The Variety of Carbon-Metal Bonds inside Cu-ZSM-5 Zeolites: A Density Functional Theory Study

Abstract
:1. Introduction
2. Computational Section
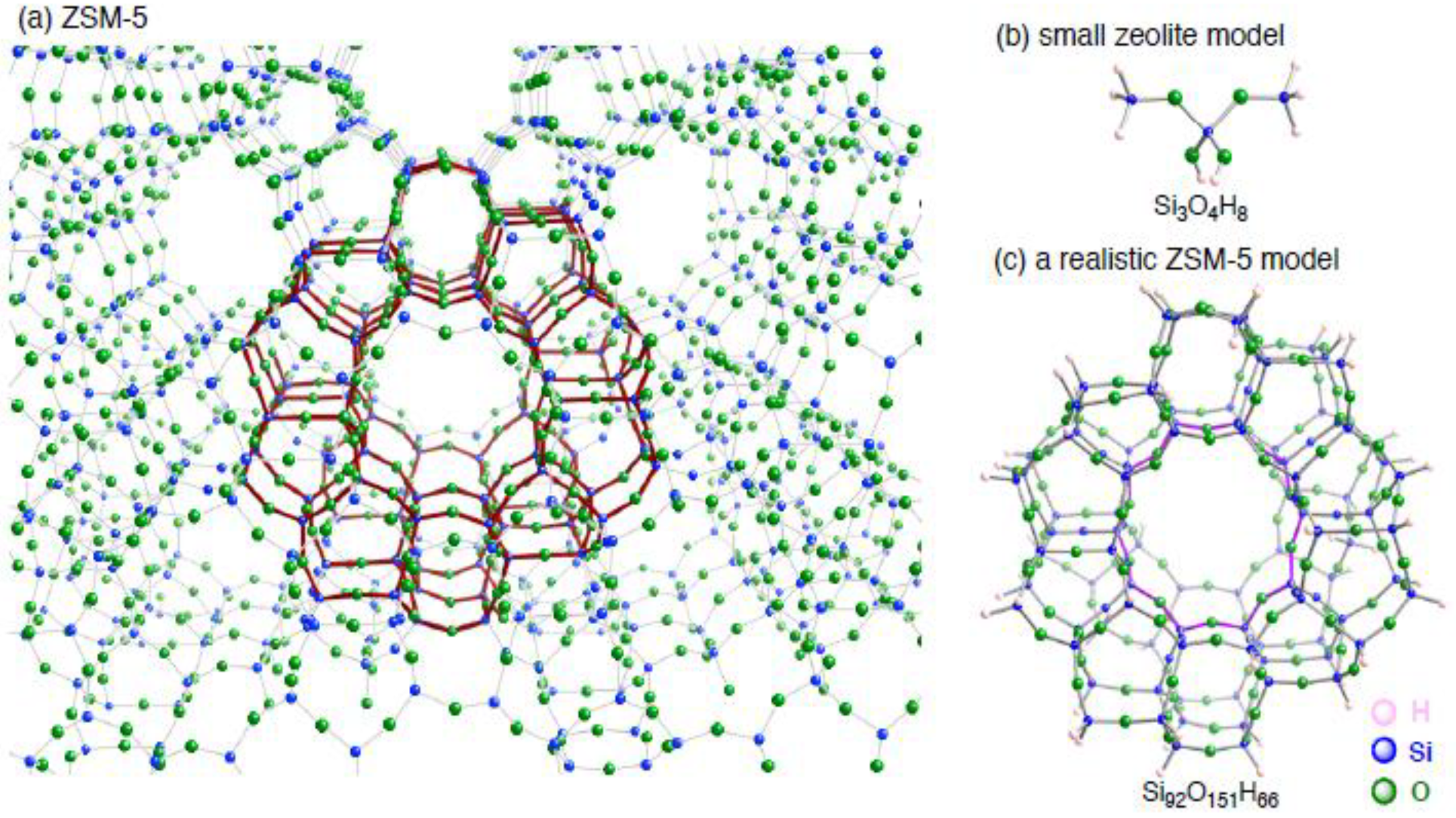
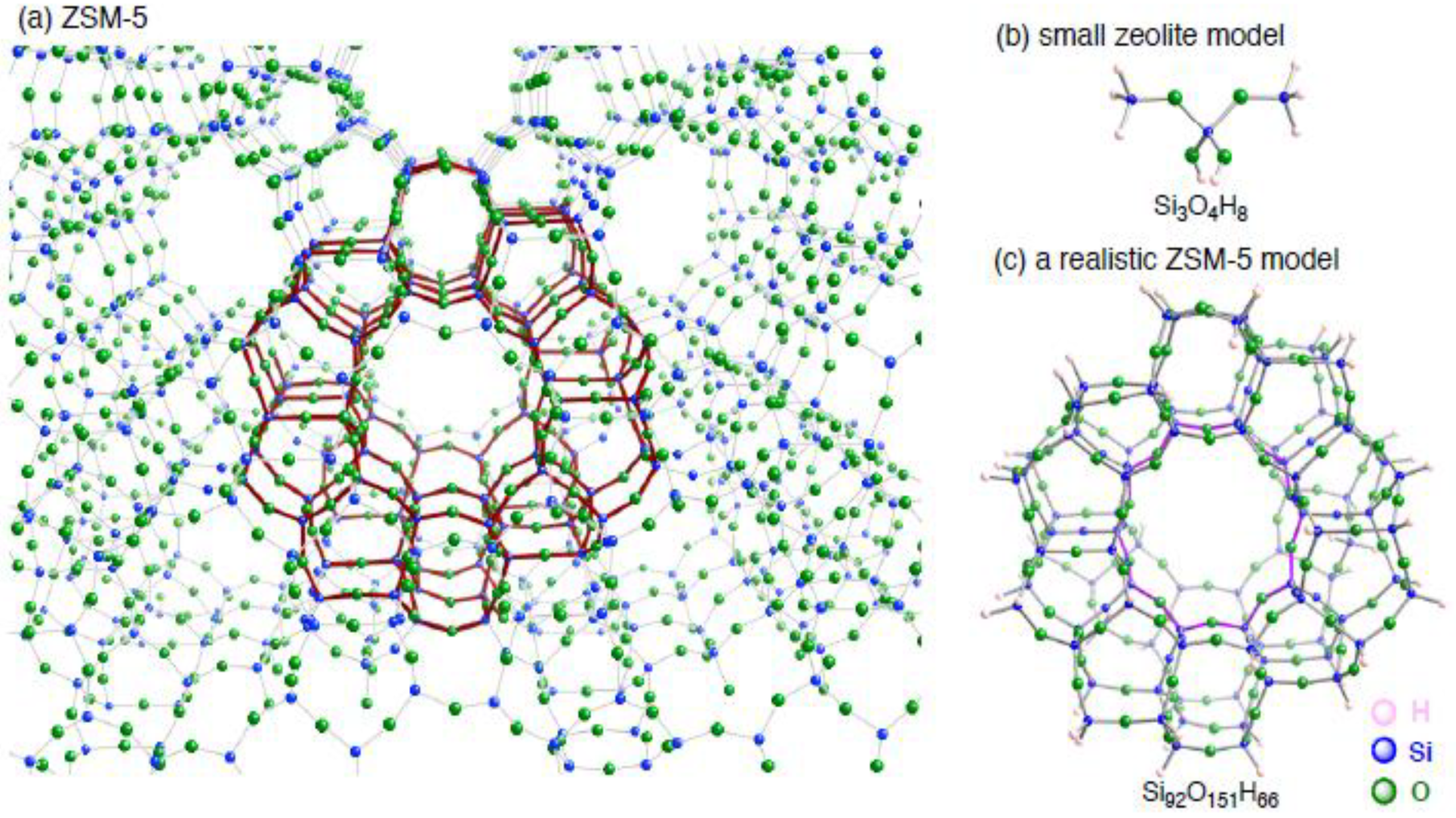
3. Results and Discussion
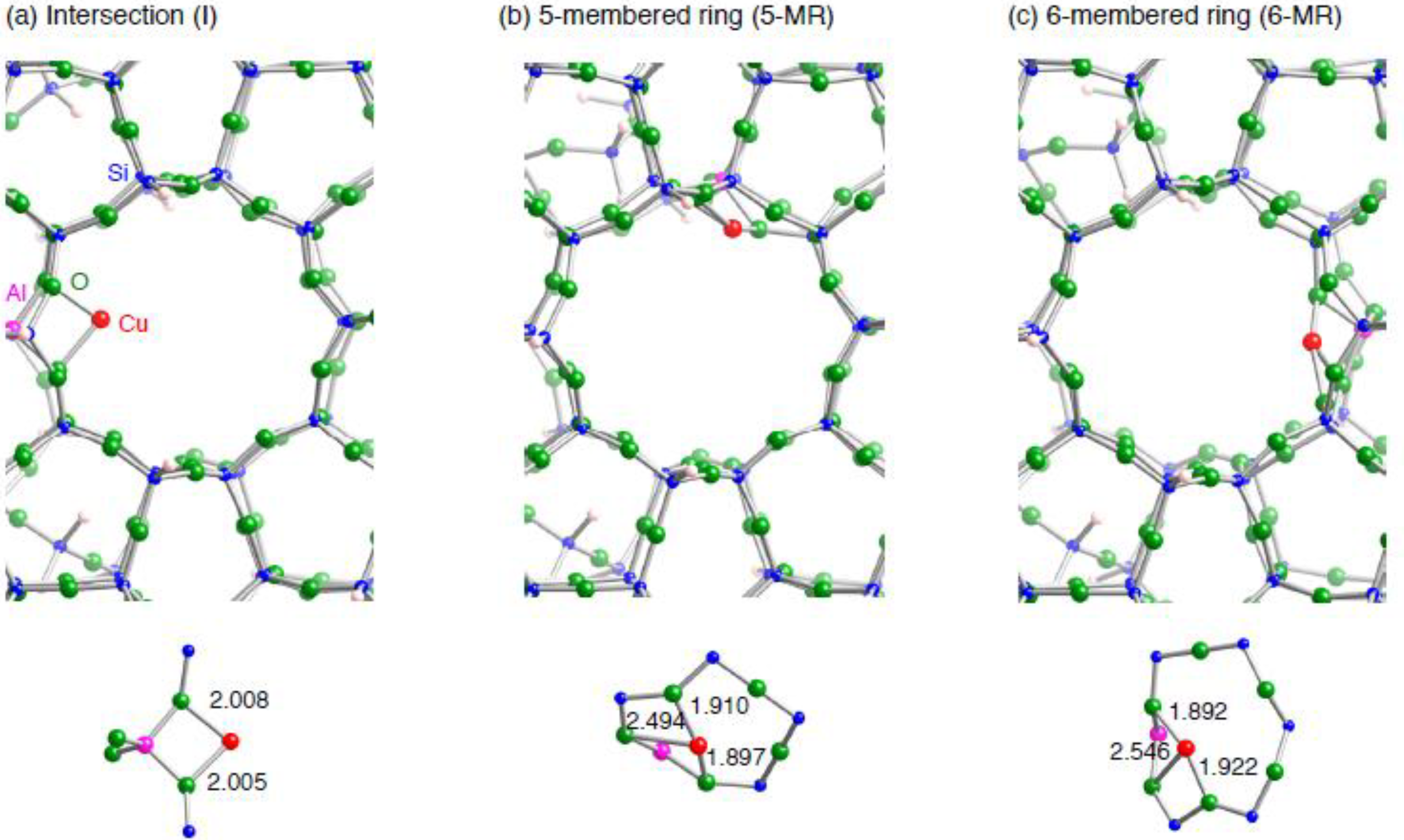
3.1. ZSM-5 containing monocopper cation (Cu1–ZSM-5)
3.1.1. The small monocopper zeolite model
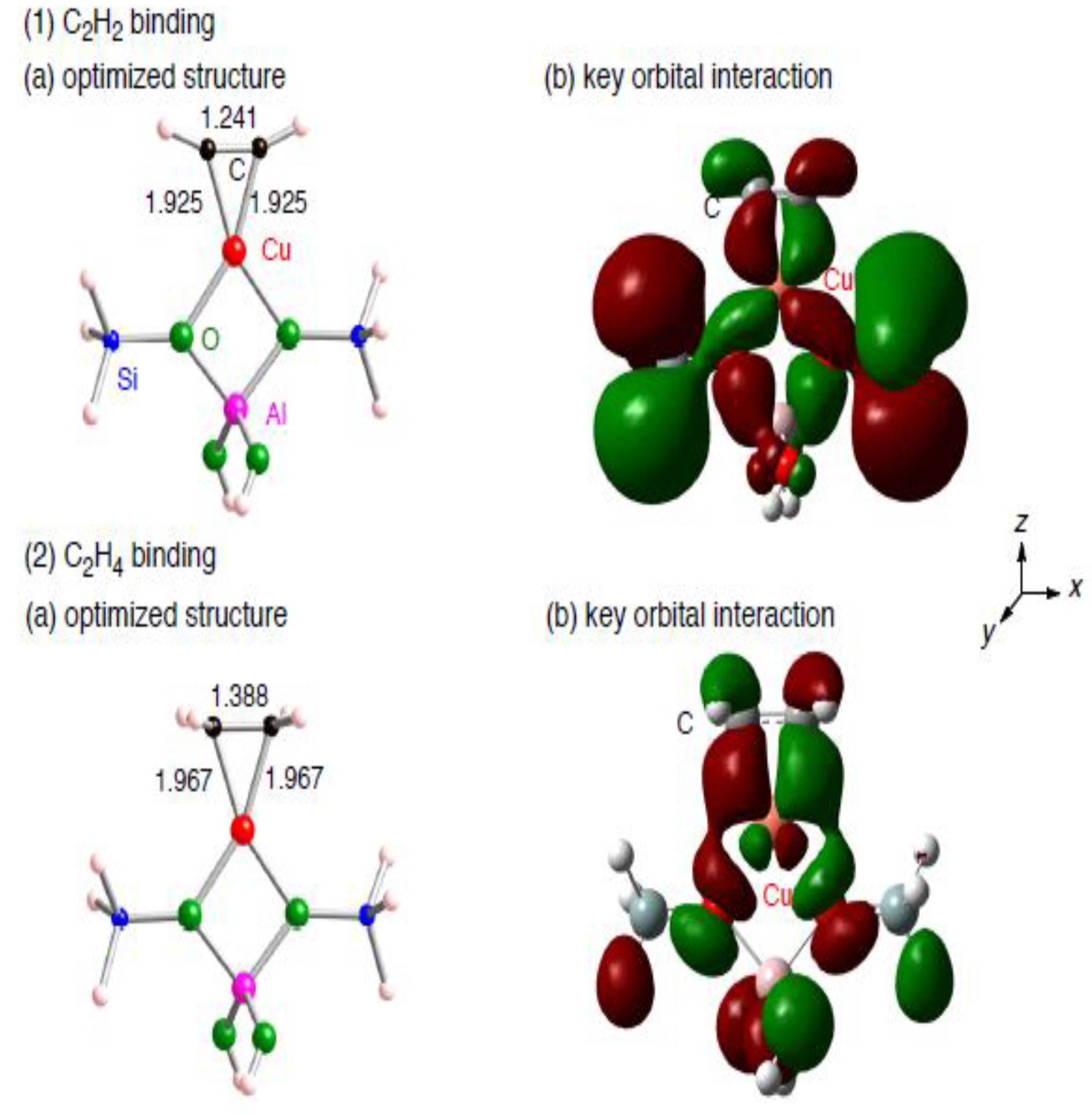

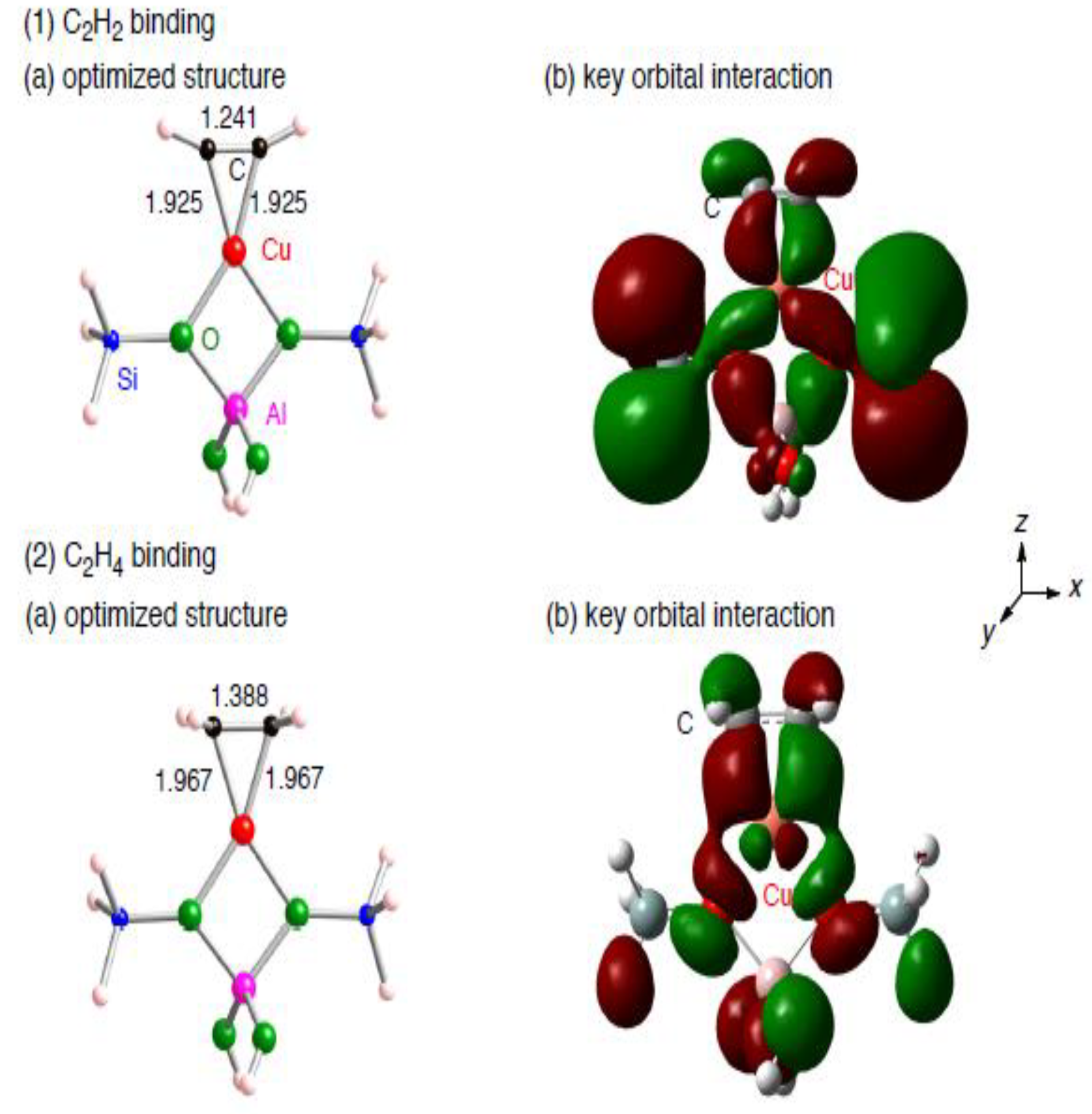
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Free C2H2 | C2H2 on Cu1-zeolite | ||||
|---|---|---|---|---|---|
| Symmetry | Frequency | IR intensity a | Frequency | IR intensity a | |
| C≡C stretch | Σg+ | 2001.4 | 0.00 | 1793.8 | 1.12 |
| C–H stretch | Σg+ | 3400.0 | 0.00 | 3273.7 | 0.40 |
| C–H stretch | Σu+ | 3300.5 | 1 | 3204.0 | 1 |
| Free C2H4 | C2H4 on Cu1-zeolite | ||||
|---|---|---|---|---|---|
| Symmetry | Frequency | IR intensity a | Frequency | IR intensity a | |
| C≡C stretch | Ag | 1645.2 | 0.00 | 1529.4 | 0.32 |
| C–H stretch | Ag | 3036.3 | 0.00 | 3023.9 | 0.00 |
| C–H stretch | B3g | 3089.8 | 0.00 | 3090.7 | 0.00 |
| C–H stretch | B1u | 3021.3 | 0.64 | 3017.7 | 0.98 |
| C–H stretch | B2u | 3117.5 | 1 | 3113.2 | 1 |
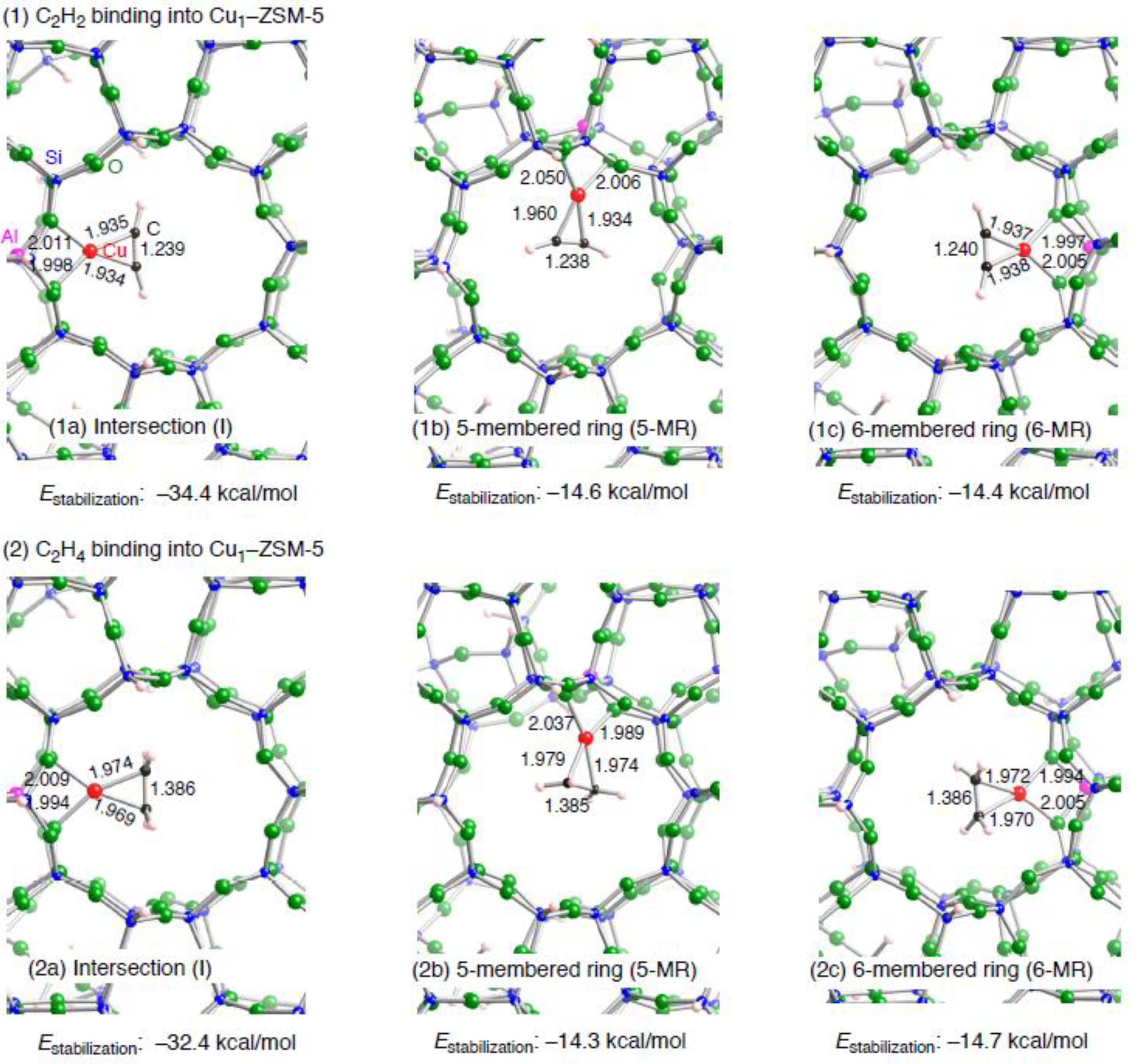
3.1.2. Realistic Cu1–ZSM-5 model
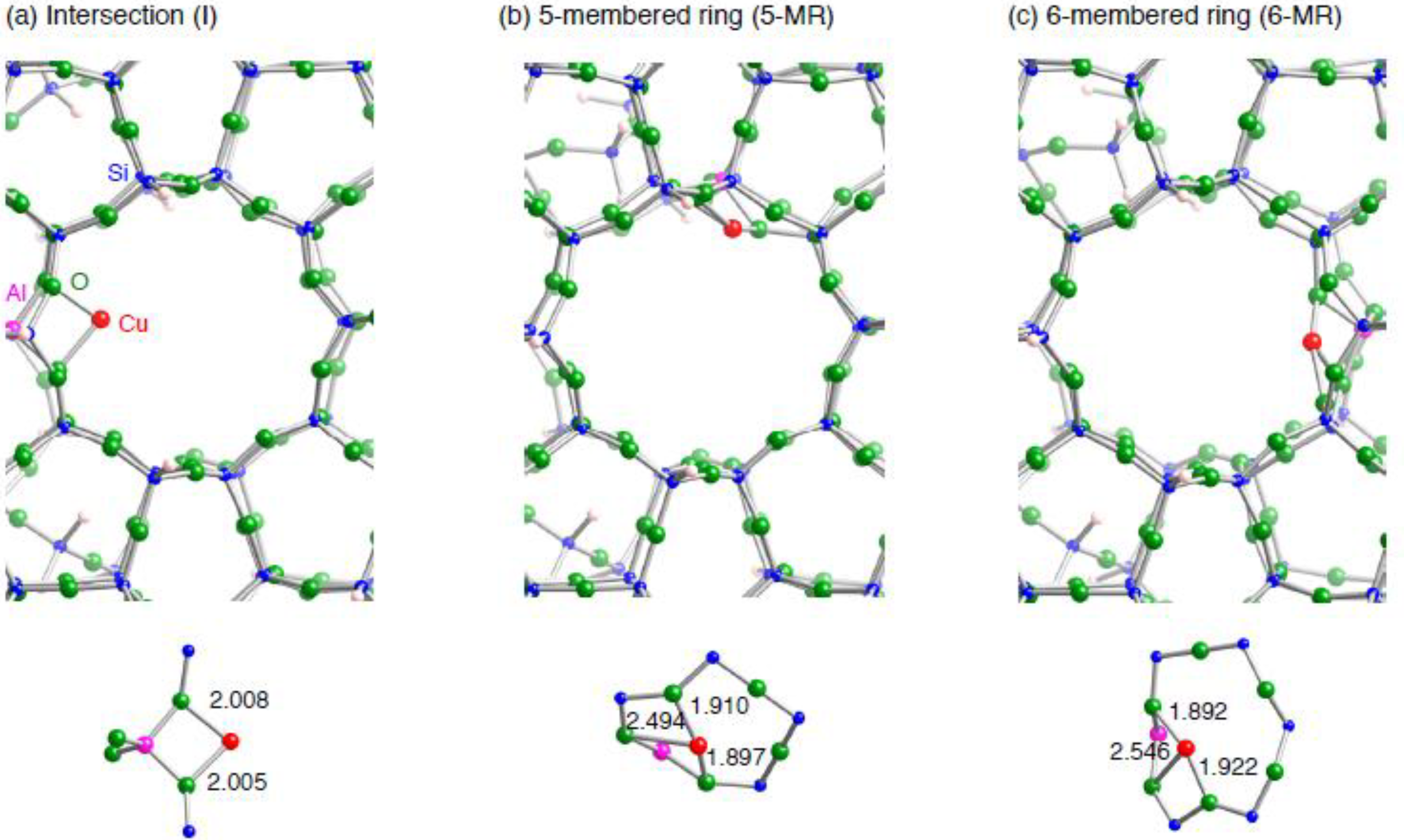
| Configuration | Adsorbent | Electronic configuration |
|---|---|---|
| I | – | 4s ( 0.25) 3d ( 9.84) |
| C2H2 | 4s ( 0.36) 3d ( 9.56) 4p ( 0.01) | |
| C2H4 | 4s ( 0.37) 3d ( 9.57) 4p ( 0.01) | |
| 5-MR | – | 4s ( 0.42) 3d ( 9.73) |
| C2H2 | 4s ( 0.36) 3d ( 9.57) 4p ( 0.01) | |
| C2H4 | 4s ( 0.36) 3d ( 9.58) 4p ( 0.01) | |
| 6-MR | – | 4s ( 0.44) 3d ( 9.71) |
| C2H2 | 4s ( 0.36) 3d ( 9.55) 4p ( 0.01) | |
| C2H4 | 4s ( 0.37) 3d ( 9.57) 4p ( 0.01) |

| Configuration a | Adsorbent | Binding mode | Cu–Cb | CCb | Estabilizationc | E(deform) d |
|---|---|---|---|---|---|---|
| I | C2H2 | η2 | 1.934, 1.935 | 1.239 | –34.4 | 2.6 |
| 5MR | C2H2 | η2 | 1.934, 1.960 | 1.238 | –14.6 | 20.4 |
| 6MR | C2H2 | η2 | 1.937, 1.938 | 1.240 | –14.4 | 22.5 |
| I | C2H4 | η2 | 1.969, 1.974 | 1.386 | –32.4 | 2.3 |
| 5MR | C2H4 | η2 | 1.974, 1.979 | 1.385 | –14.3 | 17.8 |
| 6MR | C2H4 | η2 | 1.970, 1.972 | 1.386 | –14.7 | 20.3 |
3.2. ZSM-5 containing dicopper active center (Cu2–ZSM-5)
3.2.1. The small dicopper zeolite model
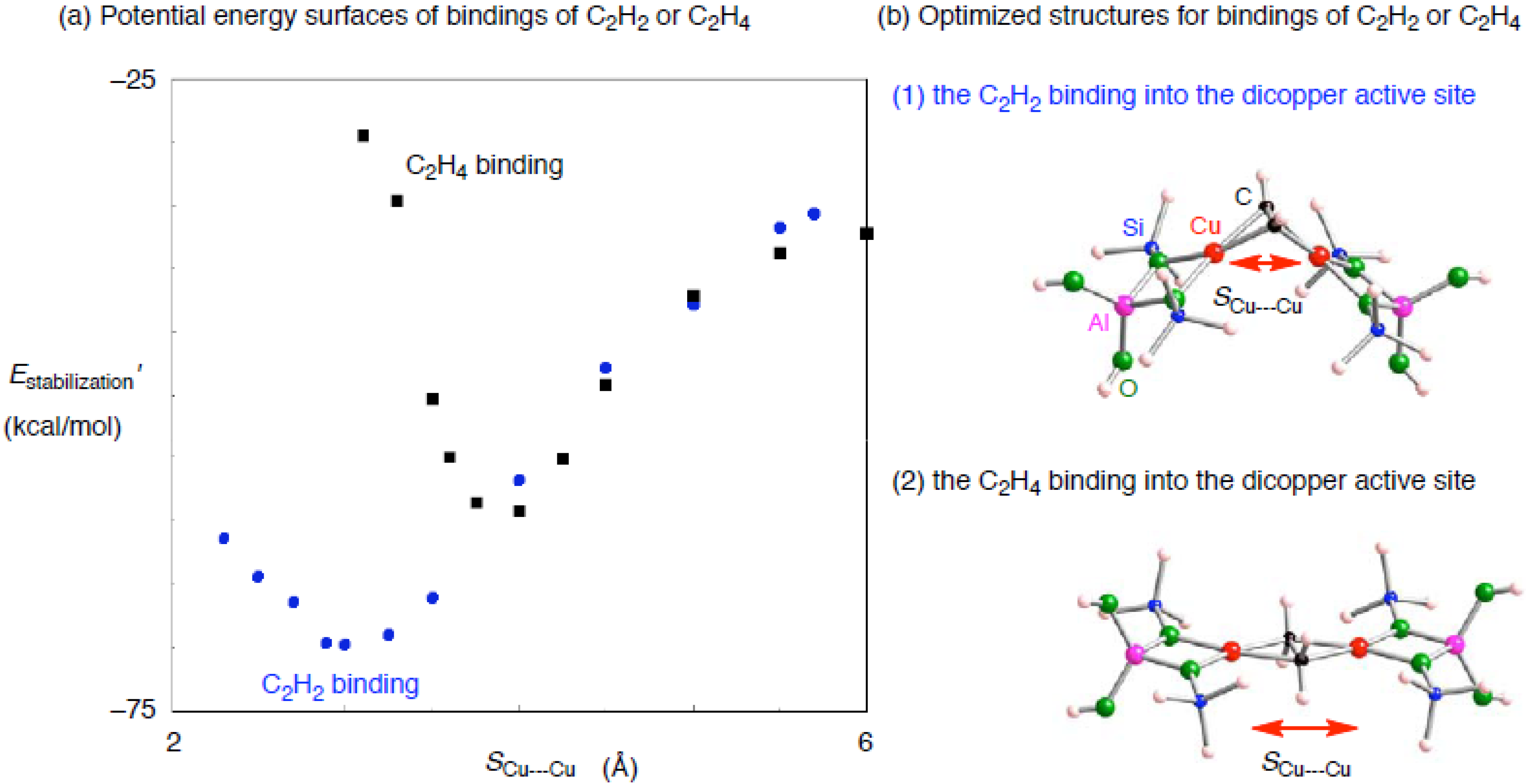
| Adsorbent | Binding mode | Cu–Ca | CCa | SCu•••Cub | Estabilization’c |
|---|---|---|---|---|---|
| C2H2 | μ−η2: η2 | 1.932, 1.933 1.933, 1.934 | 1.297 | 2.888 | –69.6 |
| C2H4 | μ−η2: η2 | 2.003, 2.003, 2.003, 2.003 | 1.449 | 3.735 | –58.6 |
| Free C2H2 | C2H2 on Cu2-zeolite | ||||
|---|---|---|---|---|---|
| Symmetry | Frequency | IR intensitya | Frequency | IR intensitya | |
| C≡C stretch | Σg+ | 2001.4 | 0.00 | 1557.1 | 1.44 |
| C–H stretch | Σg+ | 3400.0 | 0.00 | 3170.0 | 0.90 |
| C–H stretch | Σu+ | 3300.5 | 1 | 3127.8 | 1 |
| Free C2H4 | C2H4 on Cu2-zeolite | ||||
|---|---|---|---|---|---|
| Symmetry | Frequency | IR intensity a | Frequency | IR intensity a | |
| C≡C stretch | Ag | 1645.2 | 0.00 | 1469.4 | 0.00 |
| C–H stretch | Ag | 3036.3 | 0.00 | 3004.9 | 0.00 |
| C–H stretch | B3g | 3089.8 | 0.00 | 3085.4 | 0.00 |
| C–H stretch | B1u | 3021.3 | 0.64 | 3001.8 | 17.10 |
| C–H stretch | B2u | 3117.5 | 1 | 3100.5 | 1 |
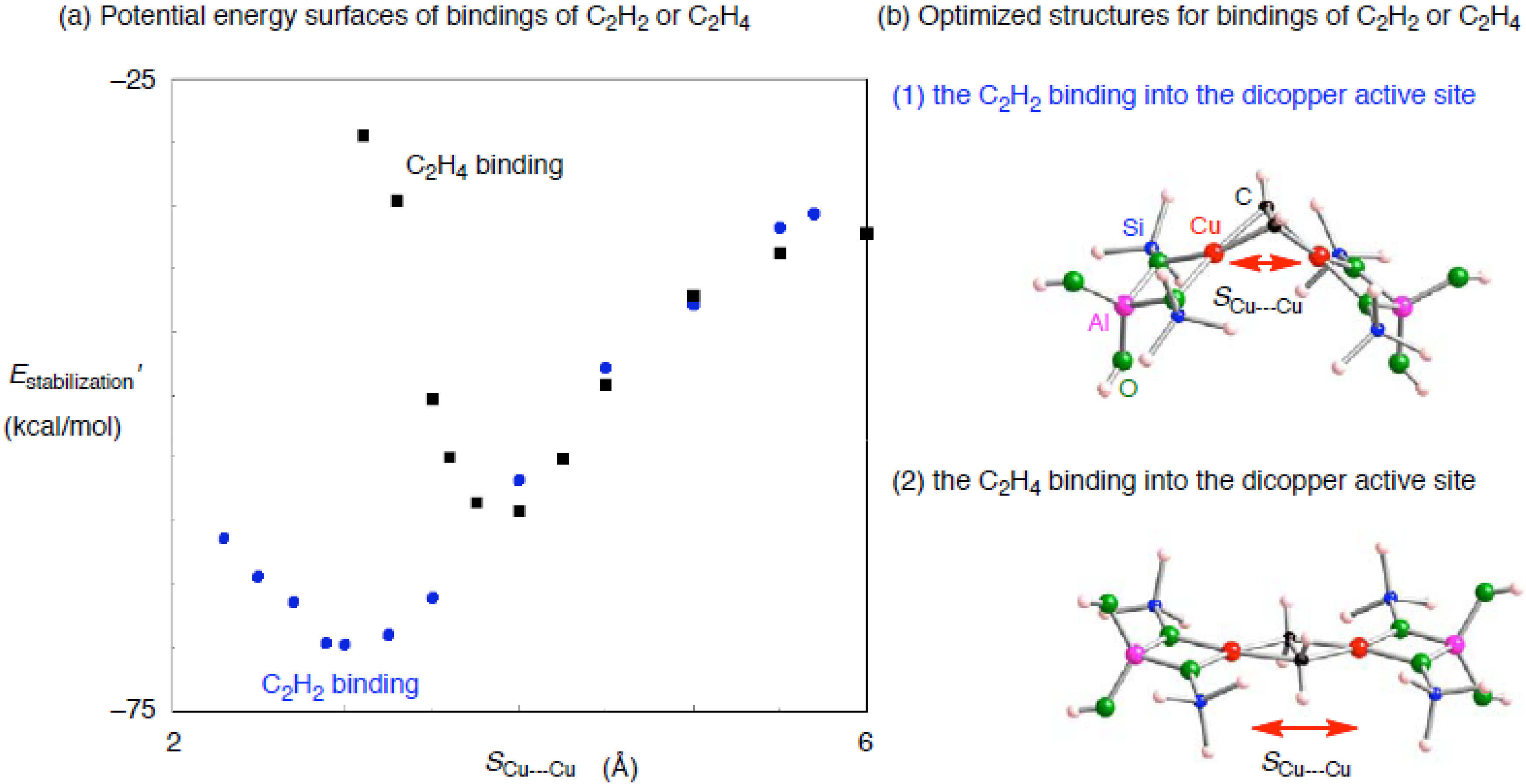
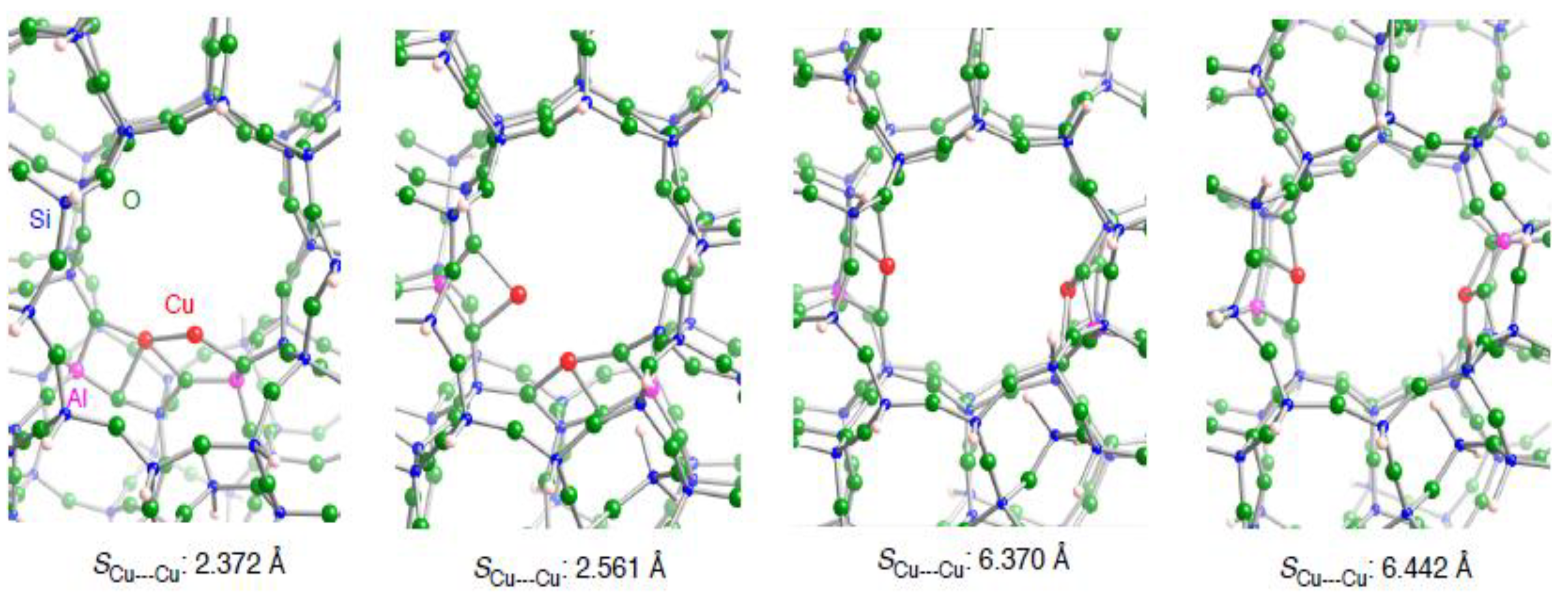
3.2.2. Realistic Cu2–ZSM-5 model

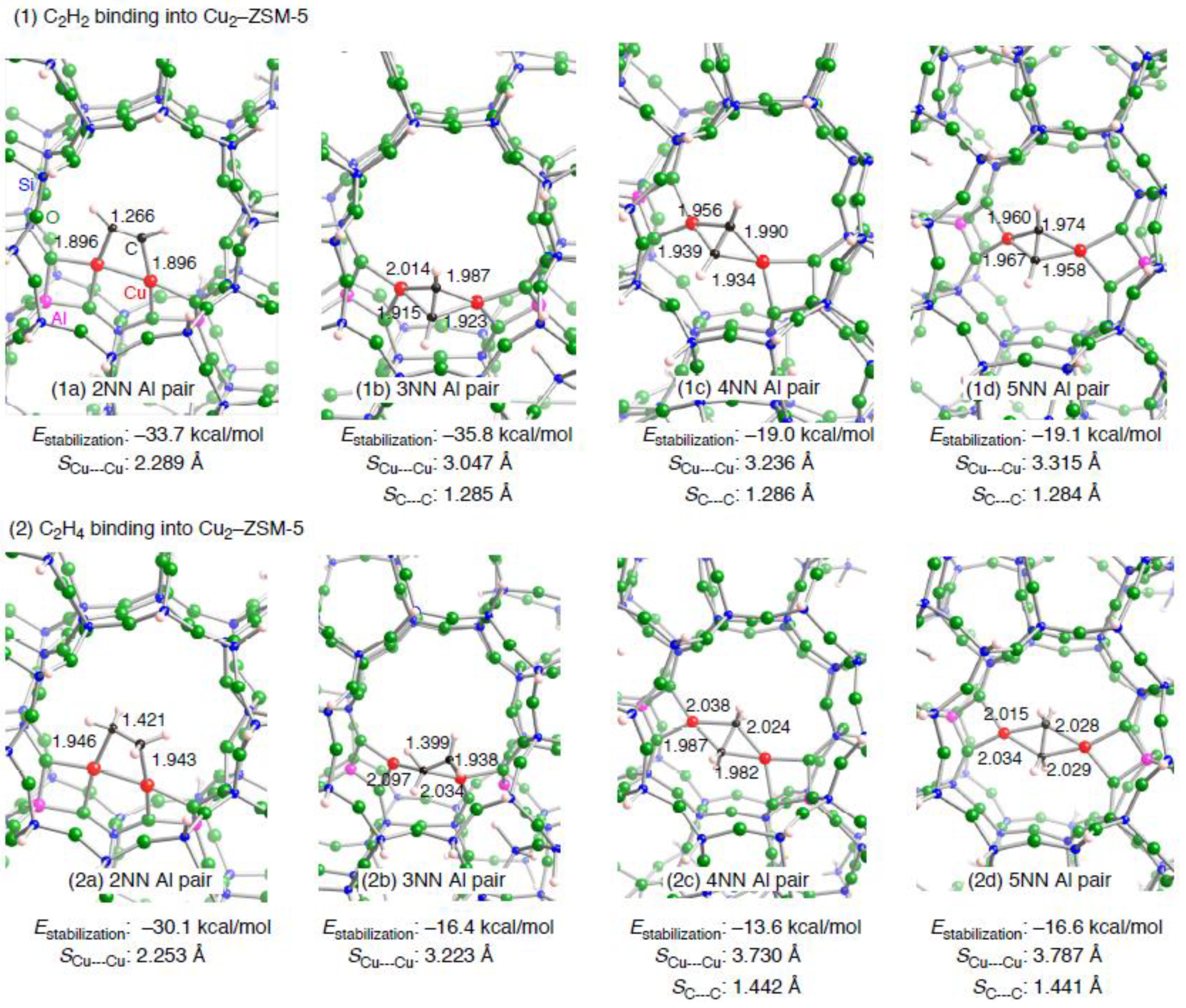
| Configuration a | Adsorbent | Binding mode | Cu–C b | CC b | SCu•••Cu c (ΔSCu•••Cud) | Estabilization e | E(deform) f |
|---|---|---|---|---|---|---|---|
| 2NN | C2H2 | μ−η1: η1 | 1.896, 1,896 | 1.266 | 2.289 (–0.083) | –33.7 | 19.0 |
| 3NN | C2H2 | μ−η2: η2 | 1.915, 1.923, 1.987, 2.014 | 1.285 | 3.047 (0.486) | –35.8 | 24.7 |
| 4NN | C2H2 | μ−η2: η2 | 1.934, 1.939, 1.956, 1.990 | 1.286 | 3.236 (–3.134) | –19.0 | 45.7 |
| 5NN | C2H2 | μ−η2: η2 | 1.958, 1.960, 1.967, 1.974 | 1.284 | 3.315 (–3.127) | –19.1 | 47.2 |
| 2NN | C2H4 | μ−η1: η1 | 1.946, 1.943 | 1.421 | 2.253 (–0.119) | –30.1 | 18.0 |
| 3NN | C2H4 | μ−η1: η2 | 1.938, 2.034, 2.097 | 1.399 | 3.223 (0.662) | –16.4 | 25.5 |
| 4NN | C2H4 | μ−η2: η2 | 1.982, 1.987, 2.024, 2.038 | 1.442 | 3.730 (–2.640) | –13.6 | 41.9 |
| 5NN | C2H4 | μ−η2: η2 | 2.015, 2.028, 2.029, 2.034 | 1.441 | 3.787 (–2.655) | –16.6 | 43.1 |
4. Conclusions
Acknowledgements
References and Notes
- Crabtree, R.H. The Organometallic Chemistry of the Transtion Metals, 2nd ed.; John Wiley & Son, Inc.: New York, NY, USA, 1994. [Google Scholar]
- Crabtree, R.H. The organometallic chemistry of alkanes. Chem. Rev. 1985, 85, 245–269. [Google Scholar] [CrossRef]
- Bergman, B.G. C–H activation. Nature 2007, 446, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Arndtsen, B.A.; Bergman, B.G.; Mobley, T.A.; Peterson, T.H. Selective intermolecular carbon-hydrogen bond activation by synthetic metal complexes in homogeneous solution. Acc. Chem. Res. 1995, 28, 154–162. [Google Scholar] [CrossRef]
- Schröder, D.; Schwarz, H. C-H and C-C bond activation by bare transition-metal oxide cations in the gas phase. Angew. Chem. Int. Ed. Engl. 1995, 34, 1973–1995. [Google Scholar] [CrossRef]
- Eller, K.; Schwarz, H. Organometallic chemistry in the gas phase. Chem. Rev. 1991, 91, 1121–1177. [Google Scholar] [CrossRef]
- Weisshaar, J.C. Bare transition metal atoms in the gas phase: reactions of M, M+, and M2+ with hydrocarbons. Acc. Chem. Res. 1993, 26, 213–219. [Google Scholar] [CrossRef]
- Trost, B.M. On inventing reactions for atom economy. Acc. Chem. Res. 2002, 35, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C. Enhancing electrophilic alkene activation by increasing the positive net charge in transition-metal complexes and application in homogeneous catalysis. Chem. Eur. J. 2004, 10, 5888–5899. [Google Scholar] [CrossRef] [PubMed]
- Shriver, D.F.; Atkins, P.W. Inorganic Chemistry, 4th ed.; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Yumura, T.; Kertesz, M.; Iijima, S. Local modifications of single-wall carbon nanotubes induced by bond formation with encapsulated fullerenes. J. Phys. Chem. B 2007, 111, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Yumura, T.; Kertesz, M. Cooperative behaviors in carbene additions through local modifications of nanotube surfaces. Chem. Mater. 2007, 19, 1028–1034. [Google Scholar] [CrossRef]
- Yumura, T.; Kertesz, M.; Iijima, S. Confinement effects on site-preferences for cycloadditions into carbon nanotubes. Chem. Phys. Lett. 2007, 444, 155–160. [Google Scholar] [CrossRef]
- Yumura, T.; Takeuchi, M.; Kobayashi, H.; Kuroda, Y. Effects of ZSM-5 zeolite confinement on reaction intermediates during dioxygen activation by enclosed dicopper cations. Inorg. Chem. 2009, 48, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Itadani, A.; Sugiyama, H.; Tanaka, M.; Ohkubo, T.; Yumura, T.; Kobayashi, H.; Kuroda, Y. Potential for C−H Activation in CH4 Utilizing a CuMFI-Type Zeolite as a Catalyst. J. Phys. Chem. C 2009, 113, 7213–7222. [Google Scholar] [CrossRef]
- Yumura, T.; Yamashita, H.; Torigoe, H.; Kobayashi, H.; Kuroda, Y. Site-specific Xe addition into Cu–ZSM-5 zeolite. Phys. Chem. Chem. Phys. 2010, 12, 2392–2400. [Google Scholar] [CrossRef]
- Iwamoto, M.; Furukawa, H.; Mine, Y.; Uemura, F.; Mikuriya, S.; Kagawa, S. Copper(II) ion-exchanged ZSM-5 zeolites as highly active catalysts for direct and continuous decomposition of nitrogen monoxide. J. Chem. Soc., Chem. Commun. 1986, 1272–1273. [Google Scholar] [CrossRef]
- Kuhn, P.; Pale, P.; Sommer, J.; Louis, B. Probing Cu-USY zeolite reactivity: design of a green catalyst for the synthesis of diynes. J. Phys. Chem. C 2009, 113, 2903–2910. [Google Scholar] [CrossRef]
- Yu, J.-S.; Kevan, L. Effects of reoxidation and water vapor on selective partial oxidation of propylene to acrolein in copper(II)-exchanged X and Y zeolites. J. Phys. Chem. 1991, 95, 6648–6653. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phy. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03. Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- The ZSM-5 structure was taken from the Cerius 2 database. Accerys, Software, Inc.: San Diego, CA, USA, 2001.
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Wachters, A.J.H. Gaussian basis set for molecular wavefunctions containing third-row atoms. J. Chem. Phys. 1970, 52, 1033–1036. [Google Scholar] [CrossRef]
- Hay, R.J. Gaussian basis sets for molecular calculations. The representation of 3d orbitals in transition-metal atoms. J. Chem. Phys. 1977, 66, 4377–4384. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Binkley, J.S.; Pople, J.A.; Hehre, W.J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J. Am. Chem. Soc. 1980, 102, 939–947. [Google Scholar] [CrossRef]
- Gordon, M.S.; Binkley, J.S.; Pople, J.A.; Pietro, W.J.; Hehre, W.J. Self-consistent molecular-orbital methods. 22. Small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 1982, 104, 2797–2803. [Google Scholar] [CrossRef]
- Pietro, W.J.; Francl, M.M.; Gordon, M.S.; Hehre, W.J.; Defrees, D.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods. 24. Supplemented small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 1982, 104, 5039–5048. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Nachtigallová, D.; Nachtigall, P.; Sierka, M.; Sauer, J. Coordination and siting of Cu+ ions in ZSM-5: A combined quantum mechanics/interatomic potential function study. Phys. Chem. Chem. Phys. 1999, 1, 2019–2026. [Google Scholar] [CrossRef]
- Spuhler, P.; Holthausen, M.C.; Nachtigallová, D.; Nachtigall, P.; Sauer, J. On the Existence of CuI Pairs in ZSM-5 - A Computational Study. Chem. Eur. J. 2002, 8, 2099–2115. [Google Scholar] [CrossRef] [PubMed]
- Davidová, M.; Nachtigallová, D.; Bulánek, R.; Nachtigall, P. Characterization of the Cu+ sites in high-silica zeolites interacting with the CO molecule: combined computational and experimental Study. J. Phys. Chem. B 2003, 107, 2327–2332. [Google Scholar] [CrossRef]
- Bulánek, R.; Silhan, M.; Nachtigallová, D.; Nachtigall, P. Calculations of site-specific CO stretching frequencies for copper carbonyls with the “near apectroscopic accuracy”: CO interaction with Cu+/MFI. J. Phys. Chem. A 2003, 107, 10381–10388. [Google Scholar] [CrossRef]
- Davidová, M.; Nachtigallová, D.; Nachtigall, P.; Sauer, J. Nature of the Cu+–NO Bond in the Gas Phase and at Different Types of Cu+ Sites in Zeolite Catalysts. J. Phys. Chem. B 2004, 108, 13674–13682. [Google Scholar] [CrossRef]
- Bludský, O.; Silhan, M.; Nachtigall, P.; Bucko, T.; Benco, L.; Hafner, J. Theoretical Investigation of CO Interaction with Copper Sites in Zeolites: Periodic DFT and Hybrid Quantum Mechanical/Interatomic Potential Function Study. J. Phys. Chem. B 2005, 109, 9631–9638. [Google Scholar] [CrossRef] [PubMed]
- Bulánek, R.; Drobná, H.; Nachtigall, P.; Rubeš, M.; Bludský, O. On the site-specificity of polycarbonyl complexes in Cu/zeolites: combined experimental and DFT study. Phys. Chem. Chem. Phys. 2006, 8, 5535–5542. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, Y.; Bell, A.T. Density Functional Theory Study of CO Adsorption on Cu(I)-ZSM-5. J. Phys. Chem. C 2007, 111, 13442–13451. [Google Scholar] [CrossRef]
- Mentzen, B.F.; Bergeret, G. Crystallographic Determination of the Positions of the Copper Cations in Zeolite MFI. J. Phys. Chem. C 2007, 111, 12512–12516. [Google Scholar] [CrossRef]
- Kumashiro, R.; Kuroda, Y.; Nagao, M. New analysis of oxidation state and coordination environment of copper ion-exchanged in ZSM-5 Zeolite. J. Phys. Chem. B 1999, 103, 89–96. [Google Scholar] [CrossRef]
- Kuroda, Y.; Kumashiro, R.; Yoshimoto, T.; Nagao, M. Characterization of active sites on copper ion-exchanged ZSM-5-type zeolite for NO decomposition reaction. Phys. Chem. Chem. Phys. 1999, 1, 649–656. [Google Scholar] [CrossRef]
- Kuroda, Y.; Yagi, K.; Horiguchi, N.; Yoshikawa, Y.; Kumashiro, R.; Nagao, M. New light on the state of active sites in CuZSM-5 for the NO decomposition reaction and N2 adsorption. Phys. Chem. Chem. Phys. 2003, 5, 3318–3327. [Google Scholar] [CrossRef]
- Hüber, G.; Rauhut, G.; Stoll, H.; Roduner, E. Ethyne adsorbed on CuNaY zeolite: FTIR spectra and quantum chemical calculations. J. Phys. Chem. B 2003, 107, 8568–8573. [Google Scholar] [CrossRef]
- Hüber, G.; Rauhut, G.; Stoll, H.; Roduner, E. FTIR measurements and quantum chemical calculations of Ethylene adsorbed on CuNaY. Phys. Chem. Chem. Phys. 2002, 4, 3112–3121. [Google Scholar] [CrossRef]
- Broclawik, E.; Rejmak, P.; Kozyra, P.; Datka, J. DFT quantum chemical modeling of the interaction of alkenes with Cu+ sites in zeolites. Catal. Today 2006, 114, 162–168. [Google Scholar] [CrossRef]
- Rejmak, P.; Mitoraj, M.; Broclawik, E. Electronic view on ethene adsorption in Cu(I) exchanged zeolites. Phys. Chem. Chem. Phys. 2010, 12, 2321–2330. [Google Scholar] [CrossRef] [PubMed]
- Calculated vibrational frequencies were scaled by 0.9659, along the following reference.: Halls, M.D.; Velkovski, J.; Schlegel, H.B. Harmonic frequency scaling factors for Hartree-Fock, S-VWN, B-LYP, B3-LYP, B3-PW91 and MP2 with the Sadlej pVTZ electric property basis set. Theor. Chem. Acc. 2001, 105, 413–421. [Google Scholar]
- Herzberg, G. Molecular Spectra and Molecular Structure: Infrared and Raman Spectra of Polyatomic Molecules; Krieger Publishing com.: Malabar, FL, USA, 1991. [Google Scholar]
- Willian, K.R.; Ewing, E. Infrared spectra and structures of ethene on NaCl(100). J. Phys. Chem. 1995, 99, 2186–2193. [Google Scholar] [CrossRef]
- Datka, J.; Kukulska-Zając, E.; Kobyzewa, W. The activation of acetylene by Cu+ ions in zeolites studied by IR spectroscopy. Catal. Today 2005, 101, 123–129. [Google Scholar] [CrossRef]
- Datka, J.; Kukulska-Zając, E. IR studies of the activation of C=C bond in alkenes by Cu+ ions in zeolites. J. Phys. Chem. B 2004, 108, 17760–17766. [Google Scholar] [CrossRef]
- Itadani, A.; Yumura, T.; Ohkubo, T.; Kobayashi, H.; Kuroda, Y. Existence of dual species composed of Cu+ in CuMFI being bridged by C2H2. Phys. Chem. Chem. Phys. 2010. submitted. [Google Scholar]
- Nachtigallová, D.; Bludsky, O.; Areán, C.O.; Bulánek, R.; Nachtigall, P. The vibrational dynamics of carbon monoxide in a confined space–CO in zeolites. Phys. Chem. Chem. Phys. 2006, 8, 4849. [Google Scholar] [CrossRef] [PubMed]
- Nachtigall, P.; Bulánek, R. Theoretical investigation of site-specific characteristics of CO adsorption complexes in the Li+-FER zeolite. Appl. Catal., A 2006, 307, 118–127. [Google Scholar] [CrossRef]
- Palomino, G.T.; Fisicaro, P.; Bordiga, S.; Zecchina, A.; Giamello, E.; Lamberti, C. Oxidation states of copper ions in ZSM-5 zeolites. a multitechnique investigation. J. Phys. Chem. B 2000, 104, 4064–4073. [Google Scholar] [CrossRef]
- Hamada, H.; Matsubayashi, N.; Shimada, H.; Kintaichi, Y.; Ito, T.; Nishijima, A. XANES and EXAFS analysis of copper ion-exchanged ZSM-5 zeolite catalyst used for nitrogen monoxide decomposition. Catal. Lett. 1990, 5, 189–196. [Google Scholar] [CrossRef]
- Grünert, W.; Hayes, N.W.; Joyner, R.W.; Shpiro, E.S.; Siddiqui, M.R.H.; Baeva, G.N. Structure, chemistry, and activity of Cu-ZSM-5 catalysts for the selective reduction of NOx in the presence of oxygen. J. Phys. Chem. 1994, 98, 10832–10846. [Google Scholar] [CrossRef]
- Borgard, G.D.; Molvik, S.; Balaraman, P.; Root, T.W.; Dumesic, J.A. Microcalorimetric and infrared spectroscopic studies of CO, C2H4, N2O, and O2 adsorption on Cu-Y zeolite. Langmiur 1995, 11, 2065–2070. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yumura, T.; Hasegawa, S.; Itadani, A.; Kobayashi, H.; Kuroda, Y. The Variety of Carbon-Metal Bonds inside Cu-ZSM-5 Zeolites: A Density Functional Theory Study. Materials 2010, 3, 2516-2535. https://doi.org/10.3390/ma3042516
Yumura T, Hasegawa S, Itadani A, Kobayashi H, Kuroda Y. The Variety of Carbon-Metal Bonds inside Cu-ZSM-5 Zeolites: A Density Functional Theory Study. Materials. 2010; 3(4):2516-2535. https://doi.org/10.3390/ma3042516
Chicago/Turabian StyleYumura, Takashi, Saki Hasegawa, Atsushi Itadani, Hisayoshi Kobayashi, and Yasushige Kuroda. 2010. "The Variety of Carbon-Metal Bonds inside Cu-ZSM-5 Zeolites: A Density Functional Theory Study" Materials 3, no. 4: 2516-2535. https://doi.org/10.3390/ma3042516