Chemoselectivity in the Dehydrocoupling Synthesis of Higher Molecular Weight Polysilanes


Abstract
:1. Introduction

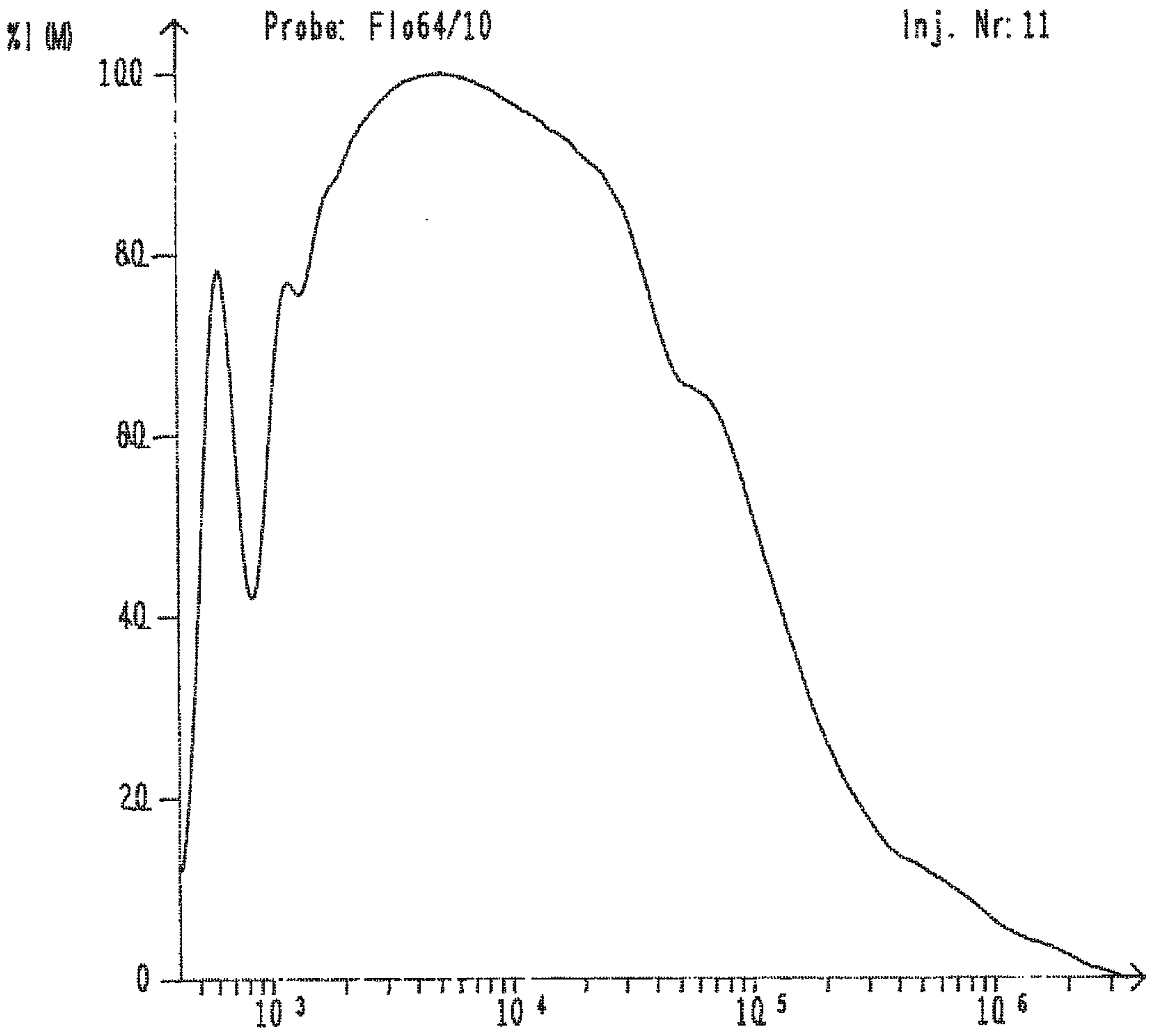
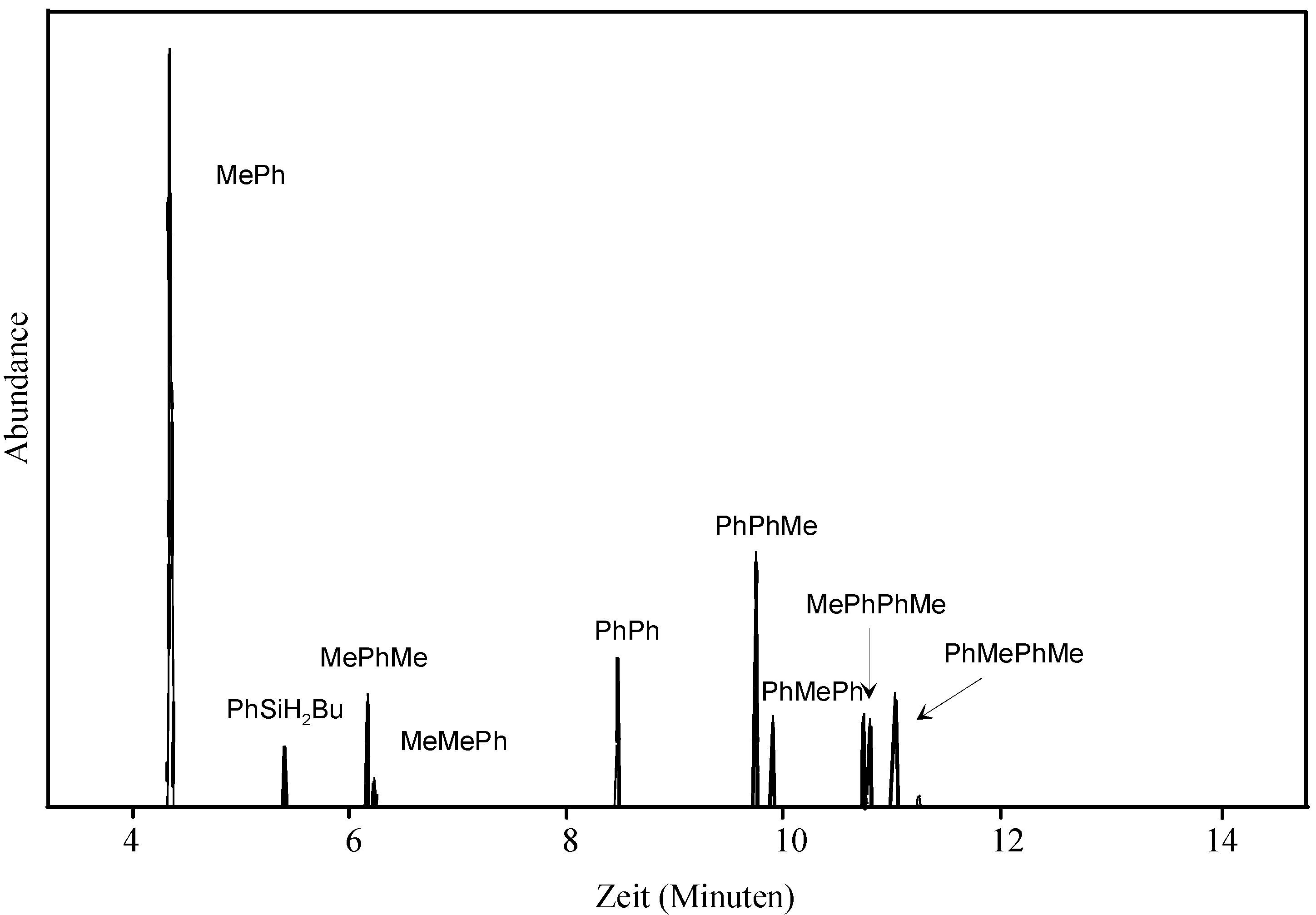
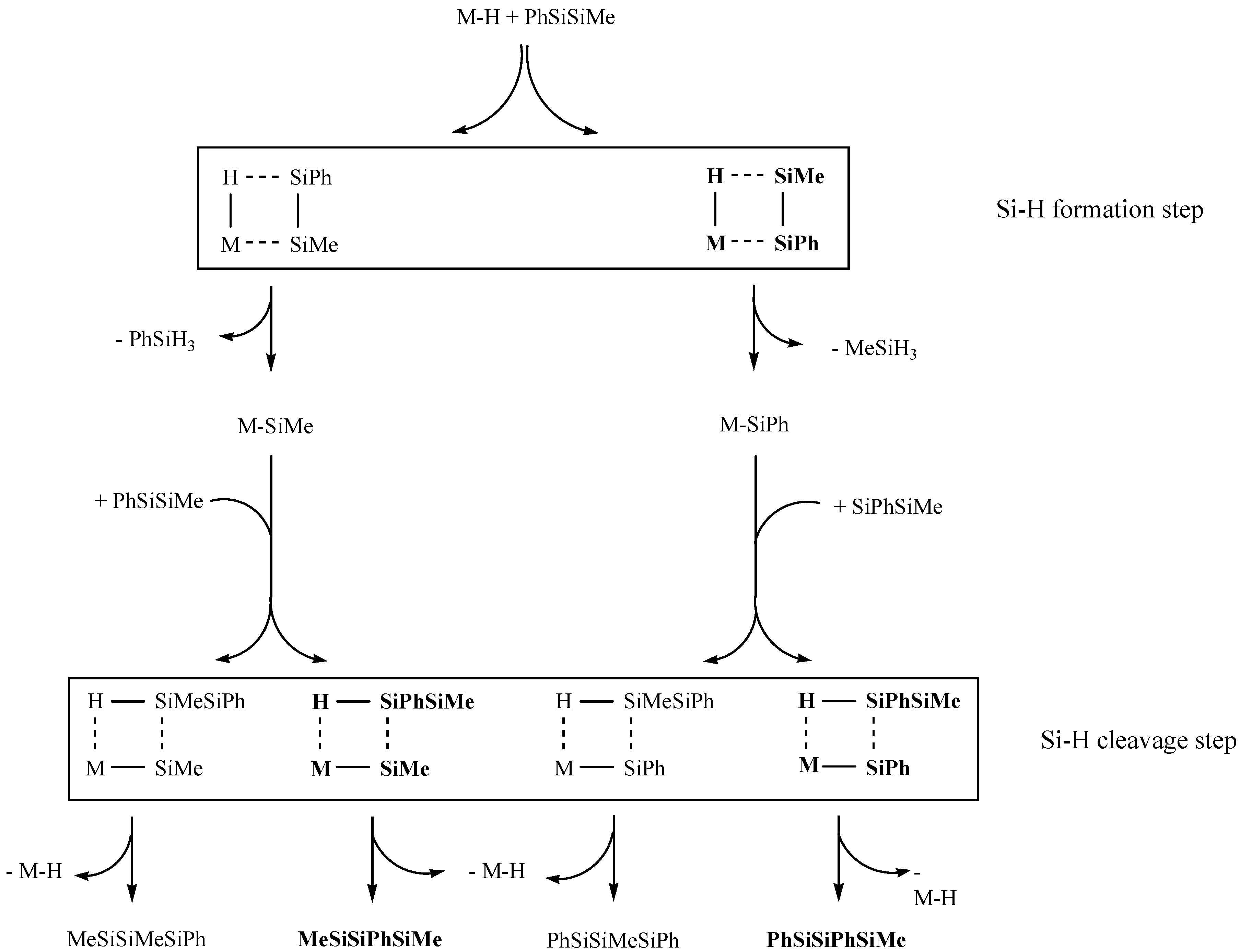
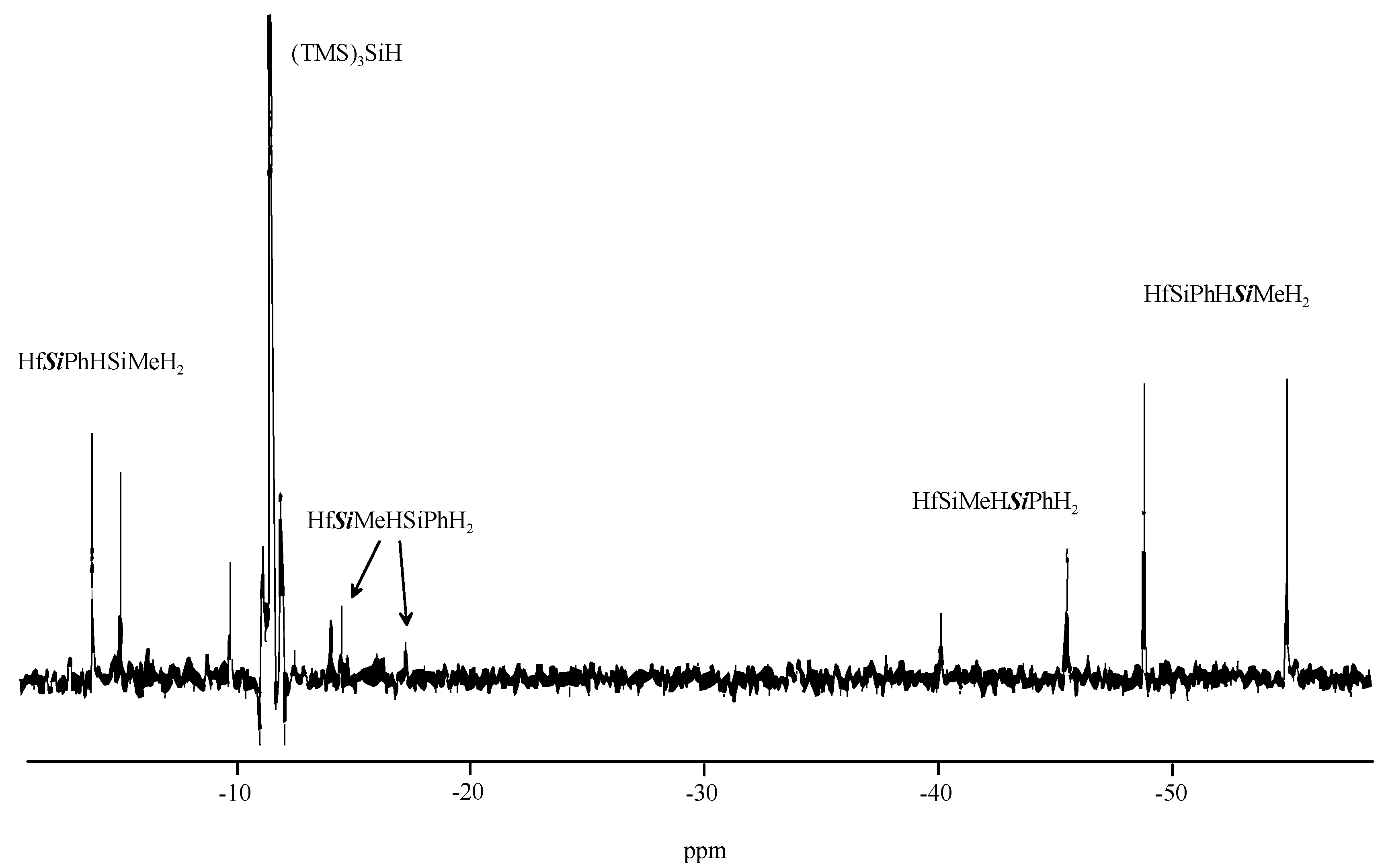
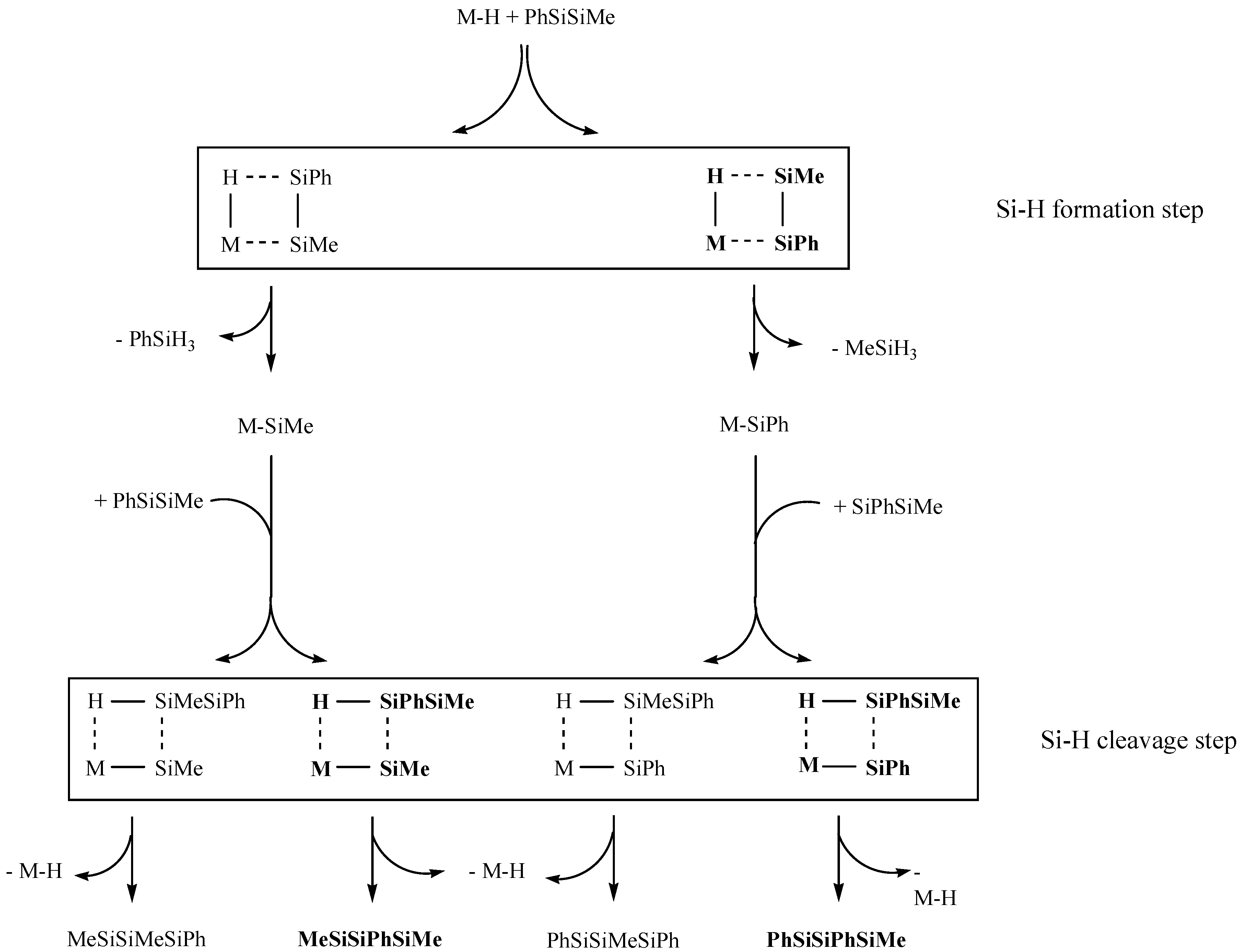
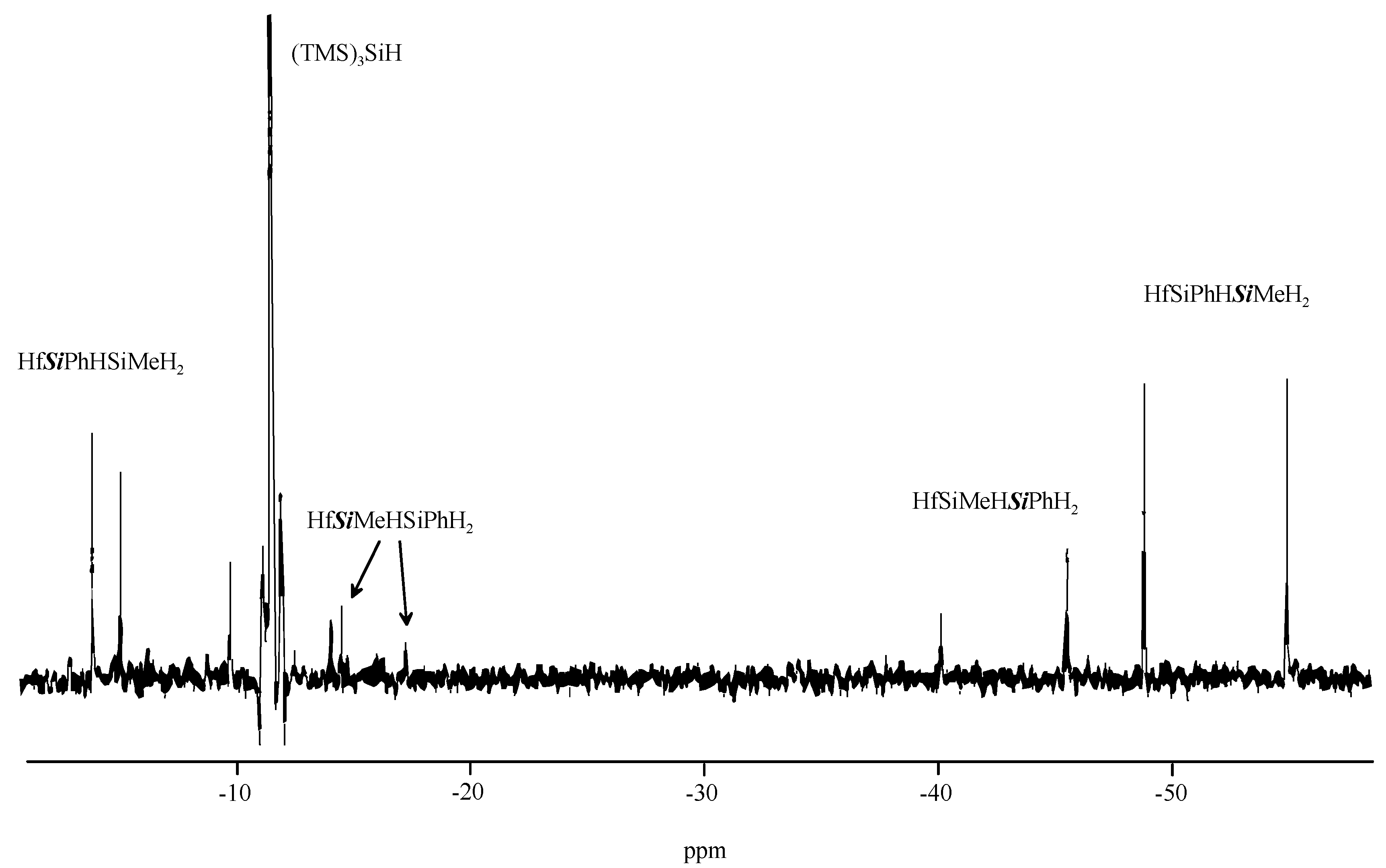
2. Results and Discussion



{kind=link}
{kind=link}
{kind=link}
{kind=link}
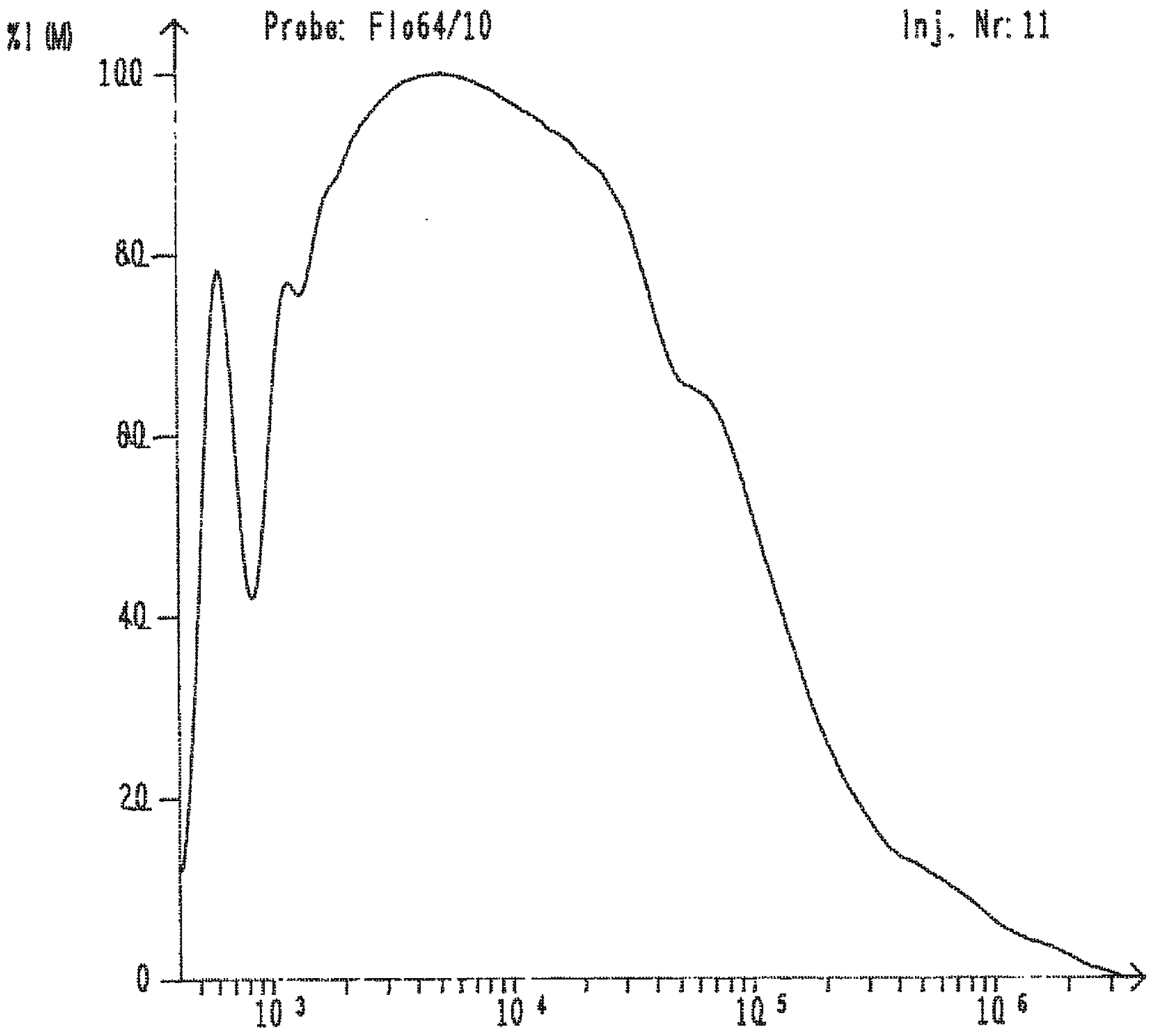
| Reaction | Mn | Mw | D |
| 2 PhSiH3 + H2MeSiSiMeH2 | 2,100 | 5,900 | 2.8 |
| PhSiH3 + H2MeSiSiMeH2 | 2,500 | 8,700 | 3.5 |
| H2MeSiSiPhH2 | 3,300 | 50,000 | 15.0 |

3. Experimental Section
3.1. Synthesis of 1-methyl-2-phenyldisilane
3.2. General polymerization procedure
3.3. Preparation of hafnocene disilanyl compounds
4. Conclusions
Acknowledgements
References and Notes
- Miller, R.D.; Michl, J. Polysilane high polymers. Chem. Rev. 1989, 89, 1359–1410. [Google Scholar] [CrossRef]
- West, R. Organopolysilanes. In Comprehensive Organometallic Chemistry II; Davies, A.G., Ed.; Pergamon: Oxford, UK, 1994; Volume 2, pp. 77–110. [Google Scholar]
- West, R. Polysilanes. In The Chemistry of Organic Silicon Compounds; Patai, S., Rappoport, Z., Eds.; Wiley: Chichester, UK, 1989; pp. 1207–1240. [Google Scholar]
- West, R. Polysilanes: Conformations, chromotropism and conductivity. In the Chemistry of Organic Silicon Compounds; Rappoport, Z., Appeloig, Y., Eds.; Wiley: Chichester, UK, 2001; Volume 3, pp. 541–564. [Google Scholar]
- Miller, R.D. Polysilanes—A new look at some old materials. Adv. Mater. 1989, 1, 433–440. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Chen, Y.L.; Kim, H.K. New synthetic routes to polysilanes. In Inorganic and Organic Polymers; Zelin, M., Wynne, K.J., Allcock, H.R., Eds.; American Chemical Society Symposium Series; American Chemical Society: Washington, DC, USA, 1988; Volume 360, pp. 78–88. [Google Scholar]
- Cypryk, M.; Gupta, Y.; Matyjaszewski, K. Anionic ring-opening polymerization of 1,2,3,4-tetramethyl-1,2,3,4-tetraphenylcyclotetrasilane. J. Am. Chem. Soc. 1991, 113, 1046–1047. [Google Scholar] [CrossRef]
- Sakamoto, K.; Obata, K.; Hirata, H.; Nakajima, M.; Sakurai, H. Chemistry of organosilicon compounds. 257. Novel anionic polymerization of masked disilenes to polysilylene high polymers and block copolymers. J. Am. Chem. Soc. 1989, 111, 7641–7643. [Google Scholar] [CrossRef]
- Aitken, C.; Harrod, J.F.; Samuel, E. Polymerization of primary silanes to linear polysilanes catalyzed by titanocene derivatives. J. Organomet. Chem. 1985, 279, C11–C13. [Google Scholar] [CrossRef]
- Kim, B.H.; Woo, H.G. Dehydrocoupling, redistributive coupling, and addition of main group 4 hydrides. Adv. Organomet. Chem. 2005, 52, 143–174. [Google Scholar]
- Corey, J.Y. Dehydrocoupling of hydrosilanes to polysilanes and silicon oligomers: A 30 year overview. Adv. Organomet. Chem. 2004, 51, 1–52. [Google Scholar]
- Gray, G.M.; Corey, J.Y. Synthesis of polysilanes by new procedures: Part 2 catalytic dehydropolymerisation of hydrosilanes. In Silicon-Containing Polymer; Jones, R.G., Ando, W., Chojnowski, J., Eds.; Kluwer Academic Publishers: Dordrecht, the Netherlands, 2000; pp. 401–418. [Google Scholar]
- Harrod, J.F. Catalysis of reactions of Si-H by titanocene and its derivatives. Coord. Chem. Rev. 2000, 206–207, 493–531. [Google Scholar] [CrossRef]
- Pukhnarevich, V.B.; Voronkov, M.G.; Kopylova, L.I. Dehydrocondensation of organylsilanes giving Si–Si bonds. Russ. Chem. Rev. 2000, 69, 137–151. [Google Scholar] [CrossRef]
- Reichl, J.A.; Berry, D.H. Recent progress in transition metal-catalyzed reactions of silicon, germanium, and tin. Adv. Organomet. Chem. 1998, 43, 197–265. [Google Scholar]
- Gauvin, F.; Harrod, J.F.; Woo, H.G. Catalytic dehydrocoupling: A general strategy for the formation of element-element bonds. Adv. Organomet. Chem. 1998, 42, 363–405. [Google Scholar]
- Imori, T.; Tilley, T.D. The influence of catalyst structure on the dehydropolymerization of phenylsilane. Polyhedron 1994, 13, 2231–2243. [Google Scholar] [CrossRef]
- Tilley, T.D. The coordination polymerization of silanes to polysilanes by a "σ-bond metathesis" mechanism. Implications for linear chain growth. Acc. Chem. Res. 1993, 26, 22–29. [Google Scholar] [CrossRef]
- Mu, Y.; Harrods, J.F.; Samuel, E. Catalytic dehydrocoupling: A general method for the formation of element-element bonds. Polyhedron 1991, 10, 1239–1245. [Google Scholar] [CrossRef]
- Corey, J.Y. Dehydrogenative coupling reactions of hydrosilanes. In Advances in Silicon Chemistry; JAi Press Inc.: Greenwich, CT, UK, 1991; Volume 1, pp. 327–387. [Google Scholar]
- Tilley, T.D. Mechanistic aspects of transition-metal catalyzed dehydrogenative silane coupling reactions. Comments Inorg. Chem. 1990, 10, 37–51. [Google Scholar] [CrossRef]
- Woo, H.G.; Walzer, J.F.; Tilley, T.D. σ-Bond metathesis mechanism for dehydropolymerization of silanes to polysilanes by d0 metal catalysts. J. Am. Chem. Soc. 1992, 114, 7047–7055. [Google Scholar] [CrossRef]
- Woo, H.G.; Tilley, T.D. Dehydrogenative polymerization of silanes to polysilanes by zirconocene and hafnocene catalysts. A new polymerization mechanism. J. Am. Chem. Soc. 1989, 111, 8043–8044. [Google Scholar] [CrossRef]
- Corey, J.Y.; Zhu, X.H.; Bedard, T.C.; Lange, L.D. Catalytic dehydrogenative coupling of secondary silanes with Cp2MCl2-BuLi. Organometallics 1991, 10, 924–930. [Google Scholar] [CrossRef]
- Dioumaev, V.K.; Harrod, J.F. A systematic analysis of the structure-reactivity trends for some ‘cation-like’ early transition metal catalysts for dehydropolymerization of silanes. J. Organomet. Chem. 1996, 521, 133–143. [Google Scholar] [CrossRef]
- Brintzinger, H.H.; Fischer, D.; Mühlhaupt, R.; Rieger, B.; Waymouth, R.M. Stereospecific olefin polymerization with chiral metallocene catalysts. Int. Ed. 1995, 34, 1143–1170. [Google Scholar] [CrossRef]
- Gauvin, F.; Harrod, J.F. Evidence for stereoregulation in the polymerization of phenylsilane by bis(indenyl)dimethylzirconium complexes. Can. J. Chem. 1990, 68, 1638–1640. [Google Scholar] [CrossRef]
- Banovetz, J.P.; Stein, K.M.; Waymouth, R.M. Stereoselectivity in the catalytic oligomerization of phenylsilane. Organometallics 1991, 10, 3430–3432. [Google Scholar] [CrossRef]
- Choi, N.; Onozawa, S.; Sakakura, T.; Tanaka, M. Dehydrocoupling of phenylsilane catalyzed by (dimethylamino)alkyl- or branched-alkyl-substituted zirconocene complexes: An alternative approach to longer chains. Organometallics 1997, 16, 2765–2767. [Google Scholar] [CrossRef]
- Grimmond, B.J.; Corey, J.Y. Catalytic dehydropolymerization of PhSiH3 to polyphenylsilane with substituted group IV metallocenes. Organometallics 1999, 18, 2223–2229. [Google Scholar] [CrossRef]
- Dioumaev, V.K.; Rahimian, K.; Gauvin, F.; Harrod, J.F. Stereostructures of linear and cyclic polyphenylsilanes produced by dehydrocoupling in the presence of group 4 metallocene catalysts. Organometallics 1999, 18, 2249–2255. [Google Scholar] [CrossRef]
- Lunzer, F.; Marschner, C.; Landgraf, S. (C5H5)2TiF2 and (C5H5)2ZrF2 as catalyst precursors for the dehydropolymersiation of Silanes. J. Organomet. Chem. 1998, 568, 253–255. [Google Scholar] [CrossRef]
- Resel, R.; Leising, G.; Lunzer, F.; Marschner, C. Structure in amorphous polysilanes determined by diffuse x-ray scattering. Polym. Commun. 1998, 39, 5257–5259. [Google Scholar] [CrossRef]
- Lunzer, F.; Marschner, C.; Winkler, B.; Peulecke, N.; Baumann, W.; Rosenthal, U. Group IV metallocene Bis(trimethylsilyl)acetylene complexes as catalysts in the dehydrocoupling polymerization of 1,2-Disubstituted Hydrodisilanes. Monatsh. Chem. 1999, 130, 215–219. [Google Scholar]
- Hengge, E.; Weinberger, M.; Jammegg, C. Dehydrogenative polymerization of disilanes catalysed by titanocene and zirconocene complexes. A novel route to oligo- and poly-silanes. J. Organomet. Chem. 1991, 410, C1–C4. [Google Scholar] [CrossRef]
- Hengge, E.; Weinberger, M. Dehydrierende polymerisation von Methyldisilanen mittels Metallocen-katalyse zu den oligomeren Me3Si[Me2Si]nSiMe2H, HMe2Si[Me2Si]nSiMe2H mit n = 3–9, H[MeSiH]nH mit n = 3–14 und hochpolymerem Poly(methylsilan) (MeSiHx)n. J. Organomet. Chem. 1992, 433, 21–34. [Google Scholar] [CrossRef]
- Hengge, E.; Weinberger, M. Dehydrierende copolymerisation zu den neuen polysilan-copolymeren poly(methyl-co-phenylsilan) H[(MeSiH)x(PhSiH)y]nH und poly(methyl-co-dimethylsilan) H[(MeSiH)x(Me2Si)y]nH. J. Organomet. Chem. 1992, 441, 397–410. [Google Scholar] [CrossRef]
- Tilley, T.D.; Woo, H.G. Dehydrogenative polymerization of silanes by σ-bond metathesis. Polym. Prepr. 1990, 31, 228–229. [Google Scholar]
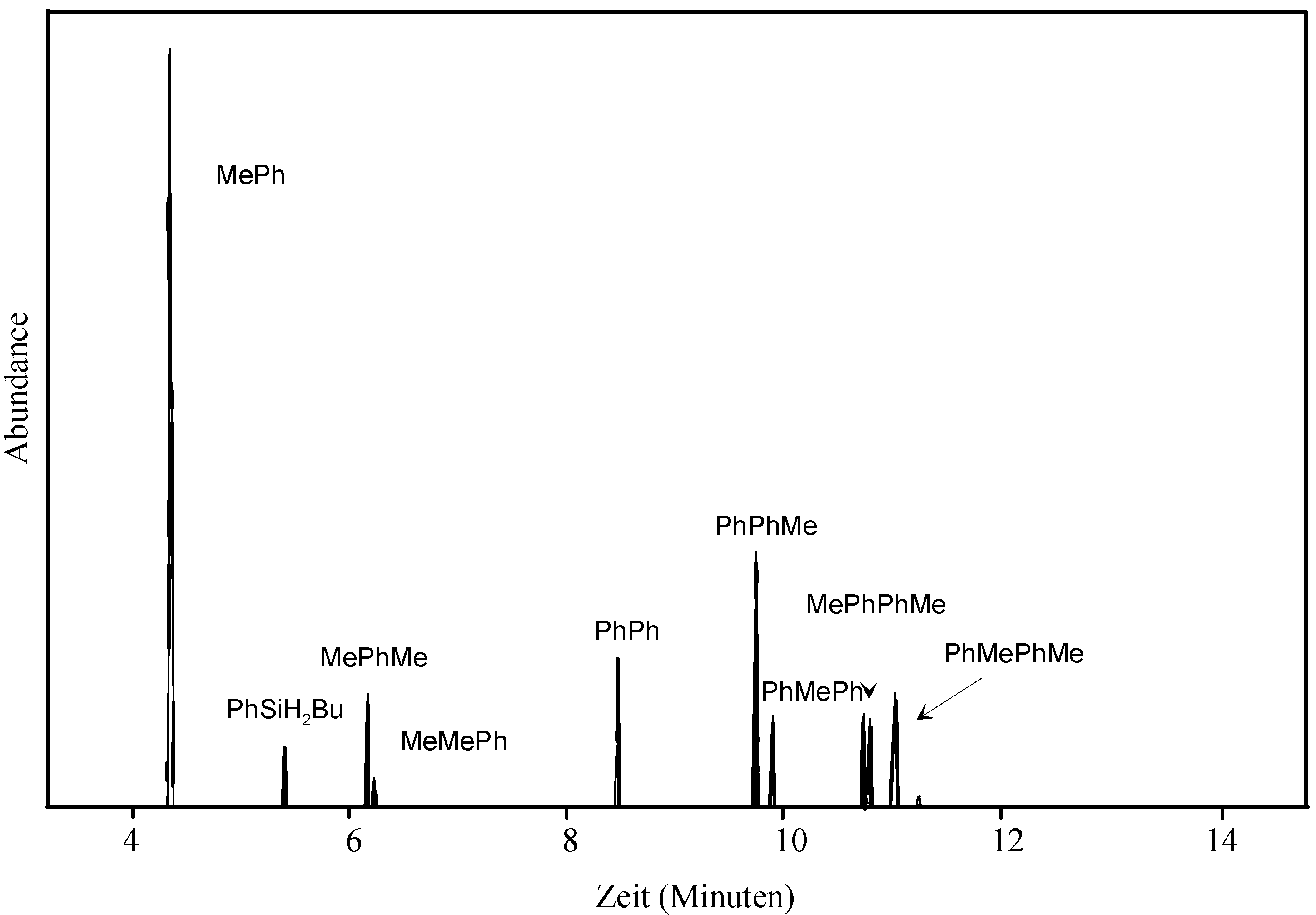
- The assignment of substitution patterns of tri- and tetrasilanes was done by fragmentation analysis of the obtained mass spectra based on earlier work (Ref. 37) and by 2D-HMQC 1H, 29Si NMR spectroscopy (a spectrum of the diastereomeric mixture 1,2 can be found in the supporting info).
- Woo, H.G.; Tilley, T.D. σ-Bond metathesis reactions of silicon-hydrogen and metal-silicon bonds. New routes to d0 metal silyl complexes. J. Am. Chem. Soc. 1989, 111, 3757–3758. [Google Scholar] [CrossRef]
- Woo, H.G.; Heyn, R.H.; Tilley, T.D. σ-Bond metathesis reactions for d0 metal-silicon bonds that produce zirconocene and hafnocene hydrosilyl complexes. J. Am. Chem. Soc. 1992, 114, 5698–5707. [Google Scholar] [CrossRef]
- Walsh, R. Bond dissociation energy values in silicon-containing compounds and some of their implications. Acc. Chem. Res. 1981, 14, 246–252. [Google Scholar] [CrossRef]
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals, 3rd ed.; Wiley-Interscience: New York, NY, USA, 2001. [Google Scholar]
- Gilman, H.; Lichtenwalter, G.D. The addition of silyllithium compounds containing methyl and phenyl groups to benzophenone in tetrahydrofuran. J. Am. Chem. Soc. 1958, 80, 607–608. [Google Scholar] [CrossRef]
- Aitken, C.; Barry, J.P.; Gauvin, F.; Harrod, J.F.; Malek, A.; Rousseau, D. A survey of catalytic activity of η5-cyclopentadienyl complexes of Groups 4–6 and uranium and thorium for the dehydrocoupling of phenylsilane. Organometallics 1989, 8, 1732–1736. [Google Scholar] [CrossRef]
- Morris, G.A.; Freeman, R. Enhancement of nuclear magnetic resonance signals by polarization transfer. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar] [CrossRef]
- Helmer, B.J.; West, R. Enhancement of 29Si NMR signals by proton polarization transfer. Organometallics 1982, 1, 877–879. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lunzer, F.; Marschner, C. Chemoselectivity in the Dehydrocoupling Synthesis of Higher Molecular Weight Polysilanes. Materials 2010, 3, 1125-1137. https://doi.org/10.3390/ma3021125
Lunzer F, Marschner C. Chemoselectivity in the Dehydrocoupling Synthesis of Higher Molecular Weight Polysilanes. Materials. 2010; 3(2):1125-1137. https://doi.org/10.3390/ma3021125
Chicago/Turabian StyleLunzer, Florian, and Christoph Marschner. 2010. "Chemoselectivity in the Dehydrocoupling Synthesis of Higher Molecular Weight Polysilanes" Materials 3, no. 2: 1125-1137. https://doi.org/10.3390/ma3021125