Structure-Composition-Property Relationships in Polymeric Amorphous Calcium Phosphate-Based Dental Composites †

Abstract
:1. Introduction
2. Results and Discussion

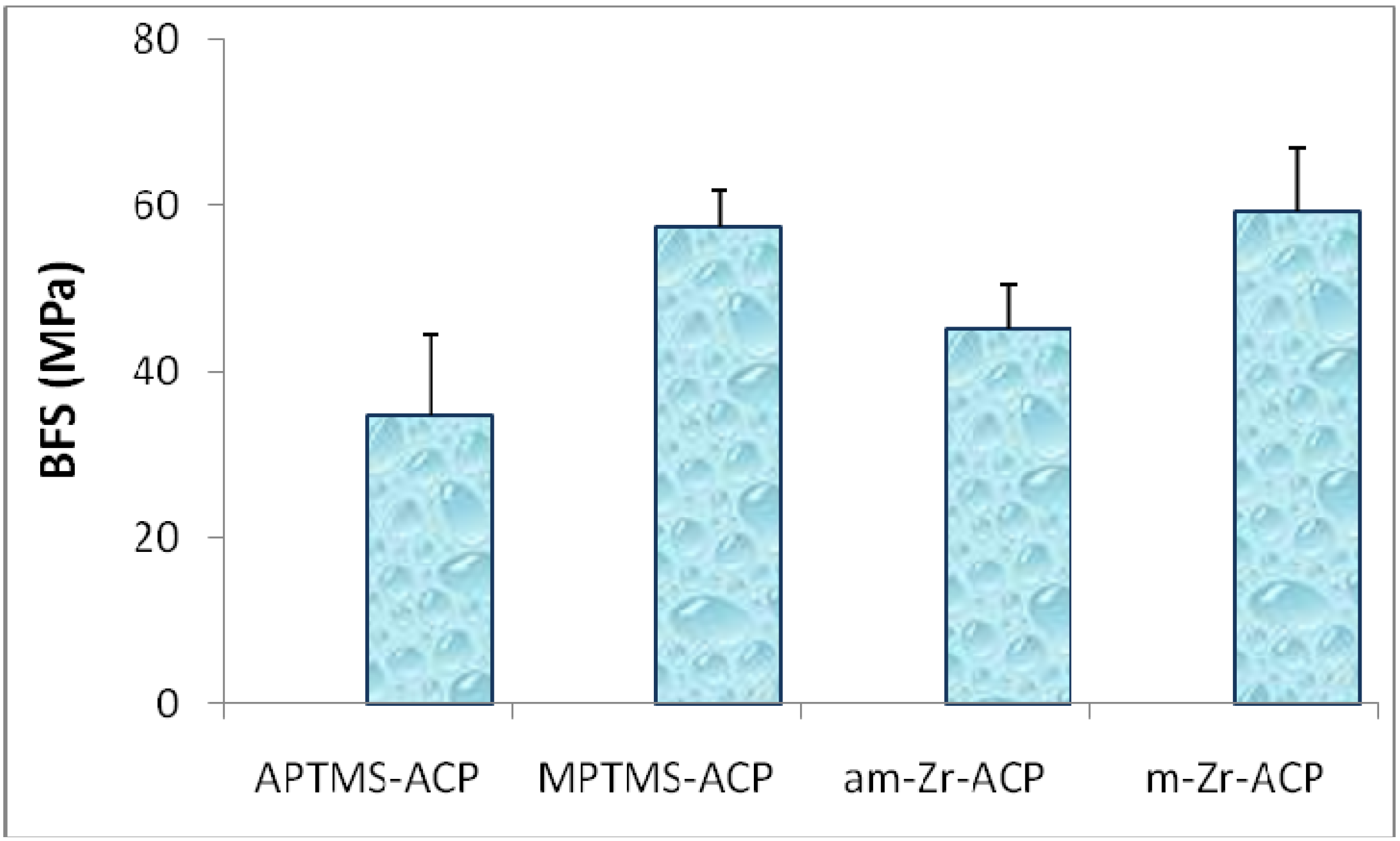{kind=link}
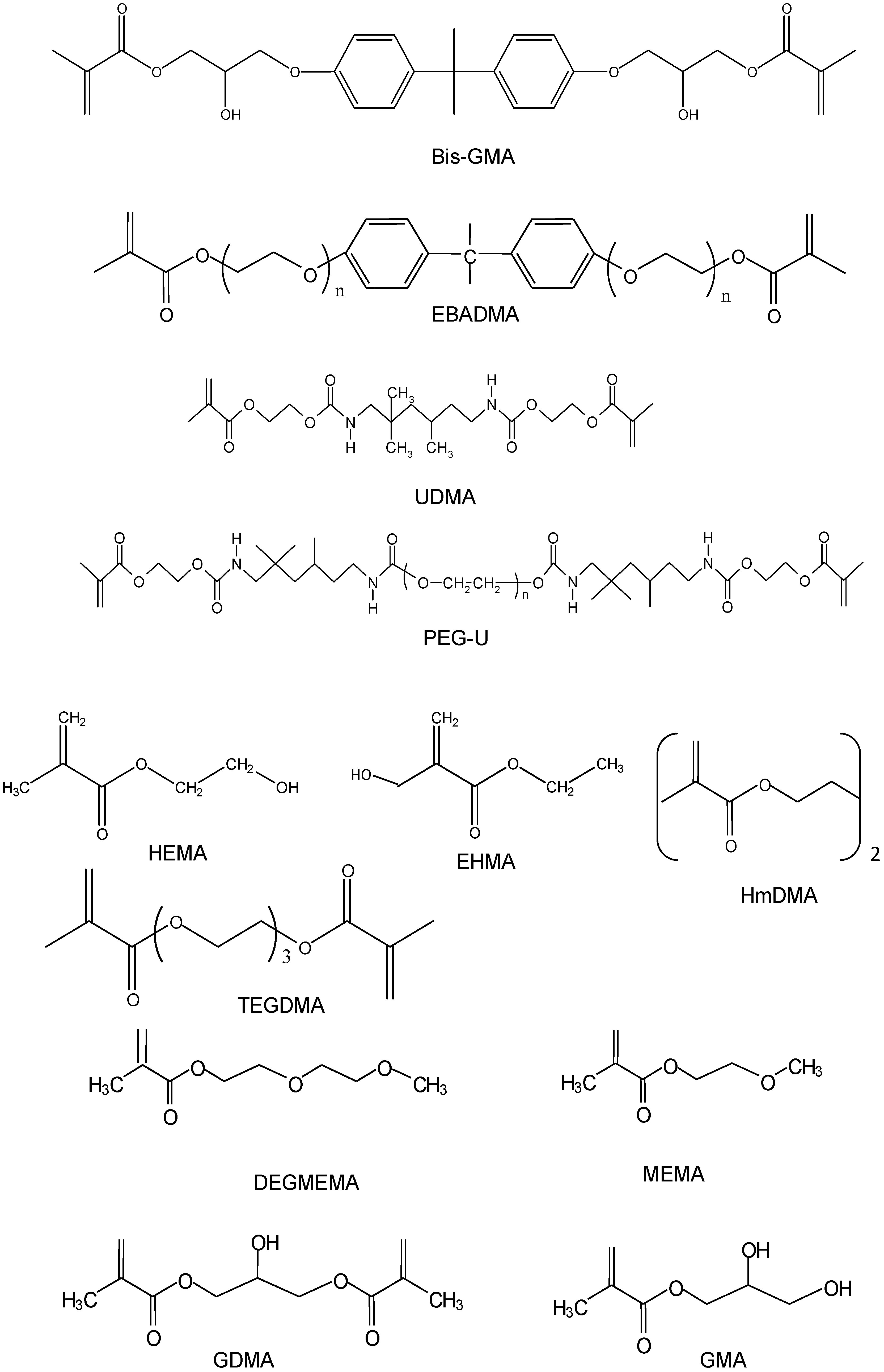{kind=link}
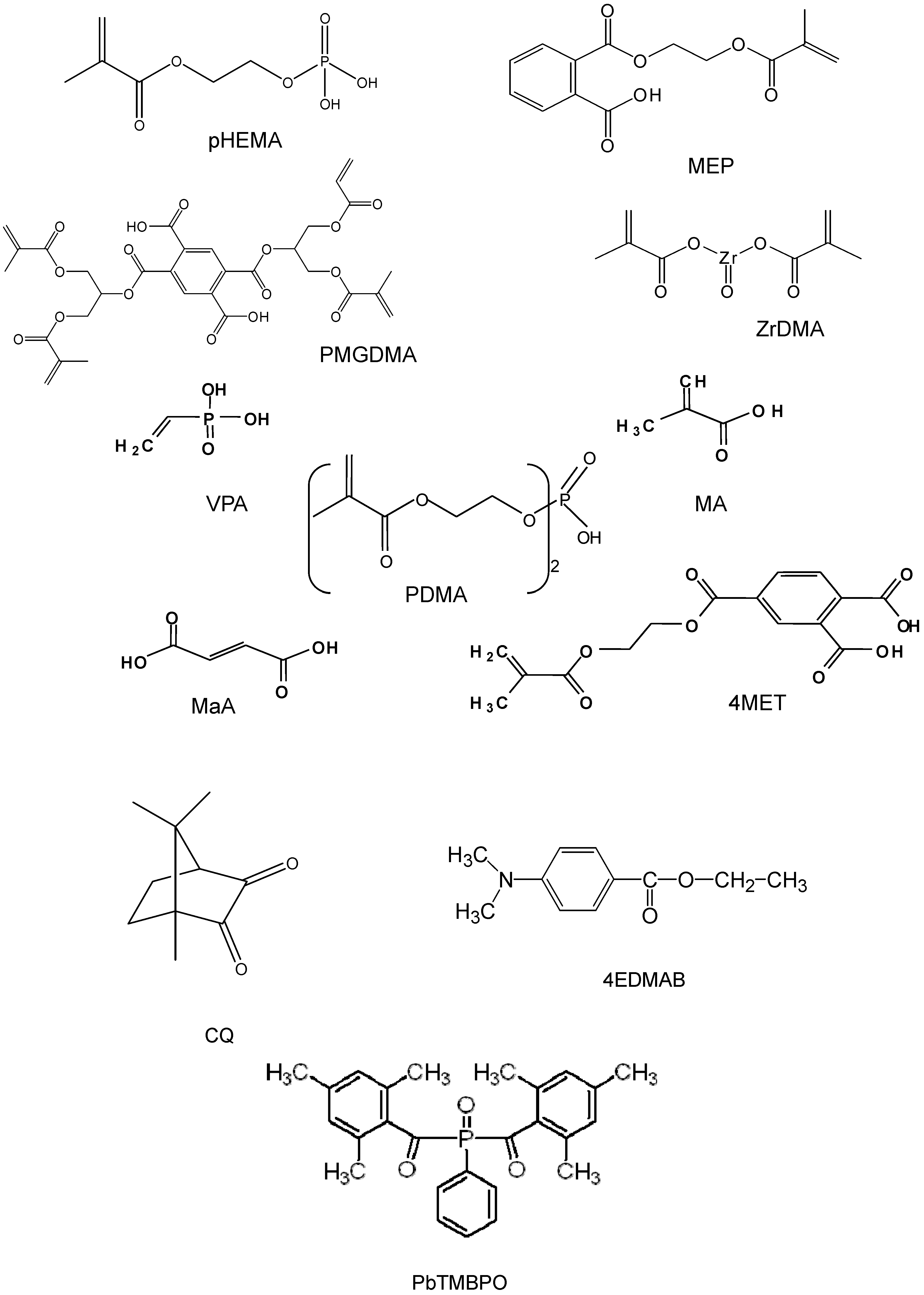{kind=link}
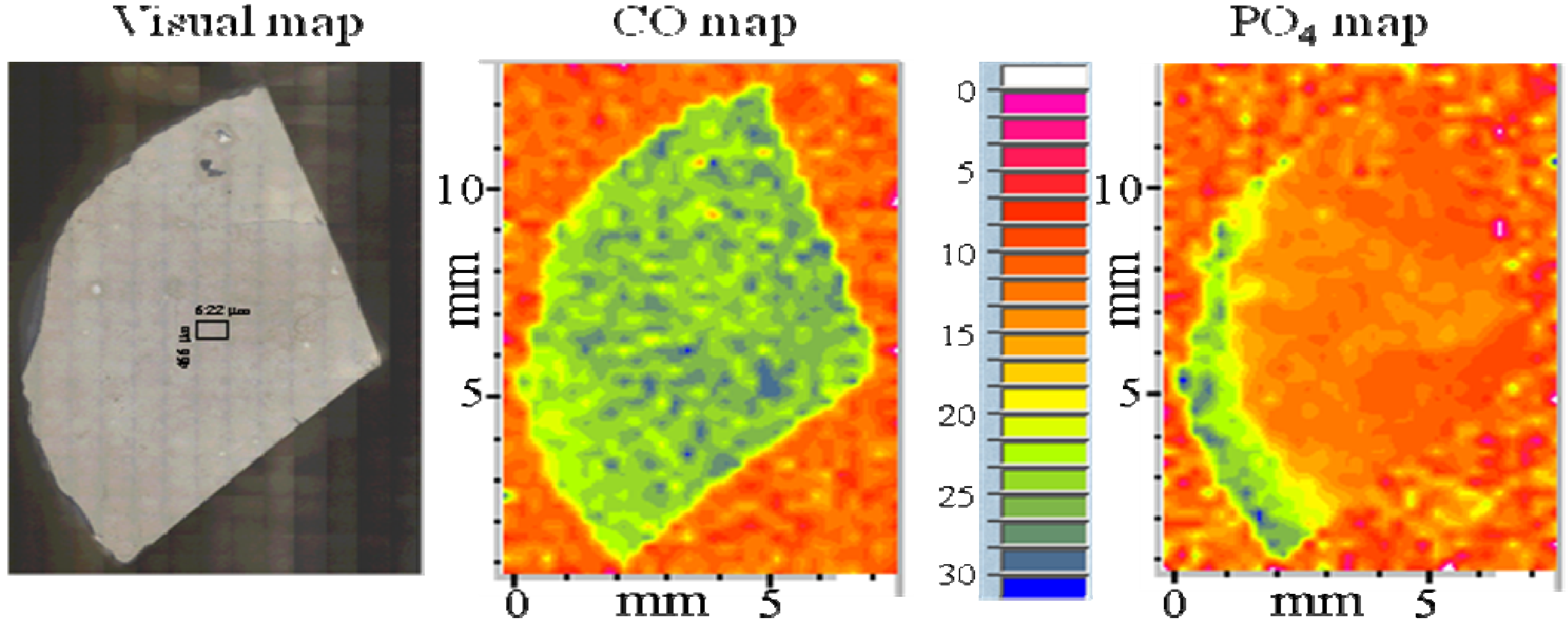{kind=link}
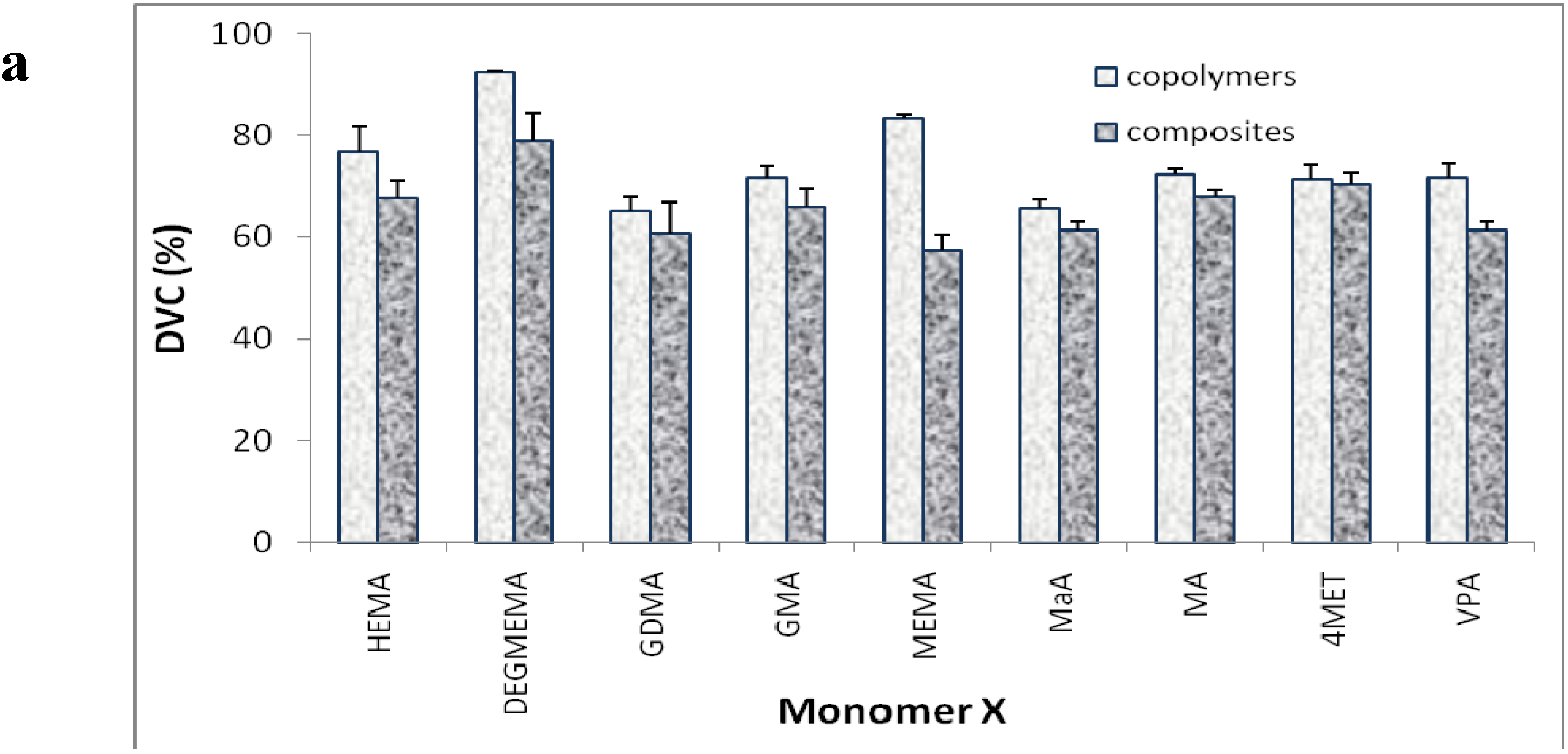{kind=link}
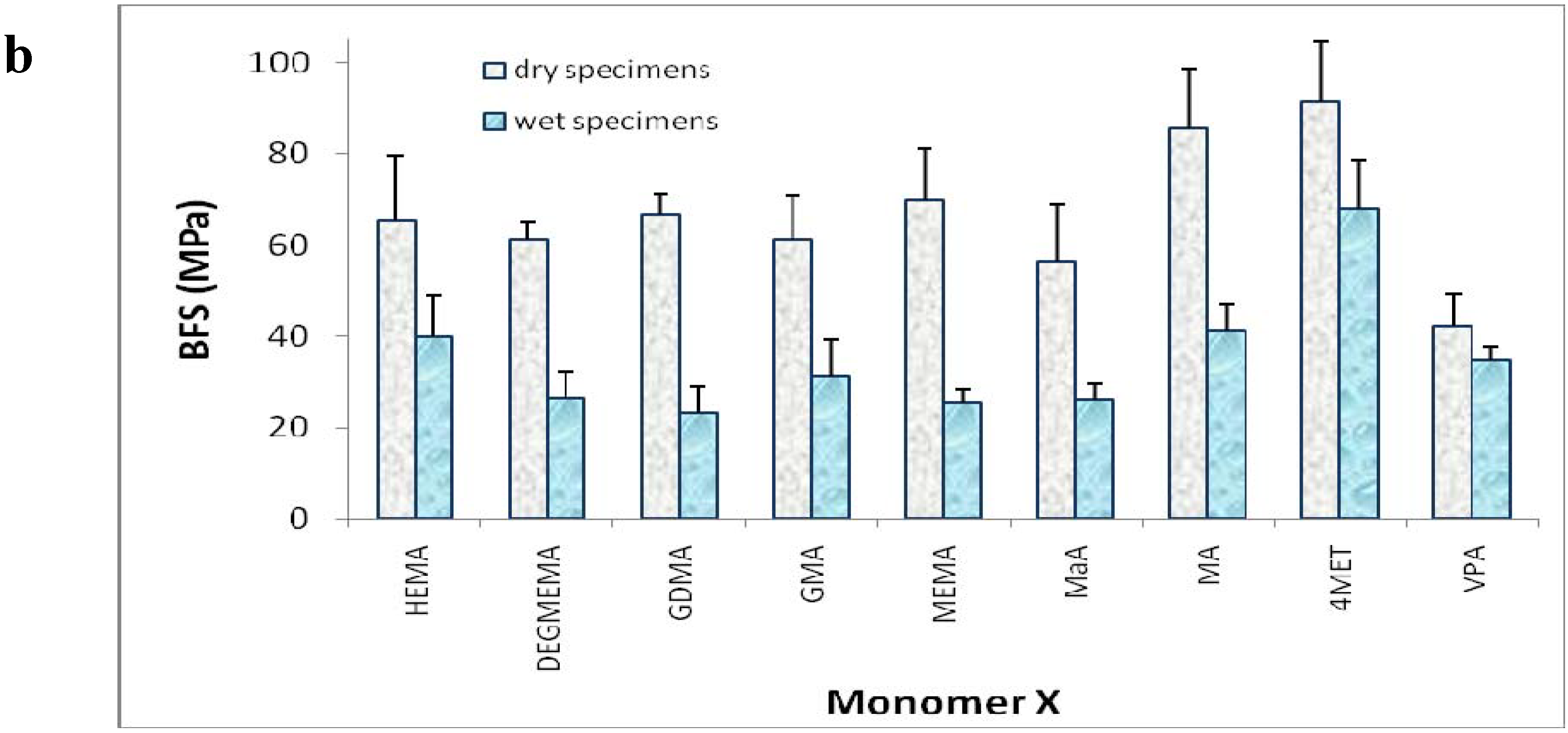{kind=link}
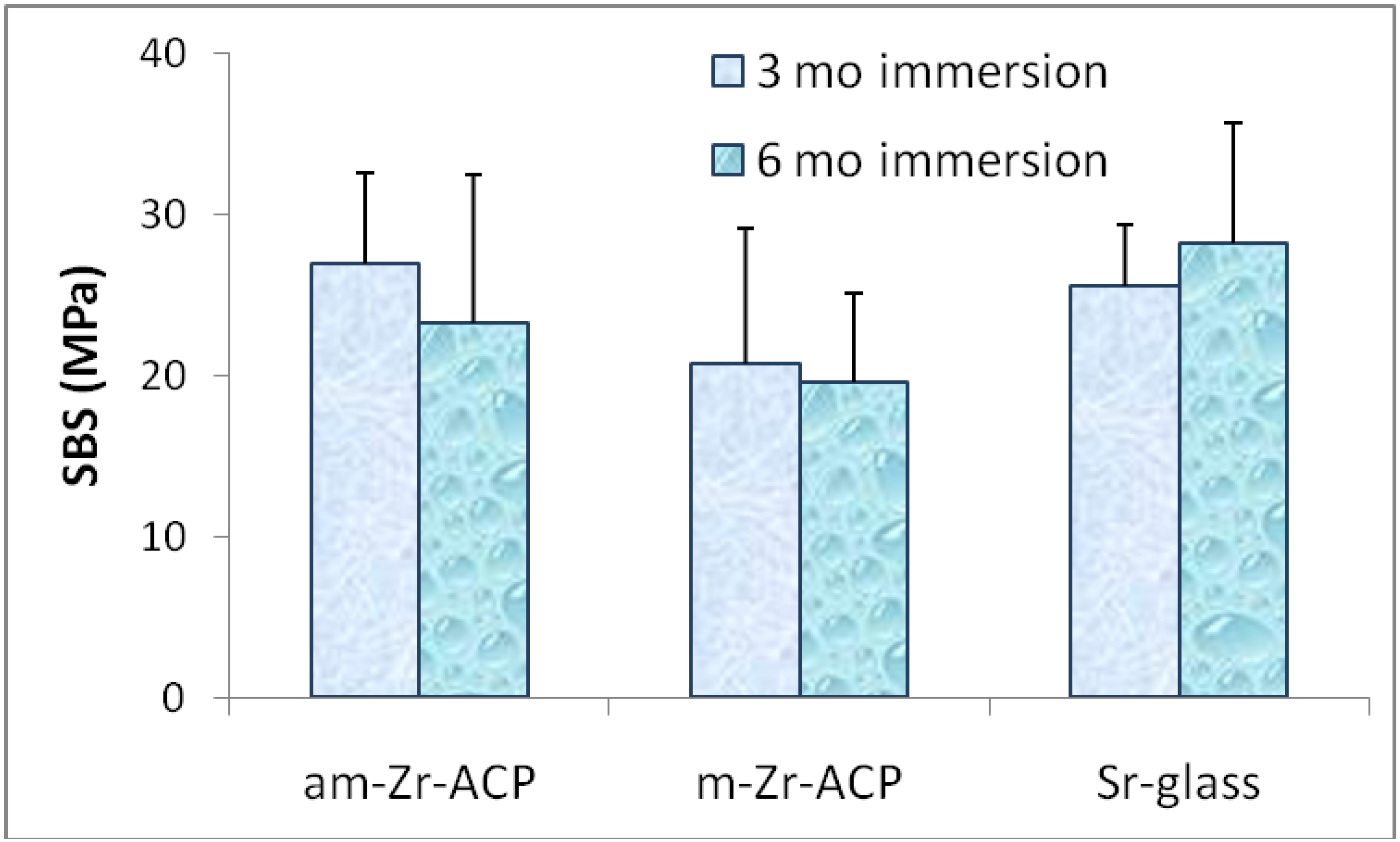{kind=link}
| Additive | dm (μm) | Structure* | SSA (m2/g) | Water content (%) | DVC (%) | BFS (MPa) |
|---|---|---|---|---|---|---|
| ilvera | 3.5 (1.9) | ACP | 1.2 (0.9) | 14.0 (2.2) | 63.3 (1.9) | disintegrated |
| Iron (II)a | 3.8 (1.8) | ACP/HAP | 1.0 (0.5) | 15.4 (1.2) | 65.7 (1.8) | disintegrated |
| Zinca | 1.4 (0.5) | ACP | 2.7 (1.1) | 16.6 (2.5) | 63.7 (2.6) | 48.4 (5.3) |
| Aluminuma | 2.2 (1.3) | ACP | 1.6 (0.8) | 14.1 (2.3) | 56.0 (3.3) | 19.8 (4.7) |
| Iron (III)a | 2.1 (0.6) | ACP/HAP | 1.6 (0.8) | 16.8 (2.8) | 56.7 (2.6) | disintegrated |
| Silica a,b,c | 5.8 (1.6) | ACP | 14.1 (1.2) | 72.5 (2.5) | 40.0 (9.0) | |
| Zirconiaa,b,c | 6.7 (1.9) | ACP | 0.5 (0.3) | 16.1 (2.0) | 80.1 (3.3) | 53.4 (12.0) |
| PAAb | 9.2 (1.9) | ACP | 0.7 (0.1) | 15.8 (1.0) | nd | 34.1 (9.9) |
| PEOb | 14.1 (4.7) | ACP | 0.5 (0.2) | 14.7 (1.2) | nd | 23.4 (4.3) |
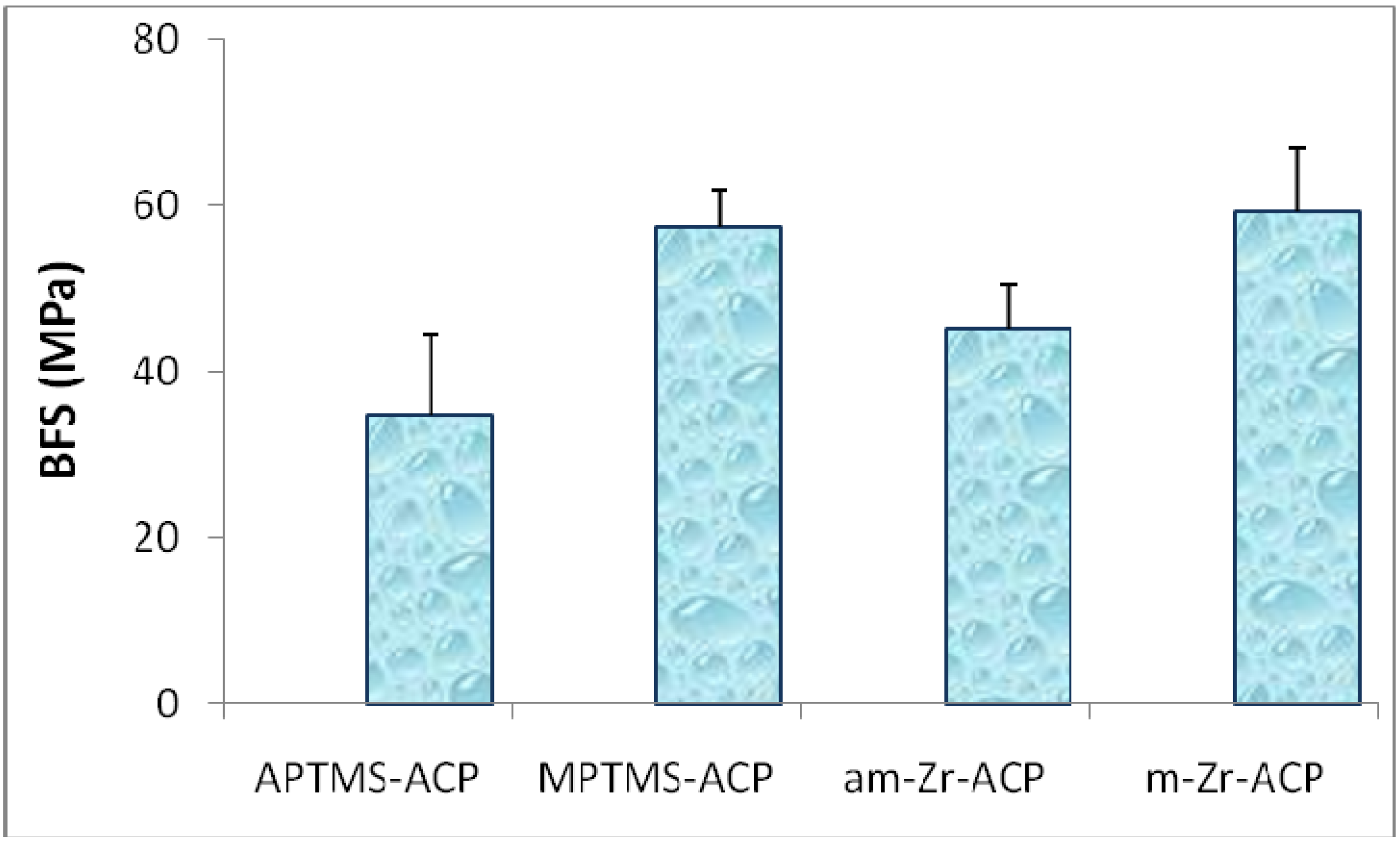
| Parameter | am-Zr-ACP | m-Zr-ACP |
|---|---|---|
| Particle size range (μm) | 0.3 to 80.0 | 0.2 to 3.0 |
| dm (μm) | 5.9 (0.7)* | 0.9 (0.2) |
| Specific surface area, SSA (m2/g) | 0.5 (0.1) | 3.8 (1.0) |
| Ion activity product, IAP | 99.26 (0.68) | 101.21 (1.02) |
| Gibbs free energy, ΔG0 (kJ/mol) | -5.66 (0.21) | -5.07 (0.31) |
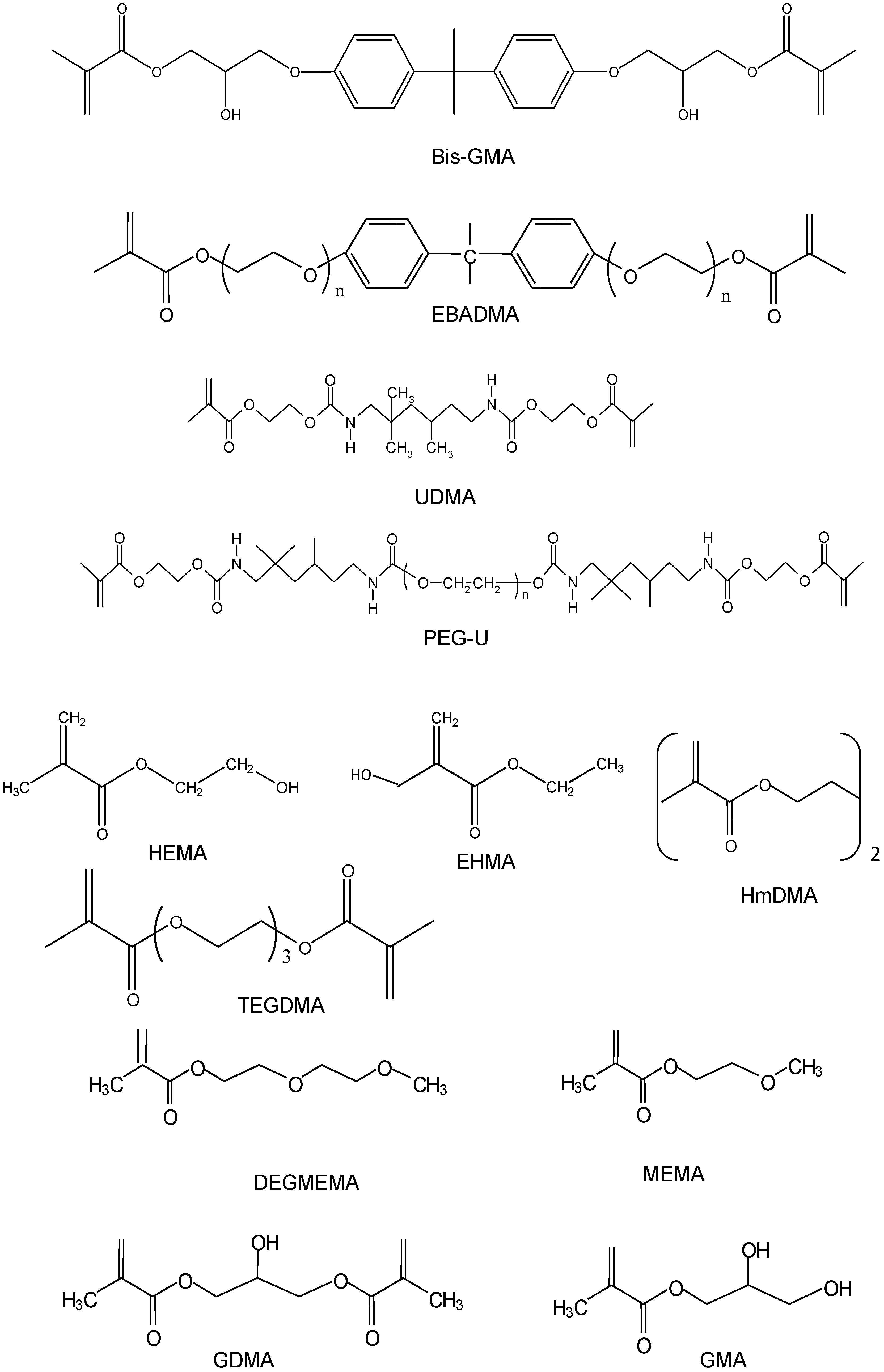
| Component | Chemical Nomenclature | Acronym |
|---|---|---|
| Base monomers | 2,2-bis[p-(2’-Hydroxy-3’-methacryloxypropoxy)phenyl]-propane | Bis-GMA |
| Ethoxylated bisphenol A dimethacrylate | EBPADMA | |
| Urethane dimethacrylate | UDMA | |
| Diluent monomers or oligomers | Di(ethyleneglycol)methyl ether methacrylate | DEGMEMA |
| Ethyl-α-hydroxymethacrylate | EHMA | |
| Glyceryl dimethacrylate | GDMA | |
| Glyceryl monomethacrylate | GMA | |
| 2-Hydroxyethyl methacrylate | HEMA | |
| Hexamethylene dimethacrylate | HmDMA | |
| 2-Methoxyethyl methacrylate | MEMA | |
| Poly(ethylene glycol)-extended UDMA* | PEG-U | |
| Triethyleneglycol dimethacrylate | TEGDMA | |
| Adhesive, surface-active monomers | Maleic acid | MaA |
| Methacrylic acid | MA | |
| Methacryloyloxyethyl phthalate | MEP | |
| 4-(Methacryloyloxy) ethyltrimellitate | 4MET | |
| Bis[2-(Methacryloyloxy)ethyl] phosphate | PDMA | |
| Ethyleneglycol methacrylate phosphate | pHEMA | |
| Pyromellitic glycerol dimethacrylate | PMGDMA | |
| Vinyl phosphonic acid | VPA | |
| Zirconyl dimethacrylate | ZrDMA | |
| Photoinitiating system | Camphorquinone | CQ |
| Ethyl-4-N,N-dimethylaminobenzoate | 4EDMAB | |
| Phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide | PbTMBPO |
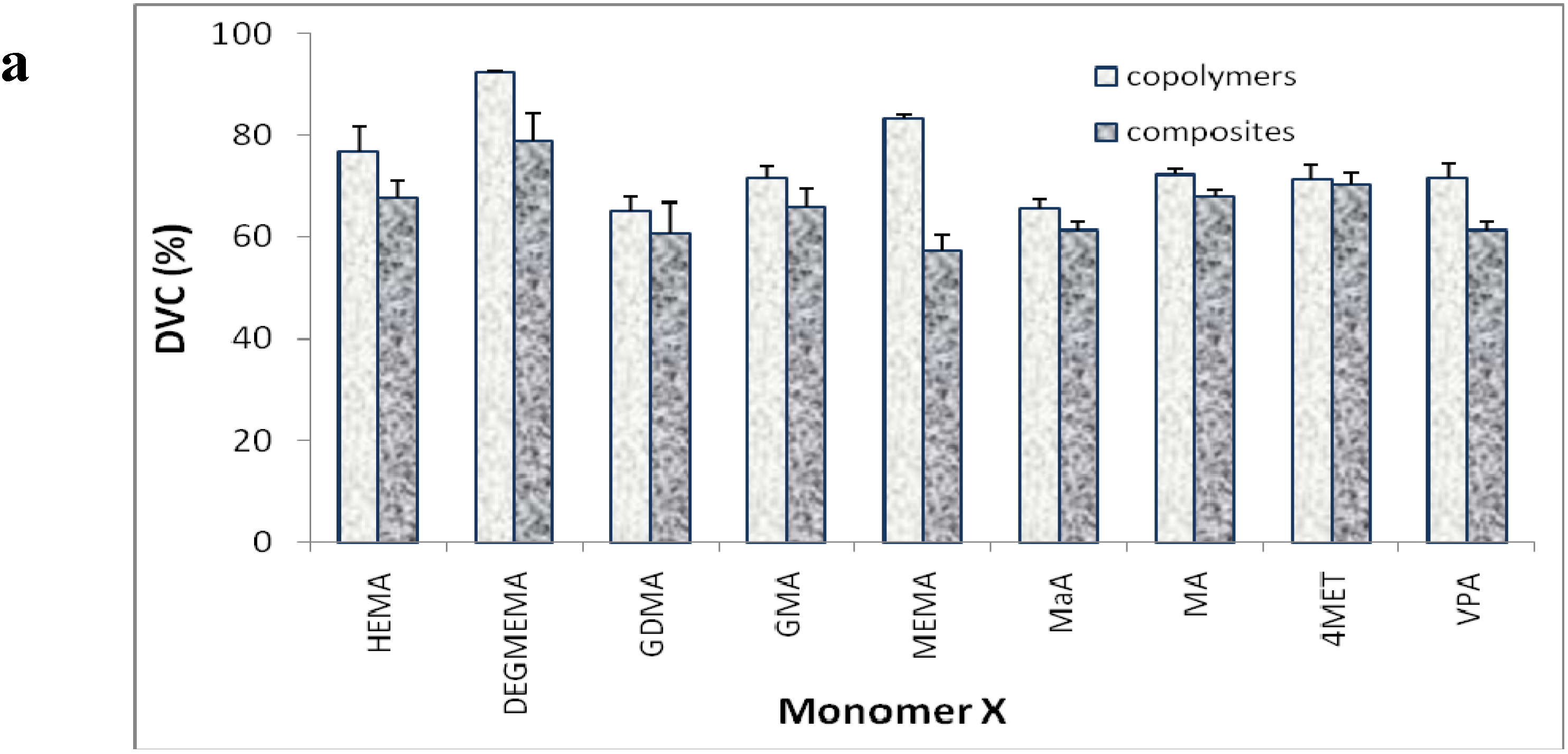
| Resin Matrix* | DVC (%) | PS (vol%) | WS (mass%) | ΔGo (kJ/mol) | BFS (mPa) | |||
| wet | ||||||||
| BH | 78 (3) | 8.6 (2.6) | 2.9 (0.3) | -4.20 (0.06) | 61 (9) | |||
| BHm | 77 (2) | 5.4 (0.6) | 2.2 (0.3) | -4.28 (0.20) | 55 (10) | |||
| BHT | 77 (5) | 7.2 (0.9) | 4.3 (0.6) | -4.43 (0.11) | 40 (9) | |||
| BHHm | 81 (4) | 4.2 (1.0) | 3.1 (0.2) | -4.70 (0.18) | 48 (7) | |||
| EH | 82 (2) | 6.4 (0.7) | 2.6 (0.2) | -5.86 (0.12) | 53 (11) | |||
| EHm | 79 (2) | 7.2 (1.5) | 2.6 (0.3) | -5.24 (0.11) | 58 (7) | |||
| EHT | 85 (2) | 7.8 (1.5) | 4.3 (0.5) | -5.75 (0.09) | 49 (8) | |||
| EHHm | 84 (2) | 8.1 (1.1) | 3.6 (0.2) | -6.58 (0.08) | 50 (9) | |||
| UH | 77 (2) | 6.9 (0.8) | 3.0 (0.4) | -4.24 (0.12) | 57 (10) | |||
| UHm | 74 (2) | 6.6 (0.5) | 2.6 (0.2) | -1.86 (0.13) | 60 (12) | |||
| UHT | 84 (2) | 7.8 (0.8) | 4.0 (0.6) | -4.36 (0.13) | 40 (10) | |||
| UHHm | 83 (2) | 7.5 (0.8) | 3.1 (0.5) | -4.48 (0.12) | 37 (11) | |||
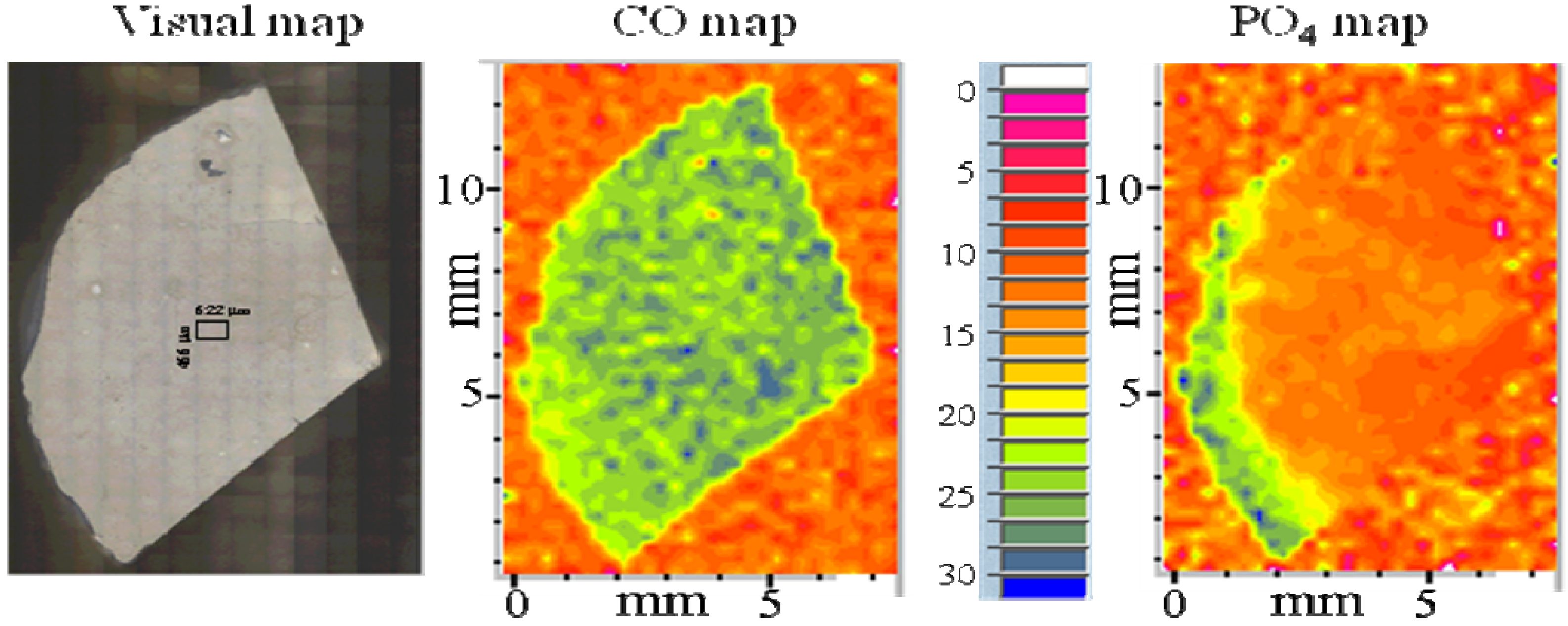
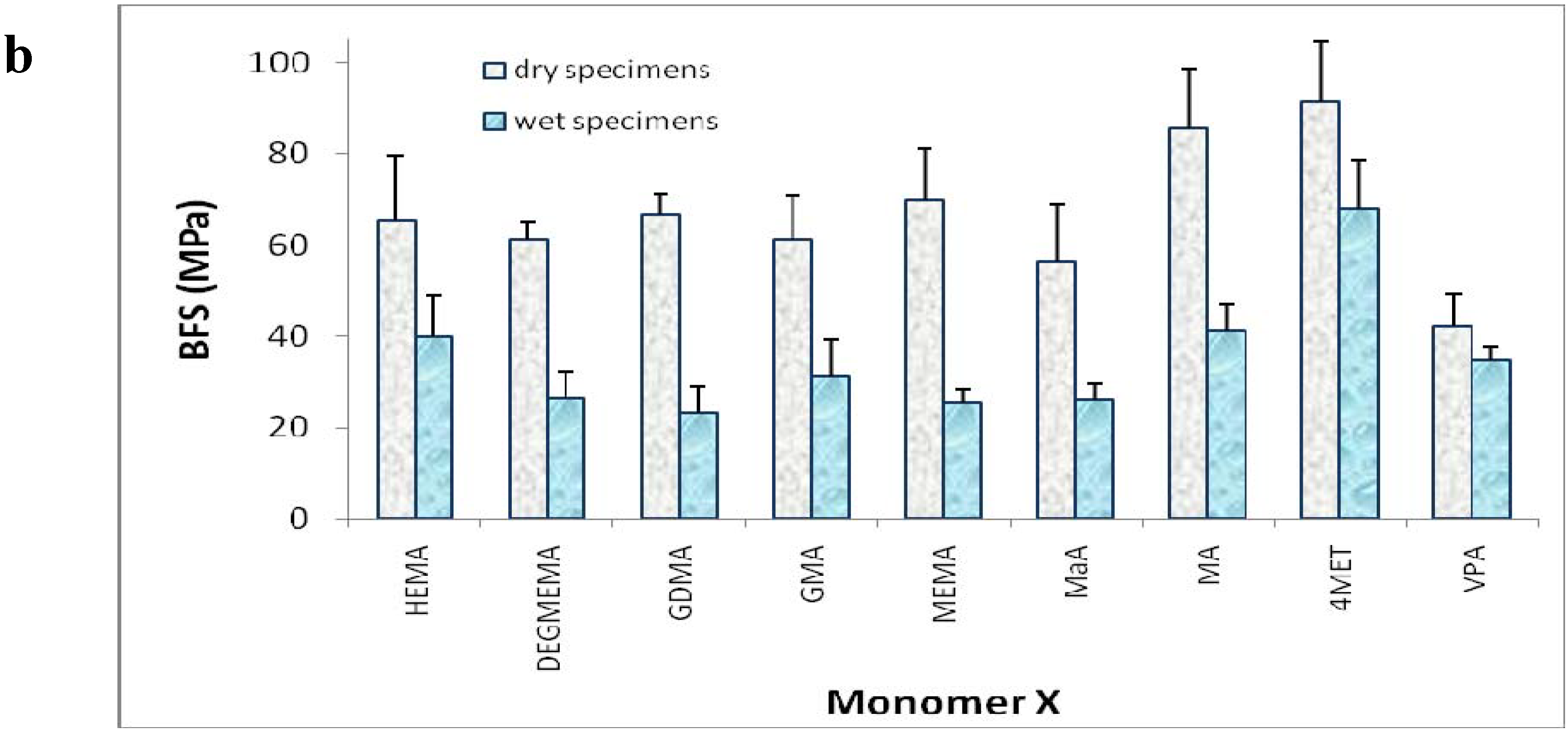
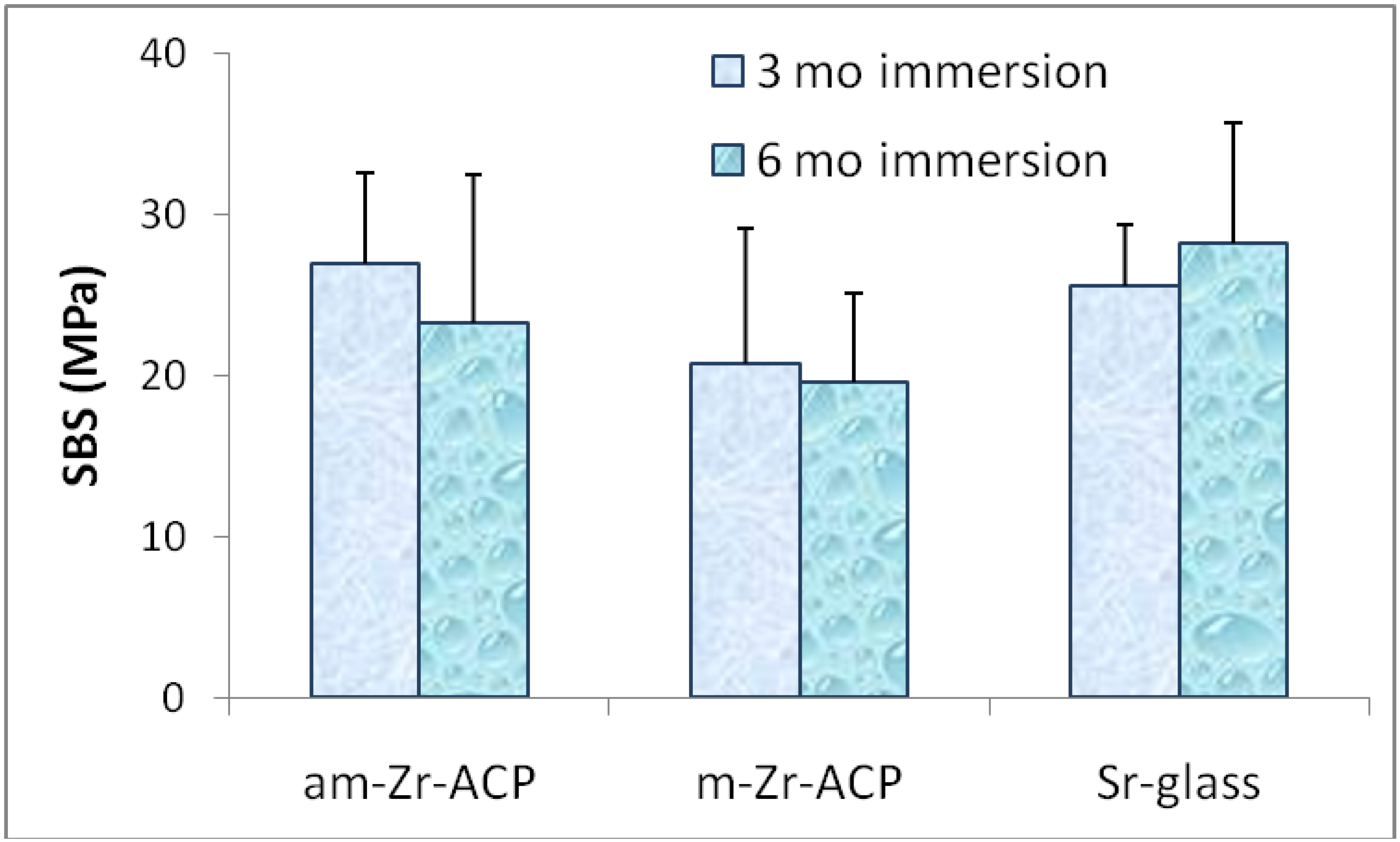
3. Experimental Section
3.1. ACP filler synthesis and characterization
3.2. Resin formulation, copolymer and composite specimen preparation
3.3. Polymerization shrinkage (PS)
3.4. Polymerization stress (PSS) measurements
3.5. Degree of vinyl conversion (DVC)
3.6. Biaxial flexure strength (BFS)
3.7. Water sorption (WS)
3.8. Ion release from composites
3.9. Shear bond strength (SBS)
3.10. Statistical data analysis
4. Conclusions
Acknowledgements
Disclaimer
Appendix. List of Acronyms Used throughout the Manuscript
| ACP | amorphous calcium phosphate |
| ADAF | American Dental Association Foundation |
| am-ACP | as made ACP |
| ANOVA | analysis of variance |
| APTMS | 3-aminopropyltrimethoxysilane |
| BFS | biaxial flexural strength |
| BH | Bis-GMA/HEMA resin |
| BHHm | Bis-GMA/HEMA/HmDMA resin |
| BHm | Bis-GMA/HmDMA resin |
| BHT | Bis-GMA/HEMA/TEGDMA resin |
| Bis-GMA | 2,2-bis[p-(2-hydroxy-3-methacryloxypropoxy)phenyl]propane |
| CQ | camphorquinone |
| DCPA | dicalcium phosphate anhydrous |
| DEGMEMA | di(ethyleneglycol)methyl ether methacrylate |
| dm | median diameter |
| DVC | degree of vinyl conversion |
| EBPADMA | ethoxylated bisphenol A dimethacrylate |
| 4EDMAB | ethyl-4-N,N-dimethylamino benzoate |
| EH | EBPADMA/HEMA resin |
| EHHm | EBPADMA/HEMA/HmDMA resin |
| EHm | EBPADMA/HmDMA resin |
| EHMA | ethyl-α-hydroxymethacrylate |
| EHT | EBPADMA/HEMA/TEGDMA resin |
| ETHM | EBPADMA/TEGDMA/HEMA/MEP resin |
| FTIR | Fourier transform infrared spectroscopy |
| FTIR-m | FTIR microspectroscopy |
| ΔG0 | Gibbs free energy |
| GDMA | glyceryl dimethacrylate |
| GMA | glyceryl monomethacrylate |
| HAP | hydroxyapatite |
| HEMA | 2-hydroxyethyl methacrylate |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| HmDMA | hexamethylene dimethacrylate |
| IAP | ion activity product |
| MaA | maleic acid |
| MA | methacrylic acid |
| m-ACP | milled ACP |
| MEMA | 2-methoxyethyl methacrylate |
| MEP | methcryloyloxyethyl phthalate |
| 4MET | 4-(methacryloyloxy) ethyltrimellitate |
| MPTMS | methacryloxypropyl trimethoxy silane |
| NIDCR | National Institute of Dental and Craniofacial Research |
| NIR | near infrared spectroscopy |
| NIST | National Institute of Standards and Technology |
| PAA | poly(acrylic acid) |
| PbTMBPO | phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide |
| PDMA | bis[2-(methacryloyloxy)ethyl] phosphate |
| PEG-U | poly(ethylene glycol) extended urethane dimethacrylate |
| PEO | poly(ethylene oxide) |
| pHEMA | ethyleneglycol methacrylate phosphate |
| PMGDMA | pyromellitic glycerol dimethacrylate |
| PRC | Paffenbarger Research Center |
| PS | polymerization shrinkage |
| PSS | polymerization stress |
| PSD | particle size distribution |
| RH | relative humidity |
| SBS | shear bond strength |
| SEM | scanning electron microscopy |
| SD | standard deviation |
| SSA | specific surface area |
| TEGDMA | tri(ethyleneglycol) dimethacrylate |
| TGA | thermogravimetric analysis |
| UDMA | urethane dimethacrylate |
| UH | UDMA/HEMA resin |
| UHHm | UDMA/HEMA/HmDMA resin |
| UHm | UDMA/HmDMA resin |
| UHT | UDMA/HEMA/TEGDMA resin |
| UPHM | UDMA/PEG-U/HEMA/MEP resin |
| VPA | vinyl phosphonic acid |
| WS | water sorption |
| XRD | X-ray diffraction |
| ZrDMA | zirconyl dimethacrylate |
References
- Pendrys, D.G.; Stamm, J.W. Relationship of Total Fluoride Intake to Beneficial Effects and Enamel Fluorosis. J. Dent. Res. 1990, 69, 529–538. [Google Scholar] [PubMed]
- Tung, M.S.; Eichmiller, F.C. Dental Applications of Amorphous Calcium Phosphates. J. Clinical Dent. 1999, 10, 1–6. [Google Scholar]
- Reynolds, E.C. Remineralization of Enamel Subsurface Lesions by Casein Phosphopeptide-stabilized Calcium Phosphate Solutions. J. Dent. Res. 1997, 76, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E.C.; Cai, F.; Cross, K.J.; Eakins, D.; Huo, N.L.; Morgan, M.V.; Novicki, A.; Perich, J.W.; Riley, P.F.; Shen, P.; Talbo, G.; Weber, F.L. Advances in Enamel Remineralization: Casein Phosphopeptide–Amorphous Calcium Phosphate. J. Clin. Dent. 1999, 10, 86–88. [Google Scholar]
- Mazzaoui, S.A.; Burrow, M.F.; Tyas, M.J.; Daspher, S.G.; Eakins, D.; Reynolds, E.C. Incorporation of Casein Phosphopeptide-Amorphous Calcium Phosphate into a Glass-ionomer Cement. J. Dent. Res. 2003, 82, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E.C.; Cai, F.; Shen, P.; Walker, G.D. Retention in Plaque and Remineralization of Enamel Lesions by Various Forms of Calcium in a Mouthrinse or Sugar-Free Chewing Gum. J. Dent. Res. 2003, 82, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D. Amorphous Calcium Phosphate-Based Bioactive Polymeric Composites for Mineralized Tissue Regeneration. J. Res. Natl. Inst. Stand. Technol. 2003, 108, 167–182. [Google Scholar] [CrossRef]
- Skrtic, D.; Stansbury, J.W.; Antonucci, J.M. Volumetric Contraction and Methacrylate Conversion in Light-Polymerized Amorphous Calcium Phosphate/Methacrylate Composites. Biomaterials 2003, 24, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D.; Eidelman, N. Dental Composites Based on Hybrid and Surface-Modified Amorphous Calcium Phosphates. Biomaterials 2004, 24, 1141–1150. [Google Scholar] [CrossRef]
- Regnault, W.F.; Icenogle, T.B.; Antonucci, J.M.; Skrtic, D. Amorphous Calcium Phosphate/Urethane Methacrylate Resin Composites. I. Physicochemical Characterization. J. Mater. Sci.: Mater. Med. 2008, 19, 507–515. [Google Scholar] [CrossRef]
- Antonucci, J.M.; O’Donnell, J.N.R.; Schumacher, G.E.; Skrtic, D. Amorphous Calcium Phosphate Composites and Their Effect on the Composite/Adhesive/Dentin Bond. J. Adhes. Sci. Technol. 2009, 23, 1133–1147. [Google Scholar] [CrossRef]
- O’Donnell, J.N.R.; Schumacher, G.E.; Antonucci, J.M.; Skrtic, D. Adhesion of Amorphous Calcium Phosphate Composites Bonded to Dentin: A Study in Failure Modality. J. Biomed. Mater. Res.: Appl. Biomater. 2009, 90B, 238–249. [Google Scholar]
- Skrtic, D.; Hailer, A.W.; Takagi, S.; Antonucci, J.M.; Eanes, E.D. Quantitative Assessment of the Efficacy of Amorphous Calcium Phosphate/methacrylate Composites in Remineralizing Caries-like Lesions Artificially Produced in Bovine Enamel. J. Dent. Res. 1996, 75, 1679–1686. [Google Scholar] [CrossRef]
- Langhorst, S.E.; O’Donnell, J.N.R.; Skrtic, D. In vitro Remineralization Effectiveness of Polymeric ACP Composites: Quantitative Micro-Radiographic Study. Dent. Mater. 2009, 25, 884–891. [Google Scholar] [CrossRef]
- Eanes, E.D. Calcium Phosphates in Biological and Industrial Systems; Amjad, Z., Ed.; Kluwer Academic Pub.: Boston, MA, USA, 1998; pp. 21–39. [Google Scholar]
- Okamoto, Y.; Hidata, S. Studies on Calcium Phosphate Precipitation: Effects of Metal Ions Used in Dental Materials. J. Biomed. Mater. Res. 1994, 28, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Hidata, S.; Okamoto, Y.; Abe, K. Elution of Metal Ions from Dental Casting Alloys and Their Effect on Calcium Phosphate Precipitation and Transformation. J. Biomed. Mater. Res. 1994, 28, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Bowen, R.L. Dental Adhesive Materials; Moskovitz, H.D., Ward, G.T., Woolridge, E.D., Eds.; Prestige Graphics Services: New York, NY, USA, 1974; pp. 205–221. [Google Scholar]
- Ito, A.; Kawamura, H.; Miyakawa, S.; Layrolle, P.; Kanzaki, N.; Treboux, G.; Onuma, K.; Tsatsumi, S. Resorbability and Solubility of Zinc-containing Tricalcium Phosphate. J. Biomed. Mater. Res. 2002, 60, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Miyamoto, Y.; Yuasa, T.; Ito, A.; Nagayama, M.; Suzuki, K. Fabrication of Zn Containing Apatite Cement and Its Initial Evaluation Using Human Osteoblastic Cells. Biomaterials 2002, 23, 423–428. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z.; Bleiwas, C.B.; Retino, M.; Rohanizadeh, R.; LeGeros, J.P. Zinc Effect on the in vitro Formation of Calcium Phosphates: Relevance to Clinical Inhibition of Calculus Formation. Am. J. Dent. 1999, 12, 65–70. [Google Scholar] [PubMed]
- Hidata, S.; Okamoto, Y.; Abe, K.; Miyazaki, K. Effects of Indium and Iron Ions on in vitro Calcium Phosphate Precipitation and Crystallinity. J. Biomed. Mater.Res. 1996, 31, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Cobb, E.N. A Silver Staining Technique for Investigating Wear of Restorative Dental Composites. J. Biomed. Mater. Res. 1981, 31, 343–348. [Google Scholar] [CrossRef]
- Wilson, A.D.; Nicholson, J.W. Chemistry of Solid State Materials. 3. Acid-Base Cements. Their Biomedical and Industrial Applications; Cambridge Univ. Press: Cambridge, MA, USA, 1993; pp. 56–102. [Google Scholar]
- Bigi, A.; Boanini, E.; Bracci, B.; Falini, G.; Rubini, K. Interaction of Acidic Poly-amino-acids with Octacalcium Phosphate. J. Inorg. Biochem. 2003, 95, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Weng, W.; Li, Z.; Cheng, K.; Song, C.; Du, P.; Shen, G.; Han, G. Effect of PEG Amount in Amorphous Calcium Phosphate on its Crystallized Products. Scand. J. Mater. Sci.: Mater. Med. 2009, 20, 359–363. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium Orthophosphate Cements and Concretes. Materials 2009, 2, 221–291. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium Orthophosphate-Based Biocomposites and Hybrid Materials. J. Mater. Sci. 2009, 44, 2343–2387. [Google Scholar]
- Wang, L.; Nancollas, G.H. Pathways to Biomineralization and Biodemineralization of Calcium Phosphates: The Thermodynamic and Kinetic Controls. Dalton Trans. 2009, 15, 2665–2672. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; Skrtic, D. Physicochemical and Mechanical Evaluation of Cation-Modified ACP Acrylic Resin Composites. Polym. Prepr. 2006, 47, 113–114. [Google Scholar] [CrossRef]
- Antonucci, J.M.; Liu, D.W.; Skrtic, D. Amorphous Calcium Phosphate Based Composites: Effect of Surfactants and Poly(Ethylene Oxide) on Filler and Composite Properties. J. Disp. Sci. Technol. 2007, 28, 819–824. [Google Scholar] [CrossRef]
- Jones, D.W.; Rizkalla, A.S. Characterization of Experimental Composite Biomaterials. J. Biomed. Mater. Res.: Appl. Biomater. 1996, 33, 89–100. [Google Scholar] [CrossRef]
- Liu, Q.; Wijn, J.R.; De Groot, K.; Vanblitterswijk, C.A. (1998): Surface-Modification of Nano-apatite by Grafting Organic Polymer. Biomaterials 1998, 19, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, N.H.; Craig, R.G. Hydrolytic Stability of Silanated Zirconia-Silica-Urethane Dimethacrylate Composites. J. Oral. Rehab. 1995, 22, 213–220. [Google Scholar] [CrossRef]
- Dupraz, A.M.P.; De Wijn, J.R.; Meer, S.A.T.; De Groot, K. Characterization of Silane-Treated Hydroxypatite Powders for Use as Filler in Biodegradable Composites. J. Biomed. Mater. Res. 1996, 30, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Venhoven, B.A.M.; De Gee, A.J.; Werner, A.; Davidson, C.L. Influence of Filler Parameters on the Mechanical Coherence of Dental Restorative Composites. Biomaterials 1996, 17, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; Skrtic, D. Polymers for Dental and Orthopedic Applications; Shalaby, S.W., Salz, U., Eds.; CRC Press: Boca Raton, FL, USA, 2007; pp. 217–242. [Google Scholar]
- Xu, H.H.K.; Weir, M.D.; Sun, L. Nanocomposites with Ca and PO4 release: Effects of reinforcement, dicalcium phosphate particle size and silanization. Dent. Mater. 2007, 23, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.R.; Kalachandra, S.; Shobna, H.K.; Gunduz, N.; Stejskal, E.O. Analysis of a Dimethacrylate Copolymer (Bis-GMA and TEGDMA) Network by DSC and 13C Solution and Solid-state NMR Spectroscopy. Biomaterials 2000, 21, 1897–1903. [Google Scholar] [CrossRef] [PubMed]
- Stansbury, J.W.; Dickens, S.H. Network Formation and Compositional Drift during Photo-Initiated Copolymerization of Dimethacrylate Monomers. Polymer 2001, 42, 6363–6369. [Google Scholar] [CrossRef]
- Antonucci, J.M.; Stansbury, J.W. Desk Reference of Functional Polymers Syntheses and Applications; Arshady, R., Ed.; ACS: Washington DC, USA, 1997; pp. 719–738. [Google Scholar]
- Antonucci, J.M.; Liu, D.W.; Stansbury, J.W. Synthesis of Hydrophobic Oligomeric Monomers for Dental Applications. J. Dent. Res. 1993, 72, 369. [Google Scholar]
- Asmussen, E.; Peutzfeldt, A. Influence of UEDMA, Bis-GMA, and TEGDMA on Selected Mechanical Properties of Experimental Resin Composites. Dent. Mater. 1988, 14, 51–56. [Google Scholar] [CrossRef]
- Morra, M. Acid-base Properties of Adhesive Dental Polymers. Dent. Mater. 1993, 19, 375–378. [Google Scholar] [CrossRef]
- Labella, R.; Lambrechts, P.; Van Meerbeek, B; Vanherle, G. Polymerization Shrinkage and Elasticity of Flowable Composites and Filled Adhesives. Dent. Mater. 1999, 15, 28–137. [Google Scholar] [CrossRef]
- Braem, M.; Davidson, C.L.; Vanherle, G.; Van Doren, V.; Lambrechts, P. The Relationship between Testing Methodology and Elastic Behavior of Composites. J. Dent. Res. 1987, 66, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.R.; Ferracane, J.L. Contraction Stress Related to Degree of Conversion and Reaction Kinetics. J. Dent. Res. 2002, 81, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Watts, D.C.; Marouf, A.S.; Al-Hindi, A.M. Photo-polymerization Shrinkage Stress Kinetics in Resin Composites: Methods Development. Dent. Mater. 2003, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Calheiros, F.C.; Braga, R.R.; Kawano, Y.; Ballester, R.Y. Relationship between Contraction Stress and Degree of Conversion in Restorative Composites. Dent. Mater. 2004, 20, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Stansbury, J.W.; Trujillo-Lemon, M.; Lu, H.; Ding, X.; Lin, Y.; Ge, J. Conversion-Dependent Shrinkage Stress and Strain in Dental Resins and Composites. Dent. Mater. 2005, 21, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Kleverlaan, C.J.; Feilzer, A.J. Polymerization Shrinkage and Contraction Stress of Dental Resin Composites. Dent. Mater. 2005, 21, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Ferracane, J.L. Developing a More Complete Understanding of Stresses Produced in Dental Composites During Polymerization. Dent. Mater. 2005, 21, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; Giuseppetti, A.A.; O’Donnell, J.N.R.; Schumacher, G.E.; Skrtic, D. Polymerization Stress Development in Dental Composites: Effect of Cavity Design Factor. Materials 2009, 2, 169–180. [Google Scholar] [CrossRef]
- Antonucci, J.M.; Regnault, W.F.; Skrtic, D. Polymerization Shrinkage and Stress Development in Amorphous Calcium Phosphate/Urethane Dimethacrylate Polymeric Composites. J. Comp. Mater. 2009, in press. [Google Scholar]
- Antonucci, J.M.; Fowler, B.F.; Weir, M.D.; Skrtic, D; Stansbury, J.W. Effect of Ethyl-α-hydroxymethylacrylate on Selected Properties of Copolymers and ACP Resin Composites. J. Mater. Sci.: Mater. Med. 2008, 19, 3263–3271. [Google Scholar] [CrossRef]
- Antonucci, J.M.; Skrtic, D. Matrix Resin Effects on Selected Physicochemical Properties of Amorphous Calcium Phosphate Composites. J. Bioact. Compat. Polym. 2005, 20, 29–49. [Google Scholar] [CrossRef]
- Schumacher, G.E.; Antonucci, J.M.; O’Donnell, J.N.R.; Skrtic, D. The Use of Amorphous Calcium Phosphate Composites as Bioactive Basing Materials. Their Effect on the Strength of the Composite/Adhesive/Dentin Bond. J. Am. Dent. Assoc. 2007, 138, 1476–1484. [Google Scholar]
- Stansbury, J.W.; Dickens, S.H. Determination of Double Bond Conversion in Dental Resins by Near Infrared Spectroscopy. Dent. Mater. 2001, 17, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Standard Test Method for Biaxial Strength (Modulus of Rupture) of Ceramic Substrates. ASTM F394–78, re-approved 1991.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
O’Donnell, J.N.R.; Schumacher, G.E.; Antonucci, J.M.; Skrtic, D. Structure-Composition-Property Relationships in Polymeric Amorphous Calcium Phosphate-Based Dental Composites. Materials 2009, 2, 1929-1954. https://doi.org/10.3390/ma2041929
O’Donnell JNR, Schumacher GE, Antonucci JM, Skrtic D. Structure-Composition-Property Relationships in Polymeric Amorphous Calcium Phosphate-Based Dental Composites. Materials. 2009; 2(4):1929-1954. https://doi.org/10.3390/ma2041929
Chicago/Turabian StyleO’Donnell, Justin N. R., Gary E. Schumacher, Joseph M. Antonucci, and Drago Skrtic. 2009. "Structure-Composition-Property Relationships in Polymeric Amorphous Calcium Phosphate-Based Dental Composites" Materials 2, no. 4: 1929-1954. https://doi.org/10.3390/ma2041929