydride and pyridine carboxylic anhydride [15].
For many applications the absorption maxima of M-PCs are best if moved near the infrared region. The strongest absorption of M-PCs in the visible region, the so-called Q band, can be attributed to the allowed highest occupied molecular orbital (HOMO)—lowest unoccupied molecular orbital (LUMO) (π−π*) transition. The Q-band of M-PCs can be moved by bathochromic effects through extension of the π conjugation system such as seen in naphthalocyanines and anthracyanines, but yields of naphthalocyanines and anthracyanines are however usually low. To solve the problem, novel M-PCs having non-peripheral S-aryl substituents have been synthesized. These novel M-PCs will show a high strain structure and no liquid crystal properties.
The authors reported a charge transfer thin film exhibiting organic electroluminescent (EL) properties that was made from an organic varnish including an aniline oligomer as a charge transfer material. Cu-PC is used as a standard charge transfer material in the field of organic EL; it is coated on a surface as a thin film using a suitable dry process such as a vacuum method because of its low solubility. There is a need for Cu-PCs having high heat and light resistance for use in wet processes such as spin-coating, spraying, and ink-jet methods because of the demand in large EL display manufacture. Cu-PCs presents the disadvantage of lower transmittance in the visible region because of their strong absorption maxima Q bands.
In the present paper, we describe the synthesis and electrochemical characteristics of some soluble M-PC derivatives and analogues which are expected to be useful as sensitizers for PDT, in photovoltaic cells and laser printing systems. Syntheses of soluble M-PCs [16], including phthalocyanine-4,4’,4’’,4’’’-tetrasulfonic acids 1 having sulfonic acid groups, phthalocyanine-2,3,9,10,16,17,23,24-octacarboxylic acids 2, 2,3,9,10,16,17,23,24-octakis-(hexoxymethyl)phthalo-cyanines 3 and a novel M-PC derivative, anthraquinocyanine (4), which has four 9,10-anthraquinone units in the molecule, have been reported [16,17]. The authors have measured cyclic voltammograms (CVs) and performed chronocoulometric analyses of 1-4 in order to estimate their electron transfer properties and the corresponding mechanisms.
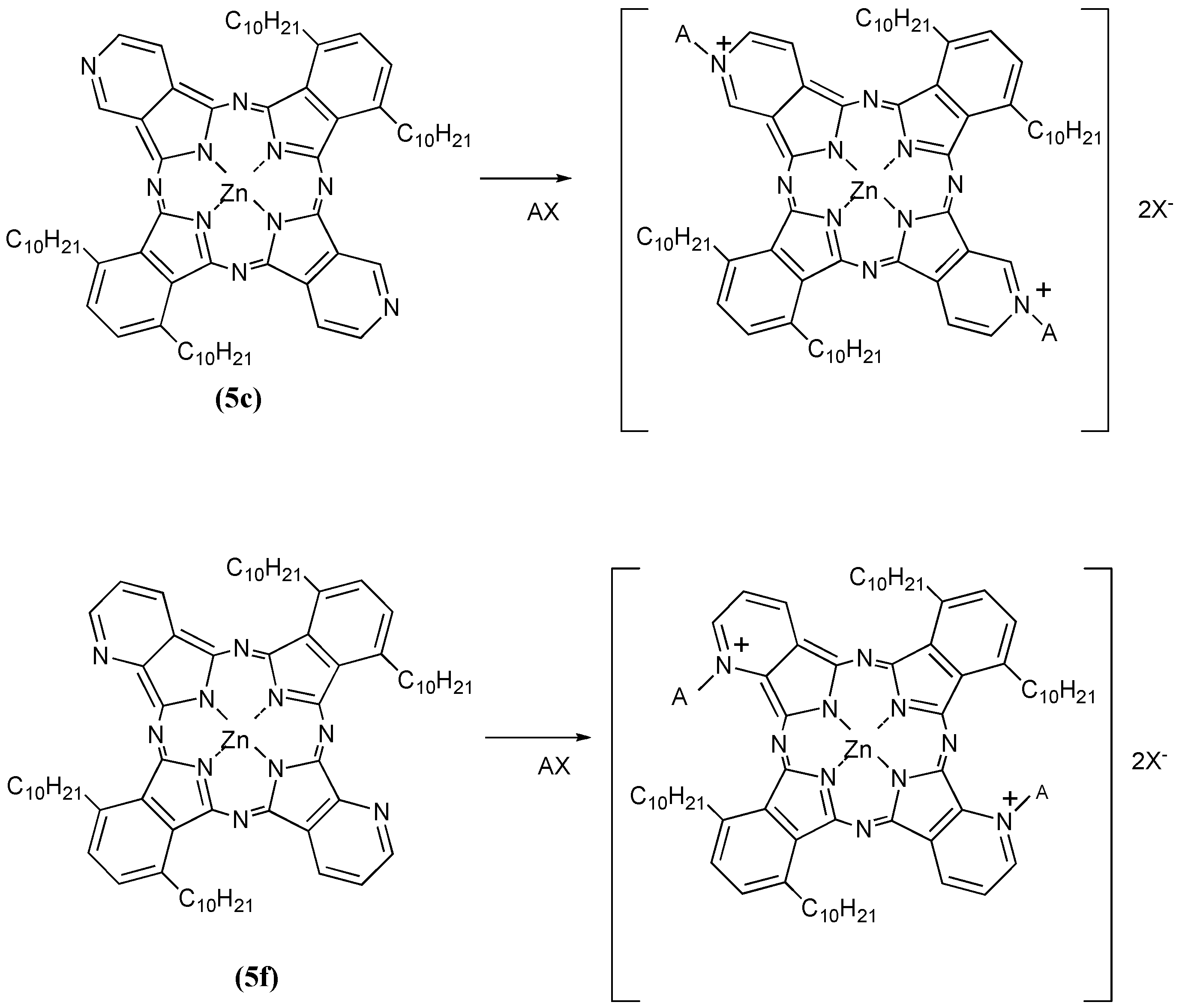
Next, we have reported the preparation and characterization of zinc alkybenzopyridoporphyrazines 5, which were synthesized by reaction of 3,6-didecylphthalonitrile [17,19] and 3,4-dicyanopyridine (or 2,3-dicyanopyridine) [18] in mole ratios of 4:0, 3:1, 1:1, 1:3 and 0:4, respectively. These compounds showed liquid crystal behavior, but incorporation of zinc into the macrocycle inhibited mesophase formation. The 1:1 mole ratio cross cyclotetramerization product has been separated, as the corresponding zinc derivative, with particular attention given to the isolation of the five possible isomers of the zinc bis(1,4-didecylbenzo)-bis(3,4-pyrido)porphyrazine (5c). Then zinc bis(1,4-didecylbenzo)-bis(2,3-pyrido)porphyrazine (5f) was also synthesized. The compounds 5c and 5f exhibit solubility in organic solvents and are expected to have a higher tumor affinity than water soluble M-PCs such as the tetrasulfophthalocyanines. Amphiphilic phthalocyanine derivatives are considered the best compounds for a new generation of photosensitizers for PDT [19] and quaternization of the Py nitrogen in zinc bis(1,4-didecylbenzo)bis(3,4-pyrido)porphyrazine is expected to confer solubility in an aqueous media [15], and lead to good bioavailability and in vivo distribution, thus the quaternization of 5c and 5f would provide amphiphilic M-PCs.
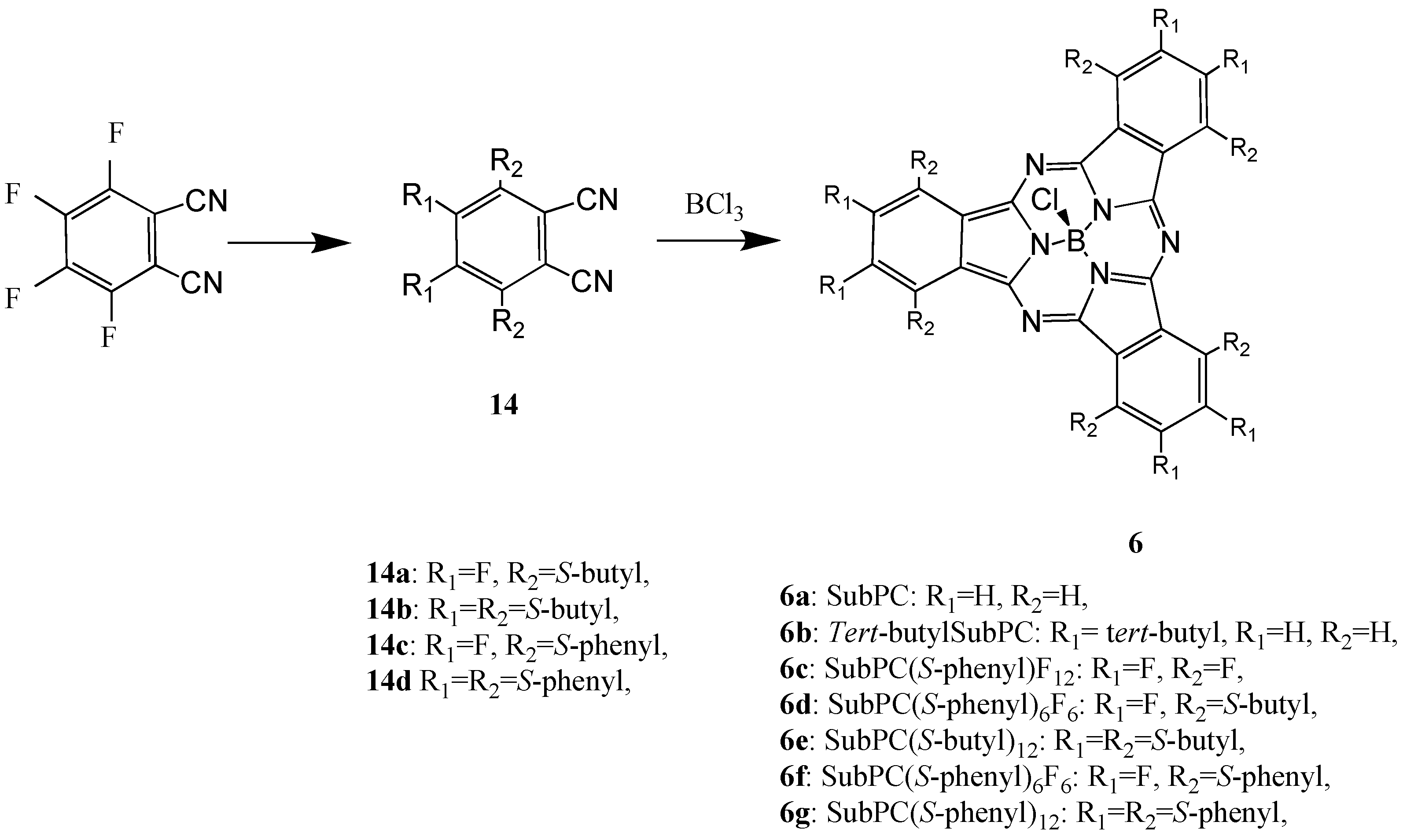
In general, M-PCs have four isoindole units and a central metal. A class of M-PC-related compounds, the subphthalocyanine (SubPC) derivatives 6, are the lowest homologues, consisting of three isoindole units and a central boron. Unsubstituted M-PC derivatives are known to have poor solubility in common organic solvents, whereas SubPC derivatives have excellent solubility since these phenomena are attributed to their non-planar crystal structures [7,22,23].
The SubPC derivatives have previously only been used as reagents for ring enlargement reactions leading to unsymmetric M-PCs. Now, another application of SubPC has been recently reported [24,25]. In order to develop new applications of SubPC, we synthesized SubPC and six of its derivatives: tris-(tert-butyl)subphtalocyaine (tert-butylSubPC; 6b), dodecylfluorosubphthalocyanine (SubPCF12; 6c), hexakis(S-butyl)hexafluoro-subphthalocyanine (SubPC(S-butyl)6F6; 6d), hexakis(S-phenyl)-hexa-fluorosubphthalocyanine (SubPC(S-phenyl)6F6; 6e), dodecylkis(S-butyl)subphthalo-cyanine (SubPC(S-butyl)12; 6f) and dodecylkis(S-phenyl)subphthalocyanine (SubPC(S-phenyl)12; 6g).
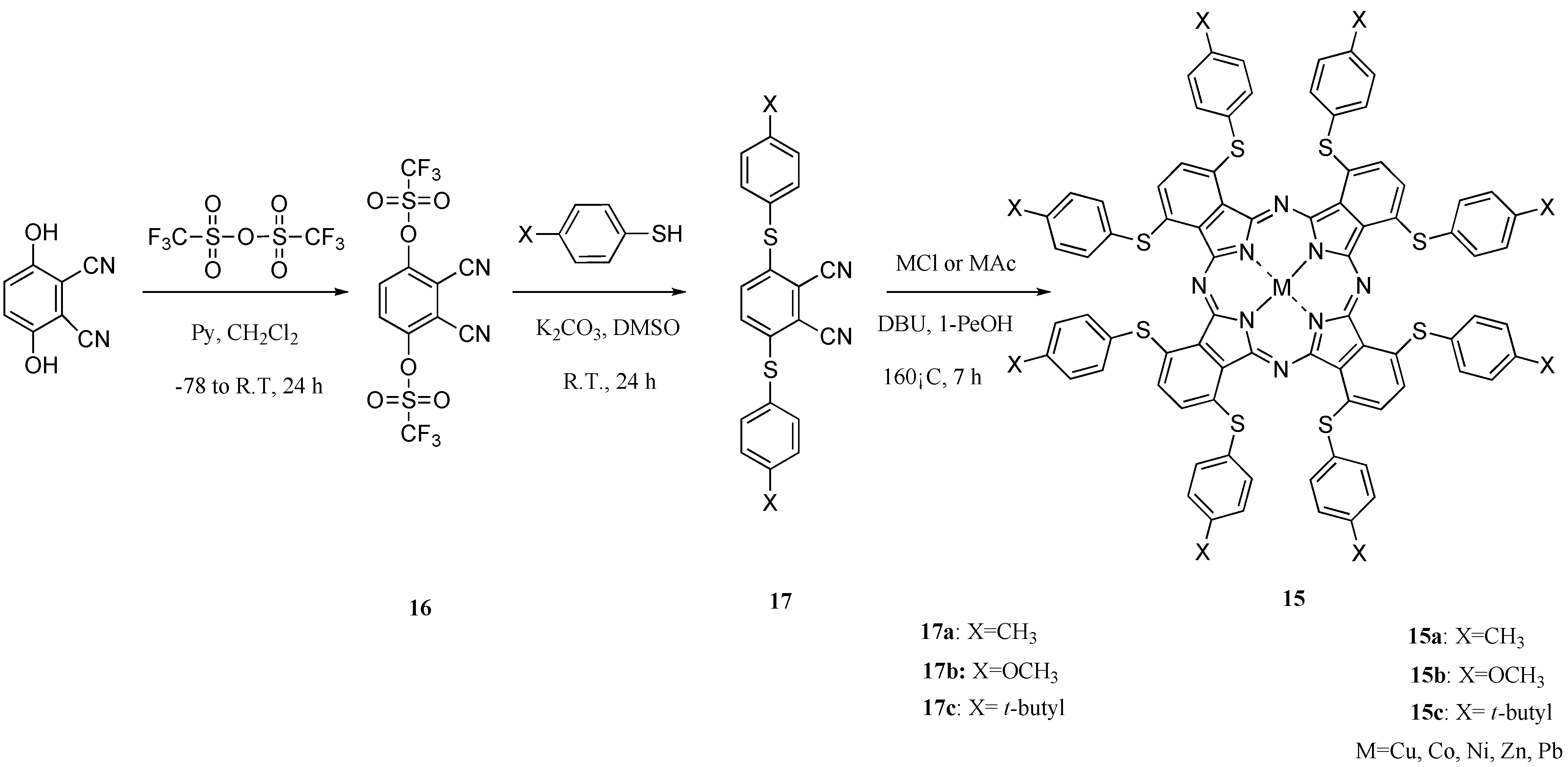
Then, to develop new charge transfer materials, we attempted the synthesis of the 1,4,8,11,15,18,22,25-octakis(thiophenyl)phthalocyanines 15, novel non-peripheral S-aryl substituted M-PCs with linking through sulfur atom electron-donating groups which show near-infrared absorption, high strain structures, no liquid crystal properties and no isomers.
2. Results and Discussion
2.1. Synthesis of phthalocyanines
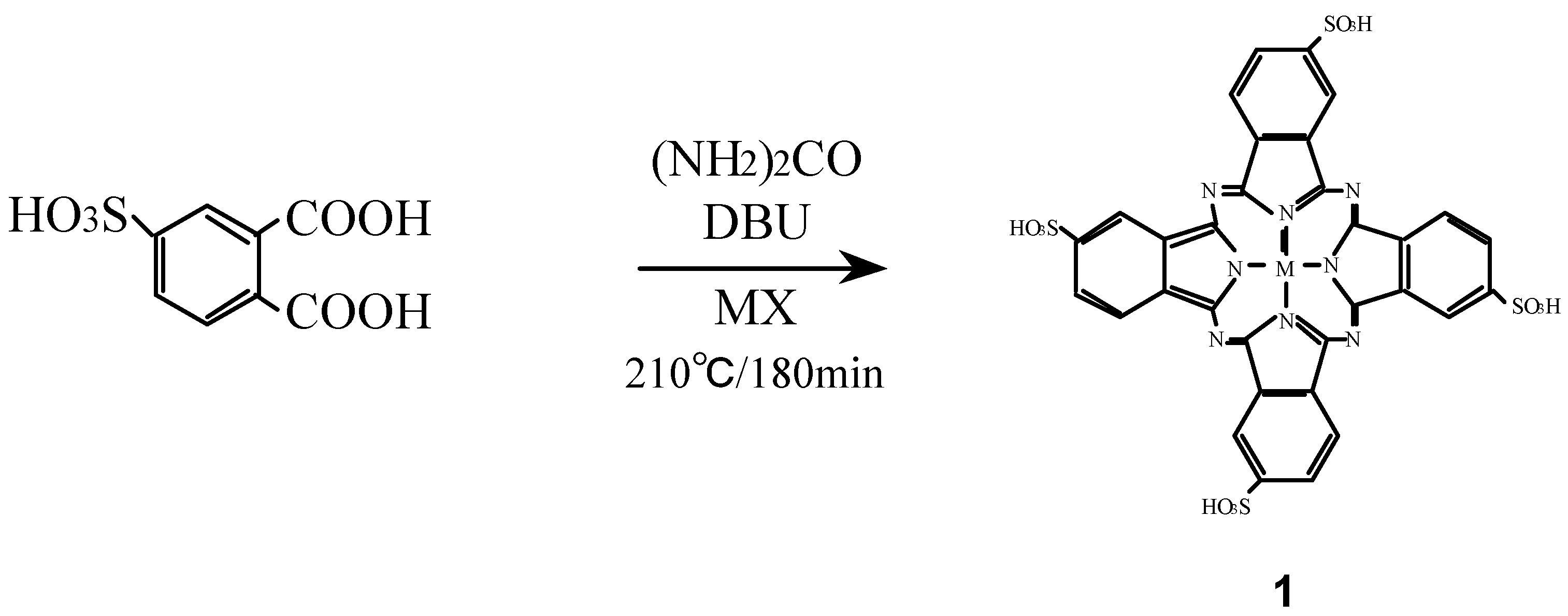
Compounds 1 were synthesized from 4-sulfophthalic acid, a metal halide, urea and 1,8-diazabicyclo-[5,4,0]undec-7-ene (DBU) as a catalyst (Scheme 1) [16,26,27].
Scheme 1.
Synthetic pathway to compounds 1.

The position of the absorption peaks in their infrared (IR) and ultraviolet-visible (UV-Vis) spectra, and elemental analytical data of 1 were all in agreement with the proposed structures and literature data [19,24].
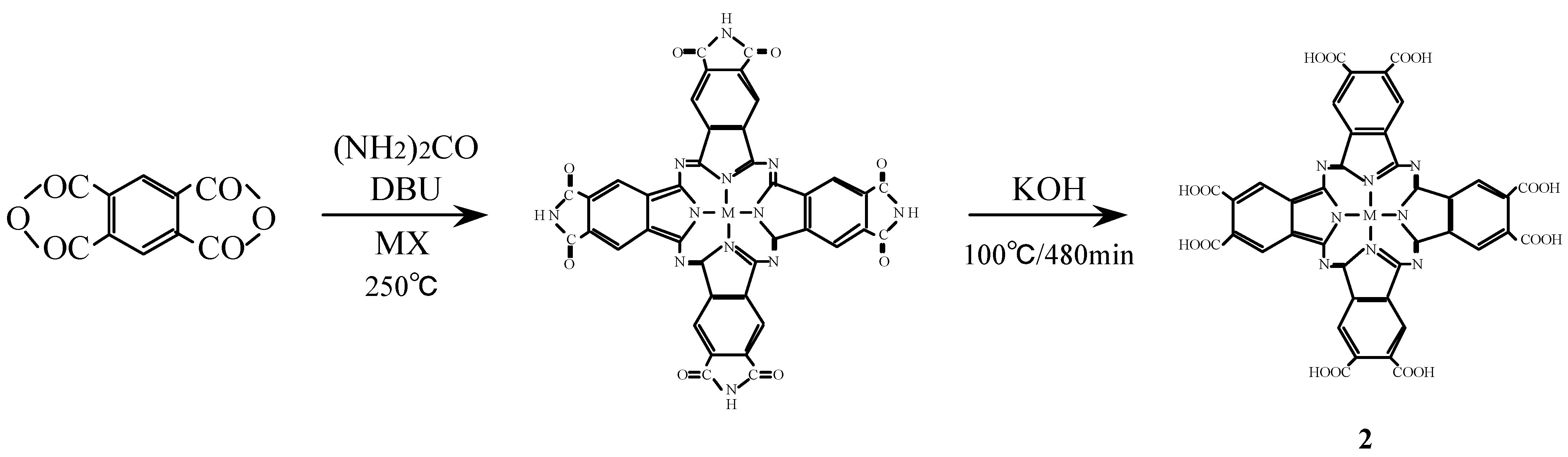
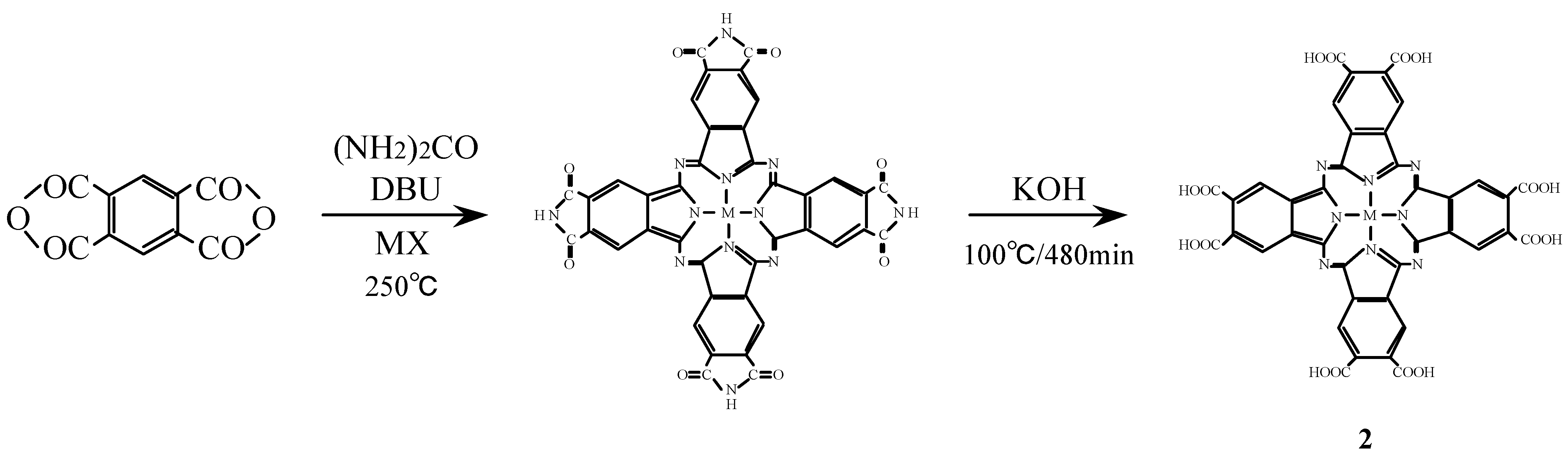
Compounds 2 were synthesized from benzene-1,2,4,5-tetracarboxylic dianhydride (pyromellitic dianhydride), a metal halide and urea under the reaction conditions used for the monomer preparation (Scheme 2) [16,26,27]. The IR spectrum of cobalt octasubstituted phthalocyanine synthesized from pyromellitic dianhydride gave a characteristic imide group pattern in the region from 1,600–1,800 cm-1. The imide functional groups in the synthesized metal octasubstituted phthalocyanine were changed to carboxylic acida by hydrolysis with potassium hydroxide (KOH). The position of the absorption peaks in the IR and UV-Vis spectra and elemental analysis data of 2 are in agreement with the proposed structure and literature data [28,29].
Scheme 2.
Synthetic pathway to compounds 2.
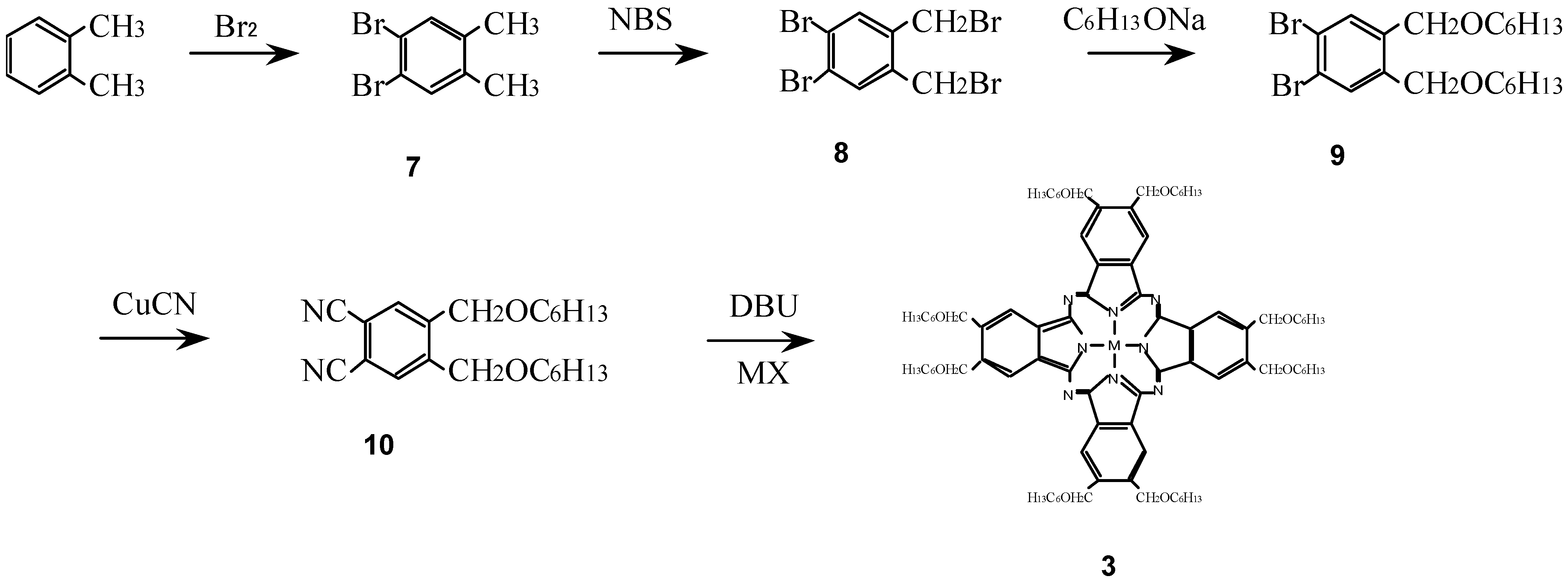
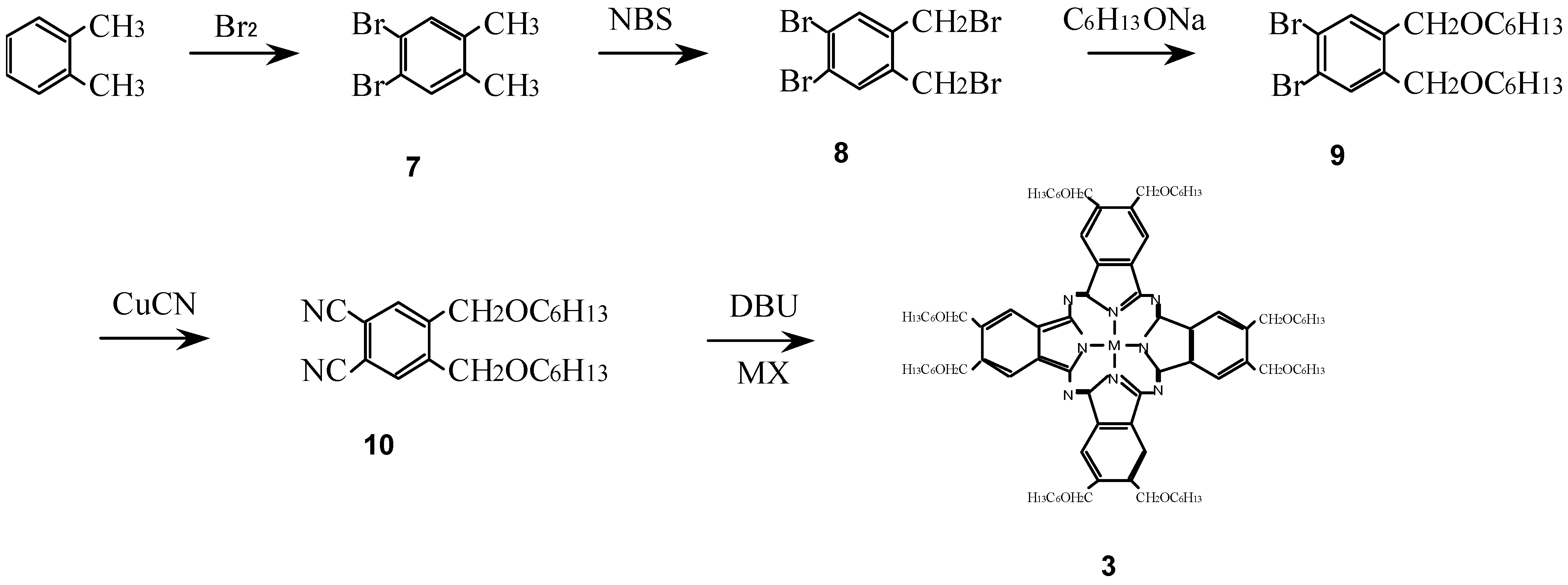
Compounds 3 were synthesized from a metal halide and 1,2-dicyano-4,5-bis(hexoxy-methyl)benzene (10) which was synthesized in turn from o-xylene via 1,2-dibromo-4,5-dimethyl-benzene (7), 1,2-dibromo-4,5-bis(bromomethylbenzene) (8) and 1,2-dibromo-4,5-bis(hexoxymethyl)-benzene (9) (Scheme 3) [16,25,27].
Scheme 3.
Synthetic pathway to compounds 3.
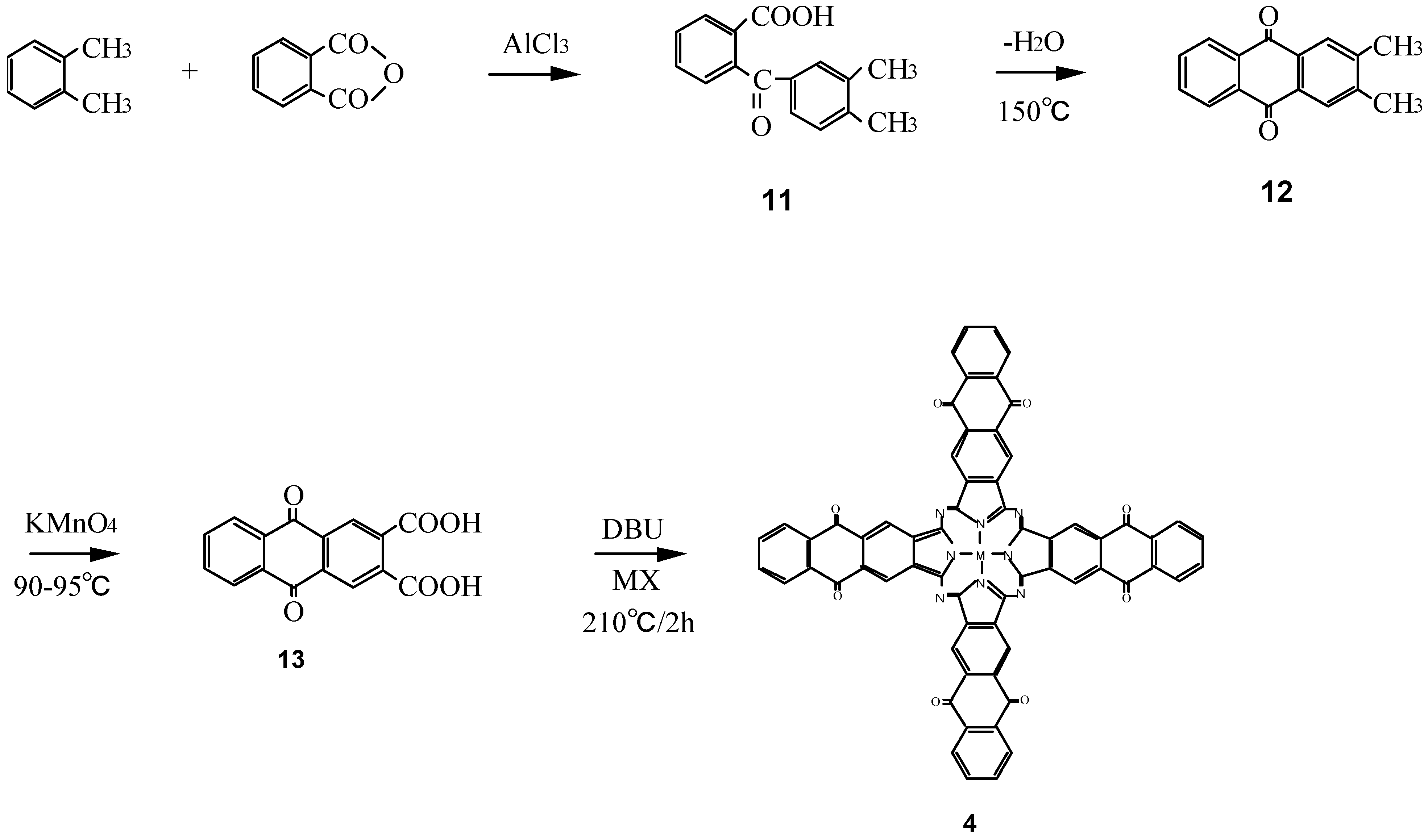
Compounds 4 are a new type of phthalocyanine derivative which was synthesized from 9,10-anthraquinone-2,3-dicarboxylic acid (13), which in turn was prepared from o-xylene and phthalic anhydride via o-(3,4-dimethybenzoyl)benzoic acid (11), 2,3-dimethyl-9,10-anthraquinone (12), and metal halide with urea (Scheme 4) [17]. The intermediates 11 - 13 of 4 have been analyzed by 1H-NMR and IR spectra, and elemental analysis and the analytical data were in good agreement with the proposed structures.
Scheme 4.
Synthetic pathway to compounds 4.

The IR spectra of 4 are similar to those of M-PC derivatives which have heterocyclic rings in the molecule and possess characteristic absorption bands. The formation of 4 was confirmed by the appearance of a pyrrole peak and the product’s IR spectrum and the electron absorption band appearing at 680 nm were consistent with the formation of the anthraquinocyanine ring. Yields of 4-M (M = cobalt, iron and zinc) were largely independent of the kind of central metal used.
The Pariser-Parr-Pople (PPP) self consistent field molecular orbital (MO) method with configuration interaction was employed to calculate the properties of the metal phthalocyanine derivatives synthesized in this work. The PPP-MO calculation was carried out with the programmed software written by Tokita and his co-workers [31].
The calculated electron absorption spectra are shown in Table 1. The Δλobs.-calc. shows the difference between the observed absorption peak λmax value in the Q band which could be attributed to the allowed π - π* transition, and the calculated one. The value of Δλobs.-calc. is in the range of 18–21 nm. The difference in value between the observed and calculated absorption peaks is attributable to the lack of estimation of the central metal in the software.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Calculated absorption spectra /nm | Δλ obs.-calcs. in Q band / nm | |||||
|---|---|---|---|---|---|---|---|
| 1 | 328 | 345 | 394 | 414 | 702 | 809 | 18 |
| 3 | 324 | 430 | 385 | 412 | 686 | 802 | 21 |
| 4 | 322 | 347 | 395 | 414 | 708 | 803 | 20 |
It is confirmed from the PPP-MO calculation that the eight alkoxyalkyl groups in metal octakis(hexoxymethyl)phthalocyanines act as electron-donating substituents, while the eight carbonyl groups in 11 act as electron-withdrawing substituents. Though the PPP-MO method is unable to estimate the effect of the central metal, it is considered that since 3 and 4 were synthesized by this method, the difference between the observed value and the calculated one was within 20 nm, even in the case of 1 and 2. These substituents produce a red shift of the absorption peak in comparison with unsubstituted M-PC.
Cobalt-1, 2, 3 and 4 have the strongest absorption peaks, around the 680 nm region. These strongest absorption peaks are assigned to the Q band, which could be attributed to the allowed π − π∗ transition [26,32]. The Q band absorption of soluble metal phthalocyanine derivatives synthesized in this work was shifted by 50-80 nm to a longer wavelength in comparison with unsubstituted metal phthalocyanines which appeared around 600 nm. The shift of absorption maxima depends upon the change in electron distribution in the phthalocyanine macrocycle by substituents (Table 2).
| Compound | λmax in Q band / nm |
|---|---|
| 1 | 649 |
| 2 | 684 |
| 3 | 665 |
| 4 | 688 |
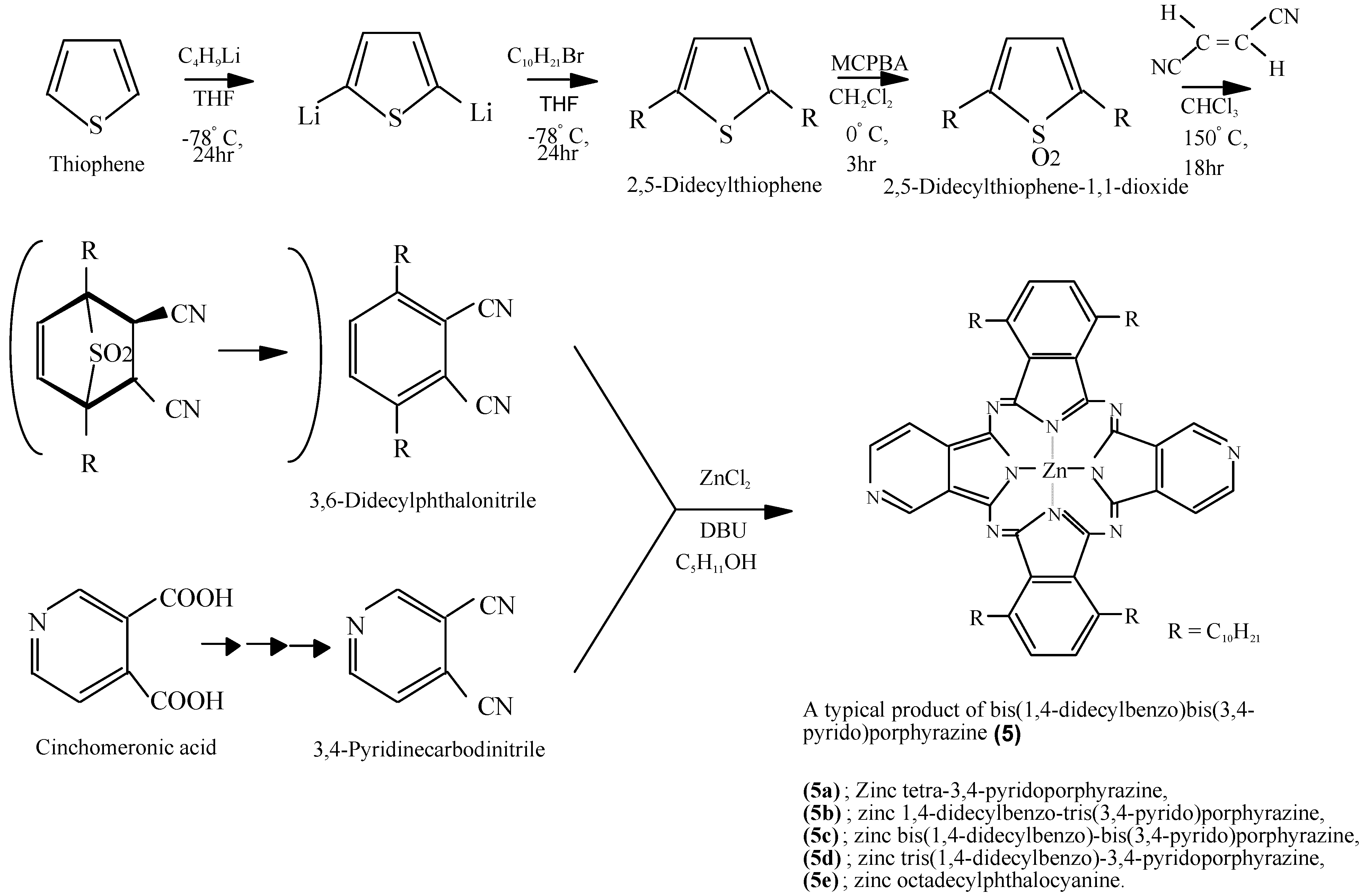
Compounds 5 were synthesized by a multi step process. One raw material, 3,6-didecylphthalonitrile, was synthesized from thiophene in three steps via 2,5-didecylthiophene and 2,5-didecylthiophene-1,1-dioxide. The other raw material, 3,4-pyridine carbodinitrile, was synthesized from cinchomeronic acid in three steps (Scheme 5) [33]. Compound 5f was synthesized from 2,5-didecylthiophene-1,1-dioxide and 2,3-pyridinecarbodinitrile, which prepared from quinolinic acid [34].
The intermediates of 5 were analyzed by IR, 1H-NMR, mass (MS) spectrometry and elemental analysis. The analytical data for all compounds were in good agreement with the proposed structures. Compounds 5 displayed strong absorption peaks around 680 nm for the Q band [18,20,33,34,35,36]. The Q band absorption of 5 shifted by 50-80 nm to a longer wavelength in comparison with unsubstituted phthalocyanines (Table 3).
Scheme 5.
Synthetic pathway to compounds 5.

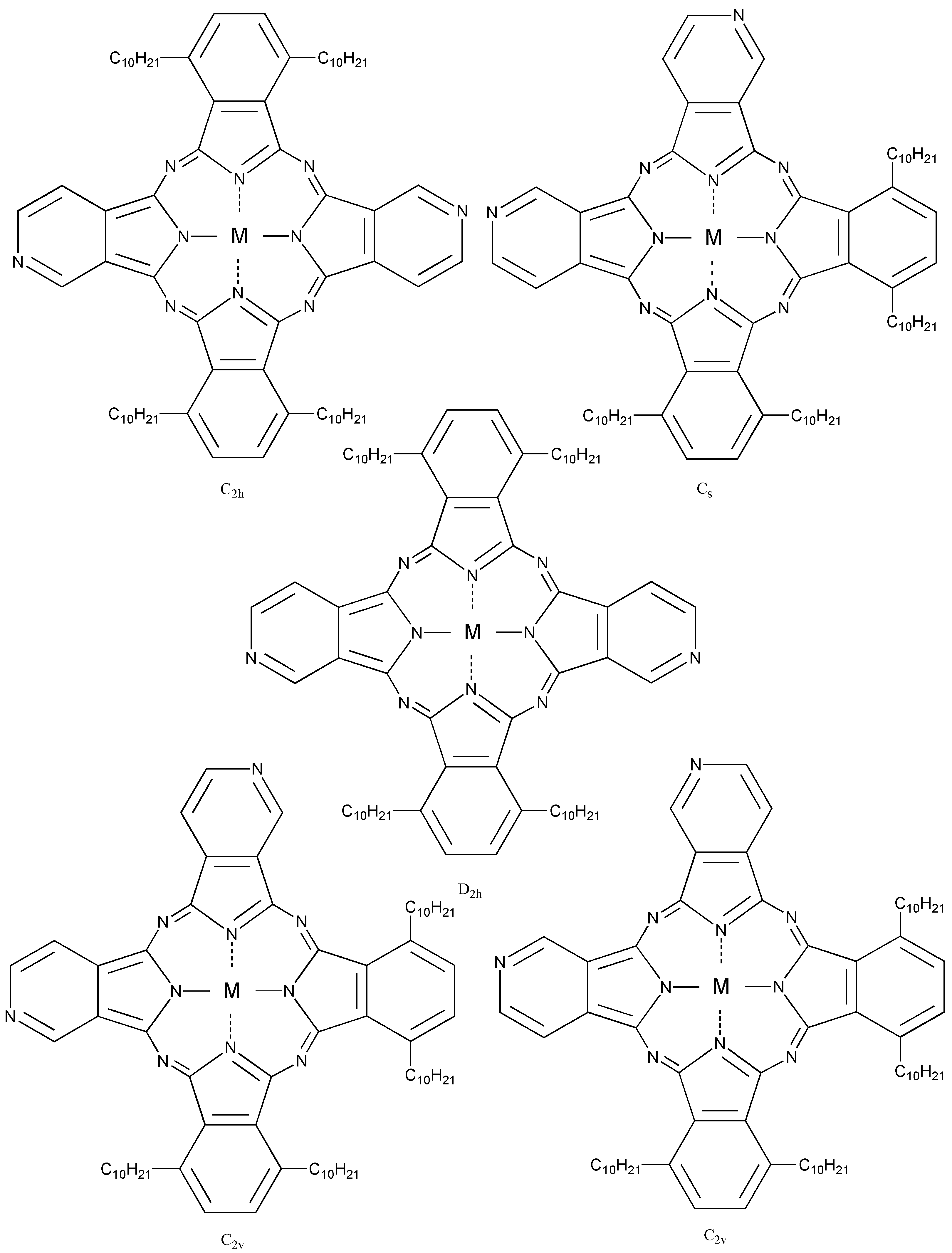
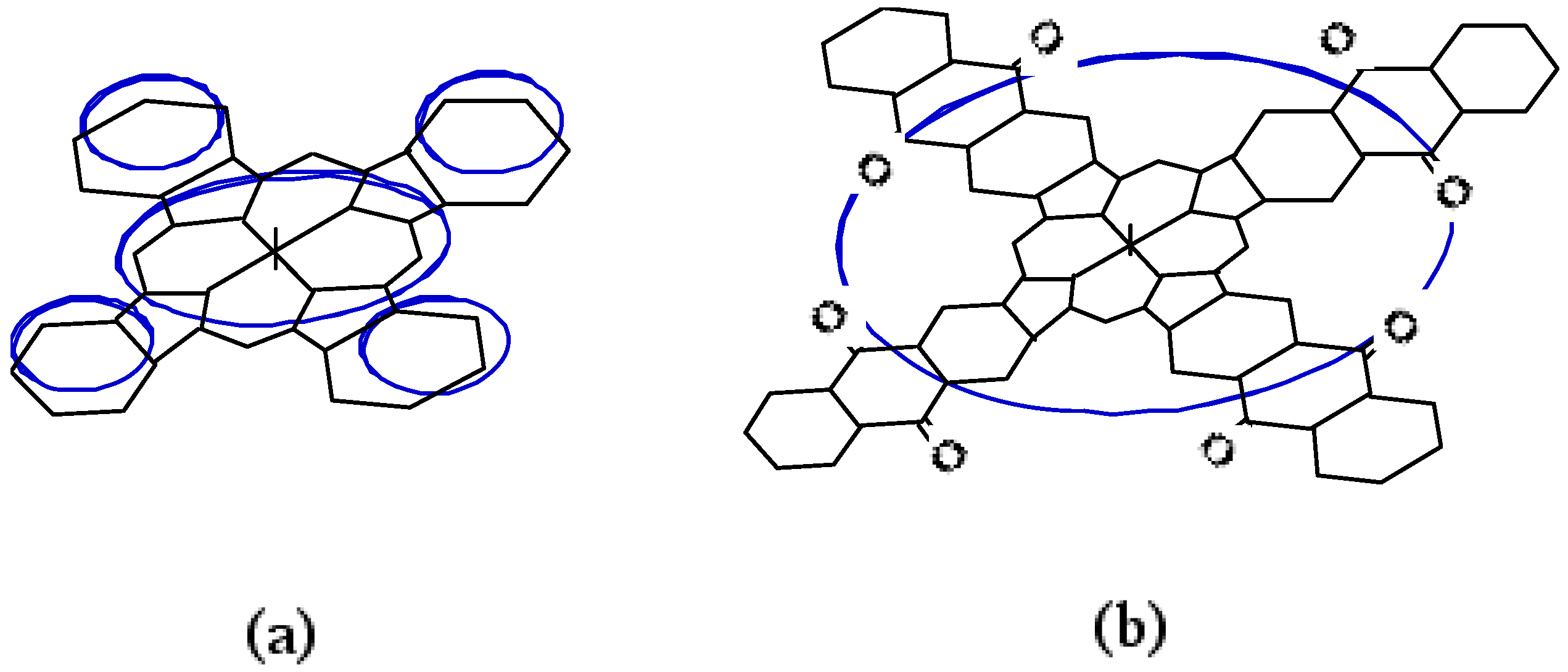
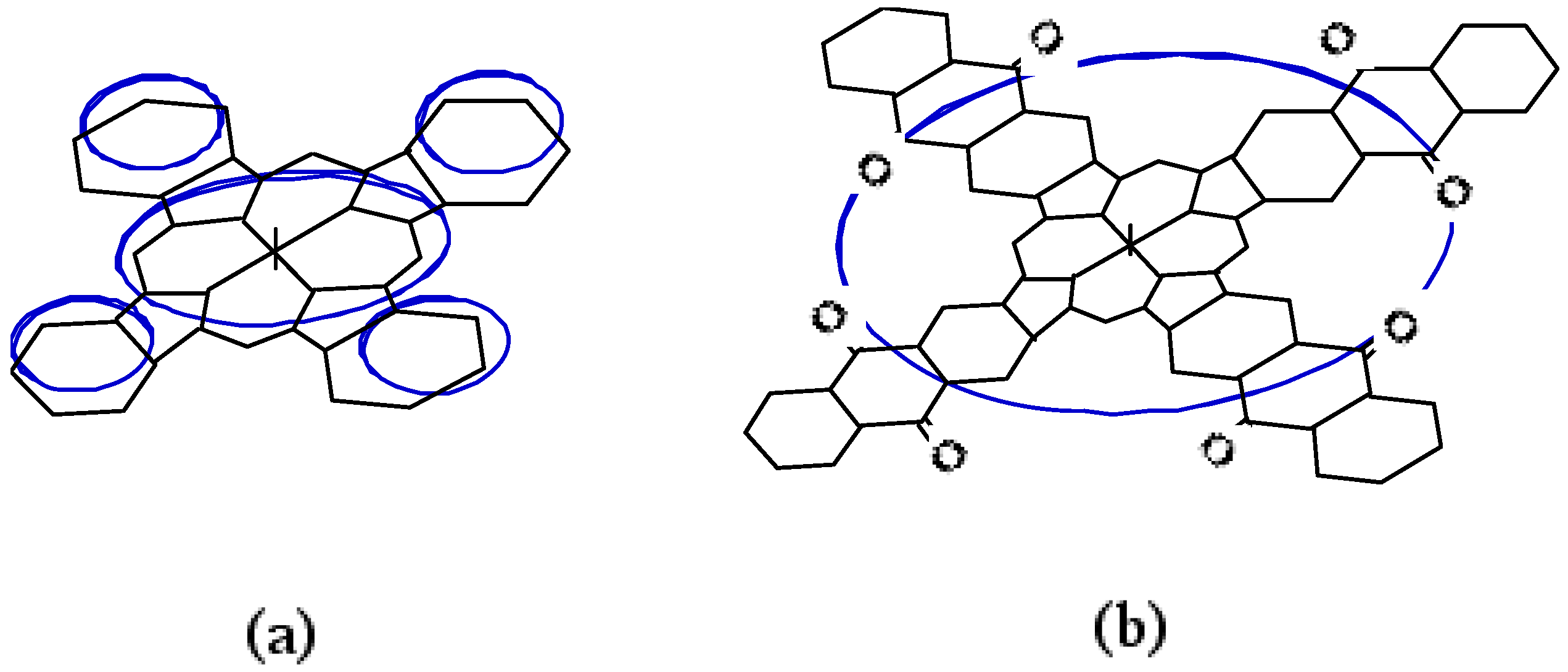
Compound 5c, synthesized from a 1:1 mol ratio of raw materials, has five regioisomers (Figure 2). It has two non-peripheral substituted benzenoid and pyridinoid rings, located at different positions. In the molecule the pyridinoid rings can be adjacent or opposite to each other. In the case of adjacent pyridinoid rings, three types of isomer exist, depending on the orientation of the ring. Meanwhile, two types of isomers exist in the case of opposite pyridinoid rings. We attempted to separate the regioisomers of the compound 5c using thin layer chromatography (TLC) (Merck silica gel 60F254 on aluminum sheet, eluent: toluene - Py, 7:3). Four green- to blue-colored fractions presenting different 1H-NMR, UV-Vis and fluorescent spectra were obtained in 26.1, 17.4, 17.4 and 39.1% yield, respectively, and can be attributed to four of five possible regioisomers of the 5c. These fractions were numbered from 1, 2, 3 and 4, according to the Rf values (0.95, 0.91, 0.75 and 0.65, respectively). Each fraction was recovered by scraping the absorbent from the TLC plate, extracting with Py, filtering and removing the solvent.
| Compound | Q band λmax toluene /nm | ||
|---|---|---|---|
| 5a | 675* | 664* | 603* |
| 5b | 686 | 635 | 617 |
| 5c | 686 | 636 | 717 |
| 5d | 686 | 635 | 617 |
| 5e | 703 | 634 | |
* in pyridine.
Figure 2.
Regioisomers of metal bis(1,4-didecylbenzo)-bis(3,4-pyrido)porphyrazine 5c.

The regioisomers of the 5c were estimated by 1H-NMR, UV-Vis Q band absorption and MO calculations. The MO calculations of the isomers were performed using the ZINDO/S semi-empirical CI configurations available in the HyperChem 5.1Pro software package in order to obtain their theoretical Q band absorptions.
Compounds 5c and 5f were next further reacted with quaternizing agents (AX) such as monochloroacetic acid (MCAA), diethyl sulfate (DES) and dimethyl sulfate (DMS) in N,N-dimethylformamide (DMF) as solvent at 140 °C (Scheme 6).
Scheme 6.
Quaternization reactions of compounds 5c and 5f with quaternizing agents (AX).
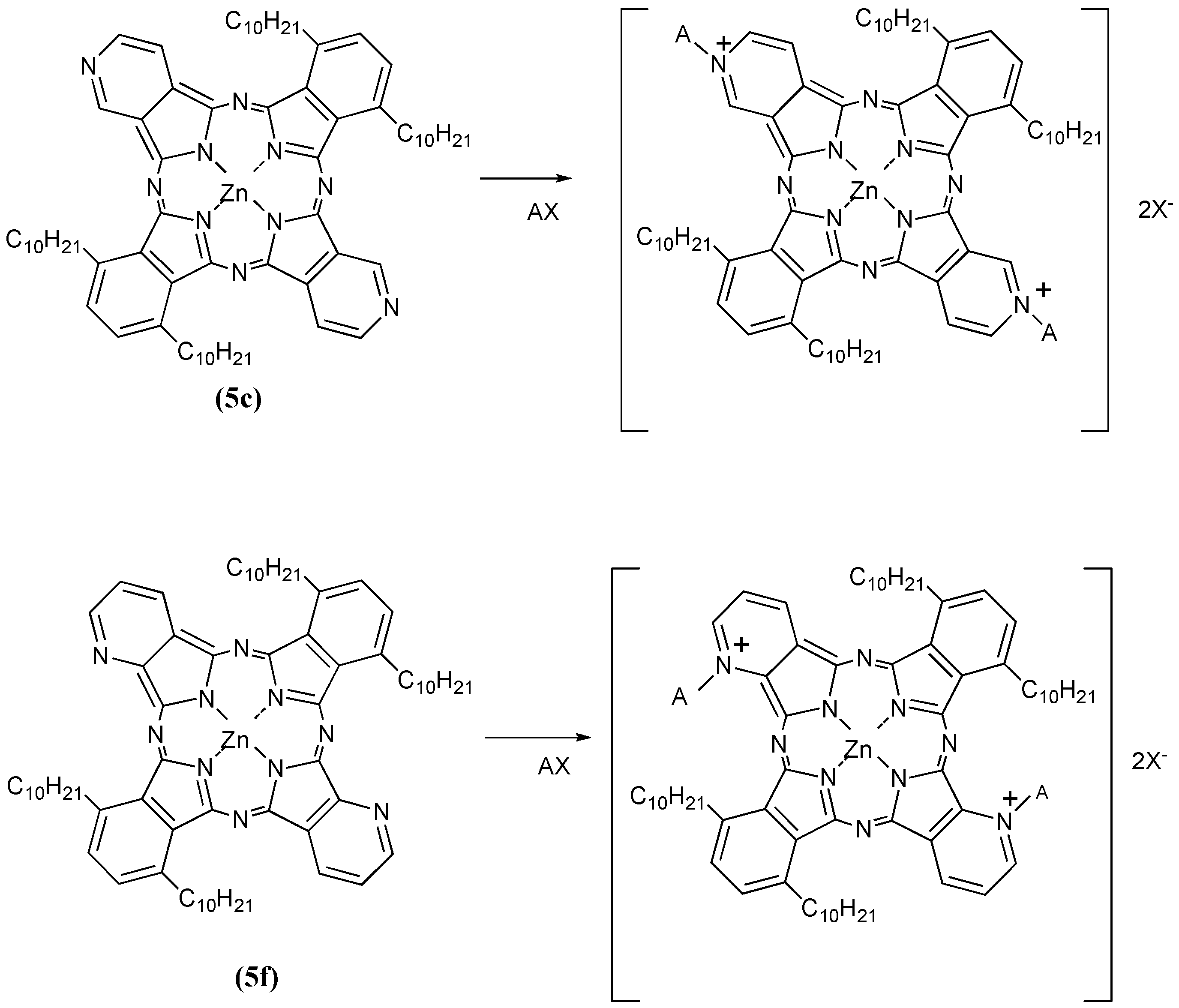
Yields of products of 5c using MCAA, DES and DMS as quaternizing agents were 23%, 17% and 28%, respectively. The corresponding yields of products with the same reagents when 5f was the substrate were 24%, 21% and 25%, respectively. After reaction with the quaternizing agents, the products of 5c and 5f were identified through spectroscopic techniques such as 1H-NMR, IR and UV-Vis spectrometry. When the quaternizing agent was MCAA or DMS, it is confirmed that N-CH2COOH or N-CH3 bonds were obtained on the pyridinoid rings, while when DES was used, we noted that quaternization was not achieved but rather sulfonation occurred. The shapes of the UV-Vis spectra in Py solution changed after quaternatization, and it is thought that interactions between the molecules are complicated when DES and DMS were used as quaternizing agents, since the quaternized products easily aggregate in aqueous media. After reaction with quaternizing agents, all compounds became water soluble, and acquired amphiphilic properties (Table 4).
| Compound | Toluene | Chloroform | Pyridine | Water |
|---|---|---|---|---|
| 5c | Soluble | Soluble | Soluble | Insoluble |
| With MCAA | Soluble | Soluble | Soluble | Soluble |
| With DES | Soluble | Soluble | Soluble | Soluble |
| With DMS | Soluble | Soluble | Soluble | Soluble |
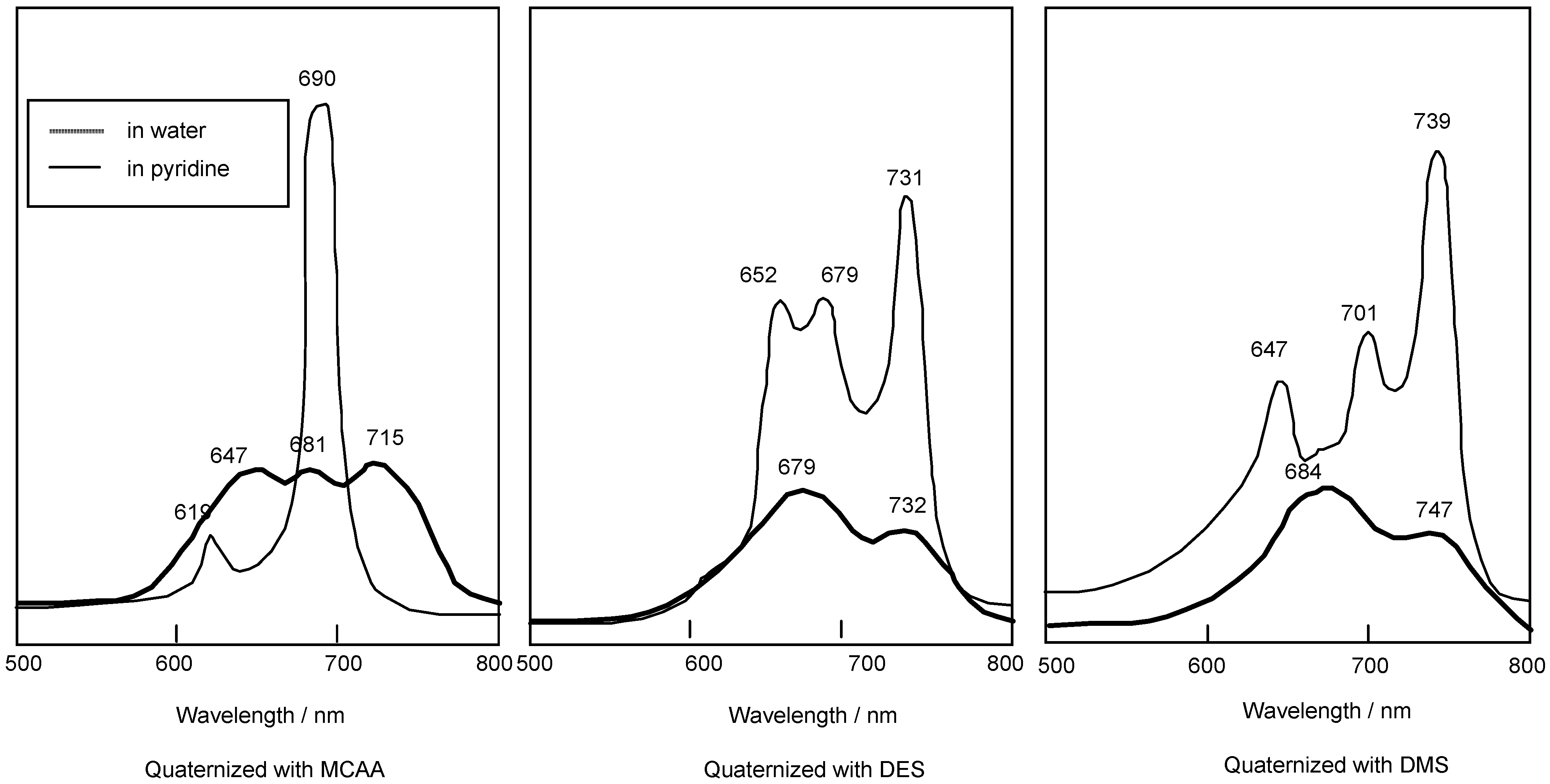
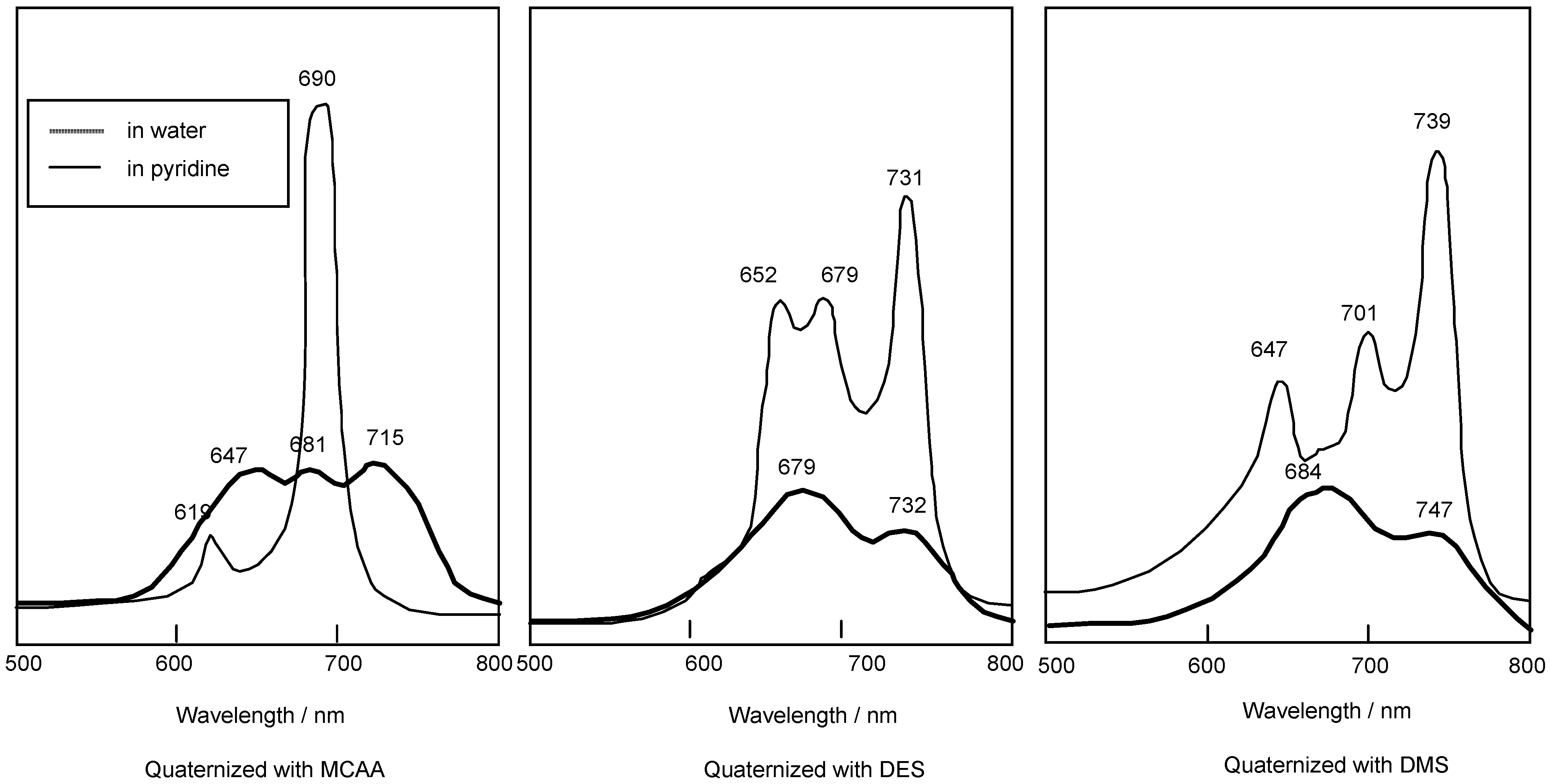
Compounds 5c and 5f are readily soluble in Py, with their strongest absorption appearing at 687 nm (log εmax 4.81). The Q band absorption of the compound quaternized with DMS showed a peak at 739 nm, shifted by 56 nm to longer wavelength in comparison with compounds reacted with MCAA and DES. After reacting with MCAA, DES and DMS, all compounds are very soluble in water, and they show strongest absorptions in the Q band at 715, 731 and 747 nm, respectively (Figure 3).
Figure 3.
UV-Vis spectra of 5c quaternized with MCAA, DES and DMS. The UV-Vis spectra of each quaternized compound was measured in pyridine solution or water.
Figure 3.
UV-Vis spectra of 5c quaternized with MCAA, DES and DMS. The UV-Vis spectra of each quaternized compound was measured in pyridine solution or water.
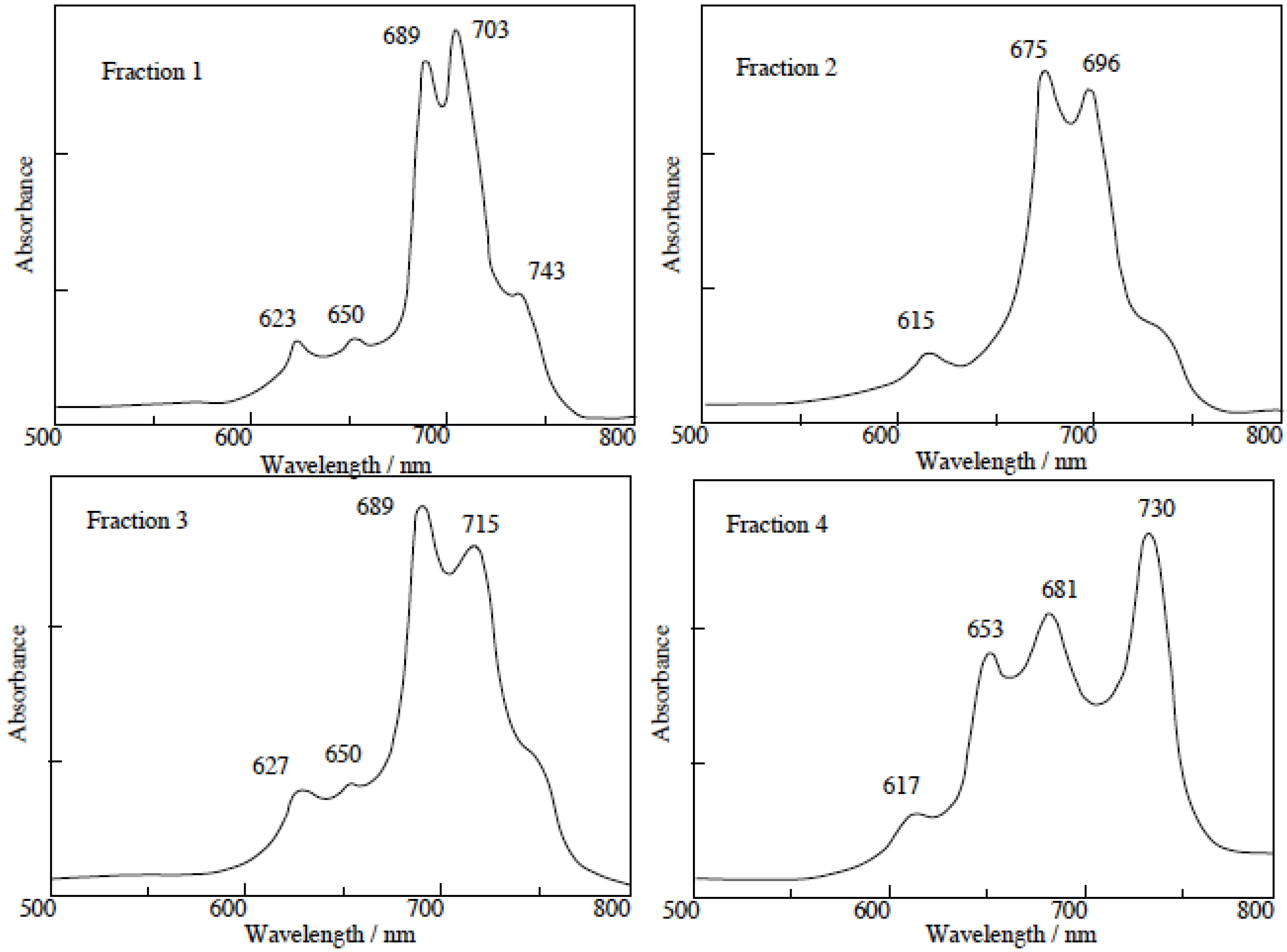
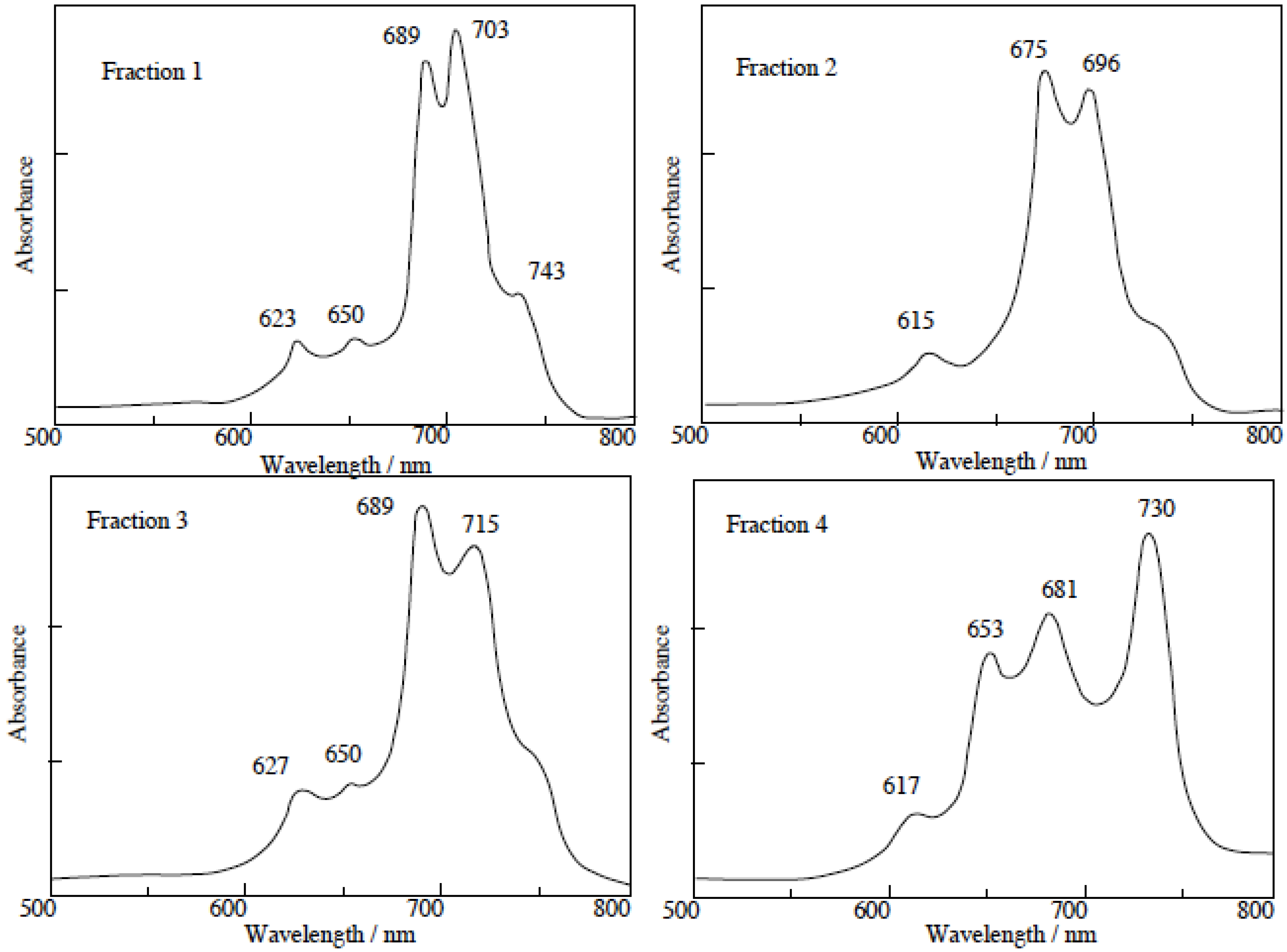
The separated regioisomers of 5c were also quaternized with DMS. Yields of products were 11%, 9%, 15% and 11% for fractions 1, 2, 3 and 4, respectively. Quaternized regioisomers were also identified using spectroscopic techniques such as 1H-NMR, IR, UV-Vis and fluorescence spectra. A comparison of the UV-Vis spectra before and after the quaternization of the regioisomers with DMS, showed that the Q band absorptions of quaternized regioisomers were split into two or more bands. After quaternization, the Q band absorptions were also moved to longer wavelengths (Figure 4).
In general, fluorescence spectra appear as the mirror images of excitation spectra. Excitation spectra show the same absorption band profiles. Fluorescence spectra show longer wavelengths than absorption and excitation spectra. For the quaternized products, except for fractions 1 and 4, the fluorescence spectra showed longer wavelengths than their corresponding absorption spectra.
In fractions 1 and 4, a degree of overlap between absorption and fluorescence spectra was observed. These phenomena result from fluorescence re-absorption or re-emission. Accordingly, the fluorescence spectra of fractions 1 and 4 showed shorter wavelength than the longest wavelength of the absorption bands.
Compound 6a was synthesized from 1,2-dicyanobenzene. Compound 6c was synthesized from 1,2-dicyano-3,4,5,6-tetrafluorobenzene (Scheme 7) [36]. Compounds 6d-g were synthesized from the precursors 1,2-dicyano-3,6-bis(thiobutyl)-4,5-difluorobenzene (14a), 1,2-dicyano-3,4,5,6-tetrakis(thio-butyl)benzene (14b), 1,2-dicyano-3,6-bis(thiophenyl)-4,5-difluorobenzene (14c) and 1,2-dicyano-3,4,5,6-tetrakis(thiophenyl)benzene (14d), respectively.
Figure 4.
UV-Vis spectra of each regioisomer in 5c quaternized with DMS. The UV-Vis spectra of each quaternized compound was measured in Py solution.
Figure 4.
UV-Vis spectra of each regioisomer in 5c quaternized with DMS. The UV-Vis spectra of each quaternized compound was measured in Py solution.
Scheme 7.
Synthetic pathway to compounds 6.
Compounds 6d-g were synthesized from the precursors 1,2-dicyano-3,6-bis(thiobutyl)-4,5-difluoro-benzene (14a), 1,2-dicyano-3,4,5,6-tetrakis(thiobutyl)benzene (14b), 1,2-dicyano-3,6-bis(thiophenyl)-4,5-difluorobenzene (14c) and 1,2-dicyano-3,4,5,6-tetrakis(thiophenyl)benzene (14d), respectively. The precursors of the SubPC derivatives were analyzed and the analytical data were in good agreement with the proposed structures.
The target SubPC and derivatives were analyzed by UV-Vis, IR and 1H-NMR spectroscopy and the analytical data were in good agreement with the proposed structures. Absorption bands in the IR spectra of SubPC and its derivatives are assigned as follows: peaks around 1,600 cm-1 are aromatic ring absorptions; those in the 1,200–1,050 cm-1 region are mainly absorption from out-of-plane bending of CH and NH in pyrrole rings, in which cyclotrimerization is found. These absorption bands in the IR spectrum are characteristic peaks of M-PCs.
The absorption maxima of SubPC and its derivatives appeared around 560 - 630 nm in benzene solution. The Q band absorption of SubPC and its derivatives is shifted by around 100 nm to a shorter wavelength in comparison with normal metal phthalocyanine derivatives, in which they appear around 650 nm. The shift of absorption maxima depends upon the change in electron distribution and the size of the macrocycle. Absorption maxima of SubPC derivatives shifted to longer wavelength with increasing molecular weight. The synthesized SubPC and its derivatives were sufficiently soluble in some organic solvents such as dichloromethane, chloroform, acetone and DMF.
Scheme 8.
Synthetic pathway to compounds 15.
The 1,4,8,11,15,18,22,25-octakis(thiophenyl)phthalocyanine compounds 15 were synthesized in three steps via the intermediates phthalonitrile-3,6-ditriflate (16) and 3,6-bis(thiophenyl)phthalonitriles (17). Intermediate 16 was synthesized from 2,3-dicyanohydroquinon and trifluoromethanesulfonic anhydride for 24 h in accordance with a description from the literature [37]. Intermediates 17 were synthesized, respectively, from 1 and thiophenols such as p-toluenethiol, 4-methoxybenzenethiol and tert-butylthiophenol at room temperature for 24 h to obtain 3,6-bis(thiophenylmethyl)phthalonitrile (17a), 3,6-bis(thiophenylmethoxy)phthalonitrile (17b) and 3,6-bis(thiophenyl tert-butyl)phthalonitrile (17c). Intermediates 16 and 17 were analyzed using IR and 1H-NMR spectroscopy, and elemental analysis. Their analytical data showed good agreement with the proposed structures (Scheme 8).
1,4,8,11,15,18,22,25-octakis(Thiophenylmethyl)phthalocyanines (15a), 1,4,8,11,15,18,22,25-octa-kis(thiophenylmethoxy)phthalocyanines (15b) and 1,4,8,11,15,18,22,25-octakis(thiophenyl tert-butyl)phthalocyanines (15c) were synthesized, respectively, from the corresponding intermediates 17a, 17b, and 17c and metal salt in 1-pentanol in the presence of DBU as catalyst for 7 h (Scheme 1) [38]. As metal salts, chloride or acetate of copper, cobalt, nickel, zinc, and lead were chosen [39]. Metal-free compounds 15 were obtained directly by refluxing 17 in 1-pentanol. The products were isolated using column chromatography on silica gel with toluene as eluent. The most readily apparent feature of the compounds 15 is their solubility in various solvents. The target compounds 15 were analyzed using elemental analysis and MS spectroscopy. The analytical data showed good agreement with the proposed structures.
2.2. Electrochemical Properties
Cyclic voltammetry (CV) is often used in electrochemistry studies. It consists of the cyclic potential of a stationary electrode immersed in a quiescent solution and measuring the resulting current. The excitation signal is a linear potential scan with a triangular waveform. This triangular potential excitation signal sweeps the potential of the working electrode. The triangle returns at the same speed and permits the display of a complete voltammogram. Therefore, if a molecular is reduced in the forward scan, it will be reoxidized on the reverse scan. The current response shows the upper half, cathodic (reduction) and the lower half, an anodic (oxidation) peak.
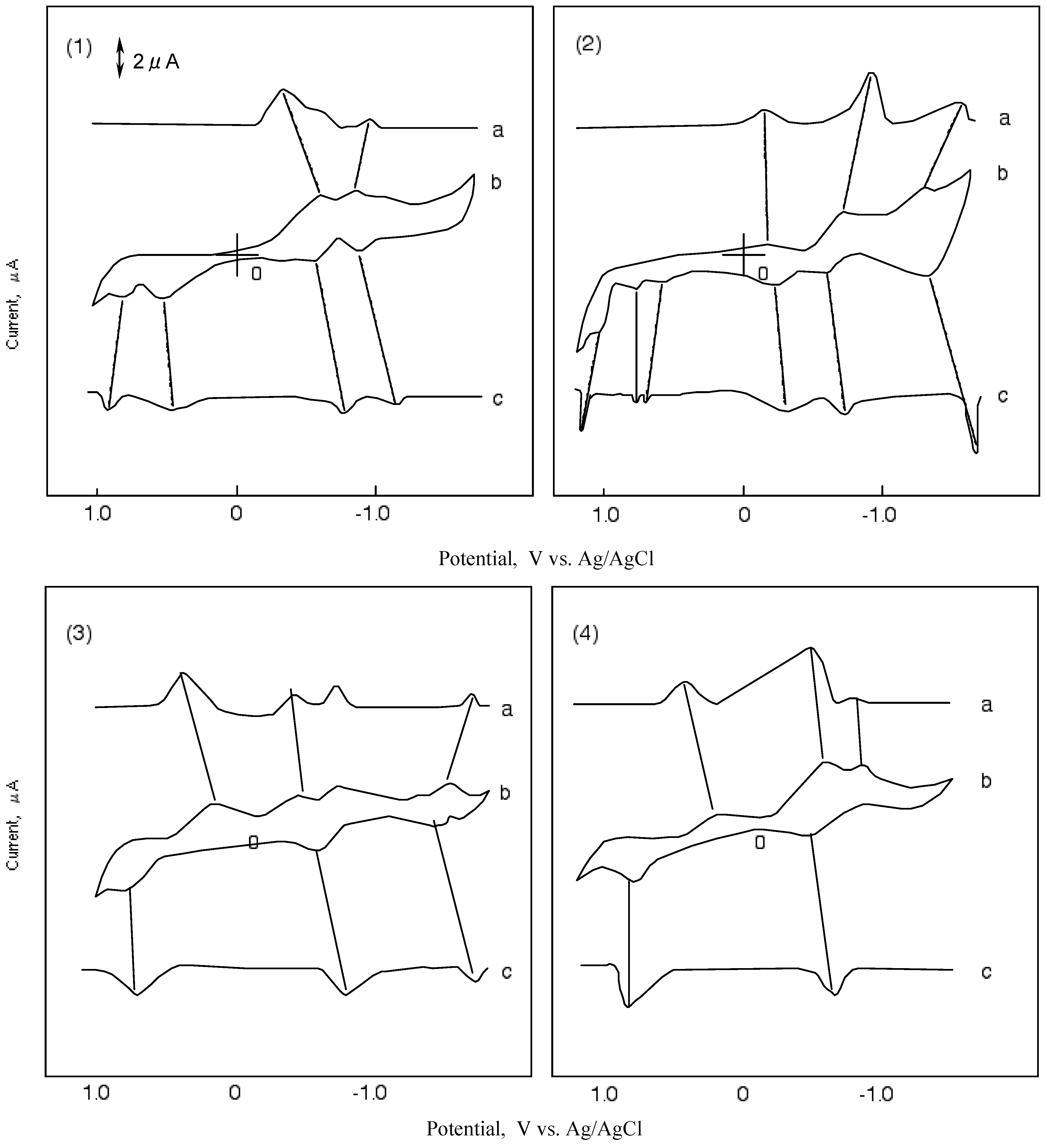
Figure 5 shows CVs and their first differential curves for compounds 1, 2, 3 and 4, respectively. The reduction and oxidation potentials of cobalt-1, 2, 3 and 4 are summarized in Table 5. The reported potentials are the midpoint potential of anodic and cathodic peaks for each couple, Emid, and the peak potential for the irreversible step.
The CV of cobalt-1 showed two cathodic peaks at -0.63 and -1.00 V vs. silver (Ag)/ silver chloride (AgCl) saturated sodium chloride, and four anodic peaks at 0.89, 0.67, -0.60 and-0.90 V vs. Ag/AgCl. The peaks of cobalt-1 are attributed to a five reduction stage. The first reversible reduction potential at -0.62 V vs. Ag/AgCl and the first oxidation potential of cobalt-1 appeared at 0.67 V vs. Ag/AgCl.
In the case of cobalt-2, three cathodic peaks and six anodic peaks appeared. The peaks were sorted into three reversible reduction couples at -0.24, -0.66 and -1.39 V vs. Ag/AgCl, and three irreversible oxidation waves at 0.67, 0.87 and 1.06 V vs. Ag/AgCl. The reduction and oxidation of metal phthalocyanine derivatives are due to the interaction between the phthalocyanine ring and the central metal [40,41]. The reduction and oxidation potential of cobalt phthalocyanine derivatives reported by Rollmann [42] and Orihashi [43] showed more negative values than in the case of cobalt-1 and 2. Carboxylic and sulfonic groups are electron-withdrawing groups so they are expected to reduce the electron charge in the phthalocyanine ring. The change in the redox potential is due to the kind and number of substituents.
Figure 5.
Cyclic voltammograms and their first differential curves of covalt phthalocyanine derivatives in dimethylsulfoxide (DMSO) with 0.1 mol·L-1 tetrabutylammonium perchlorate (TBAP), scan rate: 50 mV·s-1; a): First differential curve of upper half of cyclic voltammogram; b): Cyclic voltammogram; c): First differential curve of lower half of cyclic voltammmogram; (1) Cobalt phthalocyanine tetrasulfonic acid; (2) Cobalt phthalocyanine octacarboxylic acid; (3) Cobalt octakis(hexoxymethyl)-phthalocyanine; (4) Cobalt anthraquinocyanine.
Figure 5.
Cyclic voltammograms and their first differential curves of covalt phthalocyanine derivatives in dimethylsulfoxide (DMSO) with 0.1 mol·L-1 tetrabutylammonium perchlorate (TBAP), scan rate: 50 mV·s-1; a): First differential curve of upper half of cyclic voltammogram; b): Cyclic voltammogram; c): First differential curve of lower half of cyclic voltammmogram; (1) Cobalt phthalocyanine tetrasulfonic acid; (2) Cobalt phthalocyanine octacarboxylic acid; (3) Cobalt octakis(hexoxymethyl)-phthalocyanine; (4) Cobalt anthraquinocyanine.

The CV of cobalt-3 showed four cathodic peaks at 0.16, -0.49, -0.73 and -1.54 V vs. Ag/AgCl, and three anodic peaks at 0.73, -0.61 and -1.47 V vs. Ag/AgCl. Cobalt-3 has two irreversible oxidations at 0.16 and 0.73 V vs.Ag/AgCl, and two pairs of reversible reduction potential. Since the hexyloxymethyl substituent is an electron-donating group, the negative charge on the phthalocyanine ring is expected to increase.
The CV of cobalt-4 showed a unique shape in comparison with the other compounds cobalt-1, 2 and 3. The shape of the CV for cobalt-4 showed three cathodic peaks at 0.19, -0.69 and -0.95 V vs. Ag/AgCl, and two anodic peaks at 0.87 and -0.58 V vs. Ag/AgCl. Cobalt-4 has almost one pair of reversible potential.
The ΔΕ values in Table 5 are the anodic peak to cathodic peak separation located in the oxidation (negative) potential region. The ΔΕ values, except for cobalt-2, are around 100 mV and the redox processes are the same for these molecules.
| Compound | Potential / V vs. Ag/AgCl | ||||||
|---|---|---|---|---|---|---|---|
| Reduction | Oxidation | ||||||
| 1 | -1.63* | -0.95 | -0.62 | 0.67* | 0.89* | ||
| ΔE** | 0.98 | 0.34 | |||||
| 2 | -1.39 | -0.66 | -0.24 | 0.67* | 0.87* | 1.06* | |
| ΔE** | 1.60 | 2.27 | 0.84 | ||||
| 3 | -1.50 | -1.25* | -0.67 | -0.49* | 0.16* | 0.73* | |
| ΔE** | 0.88 | 1.27 | |||||
| 4 | -0.95* | -0.63 | 0.19* | 0.87* | |||
| ΔE** | 1.07 | ||||||
Potentials of reversible wave are midpoint of anodic and cathodic praks for each couple, E1/2.
*Irreversible peak; ** The anodic peak to cathodic peak separation for reversible couple.
Kadish et al. have suggested that the potential difference between the reduction and oxidation is expressed in the HOMO - LUMO energy gap [44]. This potential difference decreases from 1.36 V for cobalt-2 to 0.65 V for cobalt-7. The values of λmax in the Q band shown in Table 5 correlate with the potential difference between the reduction and oxidation of cobalt-1, 2, 3 and 4.
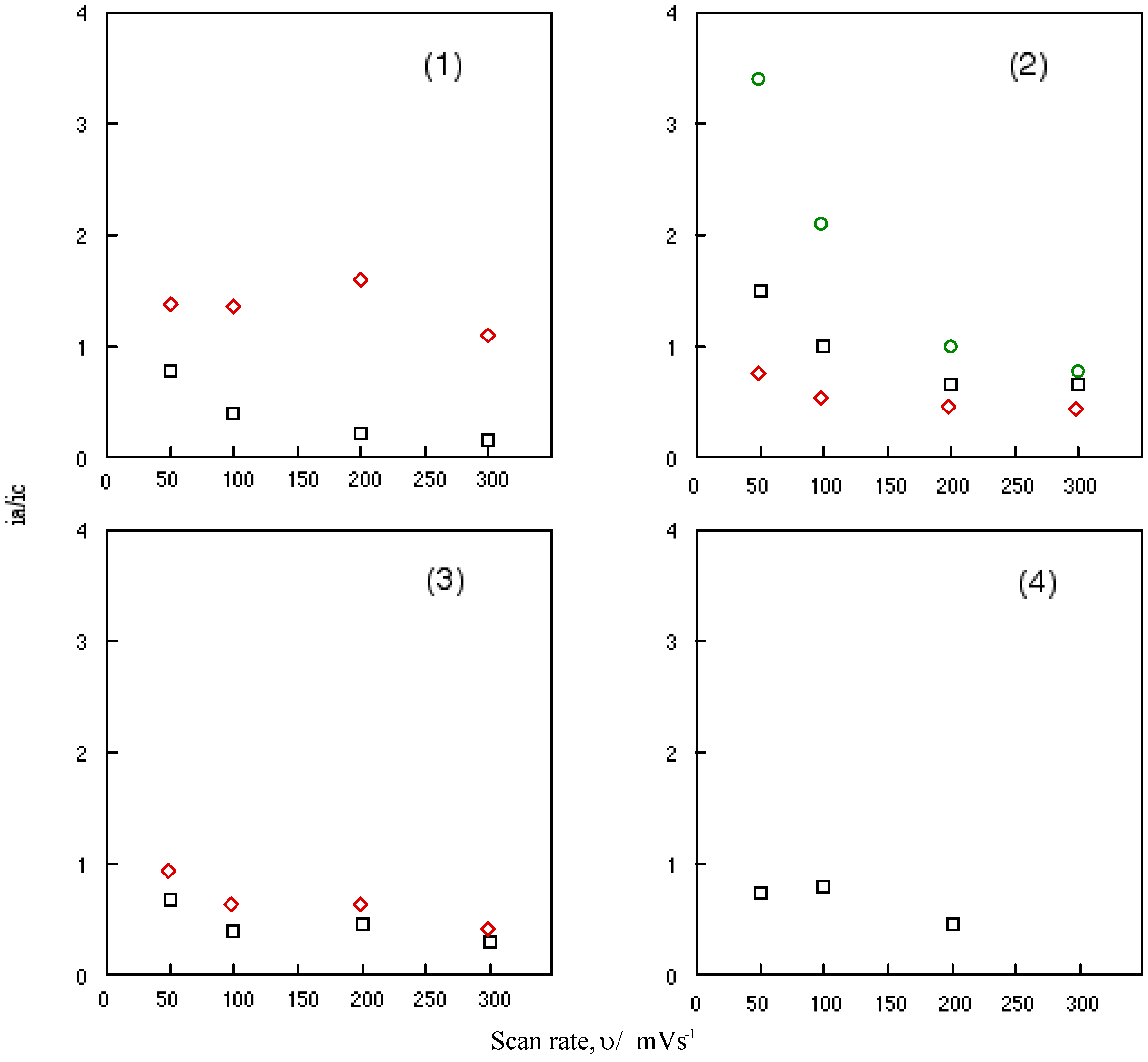
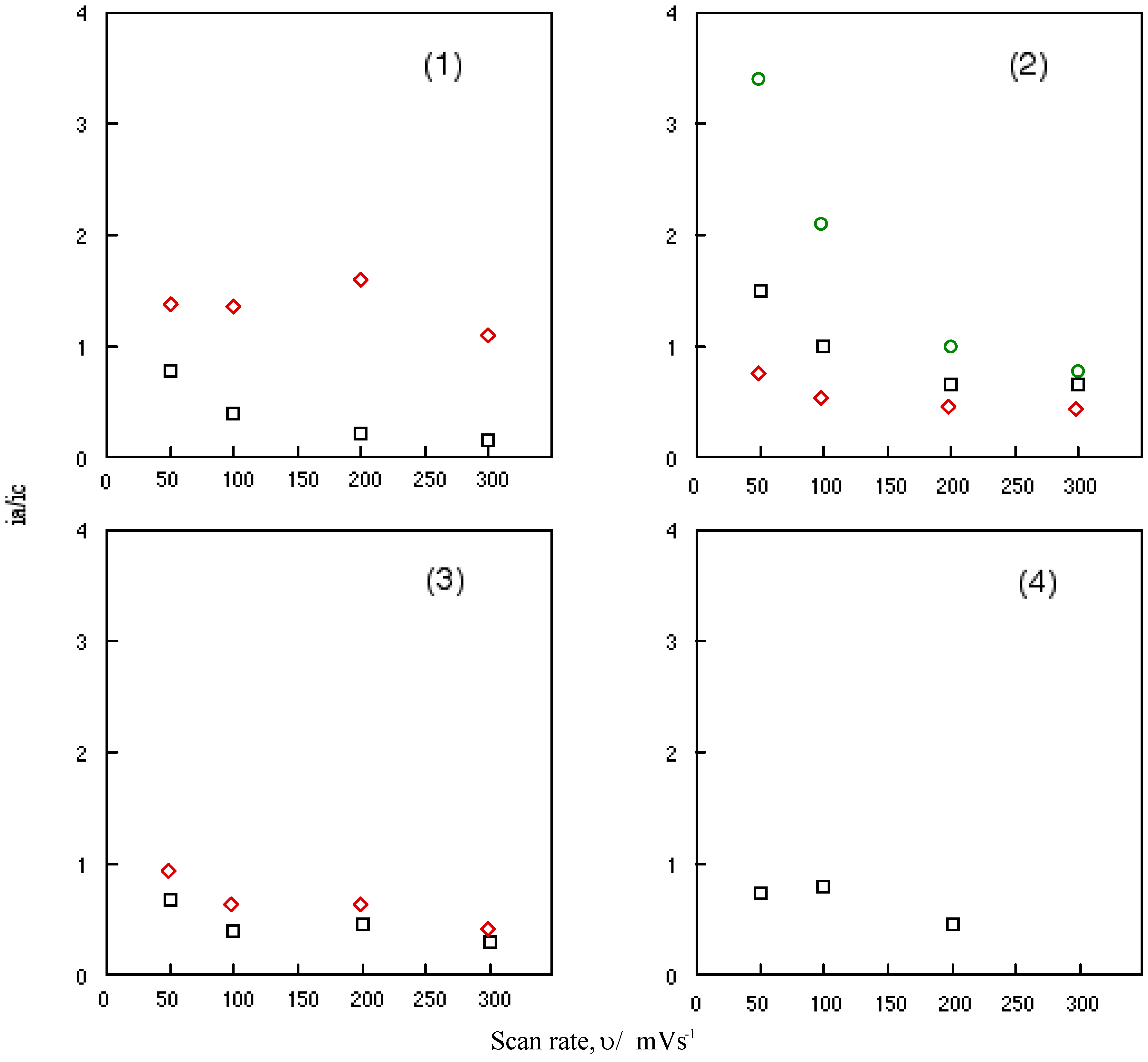
Figure 6 shows the change in the ratio of anodic peak current to the cathodic current of cobalt-1, 2, 3 and 4 with a scan rate, υ. Nicholson and his co-worker have been studied the relationship between the ratio of anodic to cathodic peak current, ia/ic and the scan rate, υ [45]. When the relationship is unity, the system involves a reversible or catalytic reaction. This relationship between the anodic to cathodic peak current ratio of a reversible couple, ia/ic and the scan rate, υ serves as a quick test for electrochemical mechanism associated with a preceding or succeeding reversible or irreversible chemical equilibrium. The scan rate varied from 0.05 to 0.3 Vs-1 in this work.
It is shown that the ratio of anodic to cathodic peak current, ia/ic decreased continuously with an increasing scan rate, υ for all reversible couples of cobalt-1, 2, 3 and 4. The reversible reduction couples of cobalt-1, 2, 3 and 4 are characterized as a fast reversible electron transfer followed by a reversible chemical reaction. The value of anodic to cathodic peak current ratio, ia/ic converges when the ratio is extrapolated to zero of scan rate, υ.
Figure 6.
Change in the anodic to cathodic current ratio with scan rate for cobalt phthalocyanine derivatives. (1) Cobalt phthalocyanine tetrasulfonic acid. (2) Cobalt phthalocyanine octacarboxylic acid. (3) Cobalt octakis(hexoxymethyl)phthalocyanine. (4) Cobalt anthraquinocyanine. □: First redox couple. ◇: Second redox couple. ○: Third redox couple.
Figure 6.
Change in the anodic to cathodic current ratio with scan rate for cobalt phthalocyanine derivatives. (1) Cobalt phthalocyanine tetrasulfonic acid. (2) Cobalt phthalocyanine octacarboxylic acid. (3) Cobalt octakis(hexoxymethyl)phthalocyanine. (4) Cobalt anthraquinocyanine. □: First redox couple. ◇: Second redox couple. ○: Third redox couple.
The potentials of anodic to cathodic peak potential ΔΕ are around 100 mV, except for cobalt-2. Extrapolated to zero scan rate, υ, the ΔΕ values approach close to 60 mV. These data suggest that the electrode processes of cobalt-1, 2, 3 and 4 take place by almost one-electron transfer. The midpoint potential of cathodic to anodic peak, Emid is independent of scan rate, υ and have constant values. Consequently, it is thought that these electrode processes are complicated diffusion-controlled electron transfers involving some weak adsorption with the oxide of cobalt-1, 2, 3 and 4.
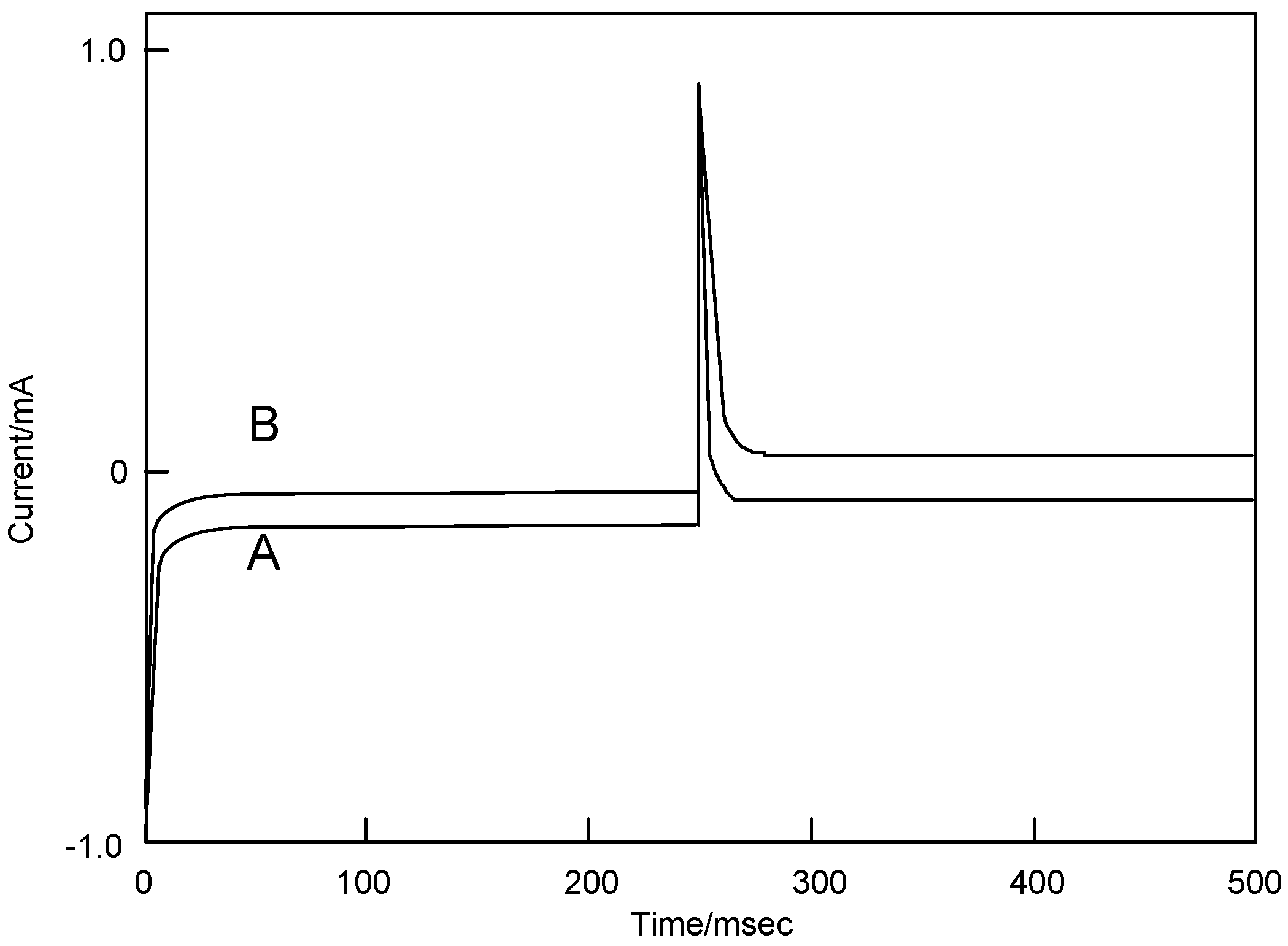
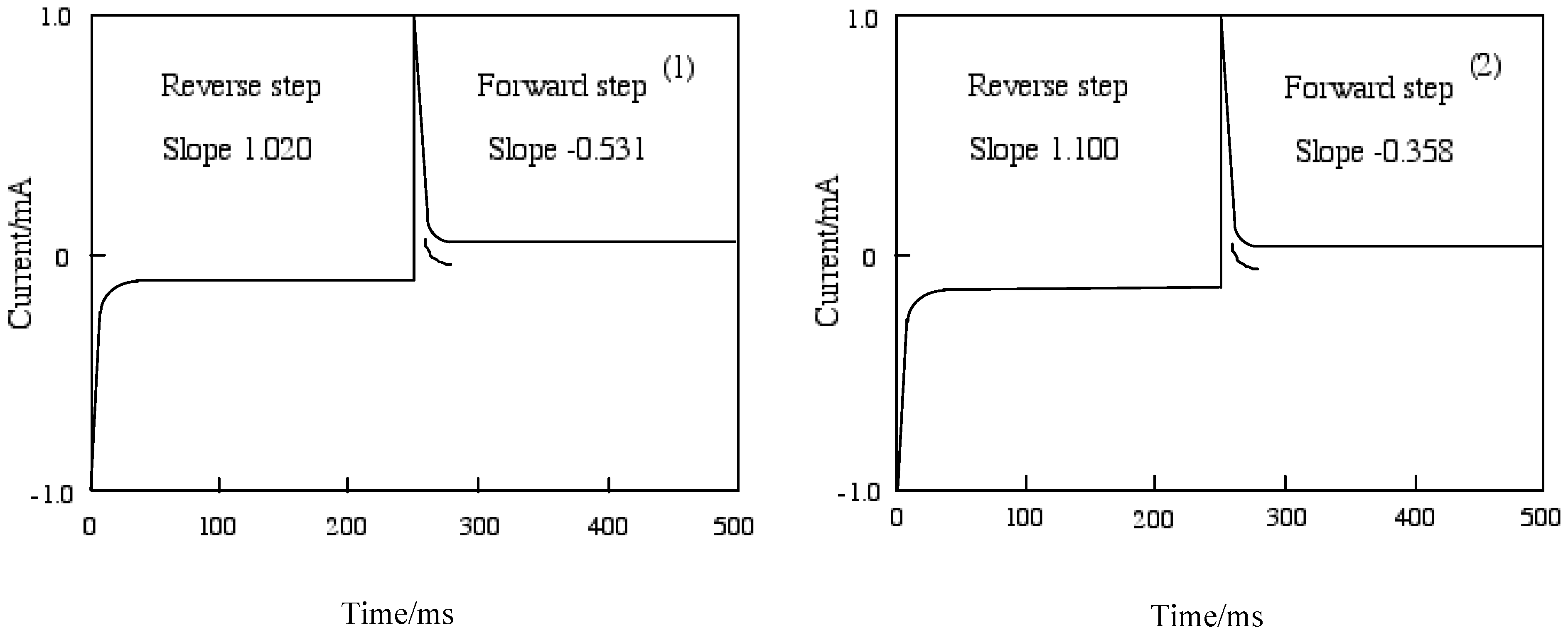
Figure 7 shows chronoamperometry and the slope calculated from Cottrell plots of cobalt-1 and 2. The current of the Cottrell plot is a measurement of the rate for electrolysis at the electrode surface. Electrolysis is controlled with a mass transfer by diffusion on the electrode so that the diffusion constant implies the rate of electrolysis. The slope means the diffusion constant in each step, and the forward step indicates the reduction reaction and the reverse step is oxidation. The oxidation processes of cobalt-1 and 2 are slower than the reduction. The oxidation of cobalt-1 was lower than cobalt-2, while cobalt-2 was lower reduction ability than cobalt-1.
Figure 7.
Chronoamperometry and the slope calculated from Cottrell plot of cobalt phthalocyanine tetrasulfonic acid. Potential step; -1.2–1.6 V vs. Ag/AgCl, time interval: 250 ms. (1) Cobalt phthalocyanine tetrasulfonic acid. (2) Cobalt phthalocyanine octacarboxylic acid.
Figure 7.
Chronoamperometry and the slope calculated from Cottrell plot of cobalt phthalocyanine tetrasulfonic acid. Potential step; -1.2–1.6 V vs. Ag/AgCl, time interval: 250 ms. (1) Cobalt phthalocyanine tetrasulfonic acid. (2) Cobalt phthalocyanine octacarboxylic acid.
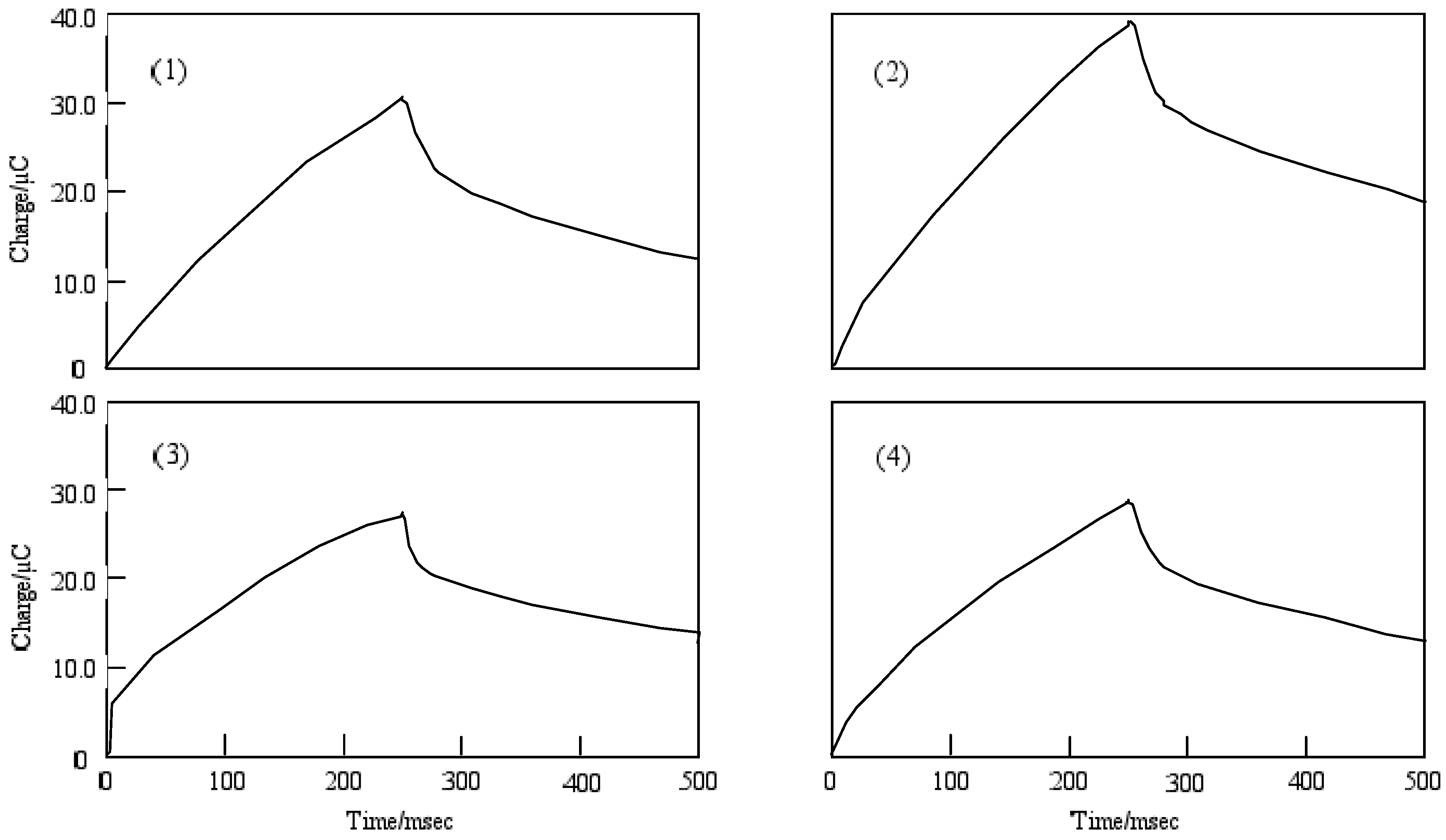
The chronocoulometry was measured by a chronoamperometry treatment in which the current response was integrated to give a response to the charge. The charge-time curve of the forward step for chronocoulometry is the chronoamperometric integral.
The extent of diffusion control increases systematically as the standard potential becomes positive for cobalt-1 and 2. In the case of cobalt-1, the electron charge was reached at about 30 μC in the forward step and was decreased to 15 μC, and reverse steps were attenuated to 0 μC with 70 ms in the reduction side from -1200 to 0 mV vs. Ag/AgCl potential. In the oxidation side from the 0 to +1,600 mV vs. Ag/AgCl step, chronocoulometry had a linear forward step and a flat reverse step indicating no faradic activity for all compounds. It is thought that reduction and oxidation take place in different pathways.
Figure 8 shows the chronoamperometry of cobalt-3 following applied voltage pulse from -1,200 to 0 mV vs. Ag/AgCl and from -1,200 to +1,600 mV vs. Ag/AgCl, and the reversible pulse. Chronoamperometry involves the measurement of the current-time response to a potential step excitation signal. A large cathodic current flows immediately when the potential is stepped up from the initial value, after that it slowly attenuates. The reduction step exhibited the same behavior in comparison with both potential steps.
The current - time curve for chronoamperometry is expressed by the Cottrell Equation (1):
where, i: current (A), n: number of electrons transferred per ion or molecule (mol-1), F: Faraday’s constant (96,485 C/mol), A: electrode area (2.0·10-2 cm2), C: concentration (mol/cm3), D: diffusion constant (cm/s), t: time (s).
Figure 8.
Chronoamperometry of cobalt phthalocyanine tetrasulfonic acid. (A) Potential step; -1.2–0.0 V vs. Ag/AgCl, step width: 250 ms. (B) Potential step; -1.2–1.6 V vs. Ag/AgCl, step width: 250 ms.
Figure 8.
Chronoamperometry of cobalt phthalocyanine tetrasulfonic acid. (A) Potential step; -1.2–0.0 V vs. Ag/AgCl, step width: 250 ms. (B) Potential step; -1.2–1.6 V vs. Ag/AgCl, step width: 250 ms.

Electron processes in the systems are the diffusion-controlled electron transfers mentioned above. Relationships between the current and square root of time are considered to respresent a finite diffusion for cobalt-1, 2, 3 and 4. Table 6 shows the slope of the relationships between the current and square root of time (Cottrell plots). The current of the Cottrell plot is a measure of the rate for electrolysis at the electrode surface. Electrolysis is controlled with a mass transfer by diffusion on the electrode so that the diffusion constant implies the rate of electrolysis. In this table, the slope means the diffusion constant in each step, and the forward step indicates the reduction reaction and the reverse step is oxidation. The oxidation processes of cobalt-1, 2, 3 and 4 are faster than the reduction. The oxidation was decreased in the following order: cobalt-2, 1, 3 and 4. On the other hand, the reduction decreased in the following order: cobalt-3, 1, 2 and 4.
| Compound | Forward step / mA | Reverse step / mA | ||
|---|---|---|---|---|
| Slop | Intercept | Slop | Intercept | |
| 1 | -0.531 | -0.0631 | 1.020 | 0.00596 |
| 2 | -0.358 | -0.106 | 1.100 | -0.00274 |
| 3 | -0.525 | -0.0360 | 0.670 | -0.000172 |
| 4 | -0.322 | -0.0632 | 0.698 | 0.00547 |
Chronocoulometry was measured by a chronoamperometry treatment in which the current response was integrated to give a response of the charge. The charge-time curve of the forward chronocoulometry step for c is the integral of Equation (1):
This is called the Anson Equation (2).
The reverse step is the following equation:
Figure 9 shows the chronocoulometry of cobalt-1, 2, 3 and 4. The initial potential in each case was -1,200 mV vs. Ag/AgCl and the step width was 250 ms. The step potential was +1,600 mV vs. Ag/AgCl.
Figure 9.
Chronocoulometry of cobalt phthalocyanine tetrasulfonic acid. Potential step; −1.2–1.6 V vs. Ag/AgCl, time interval: 250 ms. (1) Cobalt phthalocyanine tetrasulfonic acid. (2) Cobalt phthalocyanine octacarboxylic acid. (3) Cobalt octakis(hexoxy-methyl)phthalocyanine. (4) Cobalt antraquinocyanine.
Figure 9.
Chronocoulometry of cobalt phthalocyanine tetrasulfonic acid. Potential step; −1.2–1.6 V vs. Ag/AgCl, time interval: 250 ms. (1) Cobalt phthalocyanine tetrasulfonic acid. (2) Cobalt phthalocyanine octacarboxylic acid. (3) Cobalt octakis(hexoxy-methyl)phthalocyanine. (4) Cobalt antraquinocyanine.

For cobalt-1, 2, 3 and 4, the extent of diffusion control increases systematically as the standard potential becomes positive. The electron charge was reached at about 30 μC in the forward step and was decreased to 15 μC, except for cobalt-2. In the chronocoulometry of the reduction side from -1,200 to 0 mV vs. Ag/AgCl potential, reverse steps were attenuated to 0 μC with 70 ms, except for cobalt-2. In the oxidation side from the 0 to +1600 mV vs. Ag/AgCl step, chronocoulometry had a linear forward step and a flat reverse step indicating no faradic activity for all compounds.
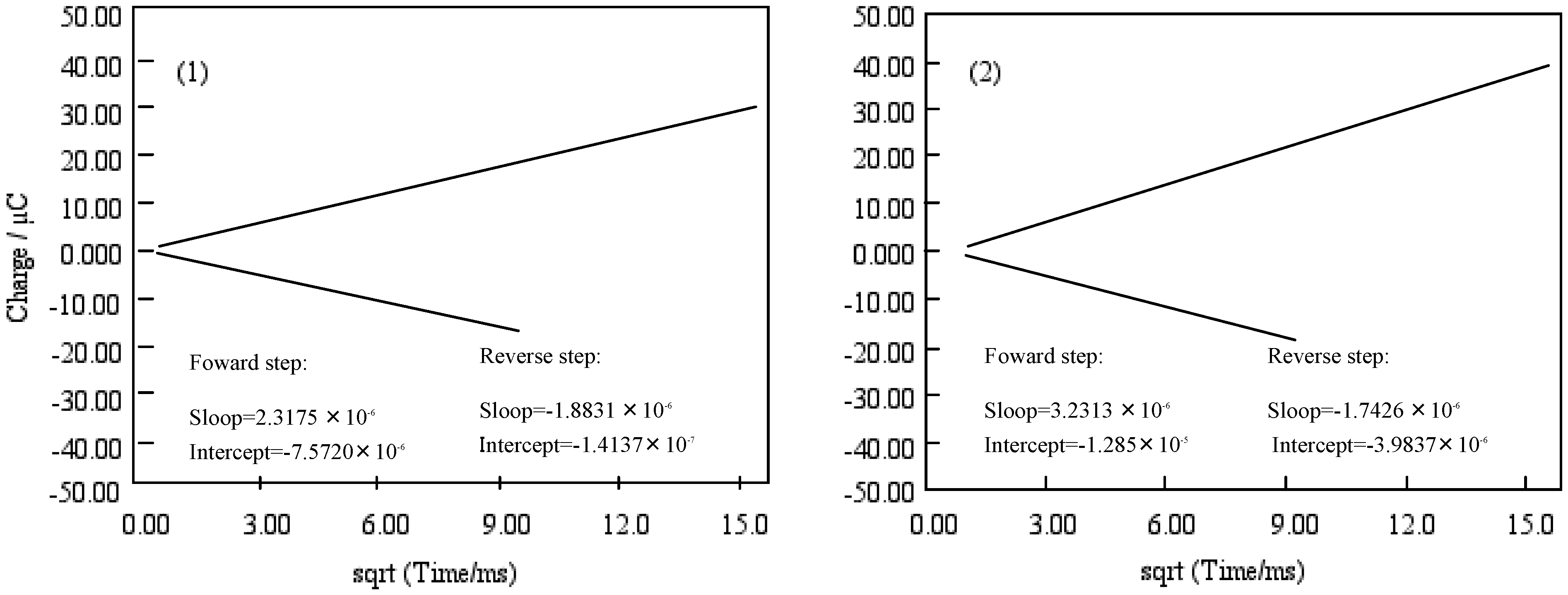
Figure 10 shows Anson plots, which are converted from the charge-time curve of chronocoulometry into the relation between charge and square root of time. Only the value of solution species (Q) in three terms depends upon the scanning time. The intercept of the Anson plot expresses the sum of a double layer charging (Qdl) and electrolysis of adsorbed (Qads). Since double step chronocoulometry is used in this work, Qads can take away Qdl which is a value of the difference of intercepts between forward and reverse steps. When no adsorption of reactant or product, the intercept of Anson plot for both forward and reverse steps are Qdl. While reactant adsorbs but product does not, the intercept of the reverse is a measure of Qdl in the presence of adsorbed reactant, and the intercept of the forward step contains both Qdl and Qads for adsorbed reactant.
Figure 10.
Anson plots of cobalt phthalocyanine tetrasulfonic acid (1) and cobalt phthalocyanine octacarboxylic acid (2).
Figure 10.
Anson plots of cobalt phthalocyanine tetrasulfonic acid (1) and cobalt phthalocyanine octacarboxylic acid (2).
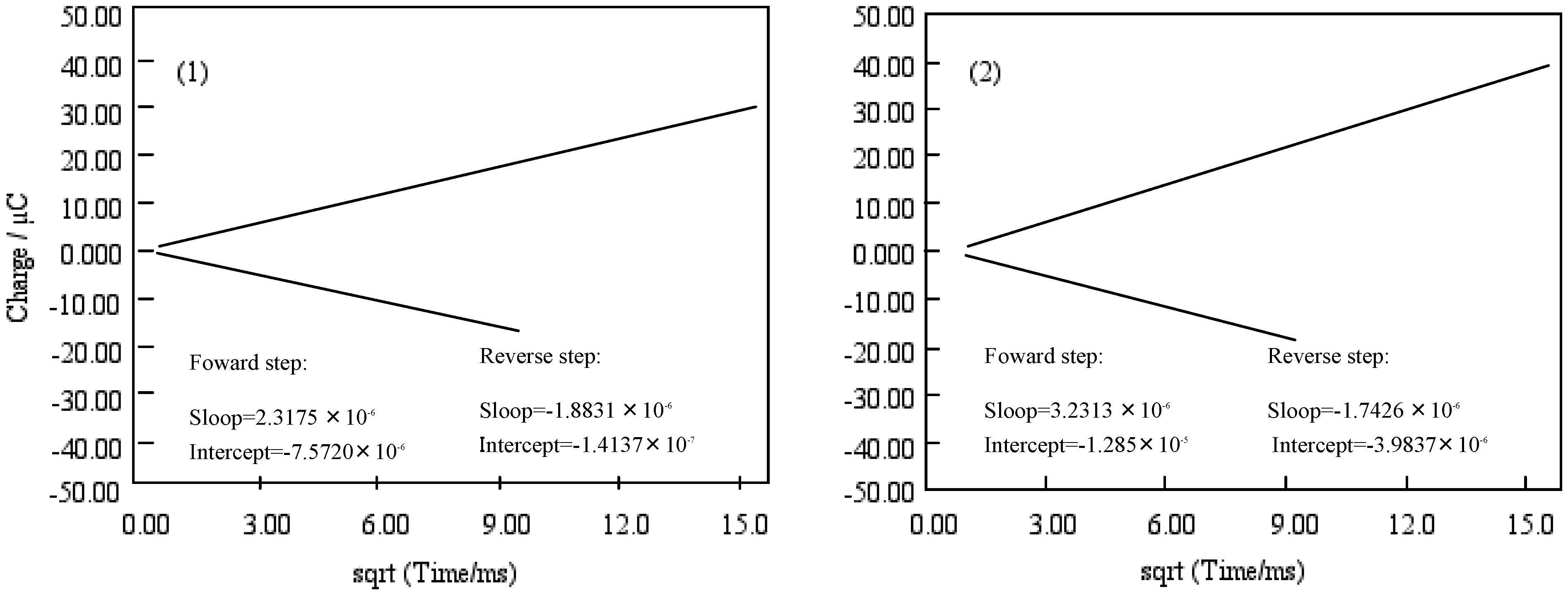
The chronocoulometry of cobalt-1 and 2 shows that the reactant is adsorbed but not the product. The Qads was found from the calculation to be 7.40 and 8.92 μC for cobalt-1 and 2, respectively. Hence, the Qdl was estimated to be 0.53 μC at 0 ms of the chronocoulometry for cobalt-1 and 2.
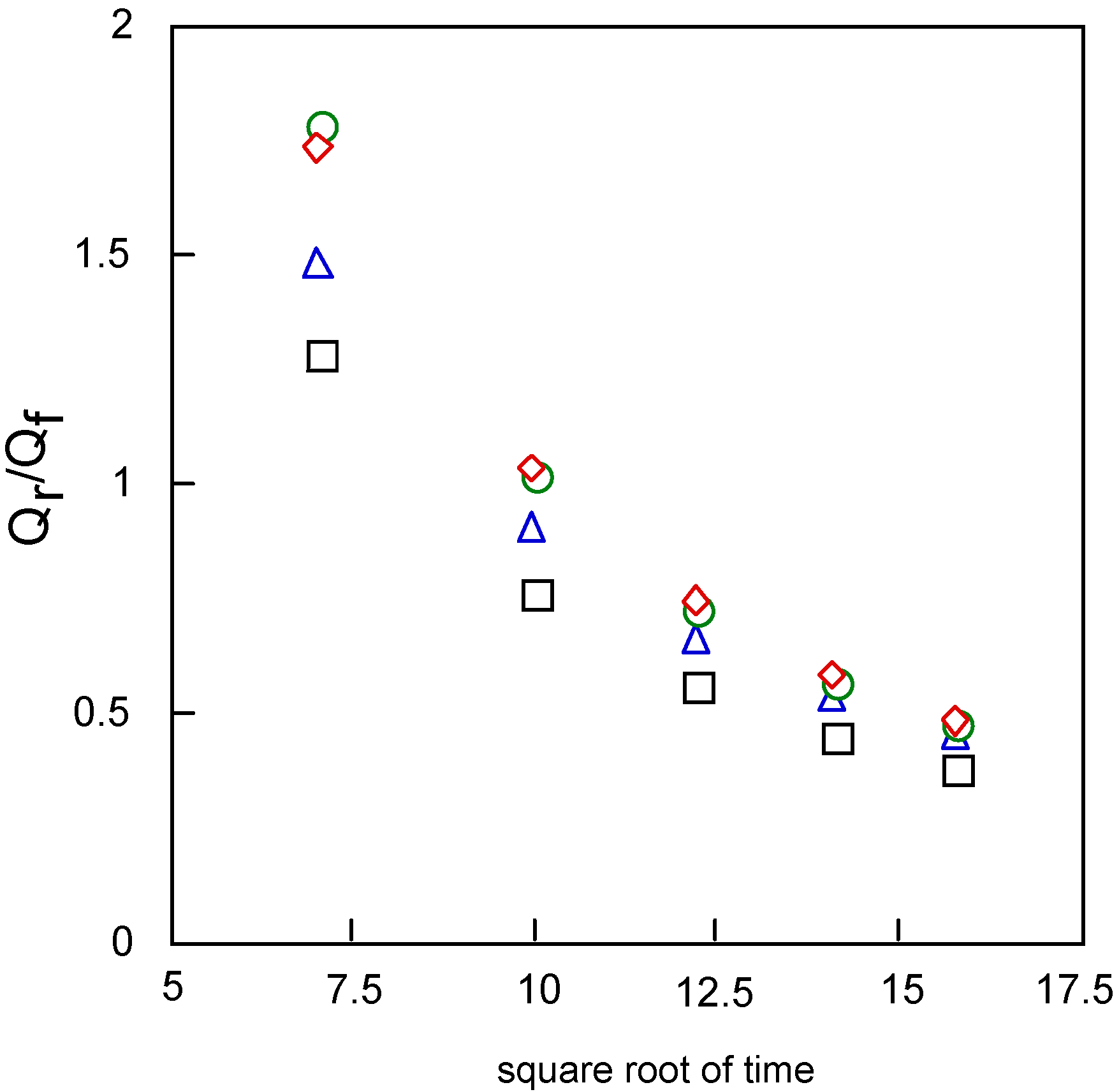
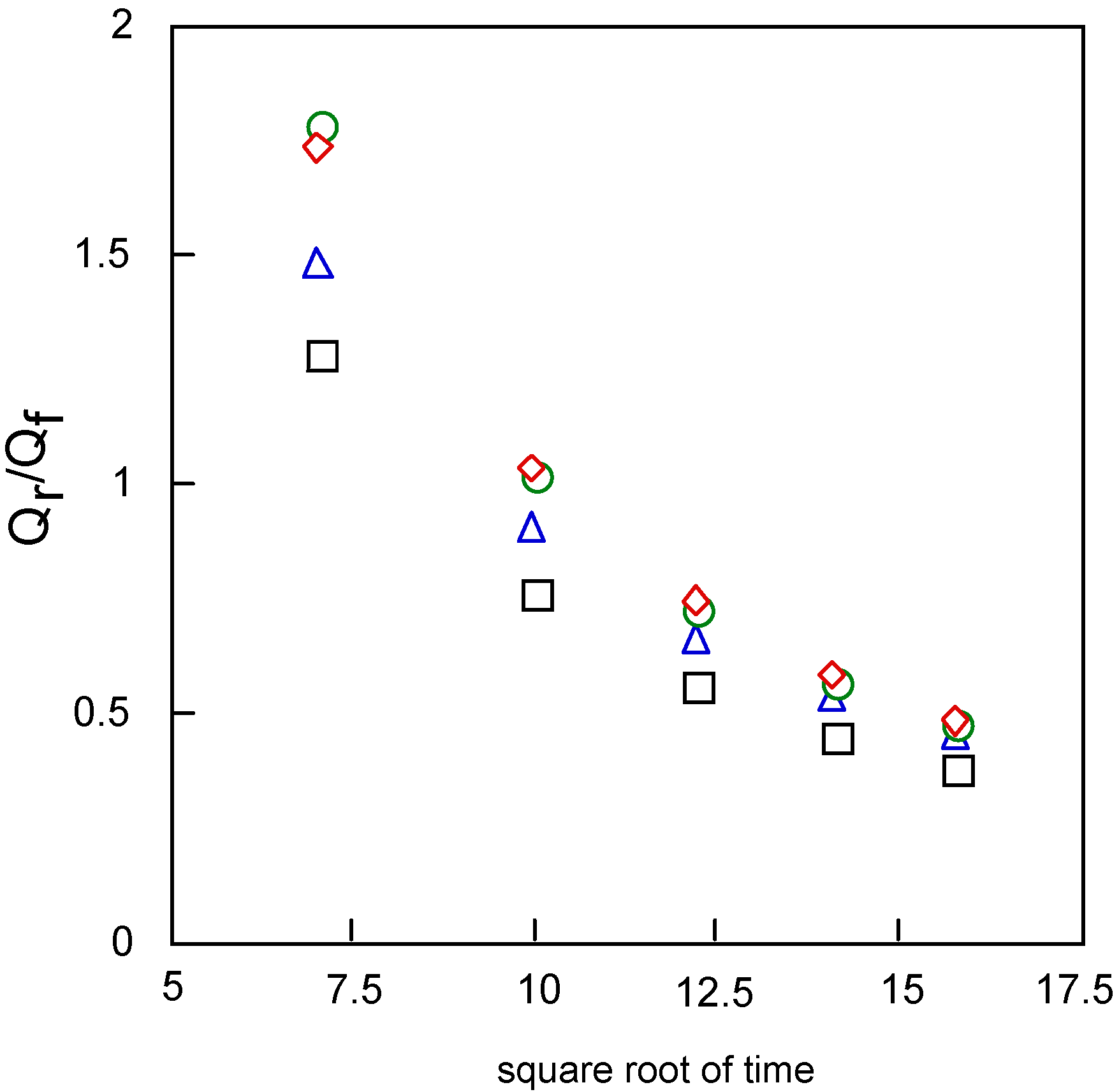
The relation between Qr/Qf and the square root of time can be estimated by the mechanism and rate of the following chemical reaction. The value Qr/Qf indicates the base line for the chronocoulometry of the reverse step charge Qr divided by the final value of forward step Qf. It was found that the following chemical reaction obeyed first-order kinetics, which found the calculation to be 0.20 and 0.26 s-1 for cobalt-1 and 2. It is thought that reduction and oxidation take place in different pathways, where especially cobalt-2 exhibits characteristic behavior.
Chronocoulometry gives rise to Qdl, and Qads and Q in the initial potential step:
where, Qdl: double layer charge (C), Qads: absorbed species charge (C), Γ: amount of adsorbed (mol/cm3).
Qabs = n F A Γ
With regards to the adsorption using equations (2) and (3), the Qabs was found from calculation to be 7.40, 8.92, 2.81 and 7.07 μC for cobalt-1, 2, 3 and 4, respectively. Hence, the Qdl was estimated to be 0.53 μC at 0 ms of the chronocoulometry for cobalt-1, 2, 3 and 4.
Figure 11 shows the relationships between Qr/Qf, the baseline for the measurement of the reverse step charge Qr divided by the final value of the forward step Qf, and the square root of time. This relationship can be estimated by the mechanism and rate of the following chemical reaction. It is found that the following chemical reaction obeyed first-order kinetics. The kinetic constants are found from calculation to be 0.20, 0.26, 0.30 and 0.30 s-1 for cobalt-1, 2, 3 and 4, respectively.
Figure 11.
Variatuon of Qr/Qf with squareroot of time for covalt phthalocyanine derivatives. Potential step; -1.2 – 1.6 V vs. Ag/AgCl, step width: 250 ms. Qf forward step of chronocoulometry; Qr forward step of chronocoulometry. □ Cobalt phthalocyanine tetrasulfonic acid; ◇ Cobalt phthalocyanine octacarboxylic acid; ○ Cobalt octakis(hexoxymethyl)phthalocyanine; △ Cobalt anthraquinocyanine.
Figure 11.
Variatuon of Qr/Qf with squareroot of time for covalt phthalocyanine derivatives. Potential step; -1.2 – 1.6 V vs. Ag/AgCl, step width: 250 ms. Qf forward step of chronocoulometry; Qr forward step of chronocoulometry. □ Cobalt phthalocyanine tetrasulfonic acid; ◇ Cobalt phthalocyanine octacarboxylic acid; ○ Cobalt octakis(hexoxymethyl)phthalocyanine; △ Cobalt anthraquinocyanine.
The oxidation of M-PCs having transition metal are electrochemically irreversible [41,46,47] and electrons are added to the orbital of phthalocyanine ring or the central metal depending on the redox potential for reduction process [47,48,49]. Consequently, it is considered that electron transfer mechanisms are proposed as the following, for cobalt-1 [50]:
- Oxidation step is:Co(II)-PC(SO3H)4 → [Co(III)-PC(SO3H)4]+ + e
- Reduction steps are:Co(II)-PC(SO3H)4+e ⇄ [Co(II)-PC(SO3H)4]-[Co(II)-PC(SO3H)4]- + e ⇄ [Co(II)-PC(SO3H)4[Co(II)-PC(SO3H)4]2- + e → [Co(I)-PC(SO3H)4]3-
For cobalt-2 [50]:
- Oxidation steps are:Co(II)-PC(COOH)8 → [Co(III)-PC(COOH)8]+ + e[Co(III)-PC(COOH)8]+ → [Co(III)-PC(COOH)8]2++e[Co(III)-PC(COOH)8]2+ → [Co(IV)-PC(COOH)8]3++e
- Reduction steps are:Co(II)-PC(COOH)8 + e → [Co(II)-PC(COOH)8]-[Co(II)-PC(COOH)8]- + e ⇄ [Co(II)-PC(COOH)8]2[Co(II)-PC(COOH)8]2- + e ⇄ [Co(I)-PC(COOH)8]3-
For cobalt-3 [51,52]:
- Oxidation steps are:Co(II)-PC(CH2OC6H13)8 → [Co(III)PC(CH2OC6H13)8]+ + e[Co(II)-PC(CH2OC6H13)8]+ → [Co(III)-PC(CH2OC6H13)8]2+ + e
- Reduction steps are:Co(II)-PC(CH2OC6H13)8 → [Co(I)-PC(CH2OC6H13)8]-[Co(I)-PC(CH2OC6H13)8]- + e ⇄ [Co(I)-PC(CH2O6H13)8]2-[Co(I)-PC(CH2OC6H13)8]2- + e ⇄ [Co(I)-PC(CH2OC6H13)8]3-
For cobalt-4 [51,52]:
- Oxidation steps are:Co(II)-AQC → [Co(III)-AQC]+ + e[Co(III)-AQC]+ → [Co(III)-AQC]2+ + e
- Reduction steps are,Co(II)-AQC + e ⇄ [Co(II)-AQC]-[Co(II)-AQC]- + e ⇄ [Co(I)-AQC]2-
The chemical reaction following the reversible redox process is an interaction between the anion of the phthalocyanine ring and a solvent molecule. It is thought that the ring current of M-PCs such as 1, 2 and 3 consists of five components, corresponding to one porphyrazine and four phenylene rings, as shown in Figure 12(a).
The molecules of 4 are also planar, symmetrical and possess a highly aromatic organic system due to the presence of its highly conjugated π electron system. For this reason, 4 can be expected to have an effect on the electron transfer for anthraquinone units in the molecules. Anthraquinone-like properties were not observed for 4, which showed reversible two step one-electron redox reactions, and were similar to 1 and 2, so the anthraquinone units in 4 apparently had no effect on the electron transfer ability in itself, so we may conclude that the ring current constituents of 4 are different from those of M-PCs. It is believed that the π electron ring current of metal anthraquinocyanines is not made up of five loops. As the 4 are also conjugated planer molecules, the π electrons of the anthraquinone units are incorporated into the macrocyclic conjugated path of the porphyrazine ring shown in Figure 12(b). As a result of the formation of a simple loop current on 4, the redox property of anthraquinone covers the macrocyclic delocalized π electrons.
Figure 12.
Ring current model of phthalocyanine ring and anthraquinocyanone ring. (a) Five-loop model. (b) Single-loop model.
Figure 12.
Ring current model of phthalocyanine ring and anthraquinocyanone ring. (a) Five-loop model. (b) Single-loop model.
The important parameters of a CV are the reduction and oxidation potentials for irreversible peaks, and the mid-point potential for a reversible couple, Emid (Table 7). Before separation of positional isomers, the reduction and oxidation potentials of 5c are sorted into six irreversible peaks.
After separation, fractions 1 - 3 have one pair of reversible oxidation potential and four irreversible peaks. Fraction 4 has one pair of reversible oxidation and three irreversible reduction waves. The reduction and oxidation of M-PCs are due to the interaction between the phthalocyanine ring and the central metal [49,53,54].
| Compound | Potential / V vs. Ag/AgCl | ||||||
|---|---|---|---|---|---|---|---|
| Reduction | Oxidation | ||||||
| 5c, before regioisomer separation | -0.97* | -0.71* | -0.45* | 0.15* | 0.37* | 0.93* | |
| Fraction 1 | -1.00* | -0.58* | -0.24* | 0.44 | 0.93* | ||
| ΔE** | 0.17 | ||||||
| Fraction 2 | -1.05* | -0.60* | -0.19* | 0.37 | 0.90 | ||
| ΔE** | 0.10 | ||||||
| Fraction 3 | -0.96* | -0.65* | -0.22* | 0.37 | 0.89* | ||
| ΔE** | 0.13 | ||||||
| Fraction 4 | -0.87* | -0.63* | -0.21* | 0.34 | |||
| ΔE** | 0.01 | ||||||
Potentials of reversible wave are midpoint of anodic and cathodic praks for each couple, E1/2.
*Irreversible peak. ** The anodic peak to cathodic peak separation for reversible couple.
The porphyrazine ring in the molecules of metal phthalocyanine derivatives or analogues is influenced by the π electrons around the closed system [16,27,32,51,55]. Although the π electron systems of 5c and fractions 1-4 consist of one porphyrazine, two pyridinoids and two didecyl- substituted phenylene rings, the locations of these rings, except for the porphyrazine, are different for each positional isomer. The irreversible peaks are attributed to the oxidation of the central metal and the reversible couples represent the redox of the phthalocyanine ring [53].
Substituents and pyridinoid rings influenced the π electron environment in the compounds 5c and fractions 1-4. It is thought that the effect of pyridinoid rings gives rise to changes of the electron density of the M-PCs. The difference of reduction and oxidation peaks between fractions 1-4 is attributed to the effect of the variation of the interaction between the central metal and the alkylbenzoporphyrazine. Then, the difference of CV between the 5c and fractions 1-4 is also the effect of the interaction, since 5c is a mixture of its positional isomers.
The ΔΕ values are the anodic peak to cathodic peak separation located in the oxidation potential region. The ΔΕ values are around 100 mV and the redox processes are the same for positional isomers, except for fraction 4. This means that the electron process of position isomers between fractions 1-3 involve approximately one electron transfer. The ΔΕ values of fraction 4 show different behavior in comparison to the others. It is thought that the different behavior for fraction 4 is attributable to the mixture of two types of C2v positional isomers. In other words, the reduction and oxidation potentials of fraction 4 are based on the interaction between two types of C2v positional isomers. No observation on the reversible couple in 5c resulted in any interaction between the positional isomers.
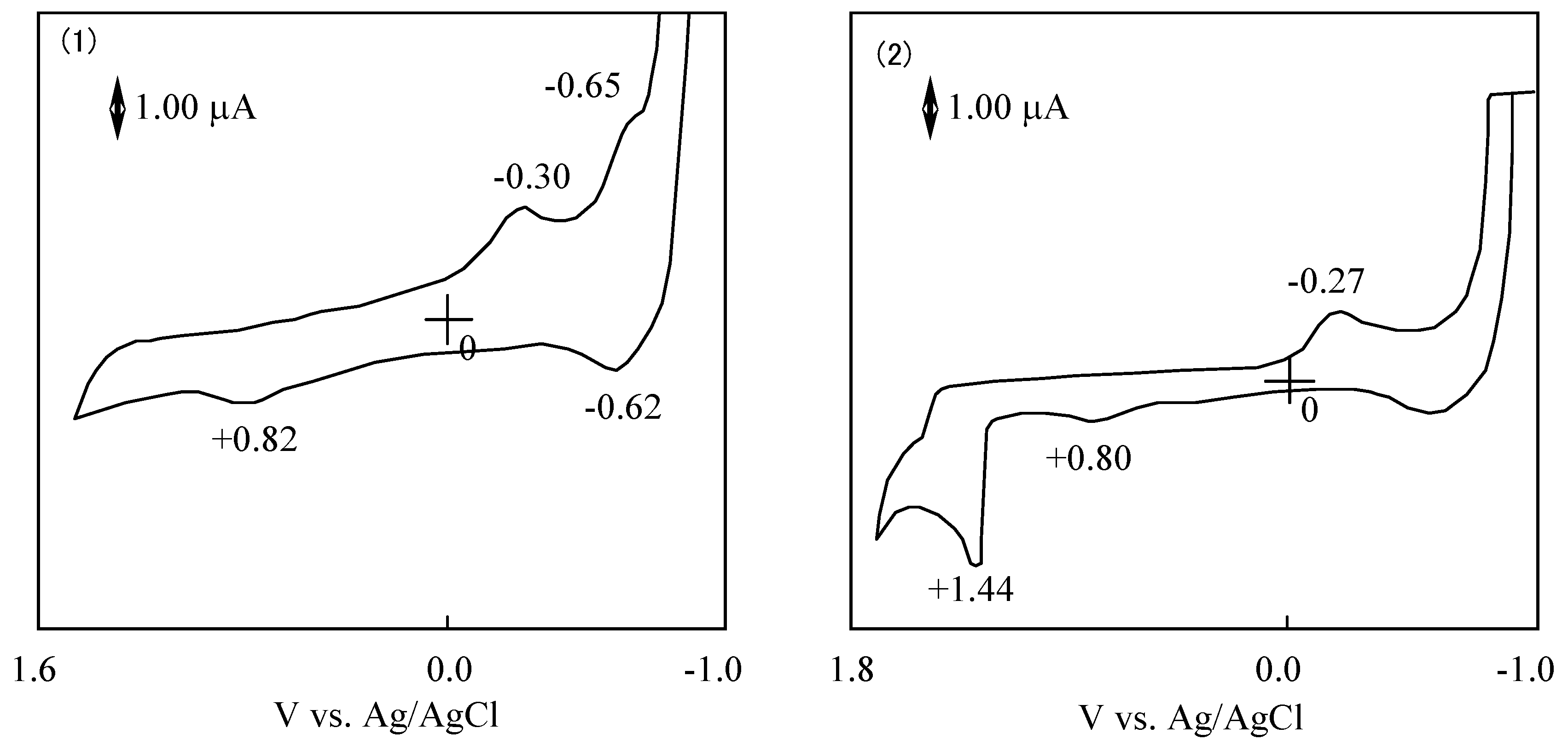
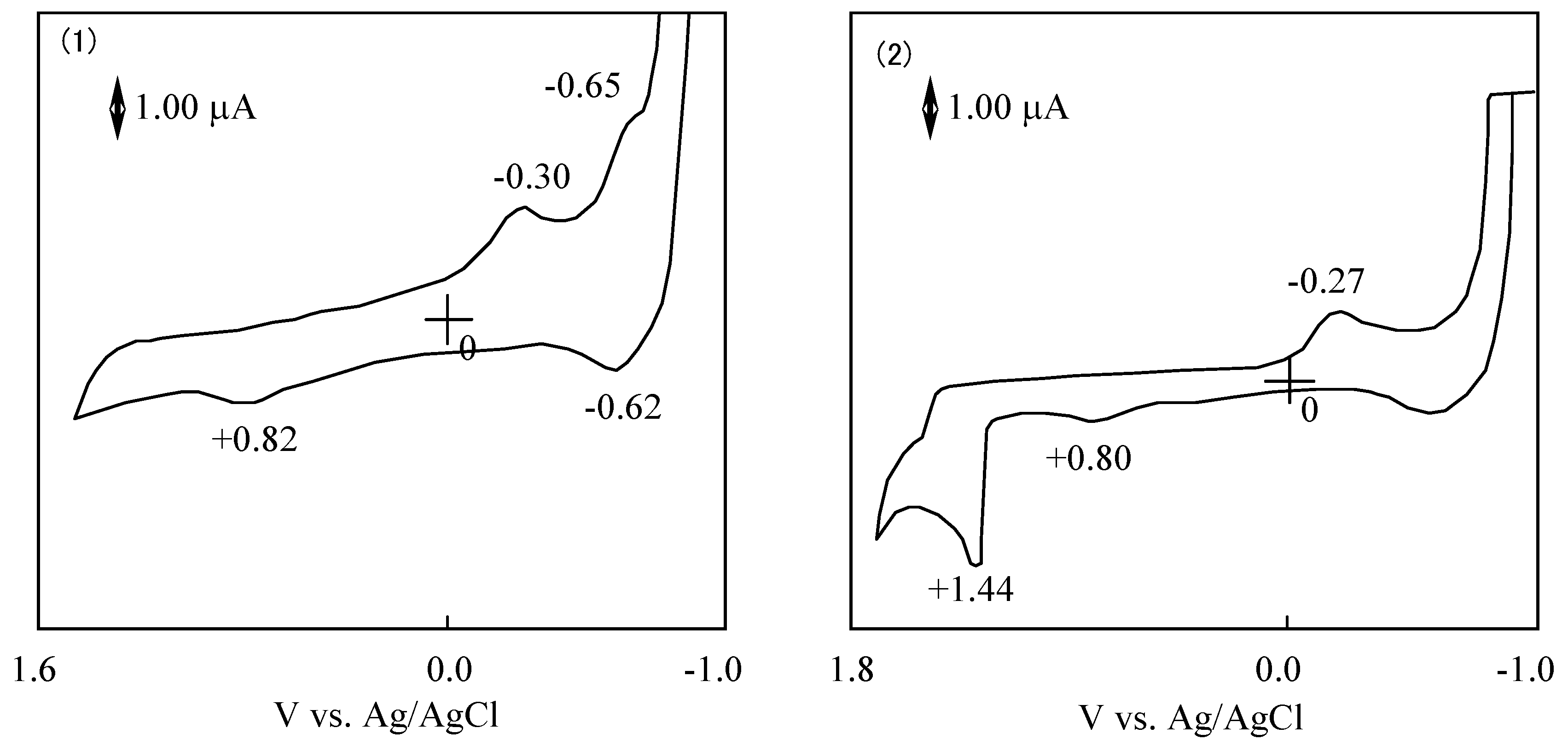
The potential difference between the reduction and oxidation is expressed in the HOMO - LUMO energy gap [41]. The values of λmax in the Q band correlated with the potential difference between the reduction and oxidation. Figure 13 shows CVs of 6a and 6f. The reduction and oxidation potentials of 6 are summarized in Table 8. The reported potentials are the midpoint potential of the anodic and cathodic peaks for each couple Emid, and the peak potential for an irreversible step.
Figure 13.
Cyclic voltammograms of subphthalocyanine derivatives in acetonitrile with tetrabutylammonium perchlorate. (1) Subphthalocyanine 6a. (2) hexakis(S-phenyl)hexa-fluorosubphthalocyanine 6d.
Figure 13.
Cyclic voltammograms of subphthalocyanine derivatives in acetonitrile with tetrabutylammonium perchlorate. (1) Subphthalocyanine 6a. (2) hexakis(S-phenyl)hexa-fluorosubphthalocyanine 6d.
| Compound | Potential / Vvs. Ag/AgCl | |||||
|---|---|---|---|---|---|---|
| Reduction | Oxidation | |||||
| 6a | -0.64 | -0.30* | 0.82* | |||
| ΔE** | 0.03 | |||||
| 6b | -0.51* | 0.25* | 0.79* | |||
| 6c | -0.56* | -0.28* | 0.83 | |||
| ΔE** | 0.03 | |||||
| 6d | -0.61* | -0.37* | 0.18* | |||
| 6e | -0.27* | 0.80* | 1.44* | |||
| 6f | -0.57* | -0.36* | 0.18* | |||
Potentials of reversible wave are midpoint of anodic and cathodic praks for each couple, E1/2. *Irreversible peak. ** The anodic peak to cathodic peak separation for reversible couple.
The cyclic voltammogram of 6a showed two cathodic peaks at −0.30 and −0.65 V vs. Ag/AgCl, and two anodic peaks at 0.82 and −0.62 V vs. Ag/AgCl. Compound 6a has two irreversible oxidation and reduction at 0.82 and −0.30 V vs. Ag/AgCl, and one pair of reversible reduction potential at −0.64 V vs. Ag/AgCl.
The reduction and oxidation behavior of M-PCs are due to the interaction between the phthalocyanine ring and the central metal [55]. In the case of M-PCs having cobalt as the central metal, phthalocyanine ring oxidation occurred after the central metal, and this reduction was followed by the reduction of the central metal [50,54,56].
In the case of M-PCs, the porphyrazine ring in the molecule is influenced by the π electrons around the closed system. The π electron system of M-PCs consists of one porphyrazine and four phynylene rings [27,32,50]. Substituents on the M-PCs influence the π electron environment in the molecule, especially the four phynylene rings. It is thought that the effect of substituents gives rise to the change of electron density of the four phynylene rings in the molecules of M-PCs. Electron transfer properties of M-PCs depend on the kind of the substituents.
In the case of 6, redox potentials had various values, but one irreversible reduction potential certainly appeared around −0.3 V vs. Ag/AgCl. In SubPC, the reduction potentials showed at −0.64 V vs. Ag/AgCl and the oxidation potential appeared at 0.82 V vs. Ag/AgCl. It is concluded that the irreversible peaks around −0.3 V vs. Ag/AgCl can be attributed to the reduction of the subphthalo-cyanine ring. The difference of cyclic voltammogram between SubPCs is attributed to the variation of the substituents that depends on the SubPC ring.
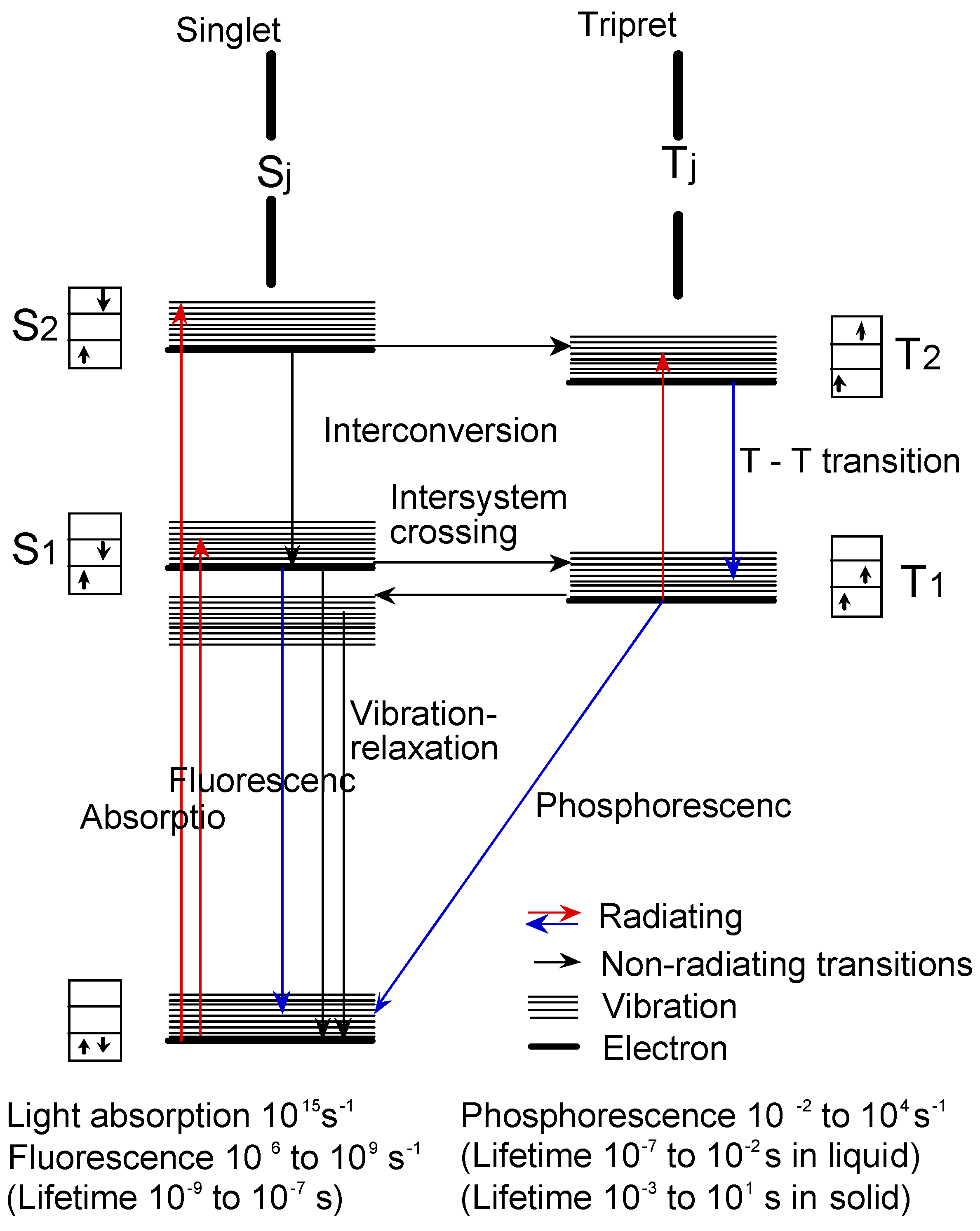
2.3. Photochemistry
Figure 14 shows the Jablonski diagram, which is a scheme of radiationless transition process and emission of radiation (fluorescence and phosphorescence). In general, photochemical reactions occur in triplet states. Triplet state lifetimes are known to be between 100 ns and 10 s. The lifetime of excited singlet states is too short for a typical chemical reactions. Photochemical properties were measured the triplet state lifetime using laser-flash photolysis.
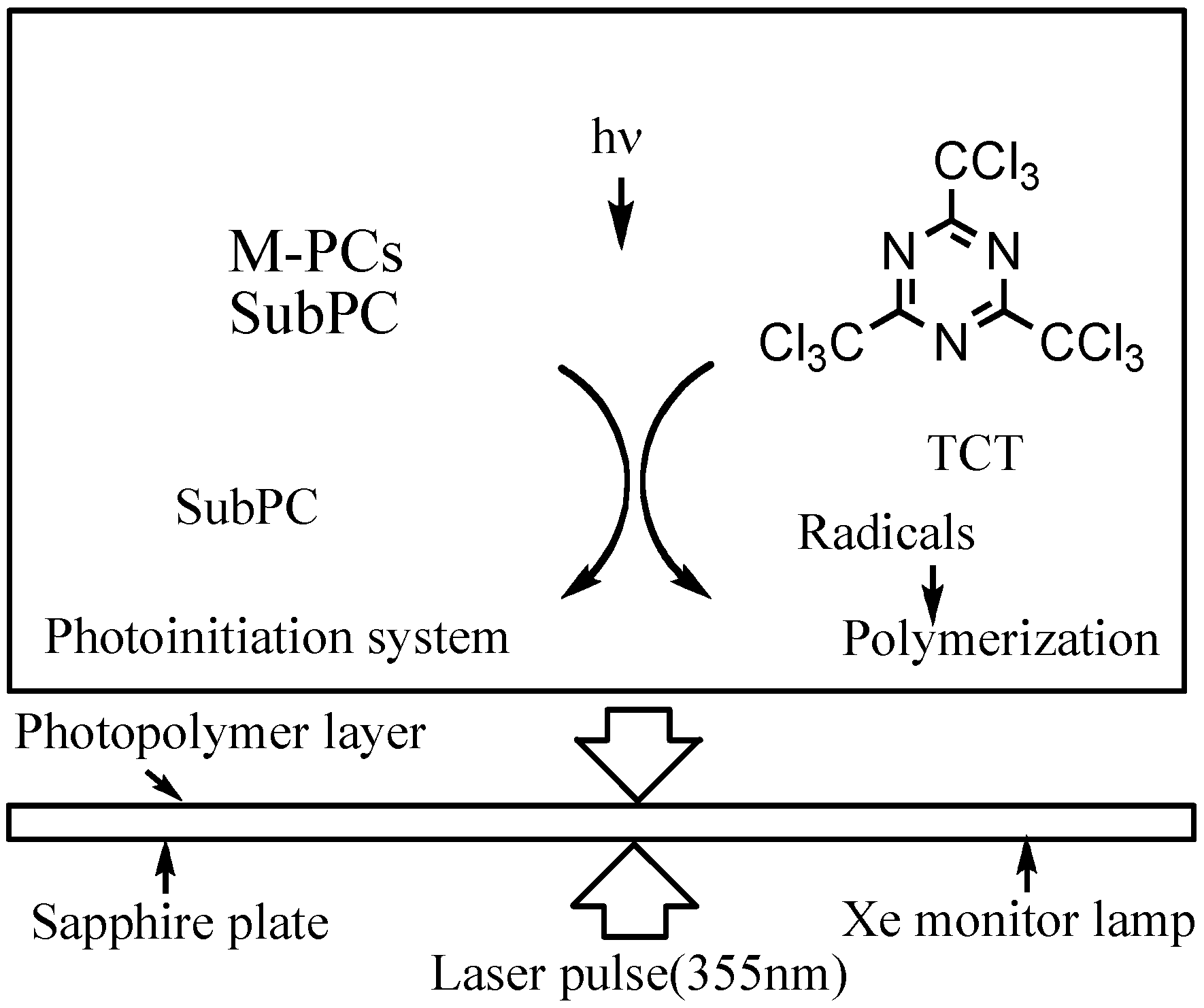
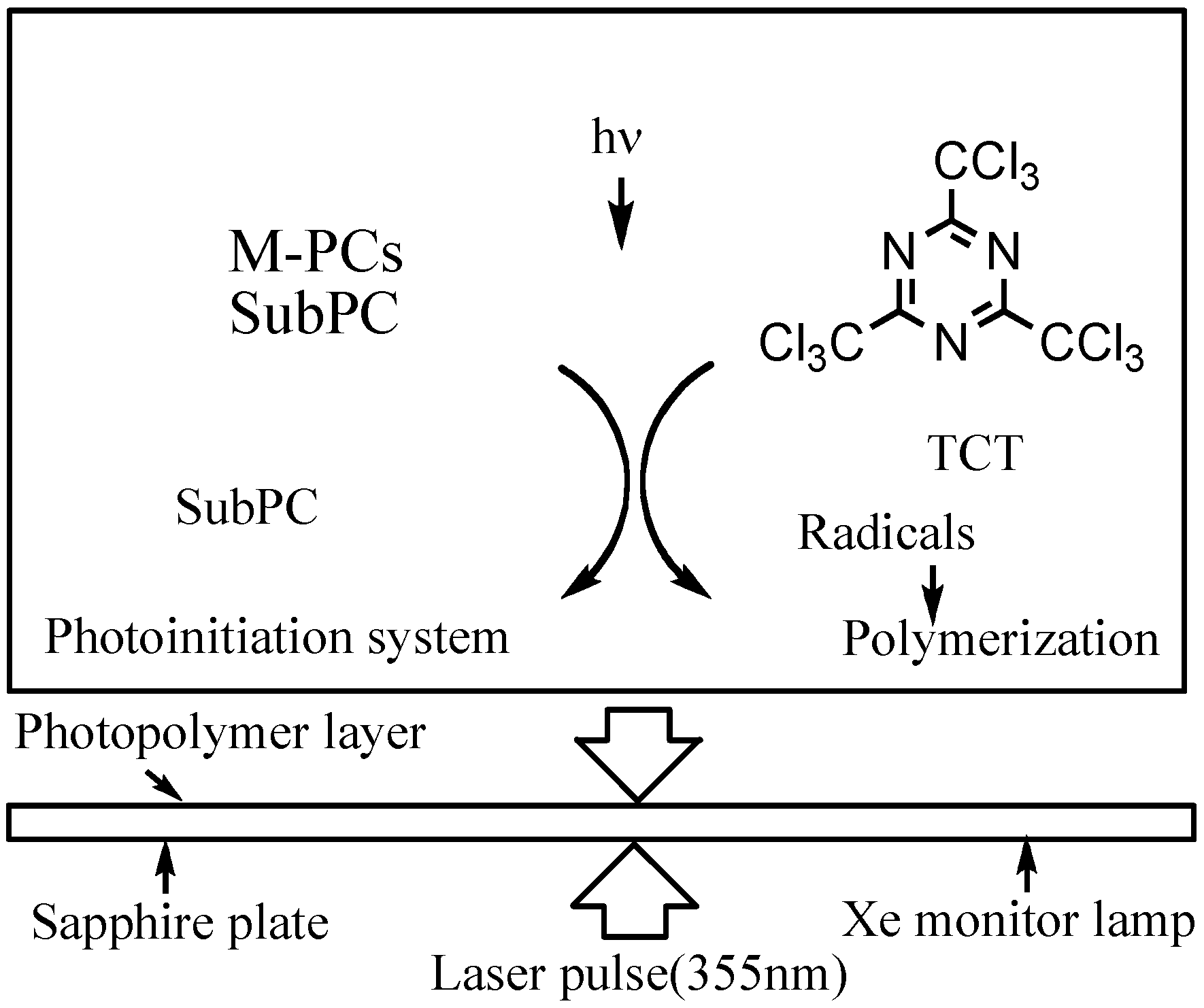
Laser-flash photolysis in film was performed using a total reflection sapphire cell (10 · 30 mm, 1 mm thickness, and both side were cut at a 45 degree angle) [57,58], which was spin-coated with a thick of 1.2 μm SubPC containing 10% poly(methylmethacrylate) (PMMA) in cyclohexane photopolymer film as shown in Figure 15. The excitation light pulse from a Spectron Laser System Model SL402 YAG laser was expanded five-fold over the entire sample cell. A xenon lamp was used as a monitoring beam [57,58].
The measurement was repeated five times with less than 3 wt% of 6 and 2,4,6-tris(trichloromethyl)-1,3,5-triazine (TCT) as a quencher; more than 90% of the 355 nm-laser light absorbed in the sample film was absorbed by 6.
Films were prepared as follows; the compounds 6 were added to this solution either by dissolving the dyes directly in polymer solution or by mixing a potion of concentrated dye solution. Films were adjusted to be 1.2 μm thick by spin-coating a solution onto a sample cell.
Figure 14.
Jablonski diagram.
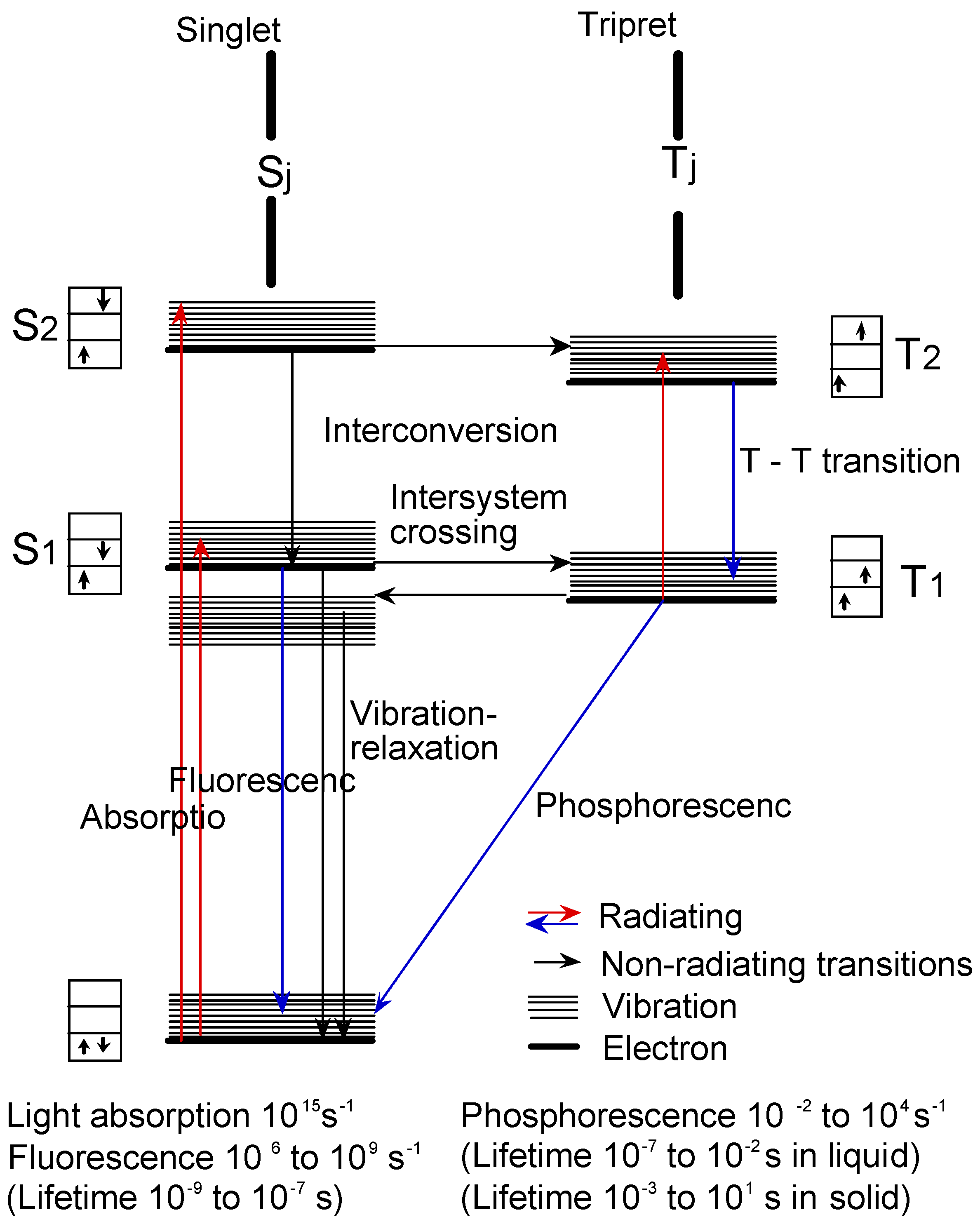
Figure 15.
Laser flash photolysis in film.
The photosensitivity of the photoinitiator system was measured as follows; the sensitive layer which was prepared by coating a cyclohexane solution of the photosensitive composition containing 5 wt% of the 6, 5 wt% of TCT, 45 wt% of trimethylrolpropane triacrylate and 45 wt% of PMMA (90:10 mol%, MW = 50,000) onto a grained aluminium plate and exposed at 550 nm by the use of a xenon lamp, which was isolated using a monochrometer.
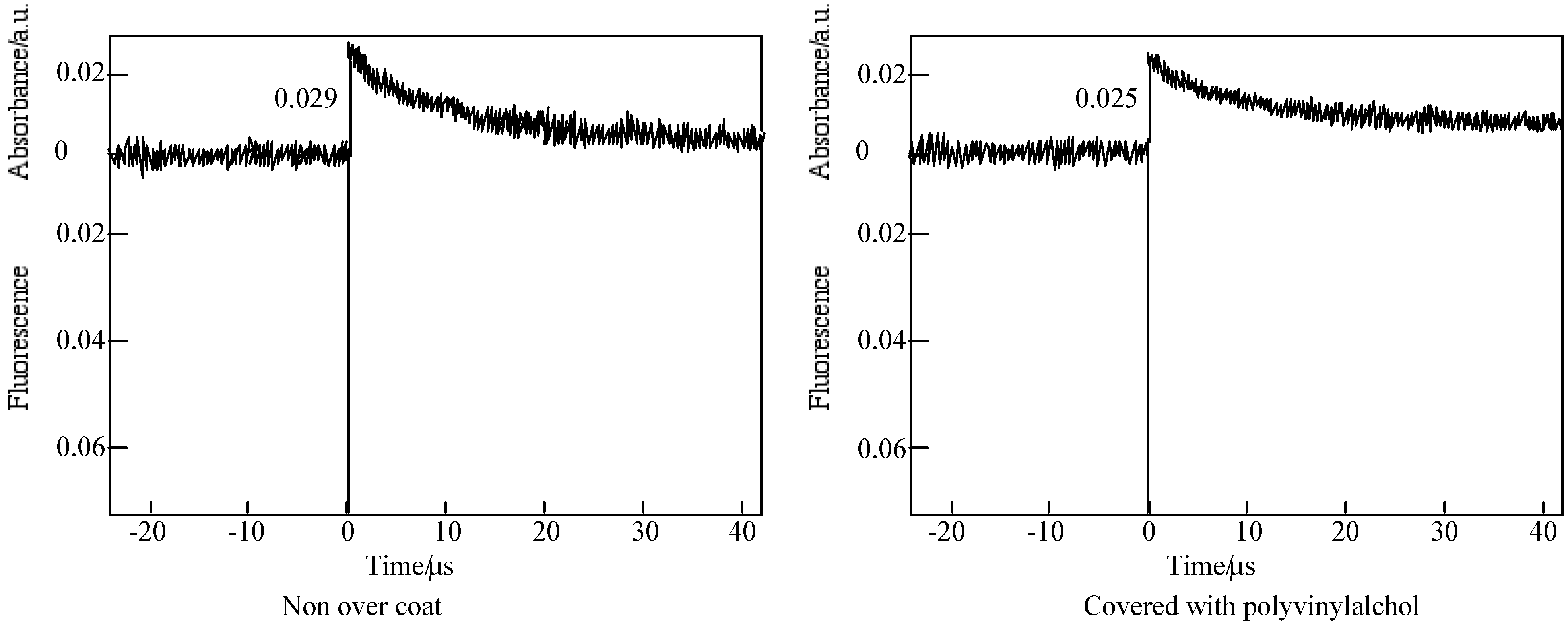
In the case of laser-flash photolysis of 5c [59], the films were prepared as follows: a 10 wt% PMMA solution was made up in cyclohexanone, allylbenzopyridoporphyrazines were added to this solution by dissolving to a thickness of 1.2 μm thick by spin-coating a solution onto a sapphire cell. After that the films were covered with a poly(vinyl alcohol) (PVA) solution. Figure 16 shows the time profiles of the triplet state for one of the alkylbenzopyridoporphyrazines, 5c in PMMA was observed using laser-flash photolysis.
Figure 16.
Decay trace of 5c in PMMA film on 560 nm. Excitation wavelength: 355 nm in the presence and absence of PVA over coatings.
Figure 16.
Decay trace of 5c in PMMA film on 560 nm. Excitation wavelength: 355 nm in the presence and absence of PVA over coatings.
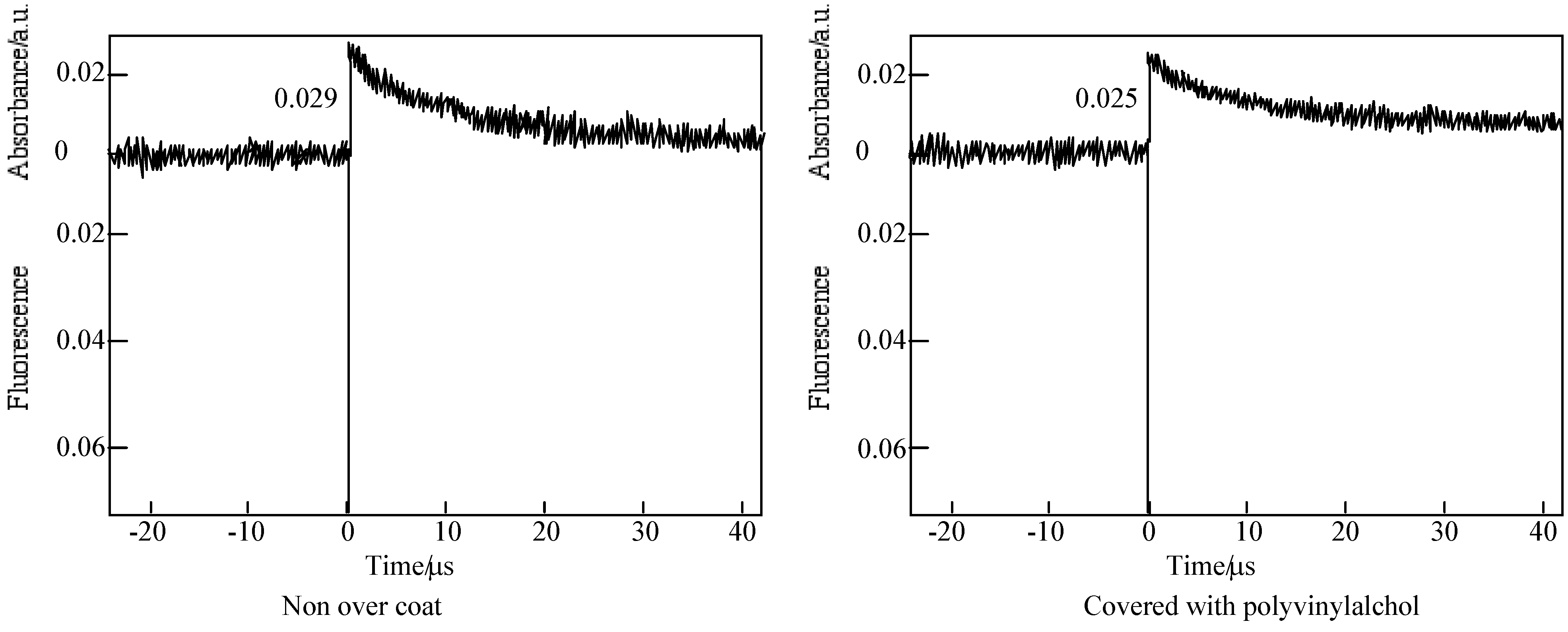
The triplet state lifetimes of alkylbenzopyridoporphyarazines, 5b - 5d and 5e are summarized in Table 9.
| Compound | Q band / nm | Lifetime / μs | |
|---|---|---|---|
| in PMMA film | Non over coat | Over coat | |
| 5b | 675.2 | 11.4 | 51.8 |
| 5c | 717.6 | 10.1 | 46.9 |
| 5d | 670.0 | 5.7 | 18.2 |
| 5e | 703.9 | 2.6 | 17.9 |
In each alkylbenzopyridoporphyrazine, it is shown that 5b and 5c have longer triplet lifetimes than 5d and 5e. The length of the triplet lifetime for alkylbenzopyridoporphyrazine depends upon its molecular structure. The triplet lifetime of alkylbenzopyridoporphyrazines increased with increasing pyridine numbers in the molecule. It seems that if tetrapyridoporphyrazine 5a can be soluble in common solvents and measured for laser-flash photolysis, its triplet lifetime will be shown the longest value.
The photoexcited triplet state lifetimes of 5b and 5c in PMMA without a PVA coating were estimation to be 11.4 and 10.1 μs, respectively. While covered with a PVA coating, the photoexcited triplet state lifetimes of 5b and 5c were estimated as 51.8 and 46.9 μs, respectively. Compared with each compound, the triplet state lifetime in PMMA covered with PVA was longer than without the PVA coating.
The Q band absorption of alkylbenzopyridoporphyrazines in PMMA films was similar to that in solution, but the profile of Q band in PMMA film became wider than that in solution and moved to a longer wavelength, except for 5e.
Non-transition M-PCs were known to be excellent photosensitizers because of their chemical stability and high absorbance in the 650–700 nm region [60]. In the presence of a photosensitizer, photooxidation progresses via singlet state oxygen [61,62,63,64,65,66,67,68,69,70,71]. M-PCs in the excited triplet state react with ground triplet state dioxygen. The triplet state dioxygen generated singlet excited state oxygen. The singlet excited oxygen reacts with a substrate to produce oxide, Equations 25 - 27 [33,72].
3M-PC + 3O2 → M-PC + 1O2
1O2 + substrate → substrate oxide
Both covered with a coating and without PVA, the photooxidation proceeded by the same mechanism. However, in the case of being covered with PVA, the photoexited triplet state lifetimes of alkylbenzopyridoporphyrazines, 5b - 5d and 5e, were longer than in the case of non-overcoating with PVA. In the case of non-coated with PVA the shorter decay time was considered due to M-PC quenching by oxygen existing in an air atmosphere. While under the coated state with PVA, they suppressed the oxygen-permeation from the air atmosphere into the photopolymer layer. As a result, grand triplet state dioxigen was not furnished from the surrounding to the system. Alkylbenzo-pyridoporphyrazines in the case of being covered with PVA behaved as a model for a practical photosensitizer in tumors or cancer cells.
In comparison with 5b and 5c, the photoexcited triplet lifetime of 5b was slightly longer than 5c, the absorption intensities for 5c were stronger than 5b. So that in these aspects, there is little to choose as a sensitizer for PDT between the two. As Py rings in the molecule of alkylbenzo-pyridoporphyrazines increased, the water-solubility is expected to increase. In the case of the N,N´,N´´,N´´´,N´´´´-tetramethylated quaternized forms of tetrapyridoporphyrazines, it was reported that the complexes do not form an aggregation in an aqueous solution [72,73,74]. Although the long alkyl-chain substituents in 5b and 5c will occur in aggregation, 5b and 5c are expected to rapidly undergo photodecomposition after the photooxidation process, similar to alkyl phthalocyanine derivatives [9].
Consequently, since 5b and 5c have the most intense absorption and a longer triplet state lifetime, we think 5b and 5c could become useful sensitizers for PDT. The photosensitizer should be made in isomerically pure form [9]. Isomers of 5b have not been reported yet, but 5c has been separated and identified [33]. Thereupon, regioisomers of 5c were examined by laser- flash photolysis, and 5b will be reported in the near future. As mentioned above, the regioisomers of the compound, 5c were separated into four green- to blue-colored fractions by TLC [33]. The four fractions have a different 1H-NMR, UV-VIS and fluorescent spectra. The four fractions separated by TLC have been attributed to four of the five possible region- isomers of 5c.
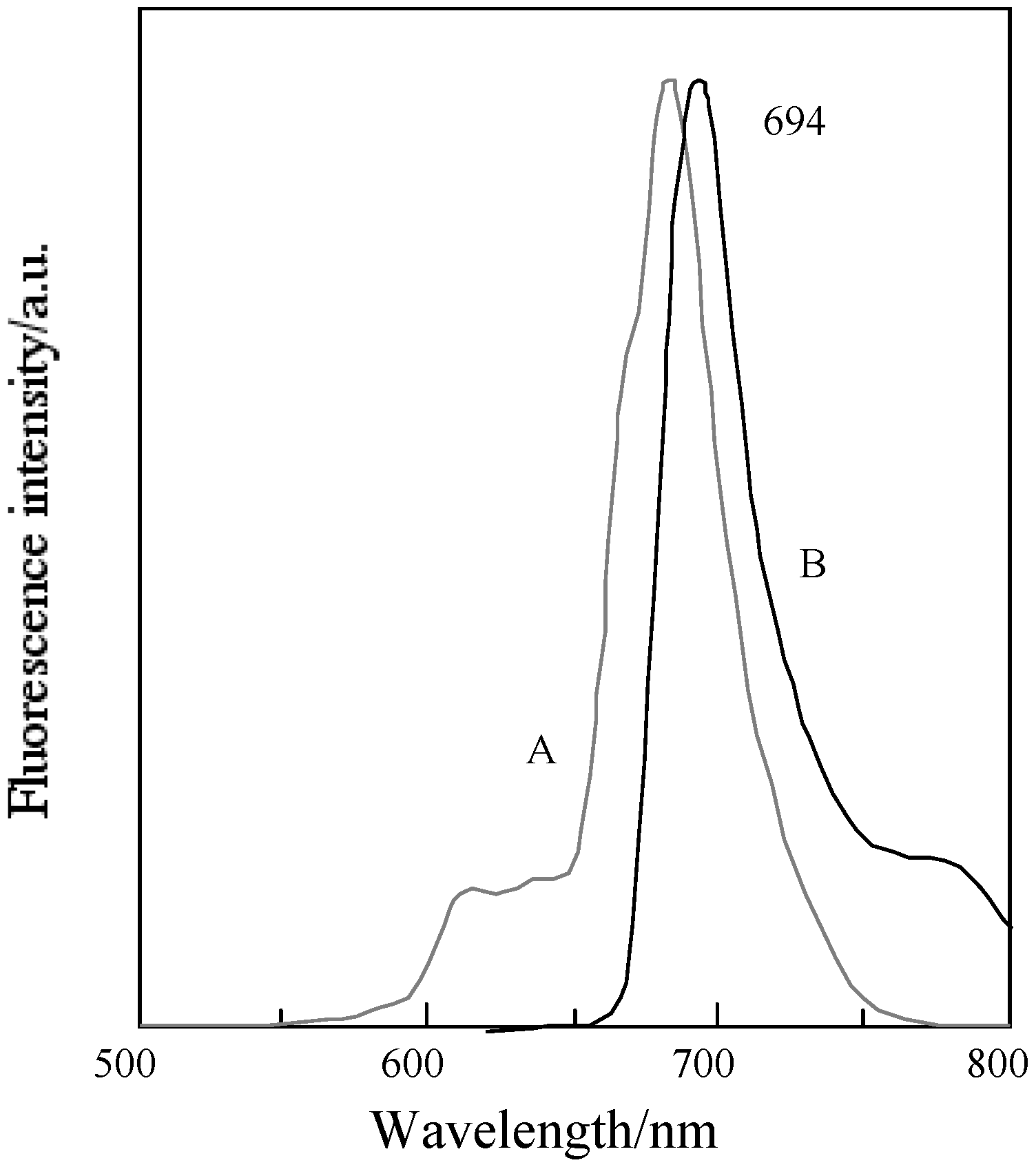
Figure 17 shows the fluorescence and excitation spectra of 5c. The excitation spectra of 5c and its fractions have almost the same profile. No significant change on the fluorescence spectra was observed for 5c and its fractions.
Figure 17.
Fluorescence and excitationspectra of 5c in DMF. A; Excitation spectrum, B; Fluorescence spectrum.
Figure 17.
Fluorescence and excitationspectra of 5c in DMF. A; Excitation spectrum, B; Fluorescence spectrum.

Table 10 shows the Q band and fluorescent maximum of fractions of 5c. The assignment of the Q band from each fraction was carried out on the theory of the relationship between symmetry and the Q band [1,4,74,75,76,77]. The Q band splits into two peaks of the highest isomer symmetry, the splitting Q band is decreased with a decreasing symmetry [78]. The symmetry of the position isomer of 5c was decreased in orders of C2h, D2h, C2v, Cs [79]. The regioisomers of 5c had molecular structures with D2h, C2h, Cs and C2v symmetry for fractions 1, 2, 3 and 4, respectively [33]. Two types of C2v isomers could not be isolated [33].
| Compound | Symmetry | Q band /nm | Fluorescence / nm |
|---|---|---|---|
| Fraction 1 | D2h | 627, 690, 705 | 704 |
| Fraction 2 | C2h | 609, 673, 708 | 706 |
| Fraction 3 | Cs | 619, 689 | 695 |
| Fraction 4 | C2v | 610, 638, 671, 688 | 701 |
Under line; mostintense peak.
Table 11 shows the photoexcited triplet lifetime of fractions separated from 5c. In spite of the presence or absence of PVA coatings, the triplet lifetime was increased with a decreasing symmetry of position isomers, which were ordered as C2h, D2h, C2v and Cs for fractions 2, 1, 4 and 3, respectively. The photoexcited triplet state lifetimes of fraction 3 in PMMA absence and presence of a PVA coating were estimated to be 14.29 and 25.97 μs, respectively. Of each fraction except for fraction 3, the length of the lifetime was shorter than 5c, and the sensitivities of triplet-triplet (T-T) absorptions were observed as very low.
| Compound | Q band / nm | Lifetime / μs | |
|---|---|---|---|
| in PMMA film | Non over coat | Over coat | |
| Fraction 1 | 782.2, 681.6, 653.5 | 7.0 | 11.3 |
| Fraction 2 | 670.0 | 0.9 | 1.6 |
| Fraction 3 | 696.4 | 14.29 | 25.97 |
| Fraction 4 | 665.5 | 6.4 | 9.2 |
Although the length of the triplet lifetime of 5c was observed to be approximately four times that seen in the absence of a PVA overcoating, the fractions of 5c were only had about 1.5 times as long as triplet lifetimes in comparison with the absence and presence of PVA coatings.
Unfortunately, a precise explanation for this cannot be provided, but it seems to be due to the following phenomenon: phthalocyanine derivatives were well known to aggregate in water and non-coordinating solvents. Zinc non-peripheral phthalocyanine derivatives having long side chains formed an aggregation at least 10−5 mol·L-1 in cyclohexane [9,78]. It is enough thought that the samples in this study for laser-flash photolysis were formed in aggregation in the experimental condition. The aggregation degrees for 5c and its isomers are different from each other, so the aggregation ability of 5c and each of its isomers is different and complicated. Since compounds 5b and 5c consisted of mixtures of their isomers, the aggregation and relationships of the energy levels between samples and the triplet state of dioxygen became a complication. For this reason, compounds 5b and 5c could give relatively long lifetimes. The molecular structure of fraction 3 is suitable to occur in the T-T absorption in the system.
In order to estimate the photoexcitation mechanism, the triplet lifetime of each fraction was measured, containing N,N’-tetramethyl-4,4’-diaminobenzophenone (Michler’s ketone) as an additional quencher. Using Micher’s ketone, the lifetimes without PVA coatings were estimated as 21.19 and 14.03 μs for fractions 3 and 4, respectively (Table 12). These values of lifetime were longer than in the absence of Micher’s ketone. In the case of fractions 1 and 2, no T-T absorption occurred. The results were thought to be that each fraction has different energy levels of grand and excited states. The T-T absorption took place via the interactions between the energy levels of grand or excited states of fractions and the triplet of dioxygen or Michler’s ketone.
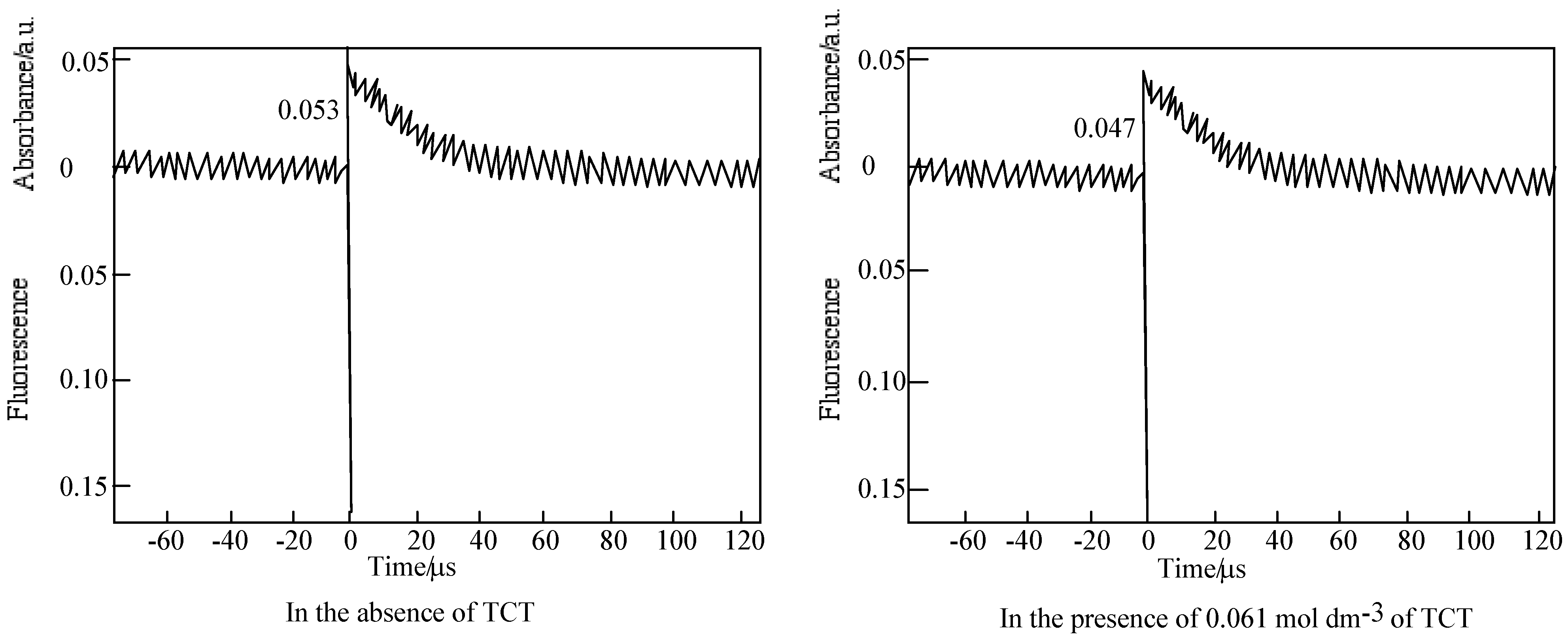
The time profiles of the triplet state for 6 in PMMA film are shown in Figure 18. The fluorescence peak and transient decay were observed upon the excitation at 355 nm pulse of a PMMA film containing 6. The photoexcited triplet state lifetime of 6 was estimated for 20 s laser pulse. The transient absorption was assigned to a triplet state [78].
| Compound | Lifetime / μs | |
|---|---|---|
| Non over coat | Over coat | |
| Fraction 1 | - | - |
| Fraction 2 | - | - |
| Fraction 3 | 21.19 | 72.72 |
| Fraction 4 | 14.03 | 47.32 |
Figure 18.
Decay trace of 6 in PMMA film on 450 nm. Excitation wavelength: 355 nm in the presence and absence of TCT. Substance concentration; 0.15 mol·L-1; Excitation wavelength; 355 nm.
Figure 18.
Decay trace of 6 in PMMA film on 450 nm. Excitation wavelength: 355 nm in the presence and absence of TCT. Substance concentration; 0.15 mol·L-1; Excitation wavelength; 355 nm.
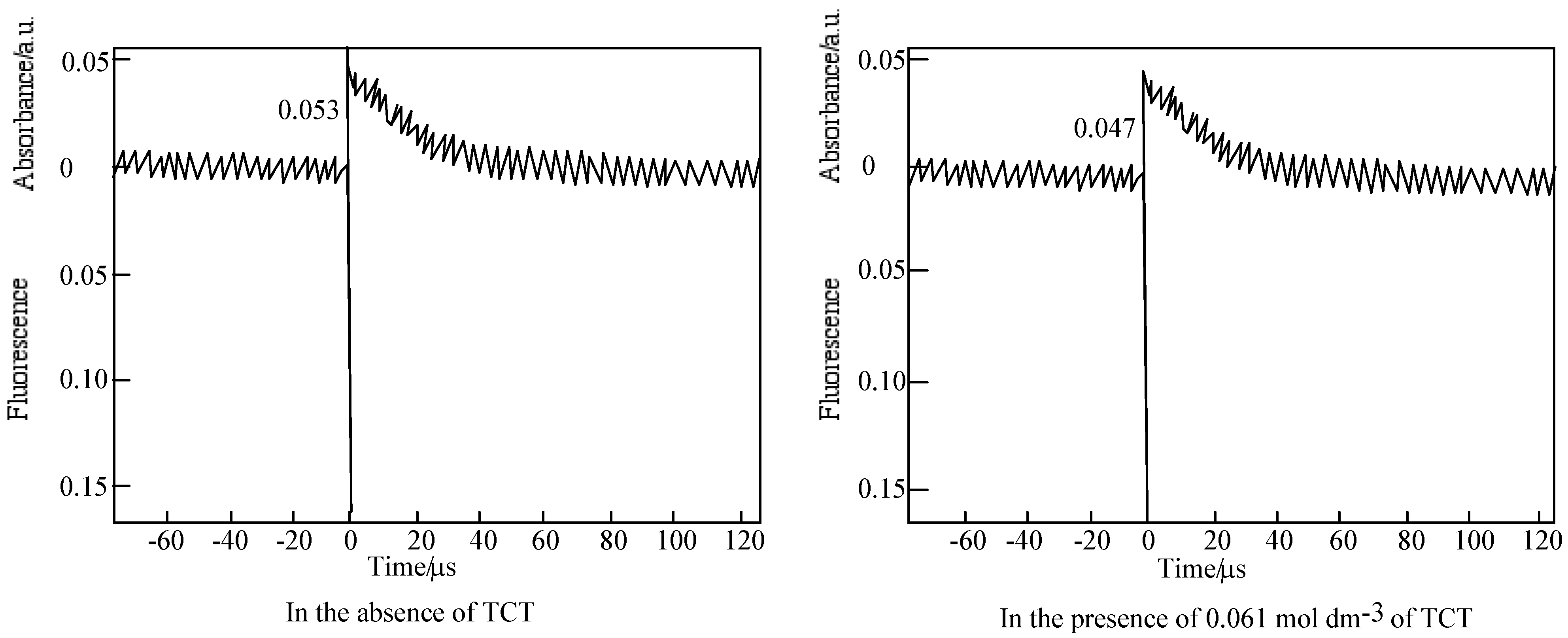
Figure 19.
Quenching of 6 by TCT in PMMA. (a) Fluorescence on 572 nm. Substance concentration: 0.03 mol·L-1; Excitation wavelength: 550 nm; (b) Triplet absorption intensity on 450 nm. Substance concentration: 0.15 mol dm−3; Excitation wavelength: 355 nm.
Figure 19.
Quenching of 6 by TCT in PMMA. (a) Fluorescence on 572 nm. Substance concentration: 0.03 mol·L-1; Excitation wavelength: 550 nm; (b) Triplet absorption intensity on 450 nm. Substance concentration: 0.15 mol dm−3; Excitation wavelength: 355 nm.
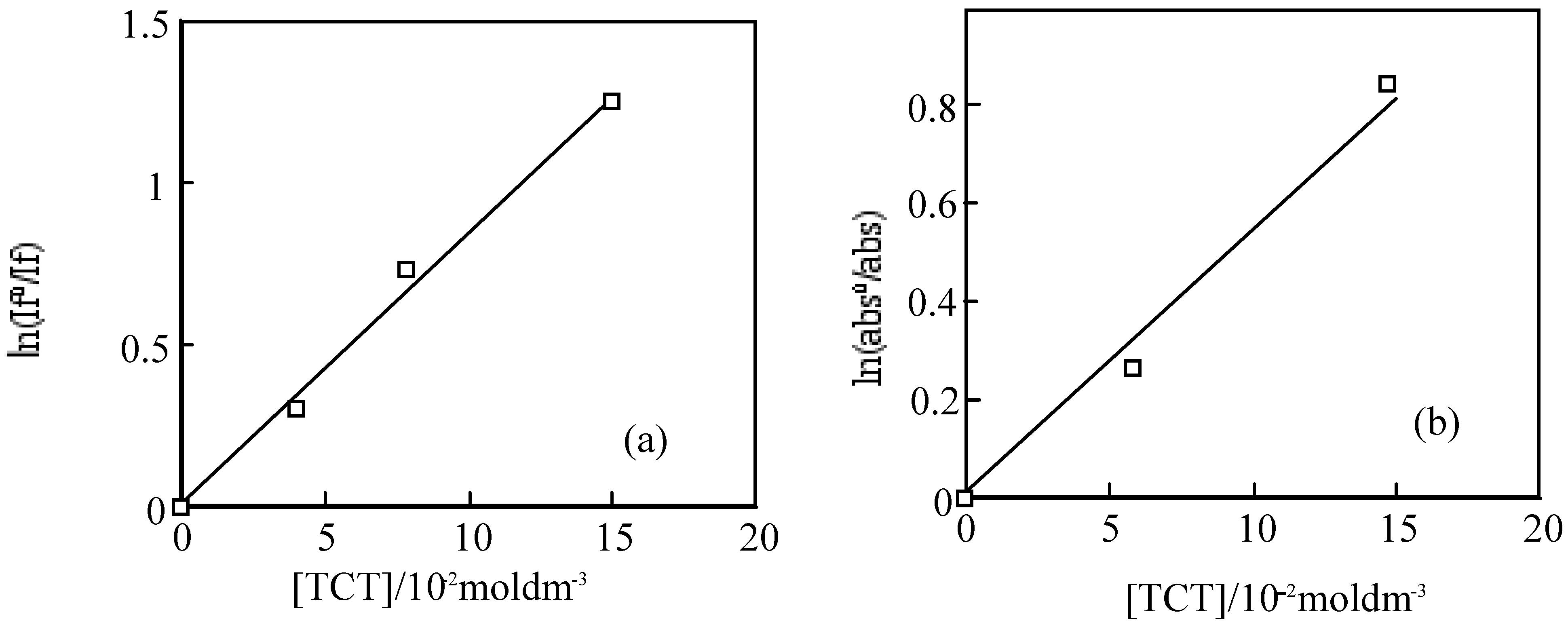
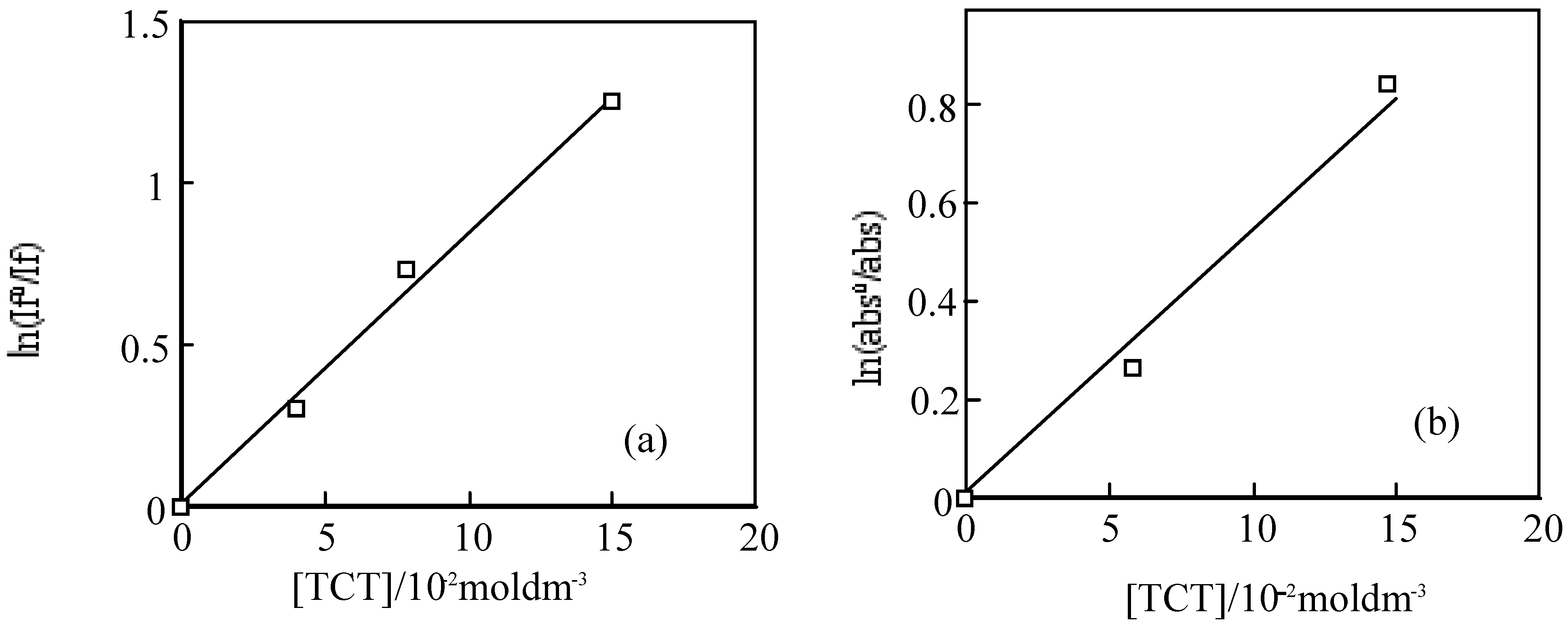
For the laser excitation of PMMA film at excitation of 550 nm using fluorescence spectrometer, the strongest fluorescence of 6 was efficiently quenched by TCT The fluorescence intensity was reduced with the concentration of TCT from 0 to 0.15 M. The fluorescence intensity is in direct proportion to the concentration of the TCT [80], the relationship gives a good straight line and it exhibits the Perrin type static quenching process [81] (Figure 19). The quenching radius (Rf) was determined from the slope of the logarithmic plot, and was estimated to be 15 nm.
The sensitization mechanisums between SubPC derivatives and TCT are thought as equations (28–41), where If0 and If are the fluorescence intensities in the absence and presence of the quencher TCT, τf0 is the lifetime in the absence of quencher, kq is the dynamic quenching rate constant, kqi is the static quenching constant, V is the quenching shere, V = 4πRf/3, N is Avogadro's number, Rf is the quenching radius, X = KiΣ[(SubPC−(i−1)TCT)] + 1. The mutistep equibrium between 6 derivatives and TCT is shown in Equations (28) and (29):
Equation (30) implies that SubPC derivatives and (SubPC–iTCT) are excited to an excited state *SubPC by absorbing laser pulse. Equations (31) and (32) mean that the *SubPC undergoes nonradiative and radiative deactivation. Then, the *SubPC reacted with TCT forms radicals such as chloro or dichloromethyl radicals as shown in Equation (33). Further, the SubPC becomes triplet state by intersystem crossing in Equation (34). The (SubPC–iTCT) also undergones (*SubPC–iTCT) by absorbing laser pulse (Equation 35). The activate state (*SubPC–iTCT) takes statically sensitized decomposition of TCT (Equation 36). Then, (*SubPC–iTCT) undergoes radiative, monoradiative deactivation and intercrossing as described in Equatons (38) and (39). The attention of fluorescence with the concentration of TCT in film is described as Equation (40). When (*SubPC–iTCT) does not emit fluorescence, meaning kqi is much larger than any decay rate. Equation (40) is described Equation (41) of the dynamic and static quenching terms.
If0 = {(1 + kq τ f0[TCT])(X + kqτf0)} / {X + kqτf0[TCT] + [Xkqiτf0 − kqτf0[TCT]exp(-VN[TCT])}
If0/If = (1 + kq τ f0[TCT]exp(VN)[TCT])
The triplet state of 6 in a PMMA films was also quenched by TCT. The initial absorptions were decreased with the concentration of TCT. The triplet life times measured almost the same. The logarithmic plots of the initial absorption at various concentration of TCT were fitted to the Perrin equation, the triplet quenching radius (Rt) were calculation to be 1.3 nm, which was almost same as Rf. The results indicate the triplet initial absorption was not quenched by TCT, since the triplet state generally came from the fluorescent singlet state, and the triplet is apparently reduced with the decrease of the singlet state.
The reduction and oxidation behavior of M-PCs are due to the interaction between the phthalocyanine ring and the central metal [40]. In the case of M-PCs, the porphyrazine ring in the molecule is influenced by the π electrons around the closed system. The 18−π electron system of M-PCs consists of one porphyrazine and four phynylene rings [27,32,50]. Substituents of M-PCs influence the 18 π electron environment in the molecule of the four phynylene rings, and are dependent on electron transfer properties of M-PCs. In the case of SubPC and its derivatives, redox potentials had various values. This implies the possibility of an electron transfer from the singlet to an excited state of 6 to the higher TCT state. The derivatives of 6 derivatives present similar phenomena to 6. It is suggested that the static-singlet-quenching process from the singlet photoexcited 6 to TCT is predominant in the sensitization.
2.4. Near infrared absorptions of 15
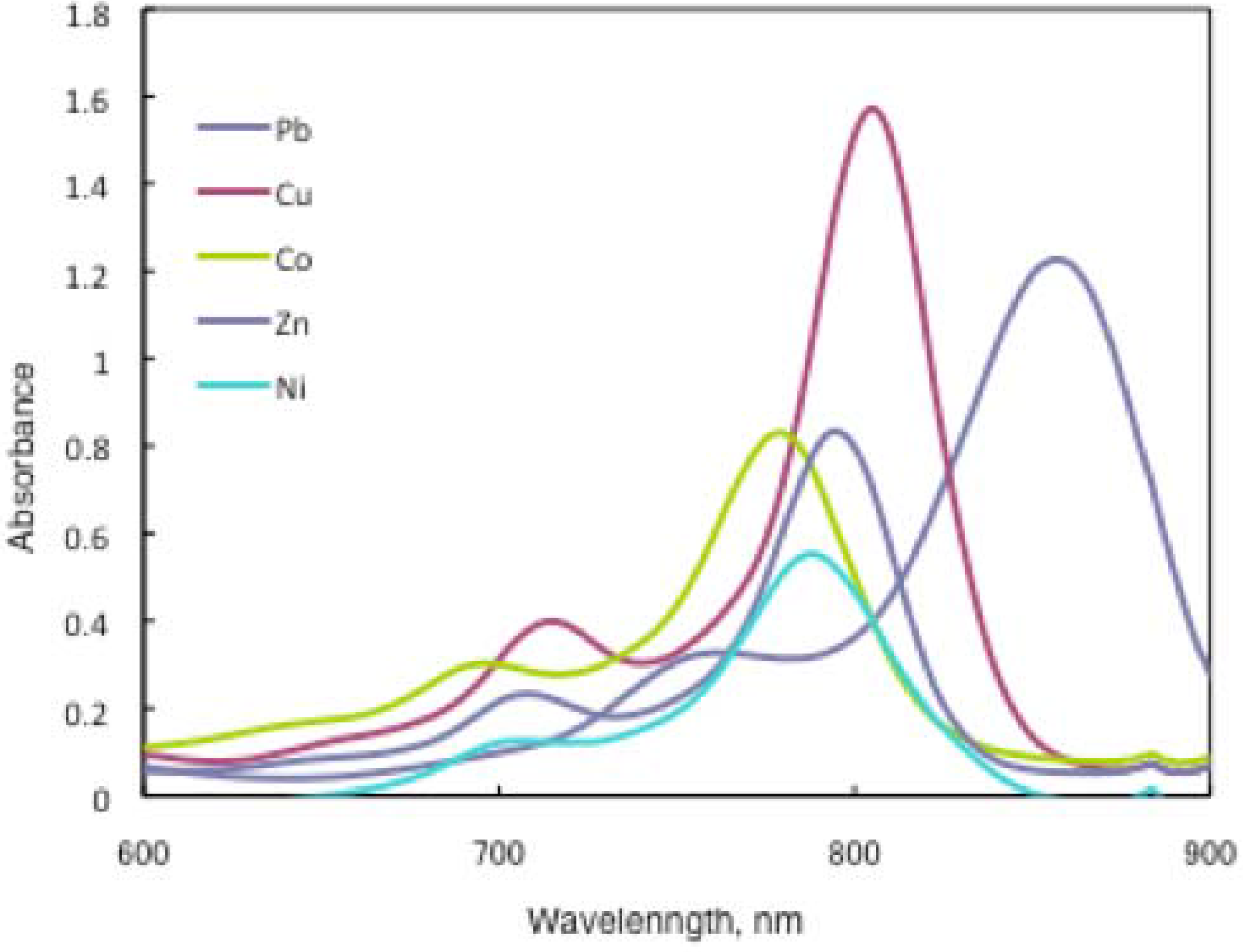
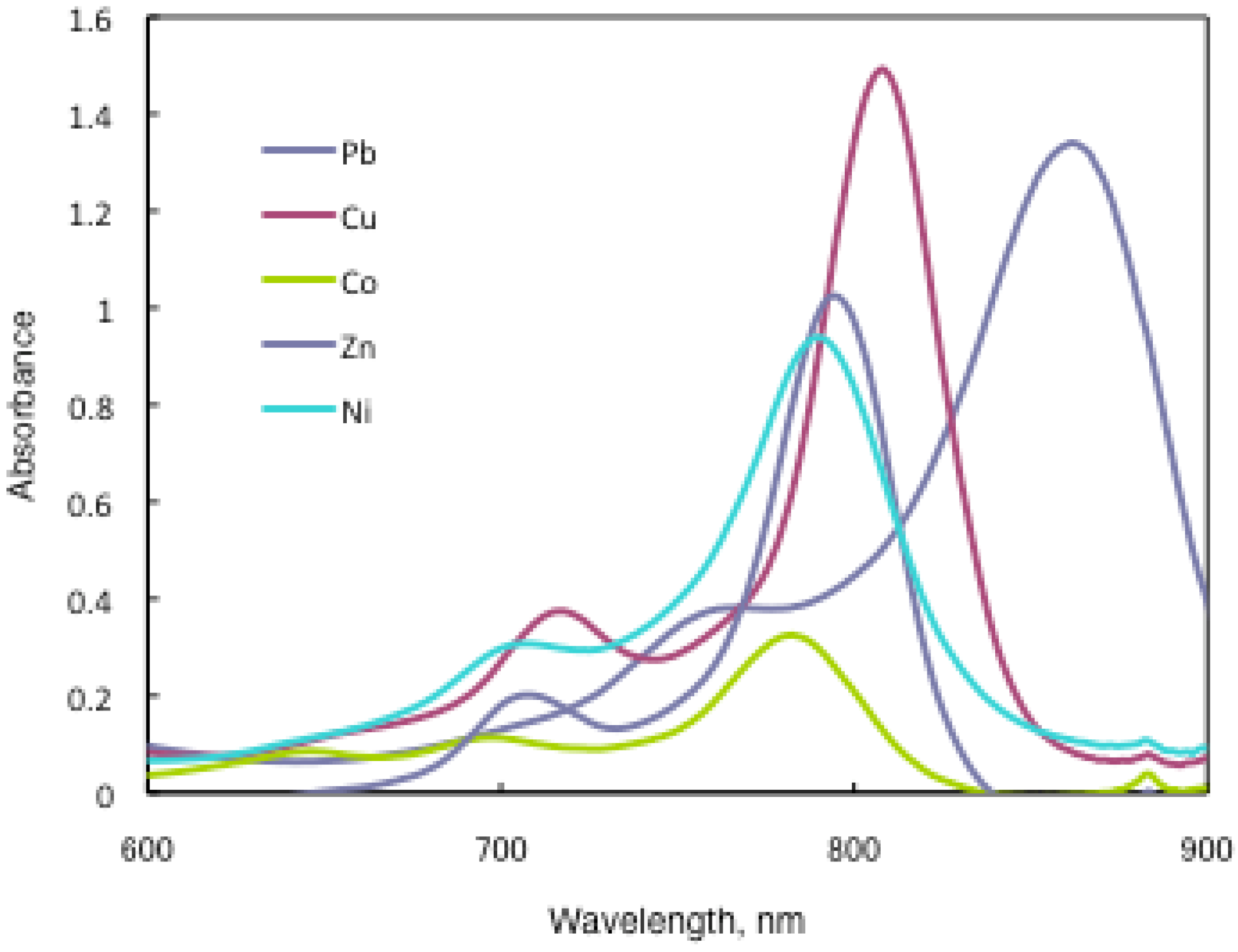
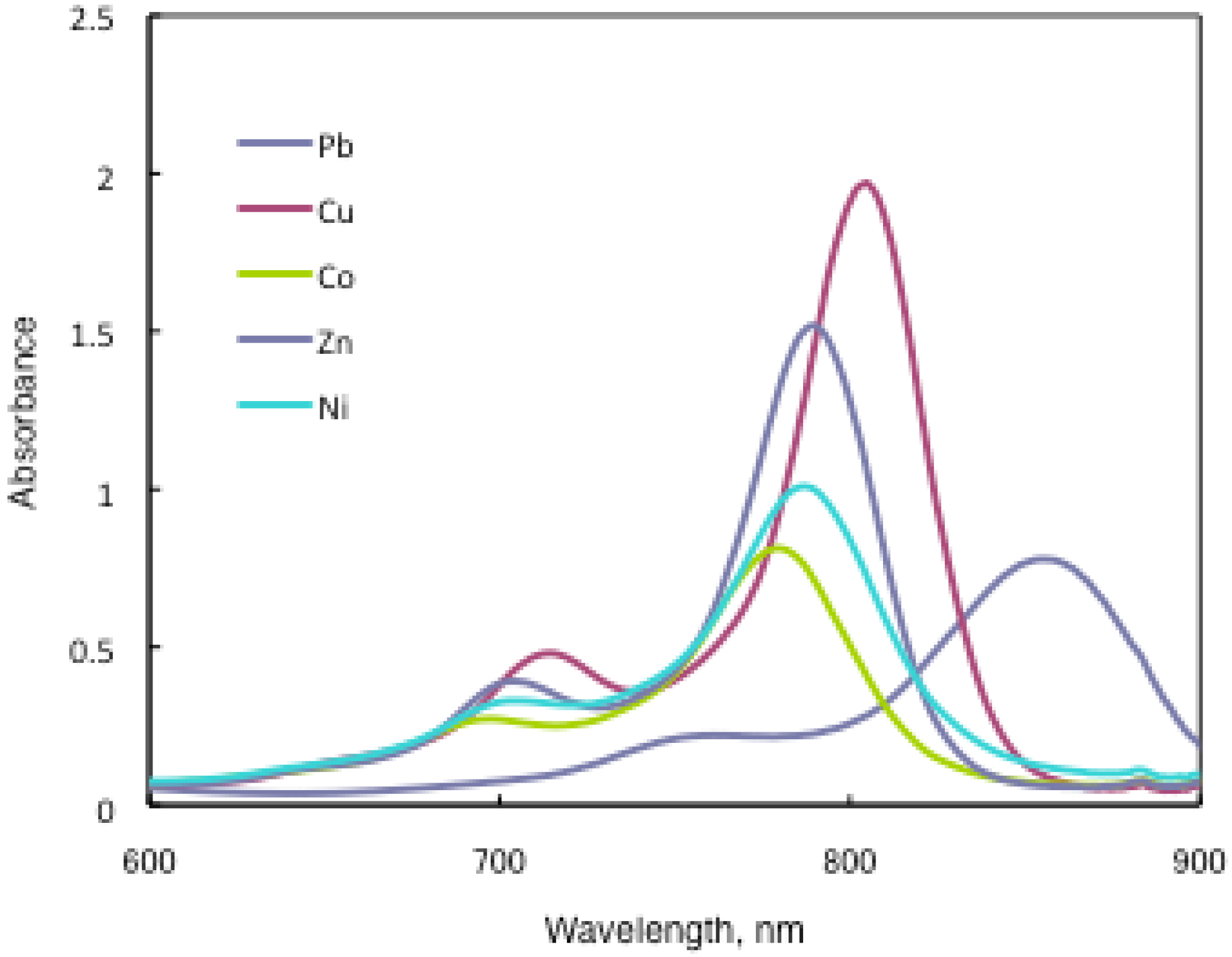
In substituted M- and H2-PCs, a strong absorption, termed the Q band, is detected in the visible region between 650 and 690 nm, and another in the UV-Vis between 320 and 370 nm, called the Soret band. A typical value for the extinction coefficient (ε) of the Q band is around 105 cm2· mol−1. The absorption spectra of the synthesized compounds 7 show typical shapes for M-PCs (Figure 20, Figure 21 and Figure 22). They also displayed strong absorption peaks in the visible region at around 800 nm. The strongest peaks in the visible region are assigned as the Q band, which were attributed to the allowed π−π∗ transition of the phthalocyanine ring. The Q band absorption of synthesized compounds 15 shifted by 100–150 nm to a longer wavelength in comparison to unsubstituted M-PCs.
Figure 20.
UV-Vis spectra of 15a.

Figure 21.
UV-Vis spectra of 15b.
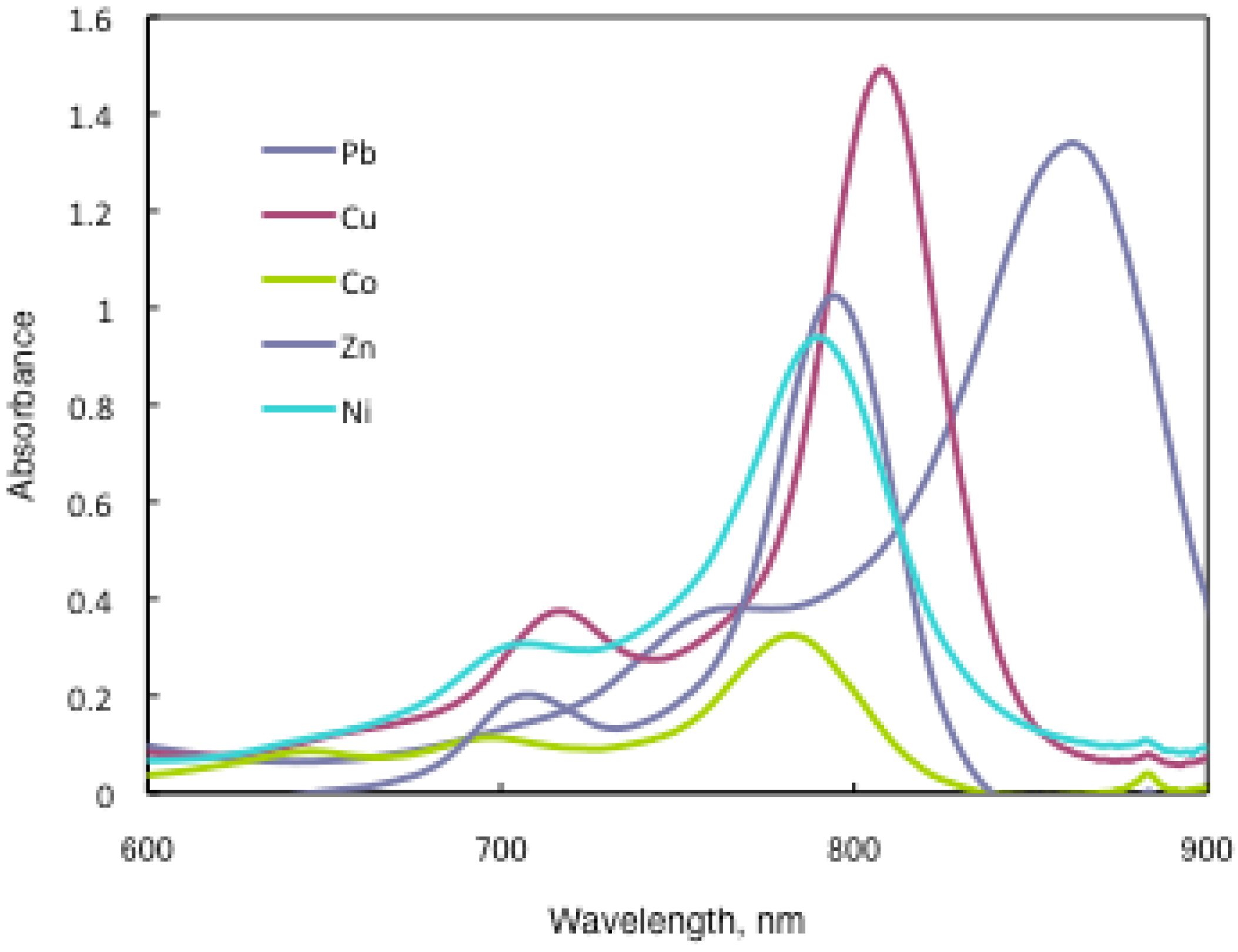
Figure 22.
UV-Vis spectra of 15c.
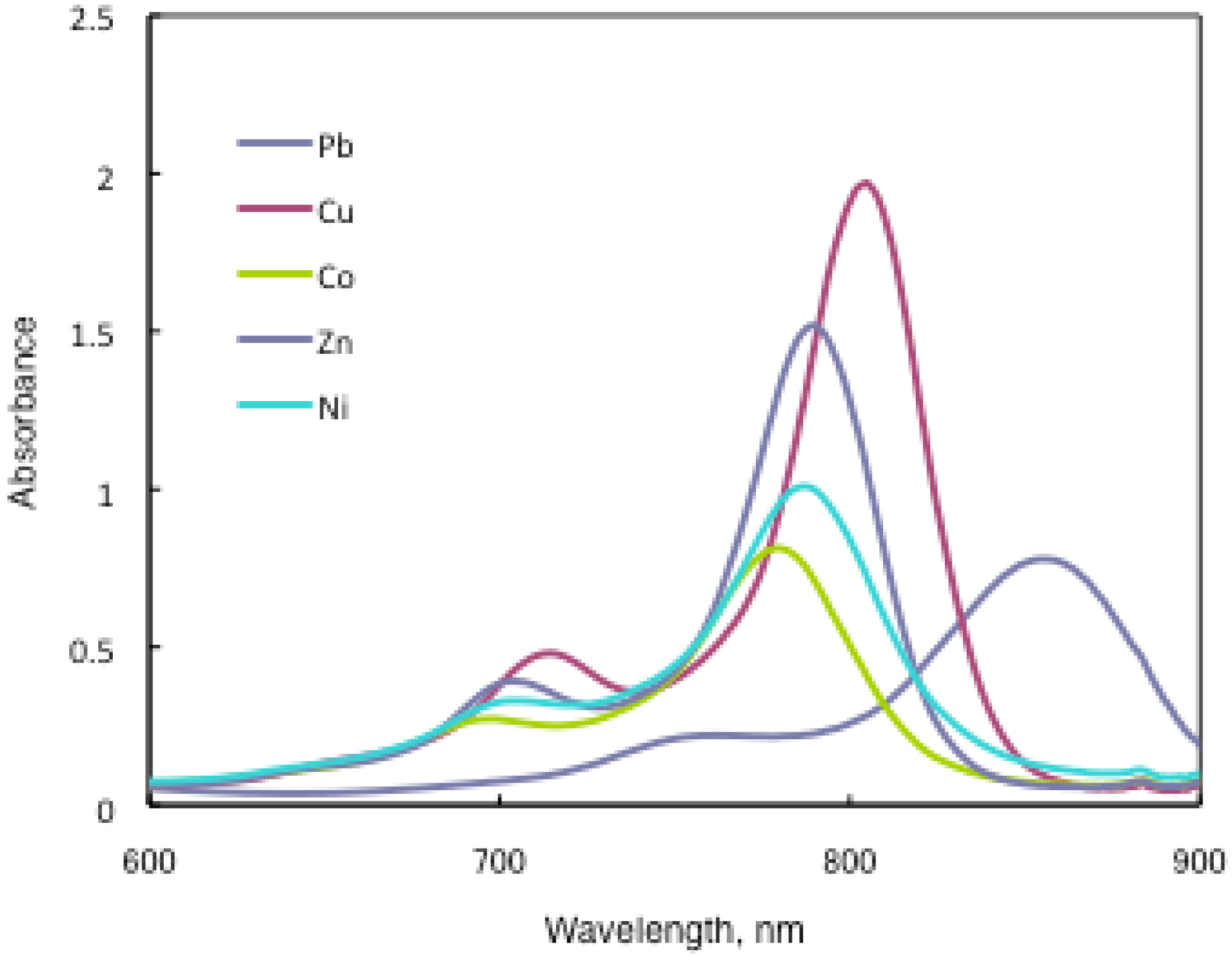
In the case of 15a, absorption maxima are moved to longer wavelengths in the order of Co, Ni, Cu, Zn, and Pb. Compounds 15b and 15c show similar phenomena. In comparison to the same central metal, non-peripheral substituted phthalocyanines, 15a, 15b, and 15c show longer wavelengths than peripheral compounds. Metal-free 15a, 15b, and 15c were also synthesized. The Q band absorption peaks of 15a, 15b, and 15c respectively appeared at longer wavelengths of 815, 820, and 816 nm. In general, H2-PCs show shorter wavelengths than corresponding M-PCs. Metal-free 15a, 15b, and 15c, which have remarkably bulky substituents, increase the distortion of the molecule because the four central cavity sizes cannot be fixed. Then, the Q band of metal-free 15a, 15b, and 15c were not split. The Q band is known to split into two peaks for high symmetry; the splitting Q band decreases with decreasing symmetry. Metal-free 15a, 15b, and 15c display decreased symmetry as a result of the molecular distortion. We leave a detailed discussion about these phenomena of Q band of metal-free 15a, 15b and 15c for another opportunity. The Q band absorption data of 15 are presented in Table 13.
| Solvent | Central metal | 15a | λmax / nm log ε 15b | 15c |
|---|---|---|---|---|
| Chloroform | Metal-free | 815 5.22 | 820 5.33 | 816 5.08 |
| Chloroform | Pb | 857 5.09 | 862 5.13 | 855 4.89 |
| Chloroform | Cu | 805 5.20 | 808 5.17 | 805 5.29 |
| Chloroform | Ni | 788 4.74 | 790 4.97 | 787 5.00 |
| Chloroform | Zn | 795 4.92 | 795 5.01 | 789 5.18 |
| Chloroform | Co | 779 4.91 | 783 4.51 | 780 4.91 |
| Toluene | Pb | 838 5.15 | 846 5.18 | 836 5.08 |
| Toluene | Cu | 791 5.01 | 796 5.06 | 790 5.01 |
| Toluene | Ni | 777 5.09 | 782 5.10 | 776 4.94 |
| Toluene | Zn | 792 4.20 | 790 4.34 | 782 4.86 |
| Toluene | Co | 775 4.71 | 778 4.89 | 774 4.89 |
The Q band shifts depend upon the change in the electron distribution in the phthalocyanine ring caused by substituents and their position. These results suggest that the steric hindrance arising from the substituted S-aryl groups appears to be as significant, as reported by Kobayashi [39]. However, a difference of the Q band in 15 is low between substituents, methyl, methoxy and tert-butyl. In this case, electron-donating substituents only slightly affect the movement to a longer wavelength. Although the effect of the central metal on the energy of Q band is usually small [20], absorption maxima of 3 are moved to longer wavelengths, and apparently increase with the ionic radius of the central metal, particularly lead. Lead complexes of 15 showed amplified structural distortion.
Because non-peripheral S-aryl substituted phthalocyanines having Pb as the central metal have an absorption band near 500 nm, 15a-Pb, 15b-Pb, and 15c-Pb give red solutions; the other central metals of 15a, 15b, and 15c give slightly reddish solutions.
3. Experimental Section
3.1. Equipment
UV-Vis spectra were measured on a Shimadzu UV-2400PC spectrometer. Each sample was prepared in toluene at 5.0 × 10−5 mol·L-1, in chloroform at 5.0 × 10−5 mol·L-1, in Py at 5.0 × 10−5 mol ·L-1. Fluorescent spectra were recorded in DMF on a Nihon Bunko Jasco FP-6600 spectrofluorometer. The 1H-NMR spectra were measured at 400 MHz on a Bruker Avance 400S and 90 MHz on a Nihon Denshi Joel EX-90 in dimethylsulfoxide-d6 (DMSO-d6), benzene-d6 or chloroform-d (CDCl3) using tetramethylsilane (TMS) as the internal standard. Elemental analysis was carried out using a Perkin-Elmer 2400CHN instrument. Mass spectra were taken with a Nihon Denshi Joel JMS-AX500 mass spectrometer. Melting points were measured with a Stanford Research Systems MPA100 Optimelt automated system.
3.2. Materials
All chemicals were purchased from Aldrich or Tokyo Chemical Industry Co. Ltd. They were used as received without further purification. For chromatographic separation, silica gel was used (60, particle size 0.063–0.200 nm, 7734-grade; Merck).
3.3. Synthesis of phthalocyanine tetrasulfonic acids 1-M (M = Fe, Co, Cu, Zn)
A mixture of 4-sulfophthalic acid (12.3 g, 50 mmol), urea (30.0 g, 0.50 mol), metal chloride (30 mmol) and DBU (1 g) as a catalyst dissolved in 150 mL of 1,2,4-trichlorobenzene as solvent was heated to 210 ºC for 180 min (Scheme 1). The product was filtered, washed with benzene until all 1,2,4-trichlorobenzene was removed and then dried under vacuum to a constant weight; 1-Fe (75%) Found: C, 43.20%; H, 1.99%; N, 12.45%. Calcd. for C32H16N8O12S4Fe: C, 43.25%; H, 1.81%; N, 12.60%. IR (KBr): νmax/cm−1 3000 (νC-H), 1700 (νC-C), 1450 (δC-H), 1380 (νS-O), 1050 (δC-H), 770 (δC-H). UV-Vis: λmax Py/nm: 688.5, 649.0, 354.0, 213.0; 1-Co (75%) Found: C, 42.92%; H, 2.08%; N, 12.67%. Calcd. for C32H16N8O12S4Co: C, 43.10%; H, 1.80%; N, 12.56%. IR (KBr): νmax/cm−1 3000 (νC-H), 1700 (νC-C), 1450 (δC-H), 1380 (νS-O), 1050 (δC-H), 770 (δC-H). UV-Vis: λmax Py/nm: 688.5, 649.0, 354.0, 213.0; 1-Cu (75%) Found: C, 42.86%; H, 1.67%; N, 12.45%. Calcd. for C32H16N8O12S4Cu: C, 42.88%; H, 1.79%; N, 12.50%. IR (KBr): νmax/cm-1 3000 (νC-H), 1700 (νC-C), 1450 (δC-H), 1380 (νS-O), 1050 (δC-H), 770 (δC-H). UV-Vis: λmax Py/nm: 688.5, 649.0, 354.0, 213.0; 1-Zn (75%) Found: C, 42.67%; H, 1.80%; N, 12.53%. Calcd. for C32H16N8O12S4Zn: C, 42.79%; H, 1.79%; N,12.47%. IR (KBr): νmax/cm-1 3000 (νC-H), 1700 (νC-C), 1450 (δC-H), 1380 (νS-O), 1050 (δC-H), 770 (δC-H). UV-Vis: λmax Py/nm: 688.5, 649.0, 354.0, 213.0.
3.4. Synthesis of phthalocyanine octacarboxylic acids 2-M (M = Fe, Co, Cu, Zn) (Scheme 2)
A mixture of pyromellitic dianhydride (2.50 g, 11.5 mmol), urea (13.0 g, 0.22 mol), metal chloride (23.5 mmol) and 0.1g of DBU was heated to 250 ºC until the reaction mixture was fused. The reaction product was washed with water, acetone and 6 M-hydrochloric acid (HCl). After drying, the solid obtained was hydrolyzed. Thirty grams of crude product, 30 g of KOH and 90 mL of water were heated for 480 min at 100 ºC. The mixture was diluted with 200 mL of water and filtered. The filtrate was acidified to pH 2 with concentrated HCl . At this point the product precipitated as a blue colored solid, which was separated from the solution by a centrifuge. The solid was washed with water three times, and dried. 2-Fe (yield: 30%) Found: C, 52.11%; H, 1.41%; N, 12.25%. Calcd. for C40H16N8O16Fe: C, 52.19%; H, 1.75%; N, 12.17%. IR (KBr): νmax/cm-1 3300 (νC-H), 2900 (νC-H), 1750 (νC-O), 1720 (νC-O), 1480 (νC-C), 1060 (δC-H), 720 (δC-H). UV-Vis: λmax Py/nm: 684; 2-Co (yield: 30%) Found: C, 52.44%; H, 1.74%; N, 12.25%. Calcd. for C40H16N8O16Co: C, 52.04%; H, 1.74%; N, 12.13%. IR (KBr): νmax/cm-1 3300 (νC-H), 2900 (νC-H), 1750 (νC-O), 1720 (νC-O), 1480 (νC-C), 1060 (δC-H), 720 (δC-H). UV-Vis: λmax Py/nm: 684; 2-Cu (yield: 30%) Found: C, 52.03%; H, 1.86%; N, 11.97%. Calcd. for C40H16N8O16Cu: C, 51.76%; H, 1.73%; N, 12.07%. IR (KBr): νmax/cm-1 3300 (νC-H), 2900 (νC-H), 1750 (νC-O), 1720 (νC-O), 1480 (νC-C), 1060 (δC-H), 720 (δC-H). UV-Vis: λmax Py/nm: 684; 2-Zn (yield: 30%) Found: C, 51.60%; H, 1.66%; N, 12.16%. Calcd. for C40H16N8O16Zn: C, 51.66%; H, 1.73%; N, 12.04%. IR (KBr): νmax/cm-1 3300(νC-H), 2900(νC-H), 1750 (νC-O), 1720 (νC-O), 1480 (νC-C), 1060 (δC-H), 720 (δC-H). UV-Vis: λmax Py/nm: 684.
3.5. Synthesis of octakis(hexoxymethyl)phthalocyanines 3-M (M=Fe, Co, Cu, Zn)
Compounds 3 were synthesized from a metal chloride and 1,2-dicyano-4,5-bis(hexoxymethyl)-benzene (10), which was synthesized from o-xylene via 1,2-dibromo-4,5-dimethylbenzene (7), 1,2-dibromo-4,5-bis(bromo-methylbenzene) (8) and 1,2-dibromo-4,5-bis(hexoxymethyl)benzene (9) (Scheme 3).
3.5.1. Synthesis of 1,2-dibromo-4,5-dimethylbenzene (7)
Bromine (500 g, 3.12 mol) was added dropwise to a mixture of o-xylene (166 g, 1.56 mol), iodine (2 g) and iron (3 g), during a 420 min period while stirring at 5-10 °C. After the reaction, the reaction mixture was poured into water and filtered. The precipitate was washed with a 3%-NaOH solution and recrystallized from methylcyclohexane; yield 49%. mp 86.6°C. Found: C, 36.18%; H, 2.64%. Calc for C8H8Br2: C, 36.66%; H, 3.08 %. IR (KBr): νmax/cm-1 2950 (νC-H), 1601 (νC-C), 1440 (νC-C), 1340 (νC-C), 1120 (νC-C), 880 (δC-H), 640 (δC-H). 1H-NMR (CDCl3): δ, ppm 2.10 (s, 6H), 7.36 (s, 2H). MS m/z = 264.
3.5.2. Synthesis of 1,2-dibromo-4,5-bis(bromomethylbenzene) (8)
A mixture of 7 (30.2 g, 0.11 mol) dissolved in 100 mL of carbon tetrachloride, 42.7 g (0.24 mol) of N-bromosuccinimide (NBS) and 0.08 g of 2,2’-azobis(isobutyro)nitrile was heated to 80 °C and refluxed for 480 min. After removing the carbon tetrachloride, the crude product was recrystallized from heptanes and dried under vacuum; yield 21%. mp 84.7°C, Found: C, 22.72%; H, 1.15%;. Calc for C10H10Br2: C, 22.78%; H, 1.43%. IR (KBr): νmax/cm-1 3000 (νC-H), 1650 (νC-C), 1460 (νC-C), 1200 (νC-C), 950 (δC-H), 620 (δC-H). 1H-NMR (CDCl3): δ, ppm 4.43 (s, 4H), 7.53 (s, 2H); MS m/z = 422.
3.5.3. Synthesis of 1,2-dibromo-4,5-bis(hexoxymethyl)benzene (9)
A piece of sodium was added to 120 g (1.18 mol) of hexanol. After the added sodium dissolved, 5.00 g (0.13 mol) of 8 was added. The temperature was gradually raised to 155 °C, and then the mixture was stirred for 90 min. The excess hexanol was removed by distillation and one hundred grams of water were added to the reaction mixture to dissolve the salt by-product. The resultant mixture was extracted with diethyl ether. The organic layer was washed with a small quantity of water and dried over calcium chloride. The diethyl ether was distilled off. The product 9 is a brown liquid; yield 88%. IR (KBr): νmax/cm-1 2900 (νC-H), 1670 (νC-C), 1240 (νC-C), 1050(δC-H), 630(δC-H). 1H-NMR (CDCl3): δ, ppm 0.89 (s, 6H), 1.28 (m, 16H), 3.52 (s, 4H), 4.45 (s, 4H ), 7.64 (s, 2H).
3.5.4. Synthesis of 1,2-dicyano-4,5-bis(hexoxymethyl)benzene (10)
A mixture of 15.2 g (33.8 mmol) of 1,2-dibromo-4,5-bis(hexoxymethyl) benzene (3), 13.6 g (96.6 mmol) of copper cyanide and 100 cm3 of DMF was refluxed for 300 minutes, then cooled, and poured into 170 cm3 of aqueous ammonia. The mixture was stirred for 10 min, and was filtered. The bluish residue was washed with water and dried. The product was extracted with diethyl ether for 1,440 min using a Soxhlet apparatus and the solvent was evaporated; yield 66%. mp 360.0°C, Found: C, 80.58%; H, 9.37%;. N, 8.26%; Calc for C22H32N2O2: C, 81.43%; H, 9.94%; N, 8.63%. IR (KBr): νmax/cm-1 3200 (νC-H), 2100 (νC-N), 1460 (νC-C), 1240 (νC-C), 1095(δC-H), 60(δC-H). 1H-NMR (CDCl3): δ, ppm 4.43 (s, 4H ), 7.53 (s, 2H). MS m/z = 357.
3.5.5. Synthesis of 3-M
Compounds 3-M were synthesized as follows: a mixture of 2.00 g (5.61 mmol) of 10, 11.8 mmol of metal chloride, 0.1 g of DBU, and 20 mL of hexanol as solvent was refluxed for 600 min at 155 ºC and then filtered; 3-Fe (yield: 10%) Found: C, 71.18%; H, 8.66%; N, 7.49%. Calcd. for C88H128N8O8Fe: C, 71.22%; H, 8.70%; N, 7.56%. IR (KBr): νmax/cm-1 3300 (νC-H), 2900 (νC-H), 1590 (νC-O), 1150 (νC-C), 1090 (δC-H), 720 (δC-H). UV-Vis: λmax Py/nm: 665. 3-Co (yield: 10%) Found: C, 71.18%; H, 8.63%; N, 7.43%. Calcd. for C88H128N8O8Co: C, 71.17%; H, 8.68%; N, 7.54%. IR (KBr): νmax/cm-1 3300 (νC-H), 2900 (νC-H), 1590 (νC-O), 1150 (νC-C), 1090 (δC-H), 720 (δC-H). UV-Vis: λmax Py/nm: 665. 3-Cu (yield: 10%) Found: C, 71.32%; H, 8.91%; N, 7.18%. Calcd. for C88H128N8O8Cu: C, 70.95%; H, 8.66%; N, 7.52%. IR (KBr): νmax/cm-1 3300 (νC-H), 2900 (νC-H), 1590 (νC-O), 1150 (νC-C), 1090 (δC-H), 720 (δC-H). UV-Vis: λmax Py/nm: 665. 3-Zn (yield: 10%) Found: C, 71.11%; H, 8.53%; N, 7.50%. Calcd. for C88H128N8O8Zn: C, 70.87%; H, 8.65%; N, 7.51%. IR (KBr): νmax/cm-1 3300 (νC-H), 2900 (νC-H), 1590 (νC-O), 1150 (νC-C), 1090 (δC-H), 720 (δC-H). UV-Vis: λmax Py/nm: 665.
3.6. Synthesis of Anthraquinocyanines 4-M (M= Fe, Co, Zn)
Compounds 4 are a new type of phthalocyanine derivatives which were synthesized from a metal chloride, urea and 9,10-anthraquinone-2,3-dicarboxylic acid (13), which was synthesized from o-xylene and phthalic anhydride via o-(3,4-dimethybenzoyl)benzoic acid (11) and 2,3-dimethyl-9,10-anthraquinone (12) (Scheme 4).
3.6.1. Synthesis of o-(3,4-dimethybenzoyl)benzoic acid (11)
Into a mixture of 16.3 g (0.11 mol) of phthalic dianhydride and 14 g of anhydrous aluminium chloride, 10.7 g of o-xylene in 25 mL of chloroform was added. The reaction mixture was refluxed for 60 min and the liquor then poured into a mixture of 30 g ice and 20 mL concentrated HCl, and then filtered. The product was recrystallized from diethyl ether; yield 81%. mp 170.6 °C. Found: C, 75.03%; H, 5.49%;. Calc for C16H14O3: C, 75.57%; H, 5.55 %. IR (KBr): νmax/cm-1 3450(νO-H), 2950 (νC-H), 1760 (νC-O), 1680 (νC-O), 1601 (νC-C), 1400 (νC-C), 1225 (νC-C), 1100 (νC-C), 900(δC-H), 710(δC-H). 1H-NMR (CDCl3): δ, ppm 2.27 (s, 6H), 7.68 (m, 7H). MS m/z = 254.
3.6.2. Synthesis of 2,3-dimethyl-9,10-anthraquinone (12)
Eighteen g of 11 and 2 mL of concentrated sulfuric acid were heated to 150 °C, then cooled and the crude product was recrystallized from benzene; yield 35%. mp 212.5 °C. Found: C, 80.89%; H, 5.02%; Calc for C16H12O3: C, 81.33%; H, 5.12 %. IR (KBr): νmax/cm-1 2900 (νC-H), 1760 (νC-O), 1675 (νC-O), 1420 (νC-C), 1300 (νC-C), 780(δC-H), 710(δC-H). 1H- NMR (CHCl3): δ, ppm 2.22 (s, 6H ), 7.46 (m, 7H). MS m/z = 236.
3.6.3. Synthesis of 9,10-anthraquinone-2,3-dicarboxylic acid (13)
A solution of 4g of 12 in 4.36 mL concentrated sulfuric acid was added to 160 g of water, and 12 g of potassium permanganate (KMnO4) was added gradually to the solution. Reaction was allowed to continue at 90–95 °C for 10 min with stirring; excess KMnO4 was then removed with oxalic acid. The crude product was filtered and washed with hot water. The product dissolved in hot dilute ammonia solution, unreacted 12 being filtered off. Product 13 was precipitated from the filtrate by addition of HCl. The material was filtered, washed with hot water and dried; yield 66%. mp 325.0 °C. Found: C, 64.26%; H, 5.03%;. Calc for C16H8O6: C, 64.87%; H, 4.72 %. IR (KBr): νmax/cm-1 3300 (νO-H), 2900 (νC-H), 1690 (νC-O), 1600 (νC-C), 1400 (νC-C), 1260 (νC-C), 980(δC-H), 780 (δC-H). 1H-NMR (CDCl3): δ, ppm 7.27 (s, 6H).
3.6.4. Synthesis of compounds 4-M
Compounds 4 were synthesized by the following procedure: a mixture of 1.00 g (3.30 mmol) of 13, 2 g (34.0 mmol) of urea, 6.75 mmol of metal chloride, and 0.1 g of DBU was heated to 210 ºC until the reaction mixture was fused. The reaction product was washed with water and was dried; 4-Fe (20%) Found: C, 70.60%; H, 2.22%; N, 10.29%. Calcd. for C64H24N8O8Fe: C, 70.96%; H, 2.28%; N, 10.13%. IR (KBr): νmax/cm-1 3300 (νC-H), 2900 (νC-H), 1690 (νC-O), 1470 (νC-C), 980 (δC-H), 780 (δC-H). UV-Vis: λmax Py/nm: 688. 4-Co (23%) Found: C, 70.46%; H, 2.22%; N, 10.27%. Calcd. for C64H24N8O8Co: C, 70.17%; H, 2.26%; N, 10.30%. IR (KBr): νmax/cm-1 3300(νC-H), 2900 (νC-H), 1690 (νC-O), 1470 (νC-C), 980 (δC-H), 780 (δC-H). UV-Vis: λmax Py/nm: 688. 4-Zn (22%) Found: C, 69.84%; H, 2.37%; N, 10.48%. Calcd. for C64H24N8O8Zn: C, 69.98%; H, 2.20%; N, 10.20%. IR (KBr): νmax/cm-1 3300 (νC-H), 2900 (νC-H), 1690 (νC-O), 1470 (νC-C), 980 (δC-H), 780 (δC-H). UV-Vis: λmax Py/nm: 688.
3.7. Synthesis of Alkylbenzopyridoporphyrazineporphyrazines 5
In the synthesis of 5, a mixture of 3,6-didecylphthalonitrile and 3,4-pyridinecarbodinitrile was dissolved in pentanol (7 mL) and zinc chloride (0.05 g) was added (Scheme 5). Zinc 1,4-didecylbenzo-tris(3,4-pyrido)porphyrazine (5b), zinc bis(1,4-didecylbenzo)-3,4-pyridoporphyrazine (5c) and zinc tris(1,4-didecylbenzo)-3,4-pyridoporphyrazine (5d) were synthesized from mixtures of 3,6-didecylphthalonitrile and 3,4-dicyanopyridine in 3 (0.18 g, 0.30 mmol):1 (0.02 g, 0.15 mmol), 1 (0.12 g, 0.29 mmol):1 (0.04 g, 0.29 mmol) and 1 (0.06 g, 0.15 mmol):3 (0.06 g, 0.45 mmol) ratios, respectively. Zinc tetra-3,4-pyridoporphyrazine (5a) and zinc octadecylphthalocyanine (5e) were synthesized from 3,4-dicyanopyridine [28,54] and 3,6-didecylphthalonitrile [33,59], respectively.
The mixtures were heated for 240 min in the presence of DBU as a catalyst. After cooling, the reaction mixture was dissolved in toluene (50 mL) and the solution filtered. The solvent was removed by evaporation. The products were separated and purified by TLC (eluent: toluene). 5a (29%) Found: C, 57.81%; H, 2.11%; N, 28.90%. Calcd. for C28H12N12Zn: C, 57.79%; H, 2.08%; N, 28.89%. IR (KBr): νmax/cm-1 2960 (νC-H), 1500 (νC-C), 1470 (νC-C), 1450 (νC-C), 1210 (δC-H), 1090 (δC-H), 790 (δC-H). UV-Vis: λmax Py/nm: 675; 5b (65%) Found: C, 68.31%; H, 6.22%; N, 17.90%. Calcd. for C49H53N11Zn: C, 68.32%; H, 6.22%; N, 17.89%. IR (KBr): νmax/cm-1 2960 (νC-H), 1600 (νC-C), 1460 (νC-C), 1440 (νC-C), 1210 (δC-H), 1080 (δC-H), 720 (δC-H). 1H-NMR (90 MHz, CDCl3): δ, ppm 7.4 (11.0H), 2.8 (4.2H), 1.3 (33.4H), 0.9 (6.7H). UV-Vis: λmax toluene/nm: 686. 5c (84%) Found: C, 73.67%; H, 8.30%; N, 12.28%. Calcd. for C70H94N10Zn: C, 73.68%; H, 8.30%; N, 12.28%. IR (KBr): νmax/cm-1 2970 (νC-H), 1600 (νC-C), 1500 (νC-C), 1470 (νC-C), 1450 (νC-C), 1210 (δC-H), 1090 (δC-H), 720 (δC-H). 1H-NMR (90 MHz, CDCl3): δ, ppm 7.4 (10.0H), 2.8 (8H), 1.3 (63.8H), 0.9 (12.3H). UV-Vis: λmax toluene/nm: 686. 5d (84%) Found: C, 76.94%; H, 9.57%; N, 8.86%. Calcd. for C91H135N9Zn: C, 76.94%; H, 9.58%; N, 8.88%. IR (KBr): νmax/cm-1 2970 (νC-H), 1600 (νC-C), 1500 (νC-C), 1470 (νC-C), 1210 (δC-H), 1100 (δC-H), 720 (δC-H). 1H-NMR (90 MHz, CDCl3): δ, ppm 7.4 (9.0H), 2.8 (12.3H), 1.3 (95.9H), 0.9 (18.8H). UV-Vis: λmax toluene/nm: 686. 5e (96%) Found: C, 79.12%; H, 10.41%; N, 6.61%. Calcd. for C112H176N8Zn: C, 79.12%; H, 10.43%; N, 6.59%. IR (KBr): νmax/cm-1 2970 (νC-H), 1600 (νC-C), 1500 (νC-C), 1470 (νC-C), 1210 (δC-H), 1100 (δC-H), 720 (δC-H). 1H-NMR (90 MHz, CDCl3): δ, ppm 7.4 (8.0H), 2.8 (16.0H), 1.3 (128.0H), 0.9 (24.1H). UV-Vis: λmax toluene/nm: 703.
The regioisomers of 5c were separated by TLC (Merk Silica gel 60 F254 on aluminium sheet, eluent: toluene - Py, 7:3) into four green- to blue-colored fractions. These fractions were numbered from 1, 2, 3 and 4, according to their Rf values of 0.95, 0.91, 0.75 and 0.65, respectively. Each fraction was recovered by scraping from the TLC plate, dissolved in pyridine, the solution filtered, and the solvent removed. Fraction 1: 1H-NMR (benzene-d6); δ ppm 0.93 (t, 12H), 1.21-1.88 (m, 48H), 1.88-2.23 (m, 8H), 2.29-2.69 (m, 8H), 4.13 (t, 4H), 4.32 (t, 4H), 7.35 (d, 2H), 7.49 (d, 2H), 8.30 (m, 6H). Fraction 2: 1H-NMR (benzene-d6); δ ppm 0.92 (t, 12H), 1.24-1.80 (m, 48H), 1.82-2.29 (m, 8H), 2.32-2.62 (m, 8H), 4.12 (t, 4H), 4.30 (t, 4H), 7.36 (d, 2H), 7.49 (d, 2H), 8.31 (m, 6H). Fraction 3: 1H-NMR (benzene-d6); δ ppm 0.92 (t, 12H), 1.16-1.95 (m, 48H), 1.95-2.20 (m, 4H), 2.20-2.36 (m, 4H), 2.36-2.70 (m, 8H), 3.99 (t, 4H), 4.23 (t, 4H), 7.36 (d, 2H), 7.49 (d, 2H), 8.30 (m, 6H). Fraction 4: 1H-NMR (benzene-d6); δ ppm 0.92 (t, 12H), 1.27-1.83 (m, 48H), 1.83-2.26 (m, 8H), 2.30-2.68 (m, 8H), 4.07 (t, 4H), 4.29 (t, 4H), 7.35 (d, 2H), 7.48 (d, 2H), 8.31 (m, 6H).
Quaternization of 5c was performed as follows: zinc 5c (0.17 g, 0.15 mmol) was reacted with quaternizing agents such as MCAA (0.57 g, 6 mmol), DES (0.1 g, 0.6 mmol) and DMS (0.2 g, 1.5 mmol), respectively, in DMF as solvent at 140°C for 120 min. The reaction mixture was dissolved in acetone (20 mL), cooled to room temperature and the solution filtered. The solvent was removed. The products were purified by TLC (eluent: THF-toluene, 8:2). Each product was recovered by scraping from the TLC plate, dissolving in Py, filtering the solution, and removal of the solvent. 5c with MCAA: dark blue solid (32 mg; yield 23%). 1H-NMR (DMSO-d6); δ ppm 0.85 (m, 12H, CH3), 1.19-1.71 (m, 48H, γ-CH2), 1.79-2.12 (m, 8H, β-CH2), 2.27-2.68 (m, 8H, β-CH2), 4.15 (m, 4H, α-CH2), 4.39 (m, 4H, α-CH2), 6.19 (s, 2H, CH2), 7.37 (m, 4H, arm), 8.32 (m, 6H, Py). IR (KBr): νmax/cm-1 3050 (νC-H), 2980 (νC-H), 1730 (νC-O), 1620 (νC-C), 1400 (νC-C), 1210 (δC-H), 1080 (δC-H), 790 (δC-H), 690 (δC-H). UV-Vis (λmaxtoluene/nm) 686: (λmaxPy/nm) 690: (λmaxwater/nm) 681; Found: C, 45.02%; H, 2.49%; N, 17.48%. Calcd. for C74H100N10O4Zn: C, 45.05%; H, 2.52%; N.17.50%. 5c with DES: blue solid (37 mg, yield 17%). 1H-NMR (DMSO-d6) δ, ppm 0.86 (m, 12H, CH3), 1.02-1.70 (m, 48H, γ-CH2), 1.88-2.11 (m, 8H, β-CH2), 2.30-2.68 (m, 8H, β-CH3), 4.11 (m, 4H, α-CH2), 4.25 (m, 4H, α-CH2), 7.38 (m, 4H, arom), 8.18 (m, 4H, Py). IR (KBr) νmax/cm-1 3050 (νC-H), 2960 (νC-H), 1500 (νC-C), 1450 (νC-C), 1400 (νC-C), 1340 (νS-O), 1180 (νS-O), 1250 (δC-H), 920 (δC-H), 760 (δC-H), 590 (δC-S). UV-Vis (λmaxtoluene/nm) 687: (λmaxPy/nm) 731: (λmaxwater/nm) 679. Found: C,37.10%; H, 1.78%; N. 18.49%. Calcd. for C70H98N10S2O6Zn: C, 37.11%; H, 1.78%; N, 18.54%. 5c with DMS: dark blue solid (53 mg, yield 28%). 1H-NMR (DMSO-d6): δ ppm 0.88 (m, 12H, CH3), 1.14-1.72 (m, 48H, γ-CH2), 1.82-2.17 (m, 8H, β-CH2), 2.29-2.62 (m, 8H, β-CH2), 4.06 (s, 6H, CH3), 4.27 (m, 4H, α-CH2), 4.51 (m, 4H, α-CH2), 7.34 (m, 4H, arom), 8.23 (m, 6H, Py); IR (KBr): νmax/cm-1 3060 (νC-H), 2980 (νC-H), 1500 (νC-C), 1450 (νC-C), 1400 (νC-C), 1250 (δC-H), 1100 (δC-H), 950 (δC-H), 830 (δC-H), 660 (δC-H). UV-Vis (λmaxtoluene/nm) 739: (λmaxPy/nm) 739: (λmaxwater/nm) 684. Found: C, 49.03%; H, 3.08%; N, 21.40%. Calcd. for C72H100N10Zn: C, 49.03%; H, 3.09%; N, 21.43%.
Quaternized regioisomers of 5c: Regioisomers of 5c (0.4 mg, 0.35 μmol) were reacted with a quaternizing agent such as DMS (0.5 mg, 3.5 μmol) in DMF as solvent at 140 °C for 120 min. The products were purified with TLC (eluent: THF-toluene, 8: 2). Each product was recovered by scraping from the TLC plate, dissolving in pyridine, filtering the solution and removal of the solvent. Fraction 1 with DMS: dark blue solid (0.053 mg, yield 13 %). 1H-NMR (DMSO-d6): δ ppm 0.86 (t, 12H, CH3), 1.15-1.70 (m, 48H, γ-CH2), 1.82-2.12 (m, 8H, β-CH2), 2.19-2.49 (m, 8H, β-CH2), 3.67 (s, 6H, CH3), 3.92 (t, 4H, α-CH2), 4.19 (t, 4H, α-CH2), 7.37 (m, 4H, arom), 8.20 (m, 6H, Py); IR (KBr): νmax/cm-1) 3070 (νC-H), 2980 (νC-H), 1500 (νC-C), 1400 (νC-C), 1250 (δC-H), 1100 (δC-H), 950 (δC-H), 820 (δC-H), 660 (δC-H); UV-Vis (λmax toluene/nm) 680: (λmax Py/nm) 703: (λmax water/nm) 690; Fluorescence (Fmax Py/nm) 687: (Fmax water/nm) 695. Found: C, 49.00%; H, 3.01%; N, 21.33%. Calcd. for C72H100N10Zn: C, 49.03%; H, 3.09%; N, 21.43%. Fraction 2 with DMS: dark blue solid (0.065 mg, yield 15 %). 1H-NMR (DMSO-d6): δ ppm 0.87 (t, 12H, CH3), 1.16-1.73 (m, 48H, γ-CH2), 1.73-2.10 (m, 8H, β-CH2), 2.10-2.55 (m, 8H, β-CH2), 3.90 (s, 6H, CH3), 3.99 (t, 4H, α-CH2), 4.17 (t, 4H, α-CH2), 7.23 (m, 4H, arom), 8.25 (m, 6H, Py). IR (KBr): νmax/cm-1 3070 (νC-H), 2980 (νC-H), 1500 (νC-C), 1400 (νC-C), 1250 (δC-H), 1100 (δC-H), 980 (δC-H), 830 (δC-H), 660 (δC-H); UV-Vis (λmax toluene/nm) 689: (λmax Py/nm) 675: (λmax water/nm) 686; Fluorescence (Fmax Py/nm) 684: (Fmax water/nm) 690; Found: C, 48.99%; H, 2.98%; N, 21.22%. Calcd. for C72H100N10Zn: C, 49.03%; H, 3.09%; N, 21.43%. Fraction 3 with DMS: dark blue solid (0.055 mg, yield 11 %). 1H-NMR (DMSO-d6): δ ppm 0.85 (m, 12H, CH3), 1.09-1.63 (m, 48H, γ-CH2), 1.70-2.11 (m, 8H, β-CH2), 2.11-2.42 (m, 8H, β-CH2), 3.86 (s, 6H, CH3), 3.97 (t, 4H, α-CH2), 4.21 (t, 4H, α-CH2), 7.23-7.33 (m, 4H, arom), 8.21 (m, 6H, Py). IR (KBr): λmax/cm-1 3080 (νC-H), 2970 (νC-H), 1500 (νC-C), 1400 (νC-C), 1250 (δC-H), 1100 (δC-H), 950 (δC-H), 830 (δC-H), 660 (δC-H). UV-Vis (λmax toluene/nm) 681: (λmax Py/nm) 689: (λmax water/nm) 689; Fluorescence (Fmax Py / nm) 690: (Fmax water / nm) 698; Found: C, 49.03%; H, 3.03%; N, 21.40%. Calcd. for C72H100N10Zn: C, 49.03%; H, 3.09%; N, 21.43%. Fraction 4 with DMS: dark blue solid (0.078 mg, yield 19 %). 1H-NMR (DMSO-d6): δ ppm 0.87 (t, 12H, CH3), 1.20-1.63 (m, 48H, γ-CH2), 1.71-2.17 (m, 8H, β-CH2), 2.17-2.47 (m, 8H, β-CH2), 3.85 (s, 6H, CH3), 4.09 (t, 4H, α-CH2), 4.27 (t, 4H, α-CH2), 7.21-7.36 (m, 4H, arom), 8.22 (m, 6H, Py). IR (KBr): λmax/cm-1 3070 (νC-H), 2980 (νC-H), 1500 (νC-C), 1400 (νC-C), 1250 (δC-H), 1100 (δC-H), 950 (δC-H), 830 (δC-H), 660 (δC-H); UV-Vis (λmax toluene/nm) 677: (λmax Py/nm) 730: (λmax water/nm) 673; Fluorescence (Fmax Py/nm) 686: (Fmax water/nm) 687; Found: C.49.02: H. 3.08: N. 21.43. Calcd. for C72H100N10Zn: C. 49.03: H. 3.09: N.21.43.
3.8. Synthesis of subphthalocyanine derivatives 6
Compound 6a was synthesized from 1,2-dicyanobenzene. Compound 6c was synthesized from 1,2-dicyano-3,4,5,6-tetrafluorobenzene. Compounds 6d-g were synthesized from the precursors, 1,2-dicyano-3,6-bis(thiobutyl)-4,5-difluorobenzene (14a) [36], 1,2-dicyano-3,4,5,6-tetrakis(thiobutyl)-benzene (14b) [36], 1,2-dicyano-3,6-bis(thiophenyl)-4,5-difluorobenzene (14c) [36] and 1,2-dicyano-3,4,5,6-tetrakis(thiophenyl)benzene (14d) [36], respectively.
Compound 6a: A solution of boron trichloride (20.5 mL, 0.02 mol, 1M in hexane) was added into a mixture of 1,2-benzenedicarbonitrile (5.0 g, 0.04 mol) and 1-chloronaphtahalene (12 mL) under an argon atmosphere at -3°C. After addition, the reaction mixture was heated to 120 °C with stirring. Hexane was distilled off. It was heated to 250°C for 10 minutes and cooled to room temperature. The product was extracted with petroleum ether for 1440 min and then toluene for 120 min on a Soxhlet extractor. The obtained product was recrystallized from ethanol (Scheme 6).
SubPC derivatives 6b-g were synthesized in accordance with above-mentioned SubPC synthetic method. 6a (71%) Found: C, 66.88%; H, 2.81%; N, 19.50%. Calcd. for C24H12N6BCl: C, 66.88%; H, 2.81%; N, 19.59%. IR (KBr): νmax/cm-1 3200 (νC-H), 3070 (νC-H), 2370 (νC-N), 1611 (νC-C), 1452 (νC-C), 1304 (νC-H), 1130 (δC-H), 950 (δC-S). 1H-NMR (CDCl3): δ, ppm 7.21 (s, 12H). MS m/z = 431. UV-Vis: λmax (toluene)/nm: 564. 6b (39%) Found: C, 72.18%; H, 6.06%; N, 14.01%. Calcd. for C36H36N6BCl: C, 72.18%; H, 6.06%; N, 14.01%. IR (KBr): νmax/cm-1 2980 (νC-H), 1720 (νC-C), 1620 (νC-C), 1450 (νC-H), 1400 (νC-H), 1360 (δC-H), 1280 (δC-H), 1190 (δC-H), 760 (δC-H). 1H-NMR (CDCl3): δ, ppm 7.80 (s, 3H), 7.49 (s, 3H), 7.32 (s, 3H), 1.35 (s, 9H). MS m/z = 599. UV-Vis: λmax toluene/nm: 566. 6c (26%) Found: C, 50.39%; N, 14.70%. Calcd. for C24N6BClF12: C, 50.39%; H, 0.00%; N, 14.70%. IR (KBr): νmax/cm-1 1490 (νC-H), 1450 (νC-N), 1270 (νC-F), 1220 (νC-F), 1120 (νC-H), 960 (δC-H), 800 (δC-H), 760 (δC-S). MS m/z = 647. UV-Vis: λmax toluene/nm: 572. 6d (8%) Found: C, 53.99%; H, 5.10%; N, 7.84%. Calcd. for C48H54N6BClF6S6: C, 53.99%; H, 5.10%; N, 7.87%. IR (KBr): νmax/cm-1 2980 (νC-H), 2150 (νC-N), 1600 (νC-C), 1500 (νC-C), 1450 (νC-C), 1150 (δC-H), 850 (δC-H). 1H-NMR (CDCl3): δ, ppm 3.15 (t, 12H), 1.51 (m, 24H), 0.90 (t,18H). MS m/z = 1068. UV-Vis: λmax (toluene)/nm: 596. 6e (5%) Found: C, 60.69%; H, 2.55%; N, 7.08%. Calcd. for C60H30N6BClF6S6: C, 60.66%; H, 2.55%; N, 7.08%. IR (KBr): νmax/cm-1 3050 (νC-H), 2250 (νC-N), 1580 (νC-C), 1450 (νC-C), 1250 (νC-F), 1090 (δC-H), 670 (δC-S). 1H-NMR (CDCl3): δ, ppm 7.26 (m, 30H). MS m/z = 1188. UV-Vis: λmax toluene/nm: 621. 6f (17%) Found: C, 58.07%; H, 7.31%; N, 5.65%. Calcd. for C72H108N6BClS12: C, 58.07%; H, 7.31%; N, 5.65%. IR (KBr): νmax/cm-1 2980 (νC-H), 2125 (νC-N), 1580 (νC-C), 1500 (νC-C), 1460 (νC-C), 1000 (νC-H), 800 (δC-H). 1H-NMR (CDCl3): δ, ppm 3.16 (t, 24H), 1.50 (m, 48H), 0.91 (t, 36H). UV-Vis: λmax toluene/nm: 612. 6g (13%) Found: C, 66.67%; H, 3.50%; N, 4.86%. Calcd. for C96H60N6BClS12: C, 66.67%; H, 3.50%; N, 4.86%. IR (KBr): νmax/cm-1 3080 (νC-H), 2190 (νC-N), 1600 (νC-C), 1500 (νC-C), 1460 (νC-C), 1000 (δC-H), 800 (δC-H). 1H-NMR (CDCl3): δ, ppm 7.26 (m, 60H). UV-Vis: λmax (toluene)/nm: 634.
3.9. Synthesis of octakis(thiophenyl)phthalocyanines 15
The target compounds 15 were synthesized in three steps via intermediates 16 and 17a-c. Intermediate 16 was synthesized by the following procedure: 2,3-dicyanohydroquinone (4.80 g, 30 mmol) in dichloromethane (100 mL) and Py (5.93 g, 75 mmol) was treated with trifluoromethanesulfonic anhydride (21.16 g, 75 mmol) under nitrogen at -78 °C. After the reaction, the mixture was allowed to warm slowly to room temperature; stirring was continued for 1,440 min. The mixture was poured into water (600 mL) and the organic layer was extracted using dichloromethane (5 × 100 mL). The extract was washed in turn with water, 2%-hydrochloric acid, water, brine and water, and dried on magnesium sulfate (MgSO4). The filtrate and the solvent evaporated. The crude product was recrystallized from dichloromethane to afford 16 (6.35 g, 50%) as colorless needles. Found: C, 28.32%; H, 0.48%; N, 6.59%. Calcd. for C10H2F6N2S2O6: C, 28.31%; H, 0.48%; F, 26.87%; N, 6.60%; O, 22.63%; S, 15.12%. IR (KBr): cm-1 3115 (νC-H), 2550 (νC-N), 1601 (νC-C), 1472 (νC-C), 1439 (νC-C), 1134 (νS=O). 1H-NMR (400 MHz, DMSO-d6): δ, ppm 8.44 (s, 2H).
Intermediates 17a-c were synthesized as follows: to a mixture of 16 (0.85 g, 2 mmol), potassium carbonate (1.16 g) and DMSO (15 mL), thiophenols such as p-toluenethiol, 4-methoxybenzenethiol and tert-butylthiophenol (4 mmol) were added; the mixture was reacted at room temperature for 1,440 min in nitrogen atmosphere. The reaction products were poured into water (300 mL), and the organic layer extracted using dichloromethane (5 × 100 mL), and dried on MgSO4. The filtrate and the solvent evaporated. The crude product was washed with methanol (3 × 50 cm3) and recrystallized from toluene to afford 17 as a yellow solid. 3,6-bis(thiophenylmethyl)phthalonitrile (2a) (0.27 g, 35%) Found: C, 70.90%; H, 4.30%; N, 7.50%. Calcd. for C22H16N2S2: C, 70.93%; H, 4.33%; N, 7.52%; S, 17.22. IR (KBr): cm-1 3050 (νC-H), 2970 (νC-H), 2218 (νC-N), 1600 (νC-C), 1535 (νC-C), 1490 (νC-C), 1435 (νC-C), 1210, 809 (δC-H). 1H-NMR (DMSO-d6): δ, ppm 7.54 (d, 4H), 7.46 (d, 4H), 7.35 (s2H), 2.66 (tt, 6H); 3,6-bis(thiophenylmethoxy)phthalonitrile (2b) (0.28 g, 34%) Found: C, 65.30%; H, 4.00%; N, 6.93%. Calcd. for C22H16N2S2O2: C, 65.32%; H, 3.99%; N, 6.93%; S, 15.82%; O, 7.91%. IR (KBr): cm-1 3050 (νC-H), 2970 (νC-H), 2216 (νC-N), 1600 (νC-C), 1540 (νC-C), 1487 (νC-C), 1430 (νC-C), 1210, 810 (δC-H). 1H-NMR (DMSO-d6): δ, ppm 7.49 (d, 4H), 7.06 (d, 4H), 7.04 (s2H), 3.79 (s, 6H); 3,6-bis(thiophenyl tert-butyl)phthalonitrile (2c) (0.38g, 42%) Found: C, 73.65%; H, 6.18%; N, 6.11%. Calcd. for C28H28N2S2: C, 73.64%; H, 6.18%; N, 6.13%; S, 14.04%. IR (KBr): cm-1 3040 (νC-H), 2960 (νC-H), 2210 (νC-N), 1600 (νC-C), 1500 (νC-C), 1460 (νC-C), 1210, 808 (δC-H). 1H-NMR (DMSO-d6): δ, ppm 7.50 (d, 4H), 7.46 (d, 4H), 7.26 (s, 2H), 1.28 (s, 18H).
The target compounds 15 were synthesized following procedure; A solution of 17 (0.25 mmol), the appropriate metal chloride or metal acetate (0.065 mmol), DBU (0.2 mL) as a catalyst and 1-pentanol (10 mL) was refluxed for 420 min. After cooling to room temperature, the reaction products were poured into methanol to form a precipitate, which was washed with water and methanol, and chromatographed on silica gel with toluene as eluent. The 1,4,8,11,15,18,22,25-octakis(thiophenyl-methyl)phthalocyanines (15a); 15a-Cu (0.02 g, 18%) Found: C, 67.98%; H, 4.51%; N, 7.01%. Calcd. for C88H65N8S8Cu: C, 67.99%; H, 4.21%; N, 7.21%; S, 16.50%; Cu, 4.09%. MS (FAB): m/z found 1554, Calcd. 1554.59: 15a-Co (0.02 g, 24%) Found: C, 68.10%; H, 4.20%; N, 7.25%. Calcd. for C88H65N8S8Co: C, 68.19%; H, 4.23%; N, 7.23%; S, 16.55%; Co, 3.80%. MS (FAB): m/z found 1550, Calcd. 1549.97: 15a-Ni (0.03 g, 28%) Found: C, 68.18%; H, 4.20%; N, 7.13%. Calcd. for C88H65N8S8Ni: C, 68.20%; H, 4.23%; N, 7.23%; S, 16.55%; Ni, 3.79%. MS (FAB): m/z found 1550, Calcd. 1549.73: 15a-Zn (0.02 g, 24%) Found: C, 67.87%; H, 4.23%; N, 7.11%. Calcd. for C88H65N8S8Zn: C, 67.91%; H, 4.21%; N, 7.20%; S, 16.48%; Zn, 4.20%. MS (FAB): m/z found 1556, Calcd. 1556.43: 15a-Pb (0.04 g, 34%) Found: C, 62.29%; H, 3.88%; N, 6.40%. Calcd. for C88H65N8S8Pb: C, 62.24%; H, 3.86%; N, 6.60%, S, 15.11%; Pb, 12.19%. MS (FAB): m/z found 1699, Calcd. 1698.24: 15a-H2 (0.04 g, 19%) Found: C, 70.85%; H, 4.66%; N, 7.51%. Calcd. for C88H66N8S8: C, 70.84%; H, 4.46%; N, 7.51%, S, 17.1%. MS (FAB): m/z found 1492, Calcd. 1492.04; 1,4,8,11,15,18,22,25-octakis(thiophenylmethoxy)phthalocyanines (15b); 15b-Cu (0.03 g, 27%) Found: C, 62.88%; H, 3.91%; N, 6.65%. Calcd. for C88H65N8S8 O8Cu: C, 62.82%; H, 3.89%; N, 6.66%; S, 15.25%; O, 7.61%; Cu, 3.77%. MS (FAB): m/z found 1682, Calcd. 1682.58: 15b-Co (0.02g, 21%) Found: C, 63.00%; H, 3.89%; N, 6.59%. Calcd. for C88H65N8S8O8Co: C, 62.99%; H, 3.90%; N, 6.68%; S, 15.29%; O, 7.63%; Co, 3.51%. MS (FAB): m/z found 1678, Calcd. 1677.97: 15b-Ni (0.03 g, 25%) Found: C, 63.01%; H, 3.92%; N, 6.67%. Calcd. for C88H65N8S8O8Ni: C, 63.01%; H, 3.92%; N, 6.67%; S, 15.29%; O, 7.63%; Ni, 3.49%. MS (FAB): m/z found 1678, Calcd. 1677.73: 15b-Zn (0.03 g, 29%) Found: C, 62.75%; H, 3.95%; N, 6.65%. Calcd. for C88H65N8S8O8Zn: C, 62.75%; H, 3.89%; N, 6.65%; S, 15.23%; O, 7.60%; Zn, 3.88%. MS (FAB): m/z found 1684, Calcd. 1684.42: 15b-Pb (0.04 g, 35%) Found: C, 57.88%; H, 3.59%; N, 6.15%. Calcd. for C88H65N8S8O8Pb: C, 57.88%; H, 3.59%; N, 6.14%; S, 14.05%; O, 7.01%; Pb, 11.33%. MS (FAB): m/z found 1827, Calcd. 1826.23: 15b-H2 (0.02 g, 8%) Found: C, 65.24%; H, 4.11%; N, 6.93%. Calcd. for C88H66N8O8S8: C, 65.24%; H, 4.11%; N, 6.92%, O, 7.90, S, 15.18%. MS (FAB): m/z found 1620, Calcd. 1620.03; 1,4,8,11,15,18,22,25-octakis-(thiophenyl tert-butyl)phthalocyanines (15c); 15c-Cu (0.02 g, 20%) Found: C, 71.13%; H, 6.05%; N, 5.87%. Calcd. for C112H113N8S8Cu: C, 61.13%; H, 6.02%; N, 5.92%; S, 13.56%; Cu, 3.37%. MS (FAB): m/z found 1891, Calcd. 1891.22: 15c-Co (0.02 g, 21%) Found: C, 71.24%; H, 6.04%; N, 5.93%. Calcd. for C112H113N8S8Co: C, 71.30%; H, 6.04%; N, 5.94%; S, 13.60%, Co, 3.12%. MS (FAB): m/z found 1886, Calcd. 1886.61: 15c-Ni (0.03 g, 23%) Found: C, 71.31%; H, 6.04%; N, 5.89%. Calcd. for C112H113N8S8Ni: C, 71.31%; H, 6.04%; N, 5.94%; S, 13.60%; Ni, 3.11%. MS (FAB): m/z found 1886, Calcd. 1886.37: 15c-Zn (0.03 g, 28%) Found: C, 71.00%; H, 6.01%; N, 5.92%. Calcd. for C112H113N8S8Zn: C, 71.06%; H, 6.02%; N, 5.92%; S, 13.55%; Zn, 3.45%. MS (FAB): m/z found 1893, Calcd. 1803.07: 15c-Pb (0.04 g, 34%) Found: C, 66.12%; H, 5.63%; N, 5.53%. Calcd. for C112H113N8S8Pb: C, 66.11%; H, 5.60%; N, 5.51%; S, 12.61%; Pb, 10.17%. MS (FAB): m/z found 2035, Calcd. 2034.88: 15c-H2 (0.02 g, 9%) Found: C, 73.55%; H, 6.26%; N, 6.13%. Calcd. for C112H114N8S8: C, 73.56%; H, 6.28%; N, 6.13%, S, 14.03%. MS (FAB): m/z found 1829, Calcd. 1828.68.
3.10. Electrochemical measurements
CV of metal octakis(hexoxymethyl)phthalocyanines and metal anthraquinocyanine were carried out with a BAS CV-50W voltammetric analyzer at room temperature in DMSO containing a 0.10 mol·L-1 solution of TBAP in a stream of nitrogen. CVs were recorded by scanning the potential at the rate of 50 mV s-1. The working and counter electrodes were platinum wires and the reference electrode was a Ag/AgCl saturated sodium chloride electrode. The chronoamperometry was measured following an applied voltage pulse from -1200 to 0 mV vs. Ag/AgCl and from 0 to +1600 mV vs. Ag/AgCl, and the reversible pulse.
3.11. Laser- flash photolysis
Laser flash photolysis in film was performed using a total reflection sapphire cell (10 · 30 mm, 1 mm thick, and both of the short side were cut at a 45 degree angle), which was spin-coated with a 1.2 μm thick photopolymer film. An excitation light pulse (20 ms, 355 nm and 10 mJ per pulse) from a YAG laser was expanded and exposed over the entire sample cell. A monitoring light from a xenon lamp passed through the multireflection cell which was connected to the head of an optical fiber attached to a monochrometer equipped with a photomultiplier or to a spectral multichannel analyzer system. The films were prepared as follows: a 10 wt% PMMA solution was made up in cyclohexanone, and allylbenzopyridoporphyrazines were added to this solution by dissolving to a thickness of 1.2 μm thick by spin-coating a solution onto a sapphire cell. After that the films were covered with a PVA solution.
4. Conclusions
M-PCs 1, 2, 3, and a new type of phthalocyanine derivative, 4 have been synthesized. Then, the cyclotetramerization products from a 1:1 mol mixture of 3,6-didecylphthalonitrile and 3,4-dicynopyridine, zinc bis(1,4-didecylbenzo)bis(3,4-pyrido)porphyrazines, 5 were synthesized. The cyclotetramerization products have position isomers. The isomers of 5c was separated into fore fractions. Moreover, metal phthalocyanine analogous, SubPCs and its derivatives which possessed substituents such as thiobutyl and thiophenyl were synthesized.
Electrochemical measurements were performed above mentioned phthalocyanine derivatives and analogous in order to examine their electron transfer abilities and electrochemical mechanism in an organic solvent.
Photochemical properties such as the triplet state lifetime were measured using laser-flash photolysis. The triplet state of zinc non-peripheral substituted alkylbenzopyridoporphyrazines, 5c and position isomers were measured, using laser-flash photolysis in a PMMA film. In generally, the triplet lifetime increased as the number of Py groups in the molecule increased. However, the lowest symmetric isomer having two Py rings in the molecule showed the longest triplet state lifetime. The compound was suitable for use as a photosensitizer in photodynamic cancer therapy.
Thiobutyl and thiophenyl substituted subPC derivatives 6 were measured sensitization properties. Sensitization properties of 6 with 2,4,6-tris(trichloromethyl)-1,3,5-triazine as radical generation reaction in a PMMA film have been investigated by laser-flash photolysis.
Phthalocyanines having S-aryl groups at non-peripheral position, i.e., 1,4,8,11,15,18,22,25-octakis(thio-phenylmethyl)phthalocyane compounds 15 can be synthesized. Their strong Q band absorption peaks appear around 800 nm. The synthesized phthalocyanines have high solubility in organic solvents, high heat resistance without discotic liquid crystal behavior, no absorptions in the visible region, and a simple structure without any regioisomers.
Acknowledgements
We thank Dr. Toshiyuki Urano (Science and Technology Research Center, Mitsubishi Chemical Corporation), Professor Dr. Michael J. Cook (University of East Anglia) and Emeritus professor Dr. Ryo Hirohashi (Chiba University) for their interest and helpful advice. We are grateful to our graduate students Mr. Taku Kato (Electronic Materials Research Laboratories, Nissan Chemical Industries), Mr. Masayoshi Kawaguchi, Ms. Tomomi Kawaguchi, Mr. Masaki Watanabe (U-Tech Corporation, Innovation Technology department), Ms. Seiko Kanazawa (Information & Communication Division, Toppan Printing Co. Ltd) and Mr. Hisashi Soga.
References
- Sakamoto, K.; Okumura, E.; Hirohashi, R. Phthalocyanine as Functional Dyes; IPC: Tokyo, Japan, 2004. [Google Scholar]
- de la Torre, G.; Vazquez, P.; Torres, T. Role of structural factors in the nonlinear optical properties of phthalocyanines and related componds. Chem. Rev. 2004, 104, 3723–3750. [Google Scholar]
- Cid, J.-J.; Yum, J.-H.; Jang, S.-R.; Nazeeruddin, M.K.; Martinez- Ferrero, E.; Palomares, E.; Ko, J.; Graetzel, M.; Torres, T. Molecular cosensitization for efficient panchromatic dye-sensitized solar cells. Angew. Chem. Int. Ed. 2007, 46, 8358–8362. [Google Scholar] [CrossRef]
- Kadish, K.M.; Smith, K.M.; Guilard, R. The Porphyrin Handbook; Academic Press: San Diego, CA, USA, 2003; Volumes 15-20. [Google Scholar]
- Pinzon, J.R.; Plonska-Brzezinska, M.E.; Cadona, C.M.; Athans, A.J.; Gayathri, S.S.; Guldi, D.M.; Herranz, M.A.; Martin, N.; Torres, T.; Echegoyen, L. Sc3N@C80-ferrocene electron-donor/acceptor conjugates as promising materials for photovoltalic applications. Angew. Chem. Int. Ed. 2008, 47, 4173–4176. [Google Scholar] [CrossRef]
- Campidelli, S.; Ballesteros, B.; Filoramo, A.; Diaz, D.; de la Torre, G.; Torres, T.; Rahman, G.M.A.; Aminur, E.C.; Kiessling, D.; Werner, F.; Sgobba, V.; Guldi, D.M.; Cioffi, C.; Prato, M.; Bourgoin, J-P. Facile decoration of functionalized single-wall carbon nanotubes with phthalocyanines via “Click Chemistry”. J. Am. Chem. Soc. 2008, 130, 11503–11509. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, T.J. Photodynamic Therapy. Phtotochem. Phthobiol. 1993, 58, 895. [Google Scholar]
- Moan, J. Properties for optimal PDT sensitizers. J. Photochem. Photobiol. B Biol. 1990, 5, 521–524. [Google Scholar] [CrossRef]
- Cook, M.J.; Chambrier, I.; Cracknell, S.J.; Mayes, D.A.; Russell, D.A. Octa-alkyl zinc phthalocyanines: Potential photosensitizers dor use in the photodynamic therapy of cancer. Phtotochem. Photobiol. 1995, 62, 542–545. [Google Scholar] [CrossRef]
- Linstead, R.P.; Noble, E.G.; Wright, J.M. Phthalocyanines IX. Derivatives of thiophene thionaphthene, pyridine and pyrazine, and a note on the nomencleature. J. Chem. Soc. 1937, 911–921. [Google Scholar] [CrossRef]
- Yokote, M.; Shibamiya, F; Shoji, S. Copper tetra-3,4-pyridinoporphyrazine from pyridine-3,4-dicarboxylic acid. J. Chem. Soc. Jpn. Ind. Chem Sec. 1964, 67, 166–168. [Google Scholar]
- Yokote, M.; Shibamiya, F; Hayakawa, H. Aza compounds XXIX. J. Synth. Org. Chem. Jpn. 1965, 23, 151–155. [Google Scholar] [CrossRef]
- de la Torre, G.; Claessens, C.G.; Torres, T. Phthalocyanines: Old dyes, new materials. Putting color in nanothechnology. Chem. Commun. 2007, 2000–2015. [Google Scholar] [CrossRef]
- Tokita, S.; Kojima, M.; Kai, N.; Kuroki, K.; Nishi, H.; Tomoda, H.; Saitoh, S.; Shiraishi, S. Synthesis and properties of 2,3,9,10,16,17,23,24-octaalkyl tetrpyrazinoporphyrazine. J. Chem. Soc. Jpn. 1990, 219–224. [Google Scholar]
- Yokote, M.; Shibamiya, F. Cu Bz-azaphthalocyanines. J. Chem. Soc. Jpn. Ind. Chem. Sec. 1959, 62, 224–227. [Google Scholar]
- Sakamoto, K.; Ohno, E. Synthesis of soluble metal phthalocyanine derivatives and their electron transfer prpperties. J. Soc. Dyers Colour. 1996, 112, 368–374. [Google Scholar] [CrossRef]
- Ohno, E.; Sakamoto, K. Synthesis of metal anthaquinocyqnine and its cyclic voltamograpy. J. Chem. Soc. Jpn. 1995, 730–735. [Google Scholar]
- Pawlowski, G.; Hanack, M. Synthesis and properties of a new kind of one-dimentional conduction Part 4. A convenient synthesis of octasubstituted phthalocyanines. Synthesis 1980, 287–289. [Google Scholar] [CrossRef]
- McKeown, N.B.; Chambrier, I.; Cook, M.J. Synthesis and Characterization of some 1,4,8,11,15,18,22,25-Octa-alkyl and 1,4,8,11,15,18-Hexa-alkyl-22,25-bis(carboxypropyl)phthalo-cyanines. J. Chem. Soc. Perkin Trans. 1 1990, 1169–1170. [Google Scholar] [CrossRef]
- Chambrier, I.; Cook, M.J.; Cracknell, S.J.; McMurdo, J. Synthesis and characterization of some non-uniformly substituted mesogenic phthalocyanines. J. Mater. Chem. 1993, 3, 841–849. [Google Scholar] [CrossRef]
- Sakamoto, K. Studies on phthalocyanine type compounds (Part 12). Solvent treatment of cobalt phthalocyanine and its pyridine adduct. J. Jpn. Soc. Colour Mater. 1995, 68, 67–74. [Google Scholar] [CrossRef]
- Weber, J.H.; Busch, D.H. Complexes derived from strong field ligands. XiX Magnetic properties of transition metal derivatives of 4,4’,4’’,4’’’-tetrasulfophthalocyanines. Inorg. Chem. 1965, 4, 469–471. [Google Scholar] [CrossRef]
- Claessens, C.G.; Gonzalez-Rodriguez, D.; Torre, T. Subphthalocyanines: Singular nonplanar aromatic compounds—Synthesis, reactivity, and physical properties. Chem. Rev. 2002, 102, 835–853. [Google Scholar] [CrossRef] [PubMed]
- Boston, D.R.; Bailar, J.C. Phthalocyanine derivatives from 1,2,4,5-tetracyanobenzene or pyromeliyic dianhydride and metal salt. Inorg. Chem. 1972, 11, 1578–1583. [Google Scholar] [CrossRef]
- Moeno, S.; Nykong, T. The photophysical studies of a mixure of CdTequantum dots and negativelycharged zinc phthalocyanines. Polyhedron 2008, 27, 1953–1958. [Google Scholar] [CrossRef]
- Ohno, E.; Sakamoto, K. Development of metal phthalocyanine derivatives fixed redox resin. J. Jpn. Soc. Colour Mater. 1998, 71, 83–90. [Google Scholar] [CrossRef]
- Hanack, M.; Beck, A.; Lehmann, H. Syntheses of liquid crystalline phthalocyanines. Synthesis 1987, 703–705. [Google Scholar]
- Sakamoto, K.; Ohno, E. Synthesis of cobalt phthalocyanine derivatives and their cyclic voltammograms. Dye. Pigment. 1997, 35, 375–386. [Google Scholar] [CrossRef]
- Freyer, W. Electornic spectra of tetra[2,3-(9,10-antraquino)]tetraazaporphyrin and related compounds. Zeitschrift Chem. 1986, 26, 216–217. [Google Scholar] [CrossRef]
- Markovitsi, D.; Lecuyer, I.; Simon, J. One-dimentional triplet energymigration in columnar liquid crystals of octasubstituted phthalocyanines. J. Phys. Chem. 1991, 95, 3620–3626. [Google Scholar]
- Tokita, S.; Matsuoka, M.; Kogo, Y.; Kihara, H. Kinouseishikiso no Bunshisekkei; [In Japanese]; Maruzen: Tokyo, Japan, 1989. [Google Scholar]
- Sakamoto, K.; Ohno, E. Synthesis and electron transfer property of phthalocyanine derivatives. Prog. Org. Coat. 1997, 31, 139–145. [Google Scholar] [CrossRef]
- Sakamoto, K.; Kato, T.; Cook, M.J. Position isomer separation of non-peripheral substituted zinc di-benzo-di(3,4-pyrido)porphyrazines. J. Porphyrin Phthalocyanine 2001, 5, 742–750. [Google Scholar] [CrossRef]
- Sakamoto, K.; Ohno-Okumura, E.; Kato, T.; Watanabe, M.; Cook, M.J. Investigation of zinc bis(1,4-didecylbenzo)-bis(2,3-pyrido)porphyrazines as an efficient photosensitizer by cyclic voltammetry. Dye. Pigment. 2008, 78, 213–218. [Google Scholar] [CrossRef]
- Rollmann, L.D.; Iwamoto, R.T. Electrochemistry, electron paramagnetic reonance, and visible spectra of cobalt, nickel and metal-free phthalocyanines in dimethyl sulfoxide. J. Am. Chem. Soc. 1968, 90, 1455–1463. [Google Scholar] [CrossRef]
- Ohno-Okumura, E.; Sakamot, K.; Kato, T.; Hatano, T.; Fukui, K.; Karatsu, T.; Kitamura, A.; Urano, T. Synthesis of subphthalocyanine derivatives and their characterization. Dye. Pigment. 2002, 53, 57–65. [Google Scholar] [CrossRef]
- Burnham, P.M.; Cook, M.J.; Gerrard, L.A.; Henney, M.J; Hughes, D.l. Structural characterization of a red phthalocyanine. Chem. Commmun. 2003, 2064–2065. [Google Scholar] [CrossRef]
- Sakamoto, K.; Ohno-Okumura, E.; Kato, T.; Soga, H. Synthesis on near infrared absorbed metal phthalocyanine with S-aryl groups at non-peripheral positions. J. Porphyrins Phthalocyanines 2009, in press. [Google Scholar]
- Kobayashi, N.; Fukuda, T.; Ueno, K.; Ogino, H. Extremely non-planer phthalocyanines with saddle or helical conformation: synthesis and structural characterization. J. Am. Chem. Soc. 2001, 123, 10740–10741. [Google Scholar] [CrossRef] [PubMed]
- Lever, A.B.P.; Licoccis, S.; Magrell, K.; Ramanamy, B.S. Mapping of the energy levels of metallophthalocyanines via electronic spectroscopy, electrochemistry and photochemistry. Adv. Chem. Ser. 1982, 210, 237–252. [Google Scholar]
- Kasuga, K.; Tsutsui, M. Some new developments in the chemistry of metallophthalocyanines. Coord. Chem. Rev. 1980, 32, 67–96. [Google Scholar] [CrossRef]
- Clack, D.W.; Hush, N.S.; Wolsey, I.S. Reduction potentials of some metal phthalocyanines. Inorg, Chem. Acta 1976, 129–132. [Google Scholar] [CrossRef]
- Orihara, Y.; Nishikawa, M.; Ohno, H.; Tsuchida, E.; Matsuda, H. Effect of substituents on the oxidation potential of phthalocyanines and electrocrystallization. Bull. Chem. Soc. Jpn. 1987, 60, 3731–3738. [Google Scholar] [CrossRef]
- Kadish, K.M.; Moninot, G.; Hu, Y.; Dubois, D.; Ibnlfassi, A.; Barbe, J-M.; Guilard, R. Double-decker actinide porphyrins and phthalocyanines. Synthesis and heteroleptic complexes. J. Am. Chem. Soc. 1993, 115, 8153–8166. [Google Scholar] [CrossRef]
- Nicholson, R.S.; Shain, I. Theory of stationaryelectrode polarography. Single scan and cyclic method applied to reversible, irreversible and kinetic system. Anal.Chem. 1964, 36, 706–723. [Google Scholar] [CrossRef]
- Lexa, D.; Reix, M. Redox behavior of metallic complexes of various tetrapyrrolic rings. Halfwave potentials of the complex. J. Chem. Phys. Phys-Chim. Biol. 1974, 71, 511–513. [Google Scholar]
- Wolberg, A.; Manassen, J. Electrochemical and electron paramagnetic resonance studies of metallophthalocyanines and their electrochemical oxidation products. J. Am. Chem. Soc. 1970, 92, 2982–2991. [Google Scholar] [CrossRef]
- Giraudeau, A.; Fan, F.-R.F.; Bard, A.J. Semiconductor electrode 30. Spectral sensitization of the semiconductors titanium oxide (n-TiO2) and tungsten oxide (n-WO3) with metal phthalocyanines. J. Am. Chem. Soc. 1980, 102, 5137–5142. [Google Scholar] [CrossRef]
- Shirai, A.; Kobayshi, N. Phthalocyanine Chemistry and Function; IPC: Tokyo, Japan, 1997. [Google Scholar]
- Sakamoto, K.; Ohno, E. Electrochemical characterization of soluble cobalt phthalocyanine derivatives. Dye. Pigment. 1998, 37, 291–306. [Google Scholar] [CrossRef]
- Sakamoto, K.; Ohno-Okumura, E. Synthesis of octasubstituted cobalt phthalocyanines and their redox properties. J. Porphyrins Phthalocyanines 1999, 3, 634–642. [Google Scholar] [CrossRef]
- Ohno, E.; Sakamoto, K. Electron transfer mechanism of metal anthraquinocyanine. J. Chem. Soc. Jpn. 1997, 58–67. [Google Scholar]
- Sakamoto, K.; Ohno-Okumura, E. Electrochemical characteristics of anthraquinocyanine and phthalocyanine derivatives. J. Jpn. Soc. Colour Mater. 1999, 72, 2–10. [Google Scholar] [CrossRef]
- Ohno-Okumura, E.; Sakamoto, K. Synthesis of metal octakis(alkoxymethyl)phthalocyanines and their redox behavior. J. Jpn. Soc. Colour Mater. 1999, 72, 345–354. [Google Scholar] [CrossRef]
- Lezonoff, C.C.; Lever, A.B.P. Phthalocyanines Properties and Applications; Wiley-VCH: New York, NY, USA, 1989-1993; Volumes 1-4. [Google Scholar]
- Wopschall, R.H.; Shain, I. Effects of adsorption of electroactive species in stationary electrode polarography. Anal. Chem. 1967, 39, 1514–1527. [Google Scholar] [CrossRef]
- Urano, T.; Nguyen, K.O.; Nagasaka, H.; Tsuchiya, M; Shimizu, S.; Itoh, H.; Senda, Y. Laser flash photolysis in high speed photopolymer coating layers. Bull. Chem. Soc. Jpn. 1994, 67, 1074–1077. [Google Scholar] [CrossRef]
- Urano, T.; Ohno-Okumura, E.; Sakamoto, K.; Itoh, H.; Yamaoka, T. Dye sensitization mechanisms in photopolymer layer for laser-printing plate. J. Photopolym. Sci. Tech. 1999, 12, 747–758. [Google Scholar] [CrossRef]
- Sakamoto, K.; Kato, T.; Kawaguchi, T.; Ohno-Okumura, E.; Urano, T.; Yamaoka, T.; Suzuki, S.; Cook, M.J. Photosensitizer efficacy of non-periperal substituted alkylbenzoporphyrazines for photodynamic therapy of cancer. J. Photochem. Photobiol. A Chem. 2002, 153, 245–252. [Google Scholar] [CrossRef]
- Darwent, J.R.; Douglas, P.; Harriman, A.; Porter, G.; Richoux, M.C. Metal phthalocyanines and porphyrins for reduction of water to hydrogen. Coord. Chem. Rev. 1982, 44, 83–126. [Google Scholar] [CrossRef]
- Ozoemen, K.; Kuznetsova, N.; Nyokong, T. Photosensitized transformation of 4-chlorophenol in the presence of aggregated and non-agregated metallophthalocyanines. J. Photochem. Photobiol. A Chem. 2001, 139, 217–222. [Google Scholar] [CrossRef]
- Murphy, S.T.; Konda, K.; Foote, C.S. Singlet-Oxygen-Sensitized delayed Fluorescence: Direct ditection of triplet phthalocyanines as an intermediate. J. Am. Chem. Soc. 1999, 121, 3751–3755. [Google Scholar] [CrossRef]
- Gerdes, R.; Wohrle, D.; Spiller, W.; Schmeider, G.; Schulz-Ekloff, G. Photo-oxidation of phenol and monochlorophenols in oxygen-saturated solutions by different photosensitizers. J. Photochem. Photobiol. A Chem. 1997, 111, 65–74. [Google Scholar] [CrossRef]
- Lang, K.; Wagnerova, D.M.; Brodilova, J. The role of excited states in the photosensitized oxidation of substrates with dioxygen. J. Photochem. Photobiol. A Chem. 1993, 72, 9–14. [Google Scholar] [CrossRef]
- Iliev, V.; Alexiev, V.; Bilyarska, L. Effect of metal phthalocyanine complex aggregation on the catalytic and photocatalytic oxidation of sulfer containing compounds. J. Mol. Catal. A Chem. 1999, 137, 15–22. [Google Scholar] [CrossRef]
- Lukyanets, E.A. Phthalocyanines as photosensitizers in the photodynamic therapy of cancer. J. Porphyrins Phthalocyanines 1999, 3, 424–432. [Google Scholar] [CrossRef]
- Kaliya, O.L.; Lukyanets, E.A.; Vorozhtsov, G.V. Catalysis and photocatalysis by phthalocyanines for technology, ecologyand medicine. J. Porphyrins Phthalocyanines 1999, 3, 592–610. [Google Scholar] [CrossRef]
- Edrei, R.; Gottfried, V.; Van Lier, J.E.; Kimel, S. Sulfonated phthalocyanines: Photophysical properties in vitro cell uptake and structure-active relationships. J. Porphyrins Phthalocyanines 1998, 2, 191–199. [Google Scholar] [CrossRef]
- Dhami, D.; Phillips, D. Comparison of the photophysics of an aggregtion and non-aggregating aluminium phthalocyanine system incorporated into unilamellar vesicles. J. Photochem. Photobiol. A Chem. 1996, 100, 77–83. [Google Scholar] [CrossRef]
- Spiller, W.; Kliesch, H.; Wohrle, D.; Hackbarth, S.; Roder, B.; Schmurpfeil, G. Singlet oxygen quantum yield of different photosensitizers in polar solvents and micellar solutions. J. Porphyrins Phthalocyanines 1998, 2, 145–158. [Google Scholar] [CrossRef]
- Tokumaru, K. Photochemical and photophysical behavior of porphyrins and phthalocyanines irradiated with violet or ultraviolet light. J. Porphyrins Phthalocyanines 2001, 5, 77–86. [Google Scholar] [CrossRef]
- Seotsanyana-Mokhoshi, I.; Kuznetsova, N.; Nyokong, T. Photochemical studies of tetra-2,3-pyridinoporphyrazine. J. Potochem. Photobiol A Chem. 2001, 140, 215–222. [Google Scholar] [CrossRef]
- Wohrle, D.; Gitzel, J.; Okura, I.; Aono, S. Photoredox properties of tetra-2,3-pyridinoporphyrazines (29H, 31H-tetrapyrido[2,3:b: 2’,3’-g:2”,3”-I:2’’’,3’’’-q]porphyrazines. J. Chem. Soc., Perkin Trans. II 1985, 1171–1178. [Google Scholar] [CrossRef]
- Yhamae, M.; Nyokong, T. Cobalt (II) porphyrazines catalyzed reduction of nitriles. J. Electroanal. Chem. 1999, 470, 126–135. [Google Scholar] [CrossRef]
- Hanack, M.; Meng, D.; Beek, A.; Sommerauer, M.; Subramanian, L.R. Separation of structural isomer of tetra-tert-butylphthalocyaninate nickel(III). J. Chem. Soc. Chem. Commun. 1993, 58–60. [Google Scholar] [CrossRef]
- Sommerauer, M.; Rager, C.; Hanack, M. Separation of 2(3), 9(10), 16(17), 23(24)-tetrasubstituted phthalocyanines with newly developed HPLC phases. J. Am. Chem. Soc. 1996, 118, 10085–10093. [Google Scholar] [CrossRef]
- Chan, W.-S.; Brasseur, N.; LaMadeleine, C.; Ouellet, R.; Lier, J. Efficacy and mechanism of alminium phthalocyanine and its sulphonated derivatives mediated photodynamic therapy on murine tumours. Eur. J. Cancer 1997, 33, 1855–1859. [Google Scholar] [CrossRef] [PubMed]
- Negrimouski, V.M.; Bouvet, M.; Lukyanets, E.A.; Simon, J. Towerd chiral 1,2-naphthalocyanines: Syntheses of the pure geometrical isomers of the magnesium and lutetium complexes. J. Porphyrins Phthlocyanines 2000, 4, 248–255. [Google Scholar] [CrossRef]
- Rihter, B.D.; Bohorquez, M.D.; Rodgers, M.A.J.; Kenny, M.E. Two new sterically hindered phthalocyanines: synthetic and photodynamic aspect. Photochem. Photobiol. 1992, 55, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.; Plenzig, F.; Rauschanbel, J.; Hanack, M.; Rey, B.D.; Satre, A.; Torres, T. Subphthalocyanines. Preparation, reactivity, and physical properties. Synthesis 1996, 1139–1151. [Google Scholar] [CrossRef]
- Birks, J.B. Photophysics of Aromatic Molecules; Wiley: New York, NY, USA, 1970. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sakamoto, K.; Ohno-Okumura, E. Syntheses and Functional Properties of Phthalocyanines. Materials 2009, 2, 1127-1179. https://doi.org/10.3390/ma2031127
AMA Style
Sakamoto K, Ohno-Okumura E. Syntheses and Functional Properties of Phthalocyanines. Materials. 2009; 2(3):1127-1179. https://doi.org/10.3390/ma2031127
Chicago/Turabian StyleSakamoto, Keiichi, and Eiko Ohno-Okumura. 2009. "Syntheses and Functional Properties of Phthalocyanines" Materials 2, no. 3: 1127-1179. https://doi.org/10.3390/ma2031127