A Green Method for Processing Polymers using Dense Gas Technology

Abstract
:1. Introduction
1.1. Polycarbonates
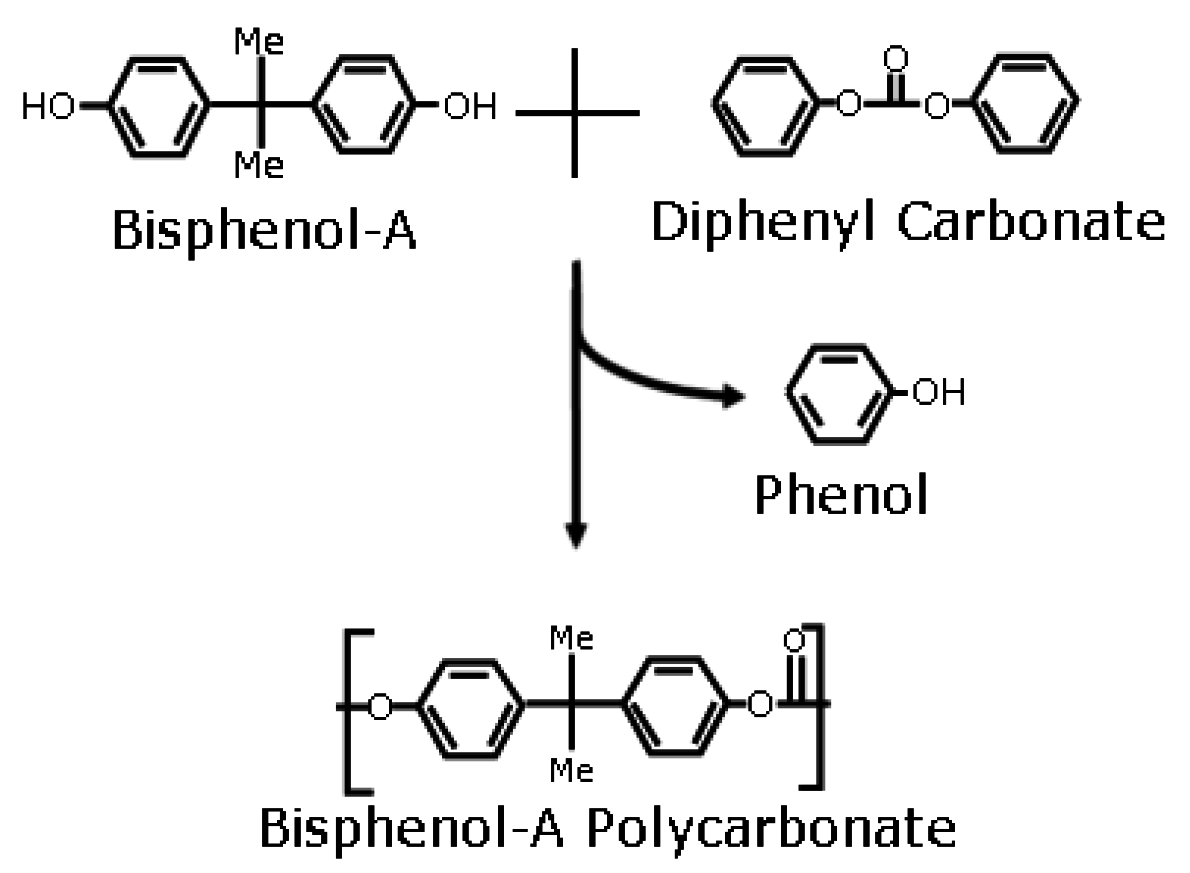
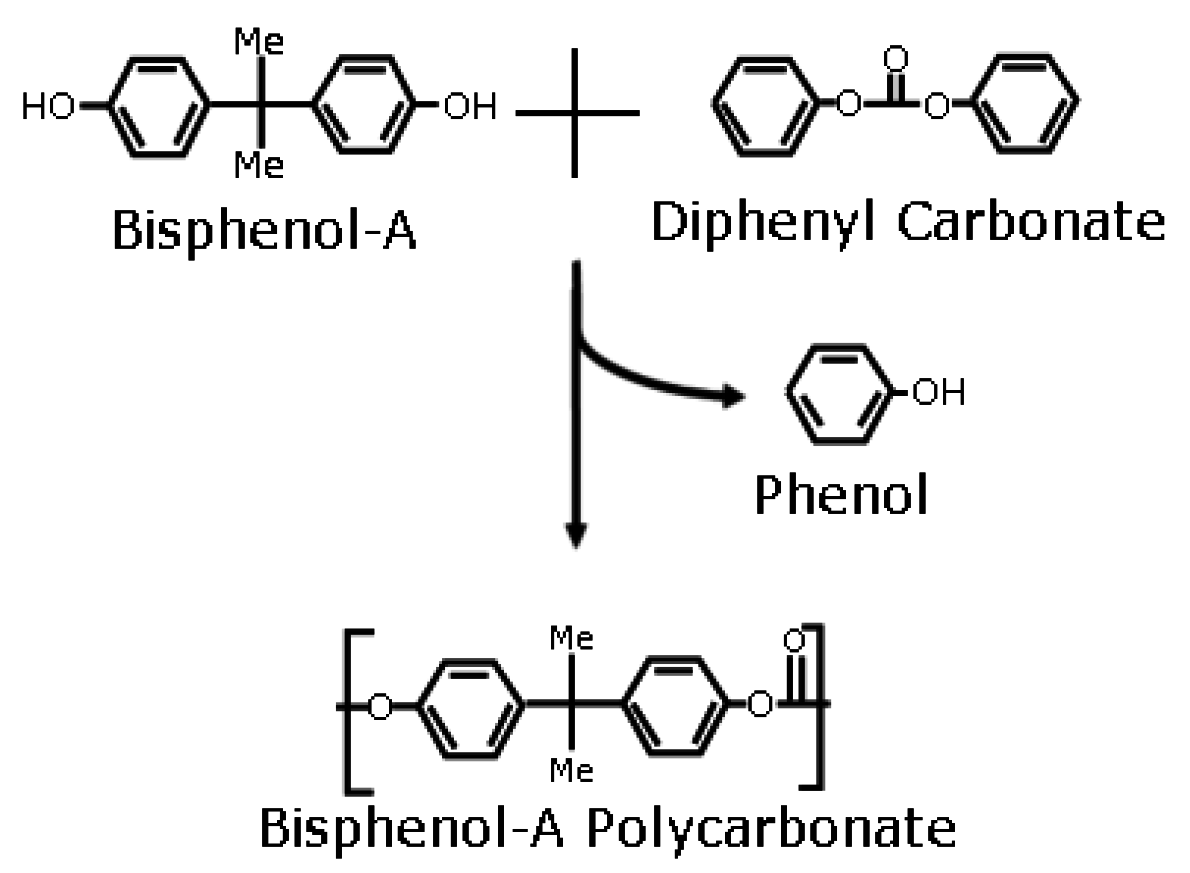
Polycarbonate Synthesis in Dense CO
1.2. Polycaprolactone
1.3. Polymer Blends
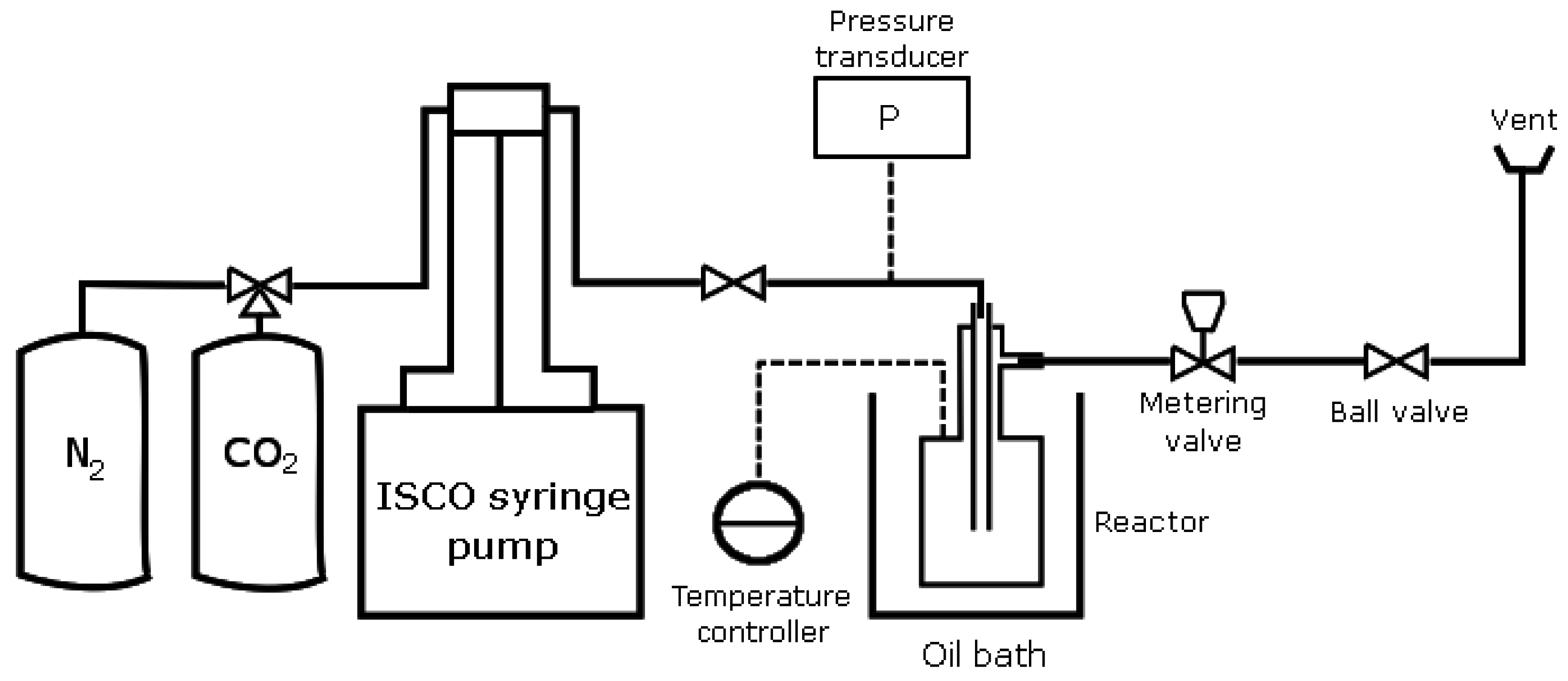
2. Materials and Methods

2.1. Two-stage PC Synthesis in N and CO
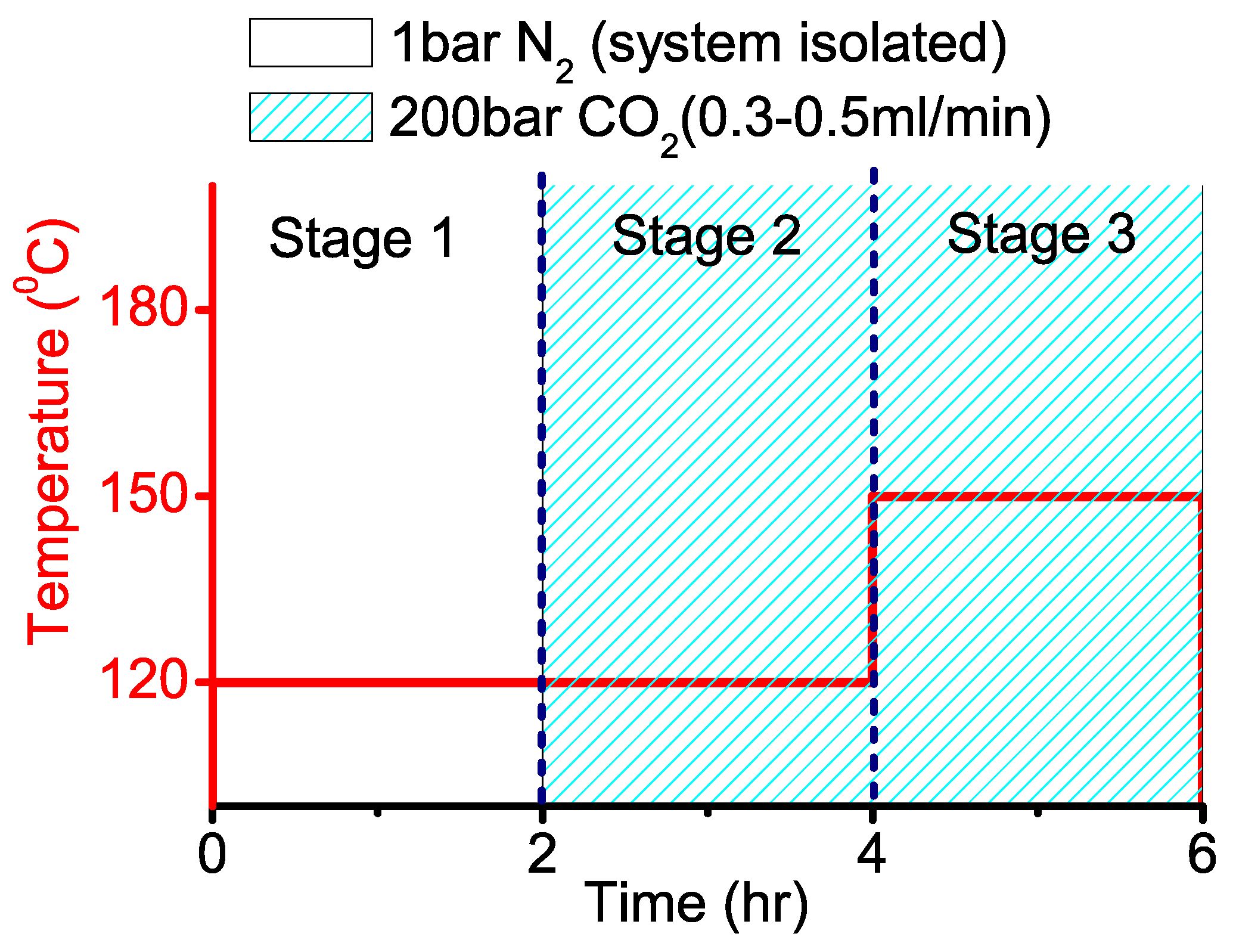
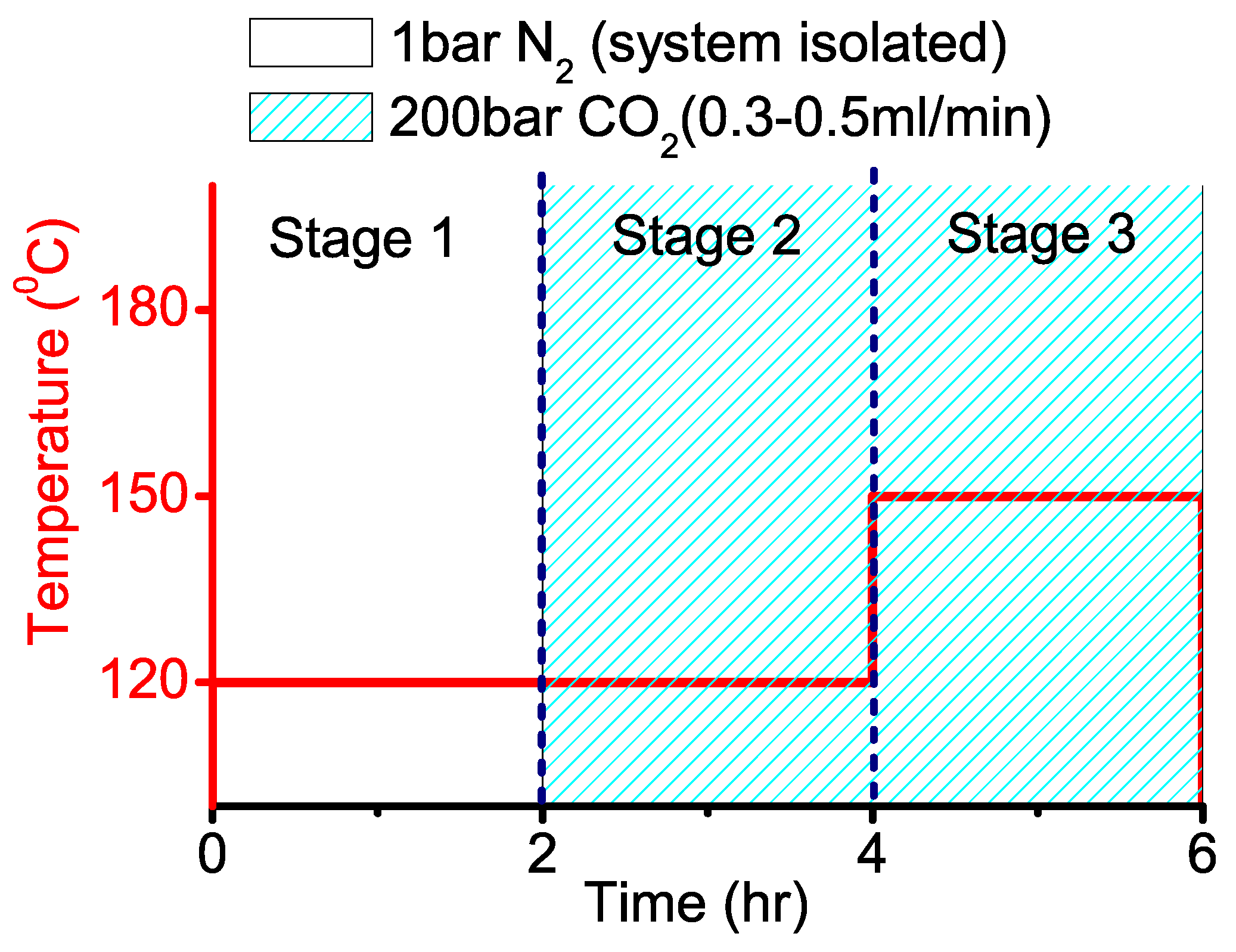
2.2. Three-stage PC Synthesis
2.3. Synthesis of PC With Dense CO-foamed PCL

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mass Ratio |
|---|---|
| PC80/PCL20 | 200mg BPA : 200mg DPC : 4mg NaOH : 100mg PCL |
| PC75/PCL25 | 150mg BPA : 150mg DPC : 3mg NaOH : 100mg PCL |
| PC67/PCL33 | 100mg BPA : 100mg DPC : 2mg NaOH : 100mg PCL |
| PCL | PCL only |
2.4. Impregnation of PC/PCL Blend
2.5. Analytical Techniques
Gel Permeation Chromatography
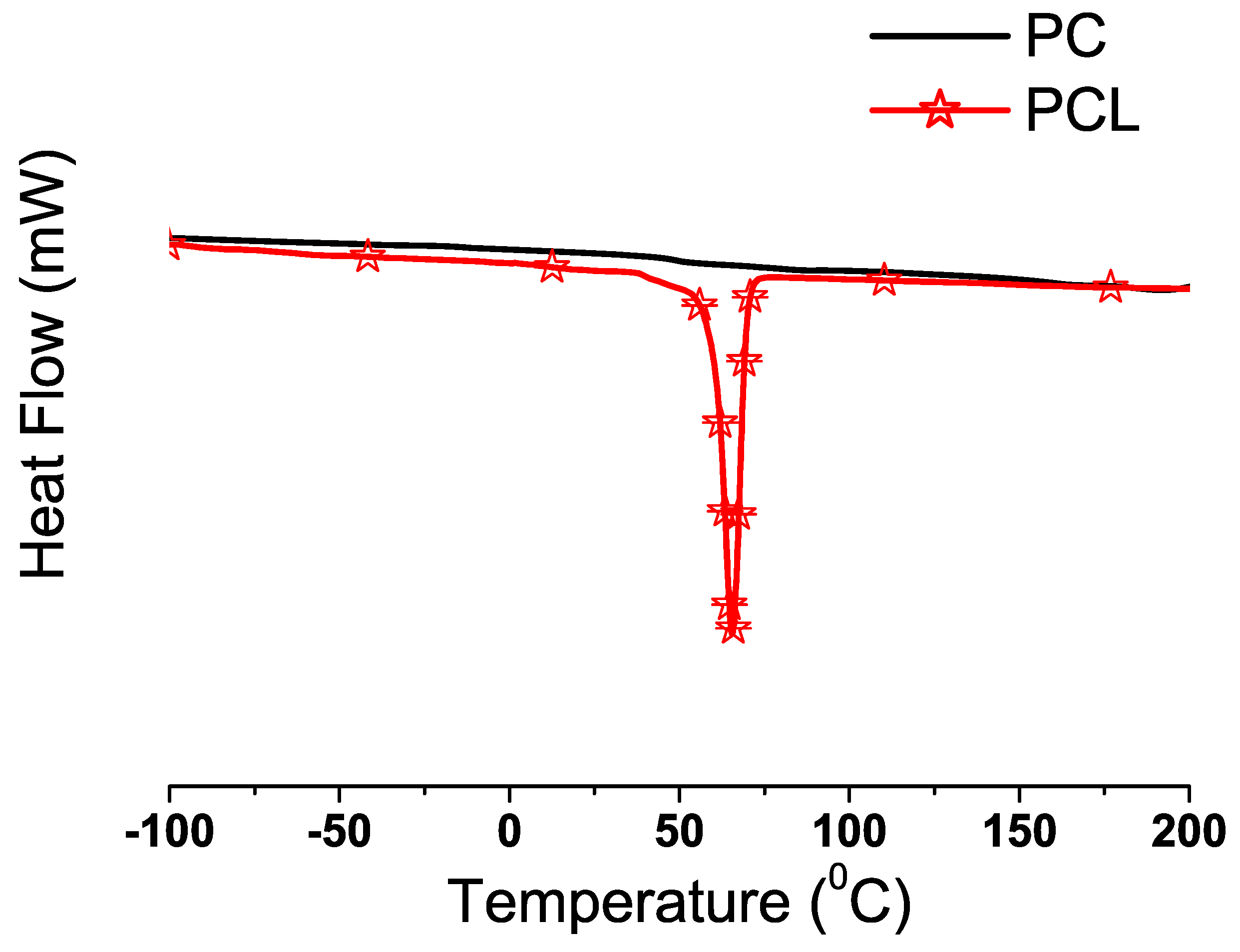
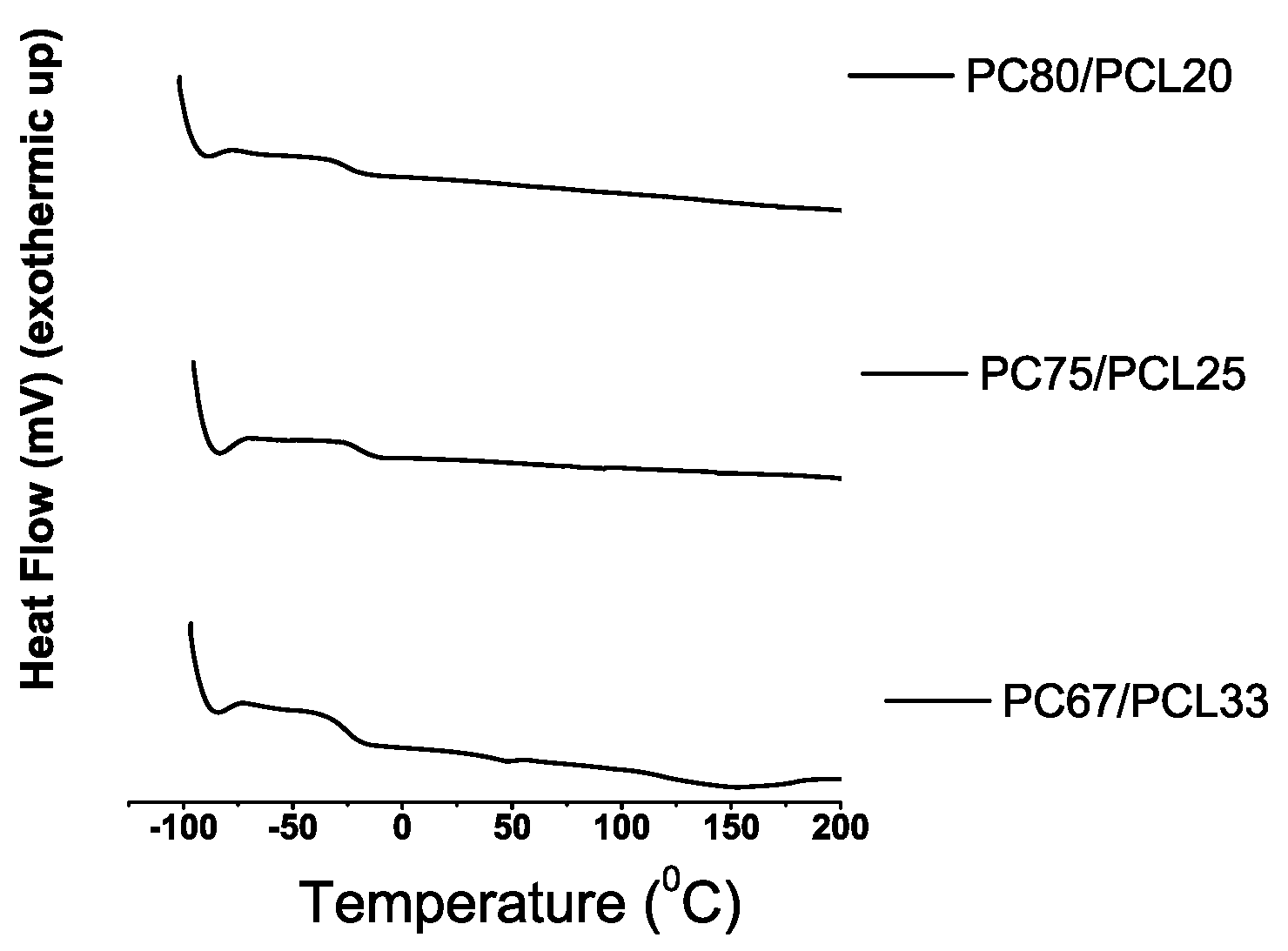
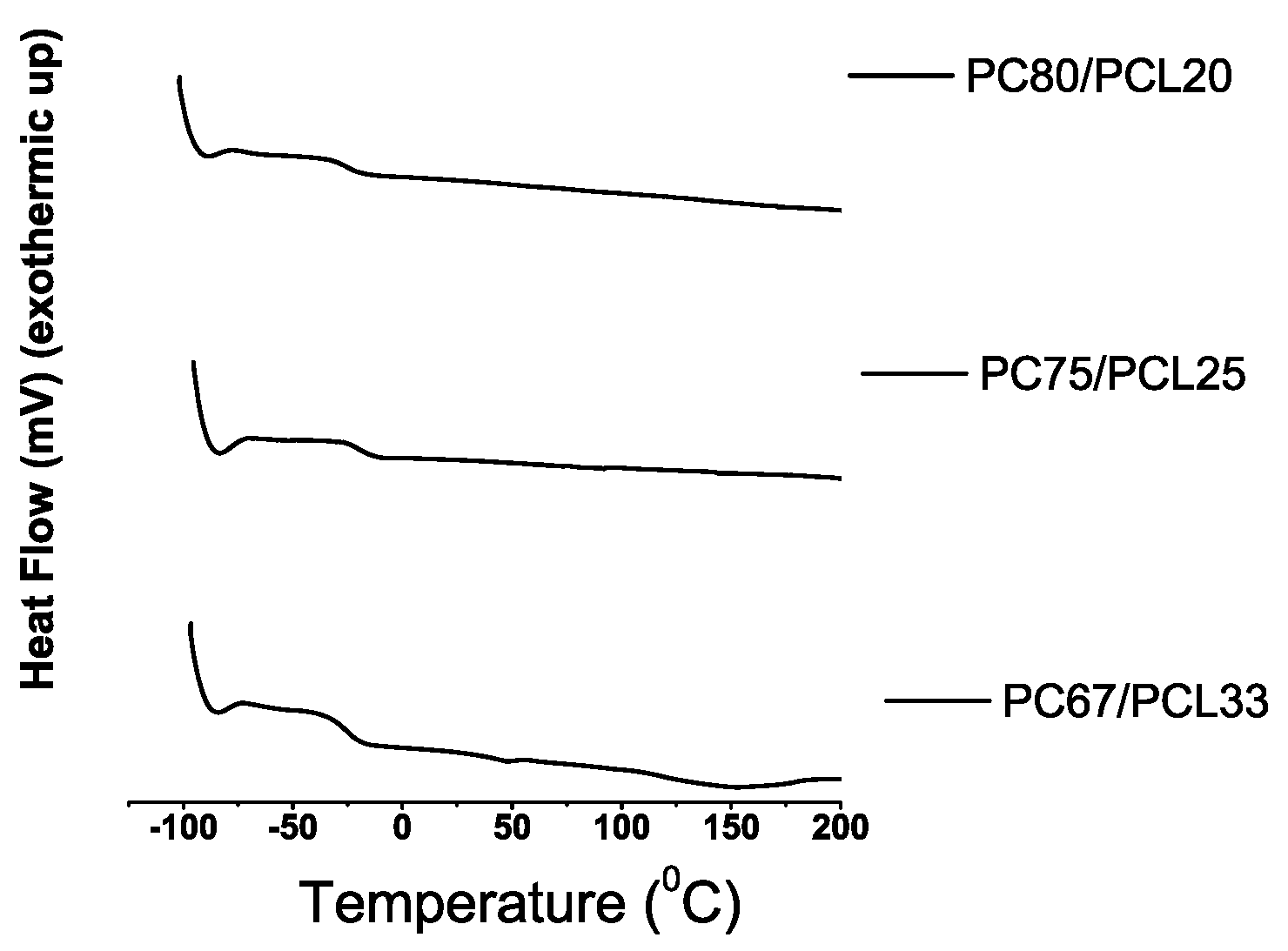
Differential Scanning Calorimetry
Scanning Electron Microscopy
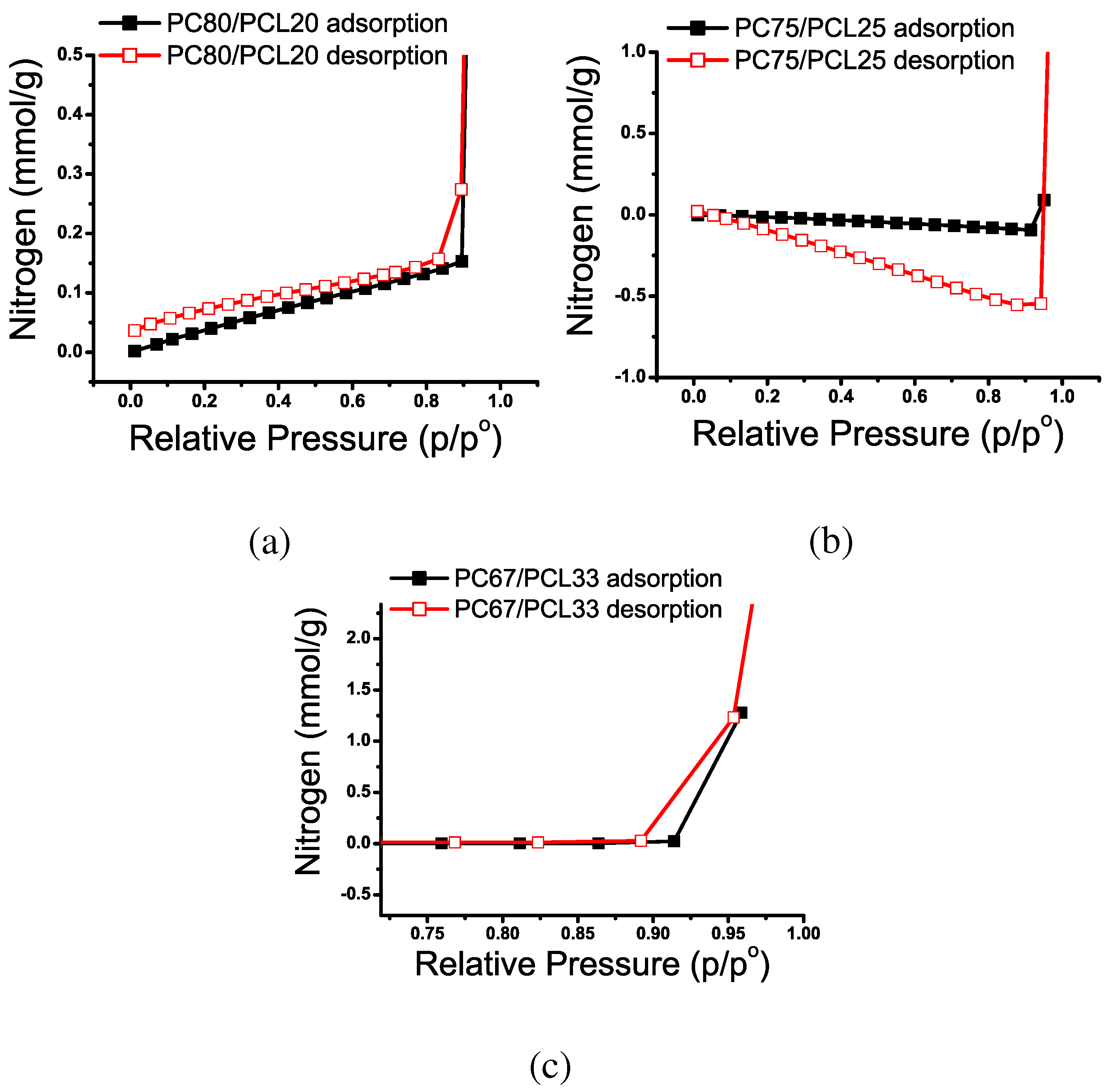
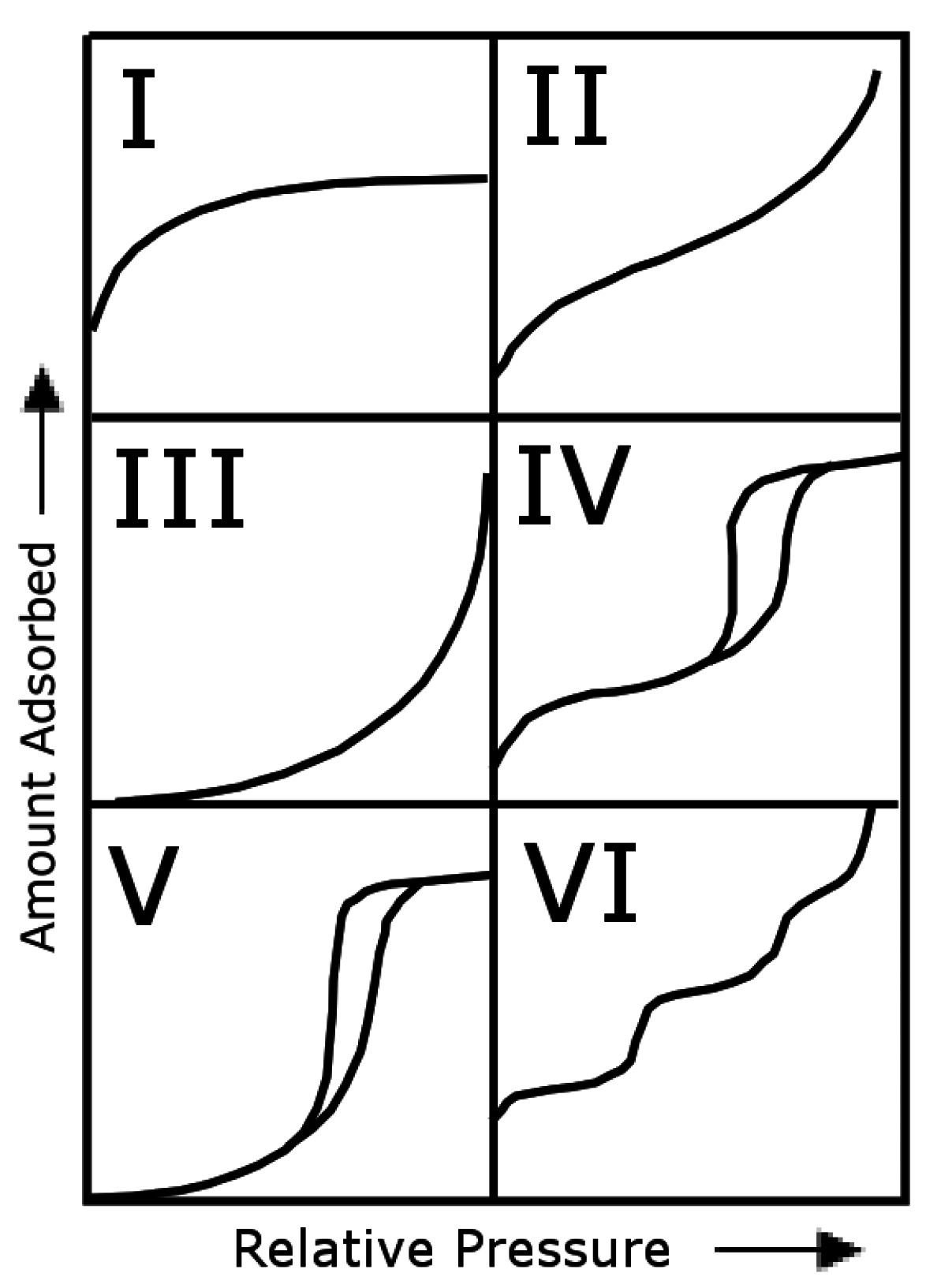
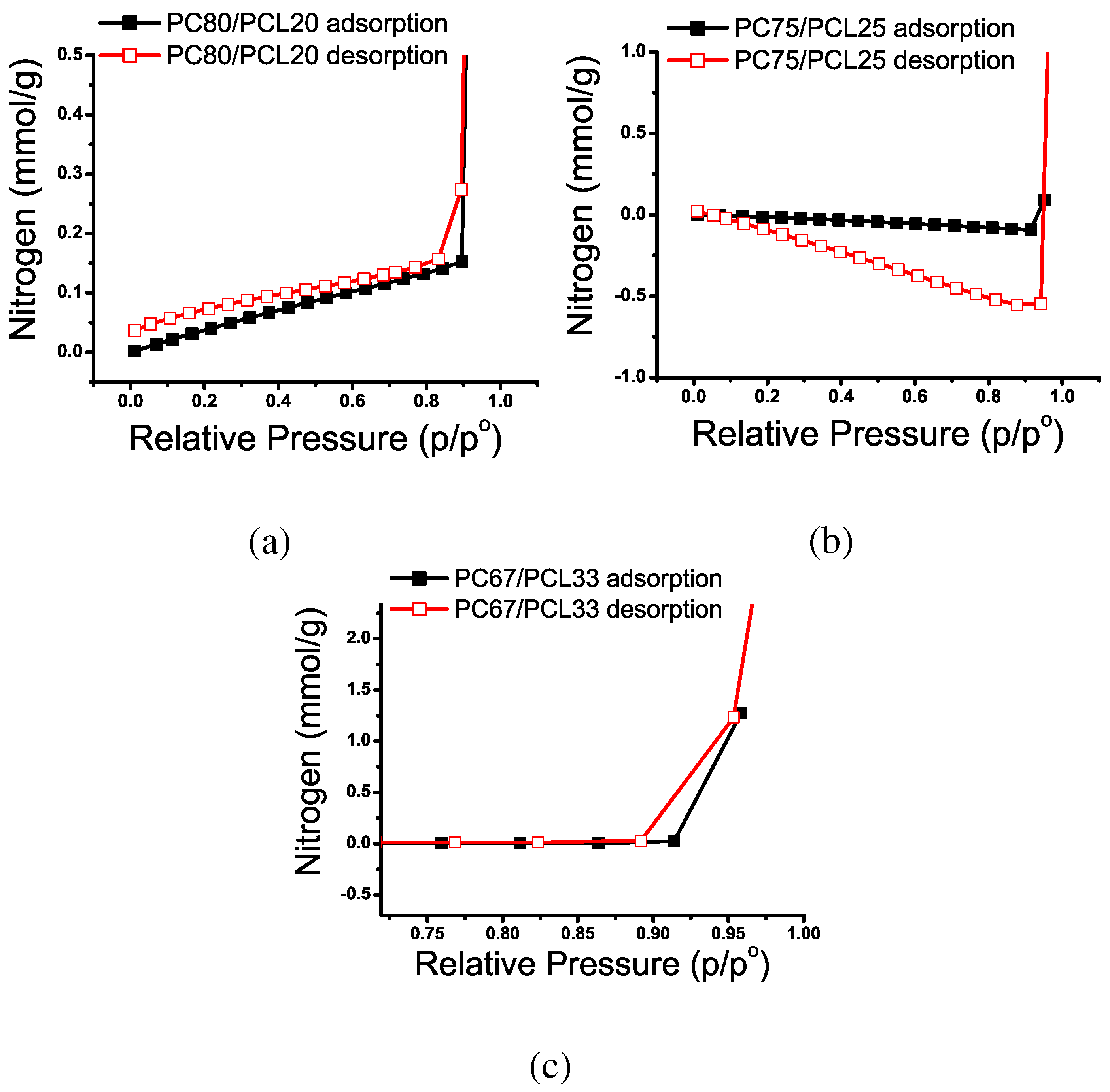
Nitrogen Adsorption
Reverse-phase High Performance Liquid Chromatography
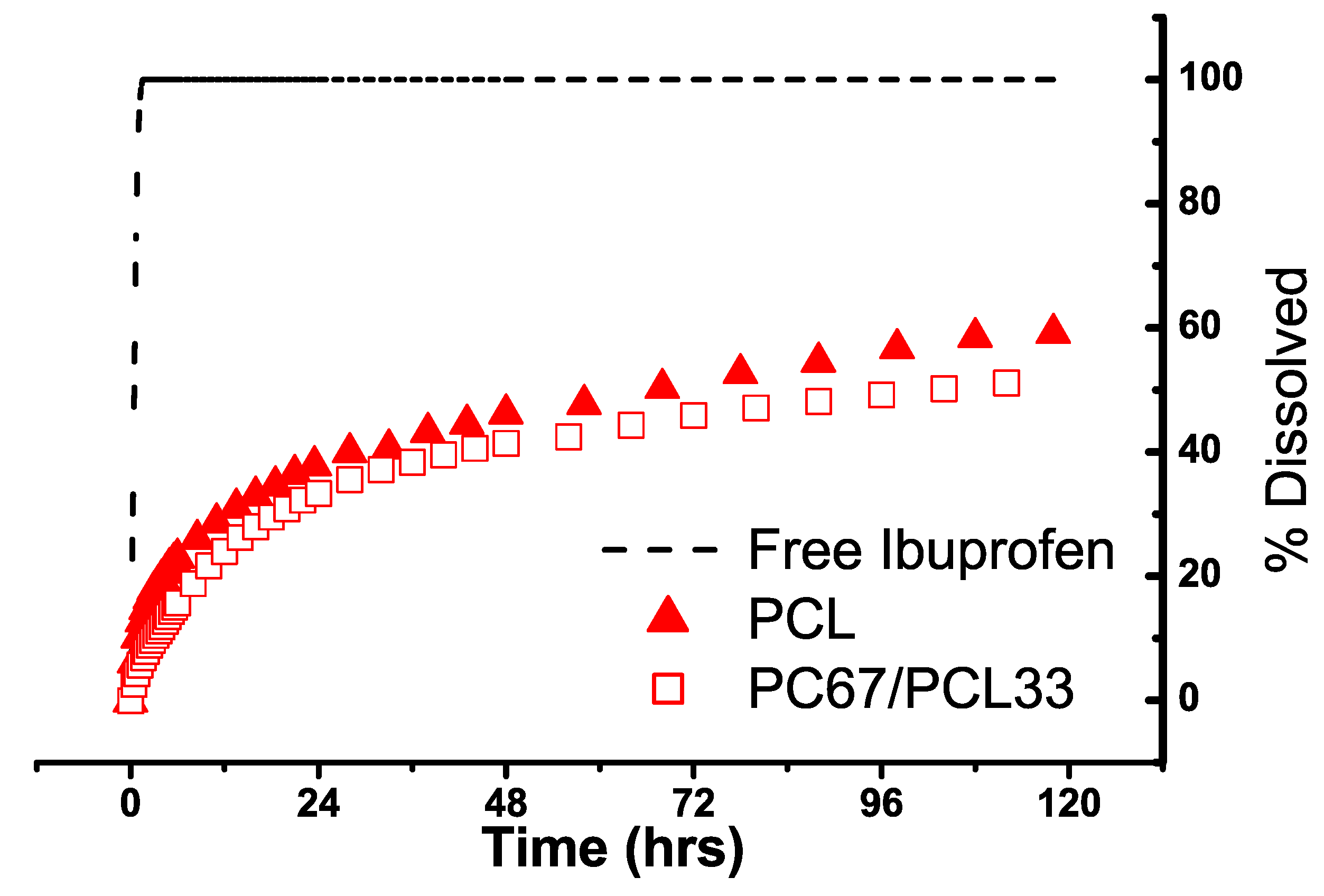
Dissolution Study
3. Results and Discussion
3.1. PC Synthesis
| Sample | Time | Temp | Mx10 | BPA Conv | DPC Conv |
| (hr) | () | (Da) | (%) | (%) | |
| N-1-120 | 1 | 120 | |||
| N-4-120 | 4 | 120 | |||
| N-12-120 | 12 | 120 | |||
| N-1-150 | 1 | 150 | |||
| N-4-150 | 4 | 150 | |||
| N-8-150 | 8 | 150 | |||
| N-24-150 | 24 | 150 | N/A | ||
| N-48-150 | 48 | 150 | |||
| N-1-200 | 1 | 200 | N/A | ||
| N-4-200 | 4 | 200 | |||
| N-48-200 | 48 | 200 |
| Sample | GPC | ESI-MS |
| M x 10 (Da) | M x 10 (Da) | |
| N-24-150 | 1.00 | 0.94 |
| N-48-150 | 0.90 | 0.92 |
| N-1-200 | 0.75 | 0.87 |
| Sample | Stage 1 time | Stage 2 time | Total | Mx10 |
| N(hrs) | CO(hrs) | (hrs) | (Da) | |
| N-2-CO-2 | 2 | 2 | 4 | 1.40 |
| N-4 | 4 | 0 | 0 | 0.99 |
| N-4-CO-1 | 4 | 1 | 5 | 1.33 |
| N-4-CO-4 | 4 | 4 | 8 | 2.60 |
| N-4-CO-8 | 4 | 8 | 12 | 1.48 |
| Sample | Stage 1 time | Stage 2 time | Total | Mx10 |
| (hrs) | (hrs) | (hrs) | (Da) | |
| N-2-120 | 2 | 0 | 2 | 1.28 |
| N-2-CO-2-120 | 2 | 2 | 4 | 1.82 |
| N-2-CO-4-120 | 2 | 4 | 6 | 2.56 |
| N-4-CO-8-120 | 2 | 8 | 10 | 4.73 |
| Sample | Stage 1 (hrs) | Stage 2 (hrs) | Stage 3 (hrs) | Mx10 (Da) |
| PC-6hrs | 2 | 2 | 2 | 3.9 |
| PC-8hrs | 4 | 2 | 2 | 3.8 |
| PC-8hrs-stage3 | 2 | 2 | 4 | 4.7 |
3.2. Synthesis of PC with Dense CO-foamed PCL

| Sample | T () |
| PC80/PCL20 | |
| PC75/PCL25 | |
| PC67/PCL33 |
Morphology

3.3. Drug Loading and Dissolution
| Sample | Drug Loading (wt%) |
| PCL | 9 |
| PC67/PCL33 | 20 |
| Pore Classification | Definition |
|---|---|
| Micropore | internal width < 2 nm |
| Mesopore | 2 nm < internal width < 50 nm |
| Macropore | internal width > 50 nm |

4. Conclusion
Acknowledgements
References
- Freitag, D.; Grigo, U.; Mueller, P.R.; Nouvertne, W. Polycarbonates. Encycl. Polym. Sci. Eng. 1987, 11, 648–718. [Google Scholar]
- Fukuoka, S.; Kawamura, M.; Komiya, K.; Tojo, M.; Hachiya, H.; Hasegawa, K.; Aminaka, M.; Okamoto, H.; Fukawa, I.; Konno, S. A novel non-phosgene polycarbonate production process using by-product carbon dioxide as starting material. Green Chem. 2003, 5, 497–507. [Google Scholar] [CrossRef]
- Pohl, L.R.; Bhooshan, B.; Whittaker, N.F.; Krishna, G. Phosgene: A metabolite of chloroform. Biochem. Biophys. Res. Comm. 1977, 79, 684–691. [Google Scholar] [CrossRef]
- Gross, S.M.; Givens, R.D.; Jikei, M.; Royer, J.R.; Khan, S.; DeSimone, J.M.; Odell, P.G.; Hamer, G.K. Synthesis and swelling of poly (bisphenol A carbonate) using supercritical carbon dioxide. Macromolecules 1998, 31, 9090–9092. [Google Scholar] [CrossRef]
- Gross, S.M.; Roberts, G.W.; Kiserow, D.J.; DeSimone, J.M. Synthesis of highmolecular weight polycarbonate by solid-state polymerization. Macromolecules 2001, 34, 3916–3920. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Song, C.-H.; Kim, J.-I.; Kim, J.-H. Preparation of aromatic polycarbonatenanoparticles using supercritical carbon dioxide. J. Nanopart. Res. 2002, 4, 53–59. [Google Scholar] [CrossRef]
- Xiao, Y.; Wu, Y.; Wang, C.; Ying, W. Synthesis of polycarbonatein supercritical carbon dioxide. J. Chem. Ind. Eng. (China) 2007, 58, 2403–2407. [Google Scholar]
- Coutsikos, P.; Magoulas, K.; Tassios, D. Solubilities of phenolsin supercritical carbon dioxide. J. Chem. Eng. Data 1995, 40, 953–958. [Google Scholar] [CrossRef]
- Greco, R.S. Implantation Biology: The Host Response and Biomedical Devices; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Busby, A.J.; Zhang, J.; Roberts, C.J.; Lester, E.; Howdle, S.M. Novel nanostructured polymeric composites of polycaprolactone and ultra-high molecular weight polyethylene via a supercritical-fluid route. Adv. Mater. 2005, 17, 364–36. [Google Scholar] [CrossRef]
- Shieh, Y.T.; Lin, Y.T. Transesterification and polymerization reactions of aliphatic polyesters in supercritical carbon dioxide fluids without the presence of a catalyst. Eur. Polym. J. 2007, 43, 1847–1856. [Google Scholar] [CrossRef]
- Mohamed, A.; Gordon, S.H.; Biresaw, G. Polycaprolactone/polystyrene bioblends characterized by thermogravimetry, modulated differential scanning calorimetry and infrared photoacoustic spectroscopy. Polym. Degrad. Stabil. 2007, 92, 1177–1185. [Google Scholar] [CrossRef]
- Yoganathan, R.; Mammucari, R.; Foster, N.R. Dense Gas Processing of Polymers. J. Polym. Rev. 2010, 50, 144–177. [Google Scholar] [CrossRef]
- Yoganathan, R.; Mammucari, R.; Foster, N.R. Impregnation of Ibuprofen into Polycaprolactone Using Supercritical Carbon Dioxide. J. Phys. Conf. Ser. 2010, 215(012087). [Google Scholar] [CrossRef]
- Paul, D.R.; Bucknall, C.B. Polymer Blends; Wiley: Chichester, UK, 1999. [Google Scholar]
- Folkes, M.J.; Hope, P.S. Polymer blends and alloys; Blackie Academic & Professional: New York, NY, USA, 1993. [Google Scholar]
- Paul, D.R.; Newman, S. Polymer blends; Vol. 2; Newman, S., Ed.; Academic Press: New York, NY, USA, 1978. [Google Scholar]
- Paul, D.R.; Newman, S. Polymer blends; Newman, S., Ed.; Academic Press: New York, NY, USA, 1978; Volume 1. [Google Scholar]
- Tomasko, D.L.; Li, H.; Liu, D.; Han, X.; Wingert, M.J.; Lee, L.J.; Koelling, K.W. A review of carbon dioxide applications in the processing of polymers. Ind. Eng. Chem. Res. 2003, 42, 6431–6456. [Google Scholar] [CrossRef]
- Watkins, J.J.; McCarthy, T.J. Chemistry in supercritical fluid-swollen polymers: Direct synthesis of polymer/polymer and polymer/metal. Polym. Mater. Sci. Eng. 1995, 73, 158–159. [Google Scholar]
- Watkins, J.J.; McCarthy, T.J. Polymerizations in supercritical fluid-swollen polymers: A new route to polymer blends and composite foams. In Polymer Preprints; American Chemical Society, Division of Polymer Chemistry: Ann Arbor, MI, USA, 1994; Volume 35, pp. 699–700. [Google Scholar] [CrossRef]
- Watkins, J.J.; McCarthy, T.J. Polymerization in supercritical fluid-swollen polymers: A new route to polymer blends. Macromolecules 1994, 27, 4845–4847. [Google Scholar] [CrossRef]
- Li, D.; Han, B. Impregnation of polyethylene (PE) with styrene using supercritical CO2 as the swelling agent and preparation of PE/polystyrene composites. Ind. Eng. Chem. Res. 2000, 39, 4506–4509. [Google Scholar] [CrossRef]
- Guo, Z.; Wingert, M.J.; Shen, J.; Lee, L.J.; Tomasko, D.L. Foaming dynamics of immiscible polymer blends. Annual Technical Conference-ANTEC, Conference Proceedings 2007, 5, 2970–2975. [Google Scholar]
- Li, H.; Lee, L.J.; Tomasko, D.L. Effect of carbon dioxide on the interfacial tension of polymer melts. Ind. Eng. Chem. Res. 2004, 43, 509–514. [Google Scholar] [CrossRef]
- Balsamo, V.; Calzadilla, N.; Mora, G.; Mueller, A.J. Thermal characterization of polycarbonate/polycaprolactone blends. J. Polym. Sci. B Polym. Phys. 2001, 39, 771–785. [Google Scholar] [CrossRef]
- Chun, Y.S.; Park, J.; Sun, J.B.; Kim, W.N. Blends of polycarbonate and poly(e-caprolactone) and the determination of the polymer-polymer interaction parameter of the two polymers. J. Polym. Sci. B Polym. Phys. 2000, 38, 2072–2076. [Google Scholar] [CrossRef]
- Gonzalez, I.; Eguiazabal, J.I.; Nazabal, J. New clay-reinforced nanocomposites based on a polycarbonate/polycaprolactone blend. Polym. Eng. Sci. 2006, 46, 864–873. [Google Scholar] [CrossRef]
- Hernandez, M.C.; Laredo, E.; Bello, A.; Carrizales, P.; Marcano, L.; Balsamo, V.; Grimau, M.; Muller, A.J. From miscible to immiscible polycarbonate/poly(e-caprolactone) blends. Macromolecules 2002, 35, 7301–7313. [Google Scholar] [CrossRef]
- Ketelaars, A.A.J.; Papantoniou, Y.; Nakayama, K. Analysis of the density and the enthalpy of poly(e-caprolactone)-polycarbonate blends: Amorphous phase compatibility and the effect of secondary crystallization. J. Appl. Polym. Sci. 1997, 66, 921–927. [Google Scholar] [CrossRef]
- Bailly, C.; Daoust, D.; Legras, R.; Mercier, J.P.; Strazielle, C.; Lapp, A. On the molecular weight determination of bisphenol-A polycarbonate. Polymer 1986, 27, 1410–1415. [Google Scholar] [CrossRef]
- Kim, J.; Roberts, G.W.; Kiserow, D.J. Effect of prepolymer molecular weight on solid state polymerization of poly(bisphenol a carbonate) with nitrogen as a sweep fluid. J. Polym. Sci. A Polym. Chem. 2008, 46, 4959–4969. [Google Scholar] [CrossRef]
- Wang, J.; Cheung, M.K.; Yl, M. Miscibility and morphology in crystalline/amorphous blends of poly(caprolactone)/poly(4-vinylphenol) as studied by DSC, FTIR, and 13C solid state NMR. Polymer 2002, 43, 1357–1364. [Google Scholar] [CrossRef]
- Lee, J.W.S.; Wang, K.; Park, C. Challenge to extrusion of low-density microcellular polycarbonate foams using supercritical carbon dioxide. Ind. Eng. Chem. Res. 2005, 44, 92–99. [Google Scholar] [CrossRef]
- Wessling, M.; Borneman, Z.; Boomgaard van den, Th.; Smolders, C.A. Carbon dioxide foaming of glassy polymers. J. Appl. Polym. Sci. 1994, 53, 1497–1512. [Google Scholar] [CrossRef]
- Fleming, G.K.; Koros, W.J. Dilation of substituted polycarbonates caused by high-pressure carbon dioxide sorption. Macromolecules 1990, 23, 1353–1360. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1402. [Google Scholar] [CrossRef]
- Lowell, S. Characterization of porous solids and powders: Surface area, pore Size, and Density; Kluwer Academic Publishers: Boston, MA, USA, 2004. [Google Scholar]
- Charoenchaitrakool, M.; Dehghani, F.; Foster, N.R.; Chan, H.K. Micronization by rapid expansion of supercritical solutions to enhance the dissolution rates of poorly water-soluble pharmaceuticals. Ind. Eng. Chem. Res. 2000, 39, 4794–4802. [Google Scholar] [CrossRef]
- Lian, Z.; Epstein, S.A.; Blenk, C.W.; Shine, A.D. Carbon dioxide-induced melting point depression of biodegradable semicrystalline polymers. J. Supercrit. Fluid. 2006, 39, 107–117. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Peppas, N.A. Analysis of Fickian and non-Fickian drug release from polymers. Pharm. Acta Helv. 1985, 60, 110–111. [Google Scholar] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license http://creativecommons.org/licenses/by/3.0/.
Share and Cite
Yoganathan, R.B.; Mammucari, R.; Foster, N.R. A Green Method for Processing Polymers using Dense Gas Technology. Materials 2010, 3, 3188-3203. https://doi.org/10.3390/ma3053188
Yoganathan RB, Mammucari R, Foster NR. A Green Method for Processing Polymers using Dense Gas Technology. Materials. 2010; 3(5):3188-3203. https://doi.org/10.3390/ma3053188
Chicago/Turabian StyleYoganathan, Roshan B., Raffaella Mammucari, and Neil R. Foster. 2010. "A Green Method for Processing Polymers using Dense Gas Technology" Materials 3, no. 5: 3188-3203. https://doi.org/10.3390/ma3053188