Molecular Dynamics in Two-Dimensional Supramolecular Systems Observed by STM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
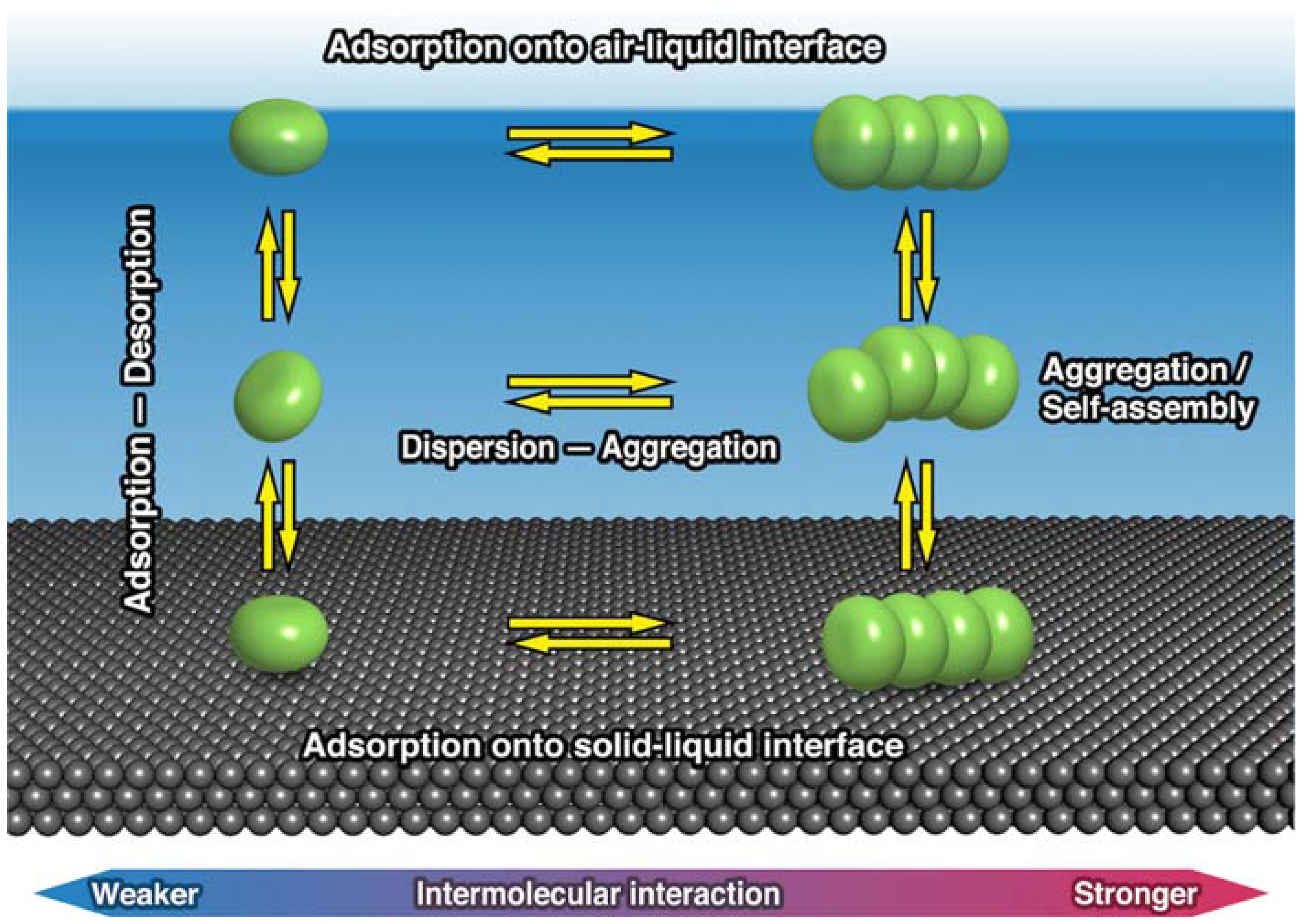
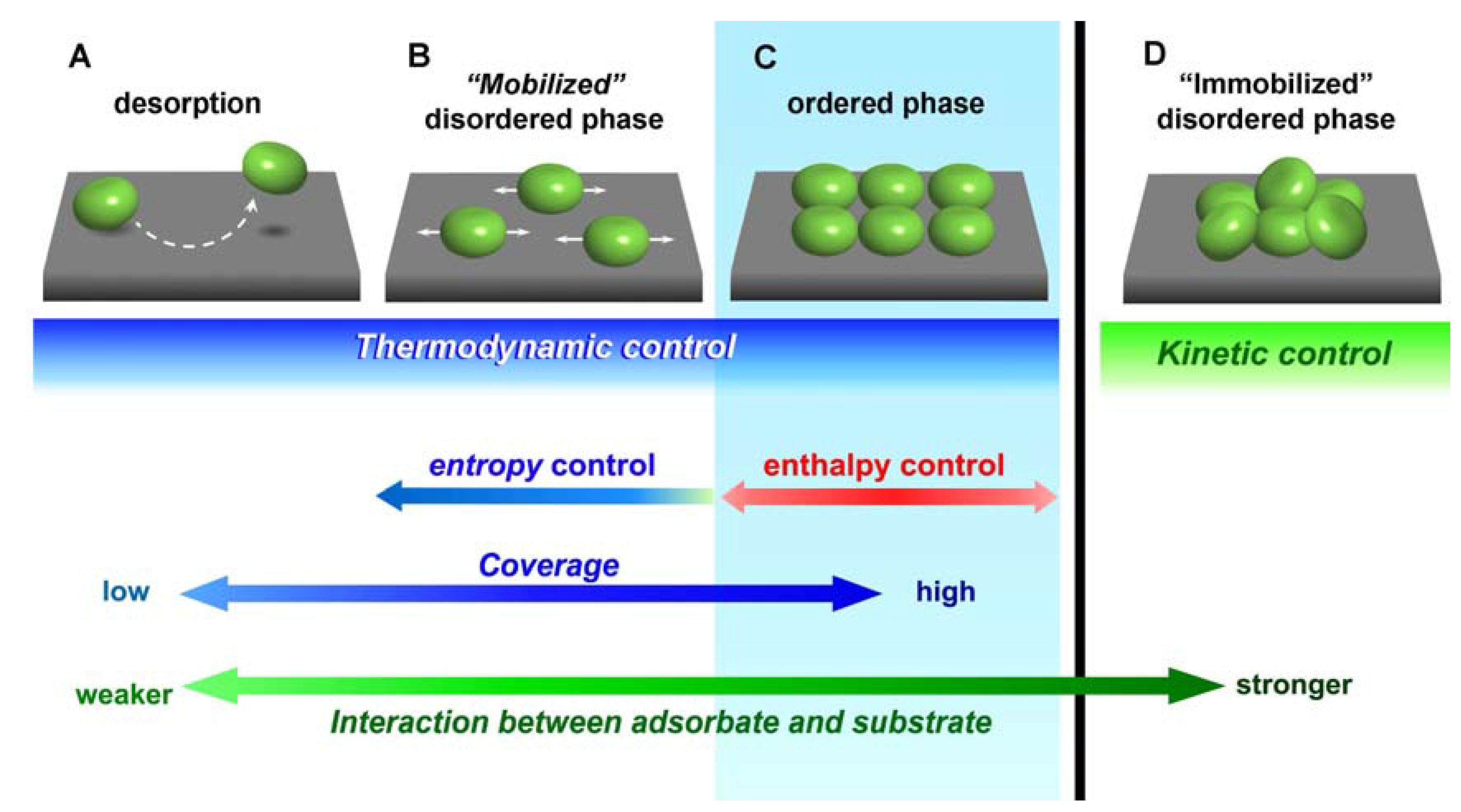
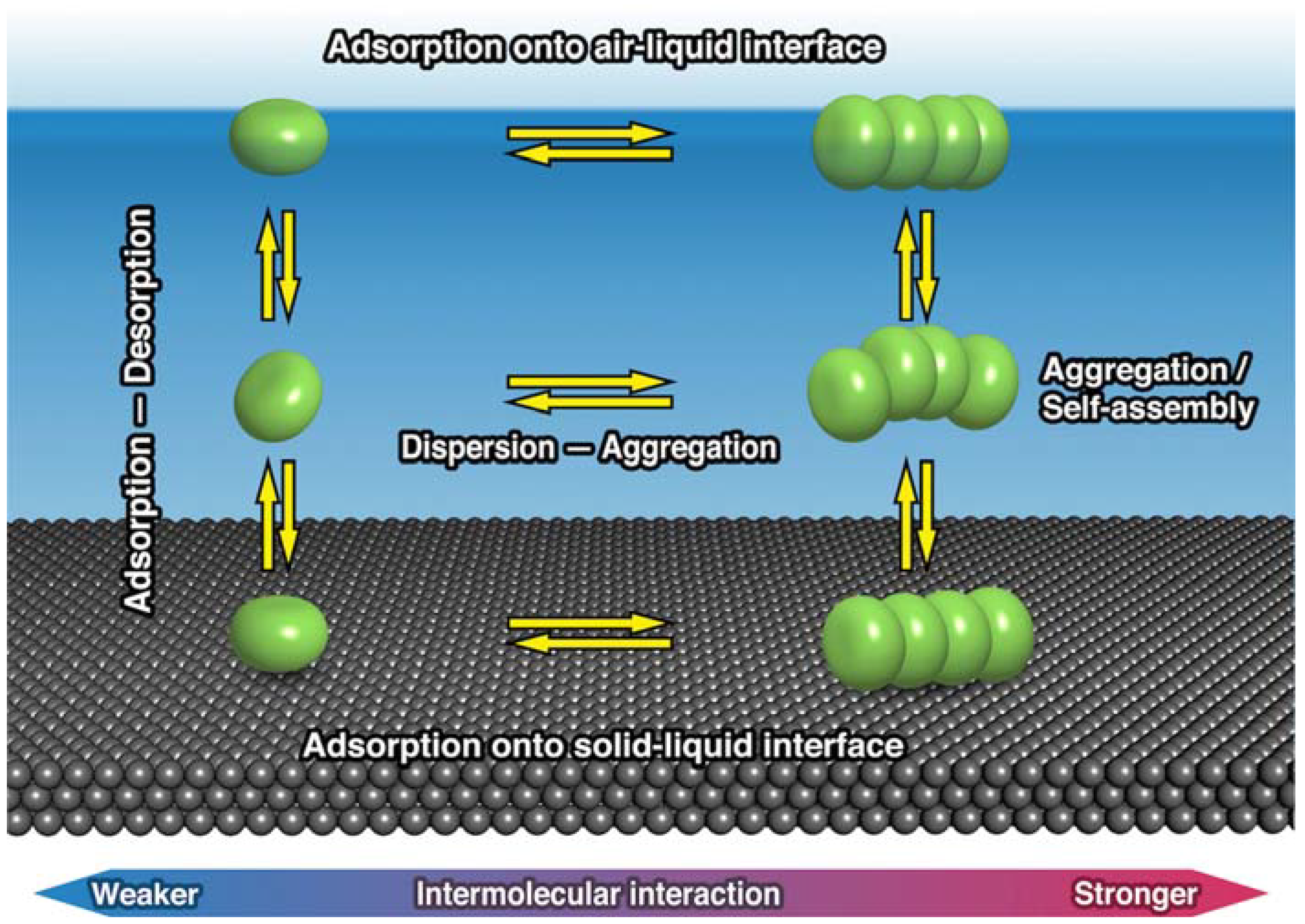
2. Thermodynamic Equilibrium for Molecules on a Substrate
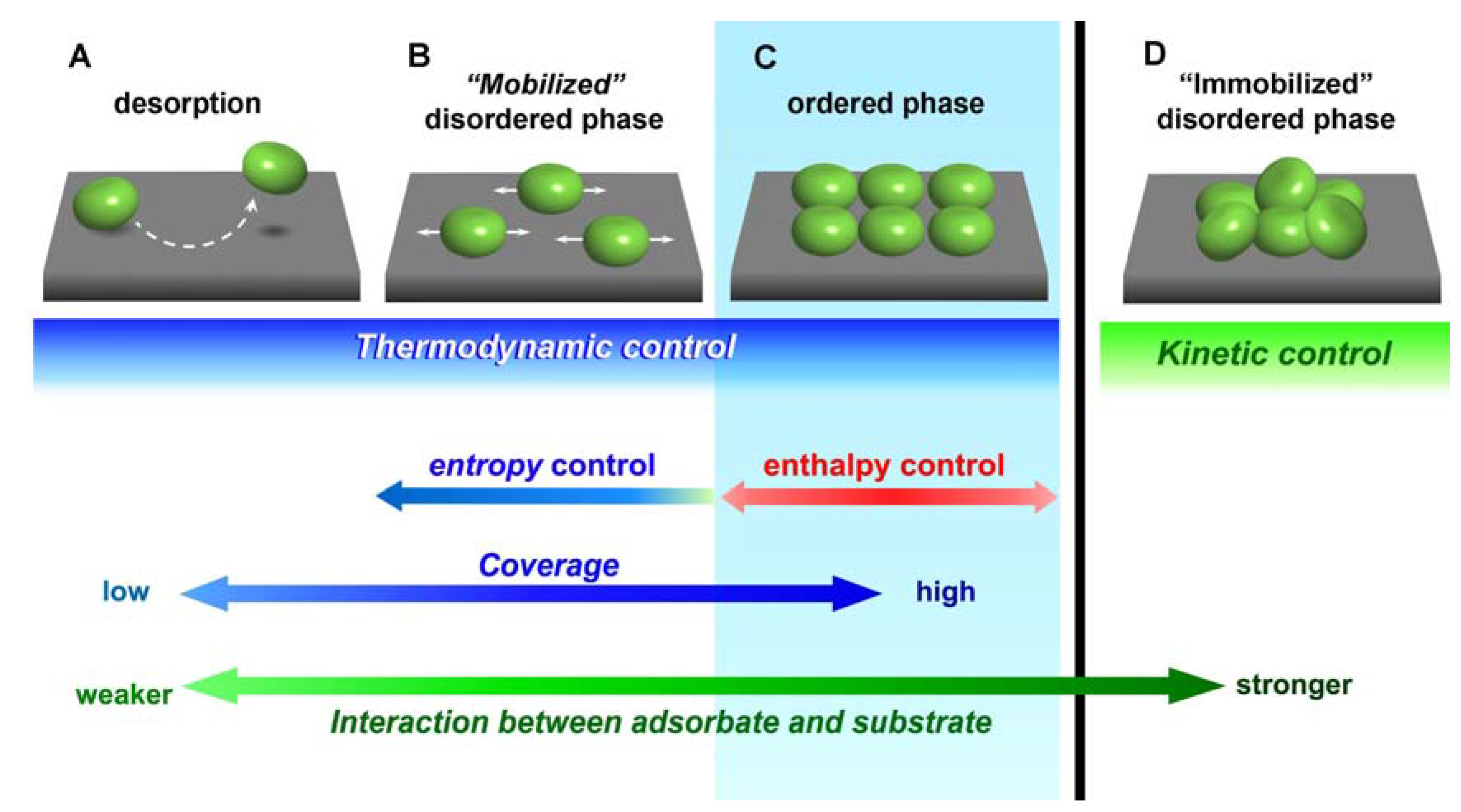
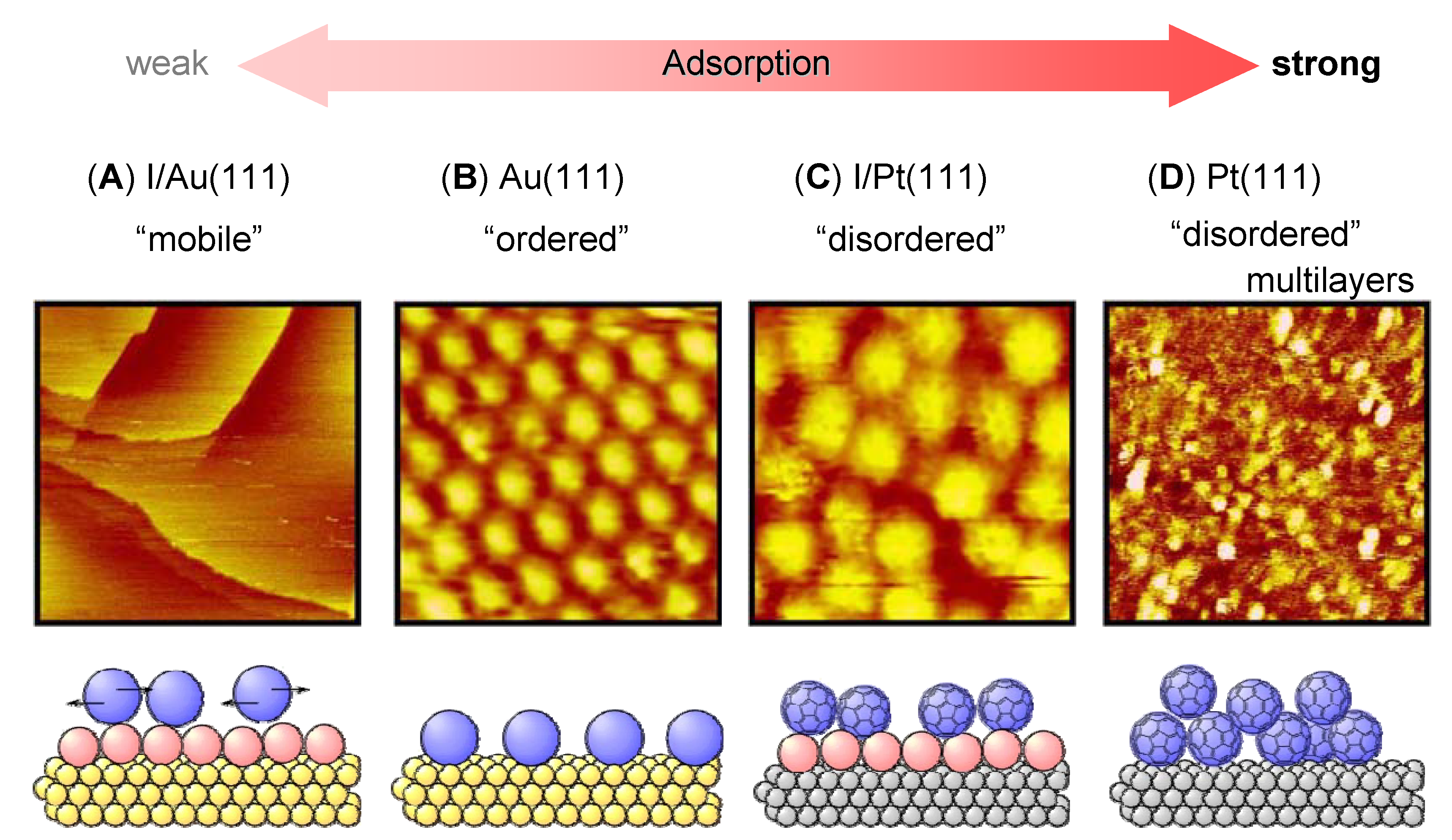
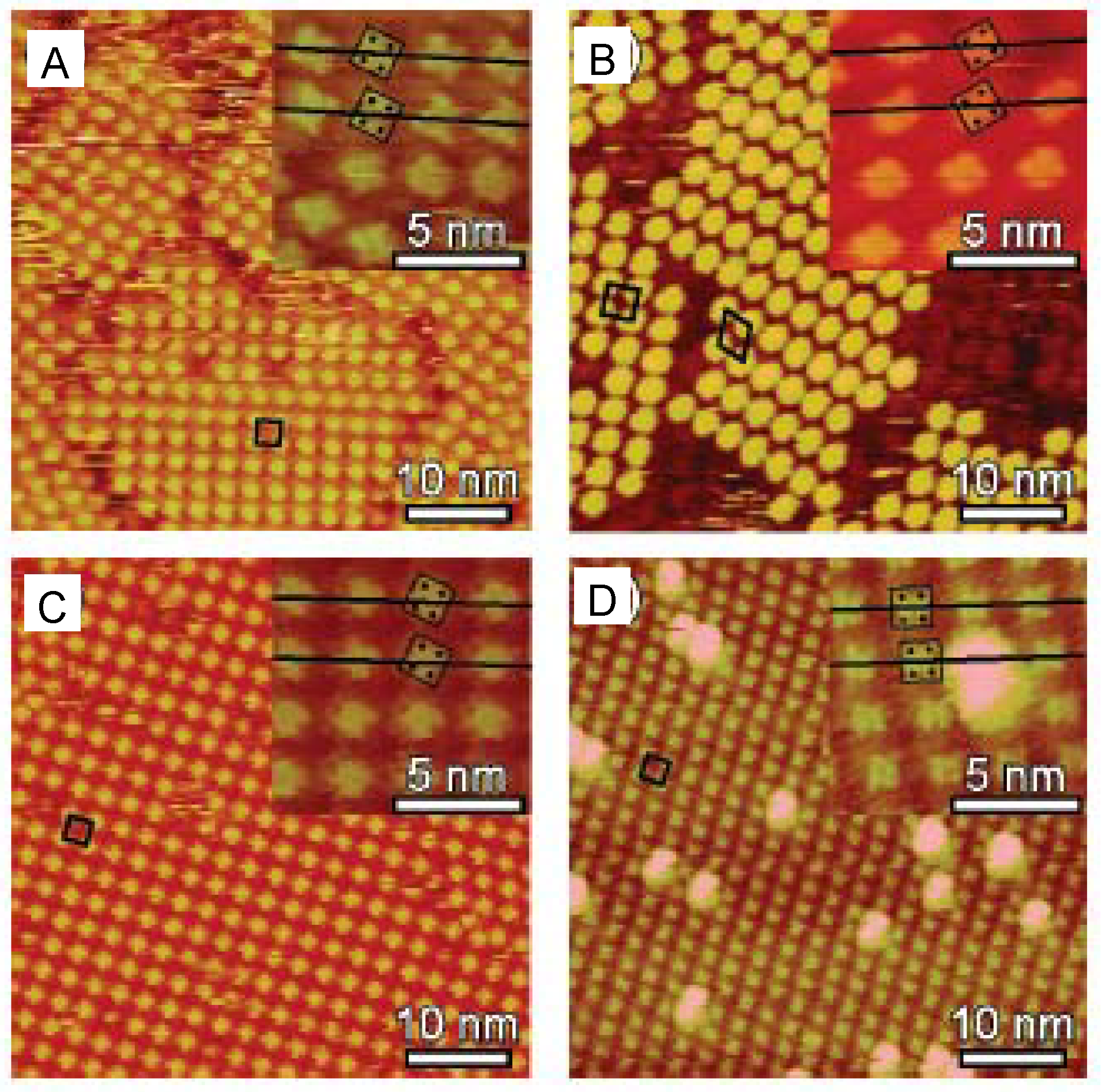
3. Relation between Molecular Motion and Adsorption Strength
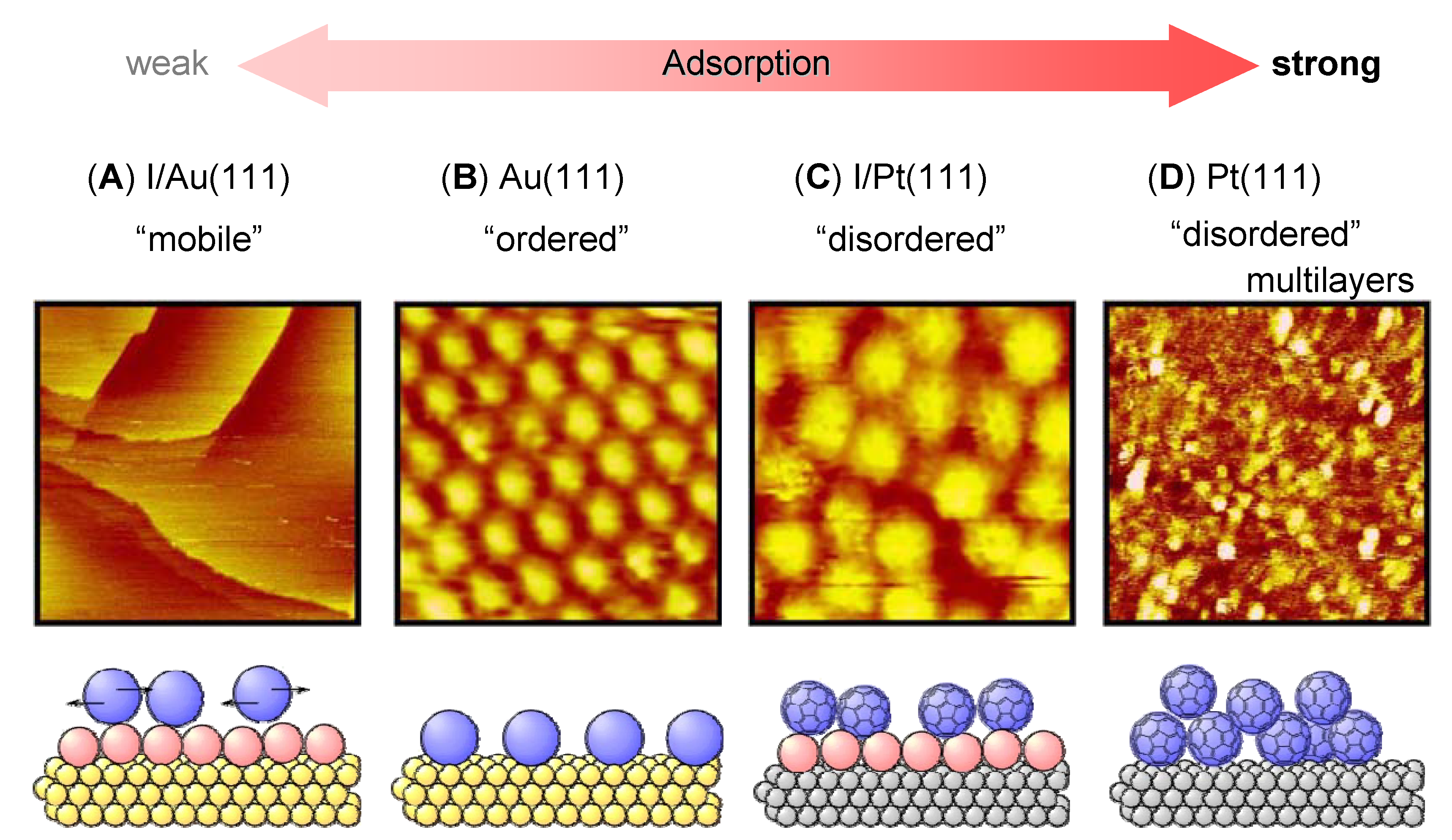
4. Dynamics of 2D Supramolecular Structures
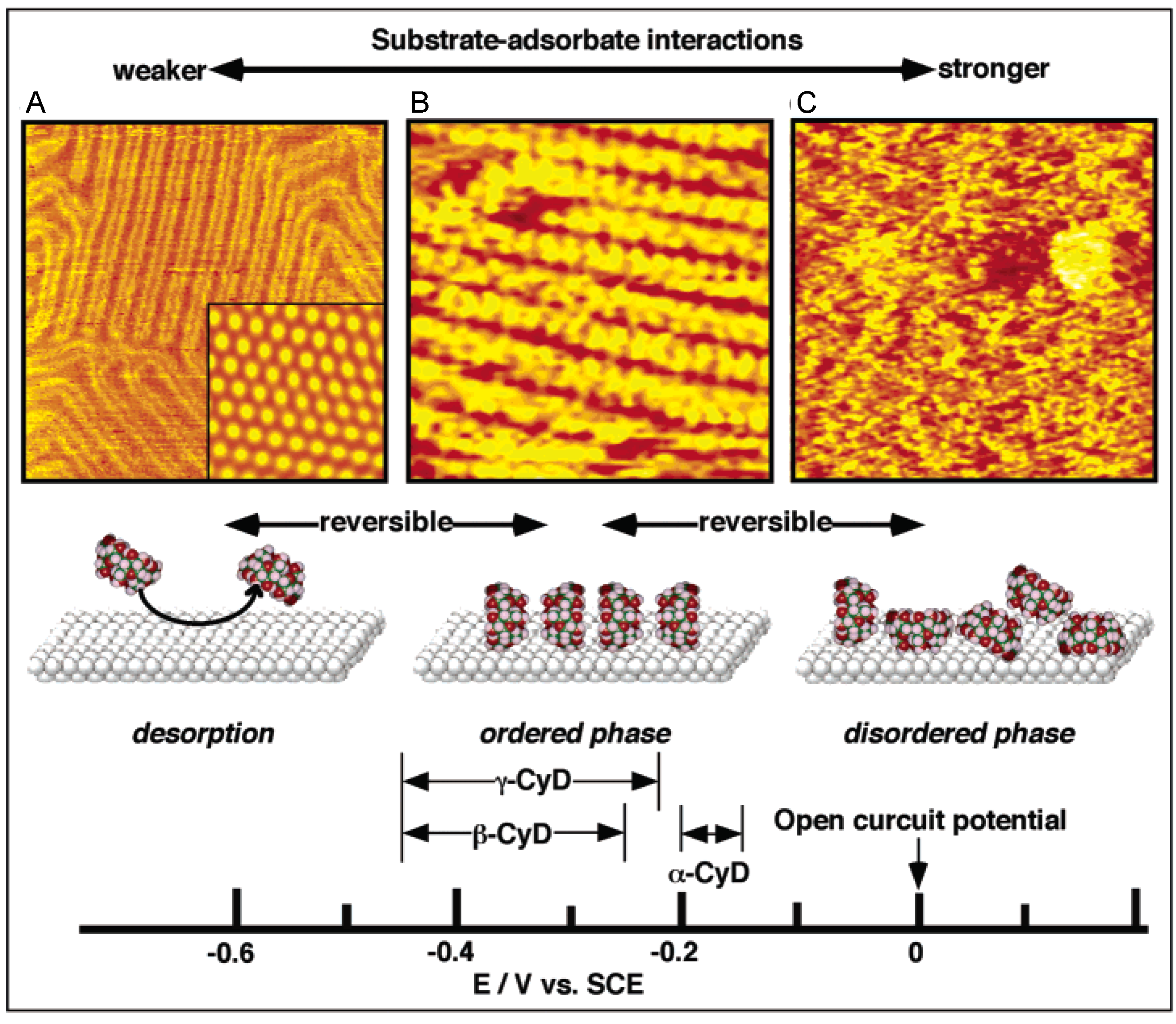
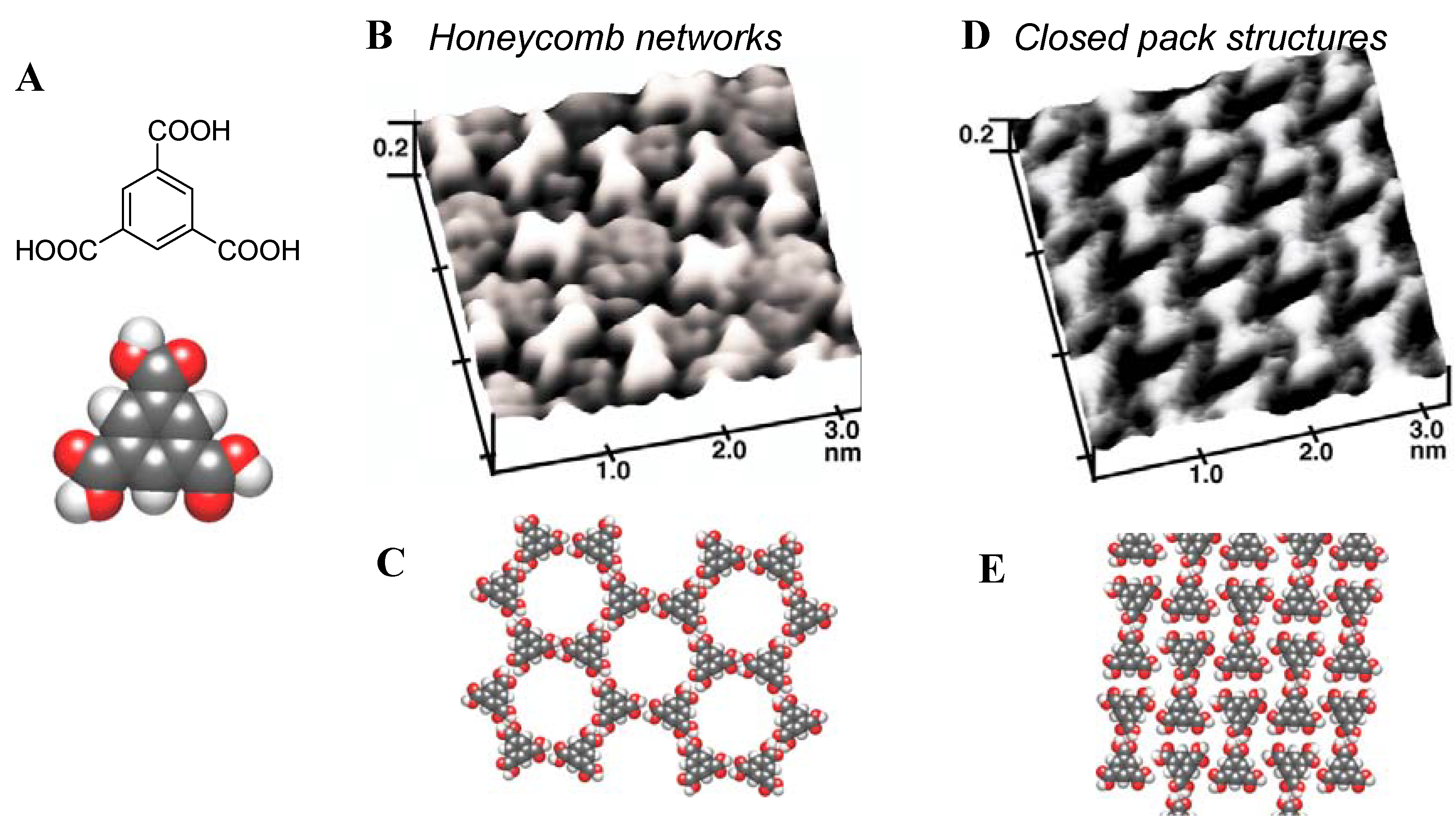
4.1. Self-ordering Processes Induced by Adsorption from Solution [12,23]
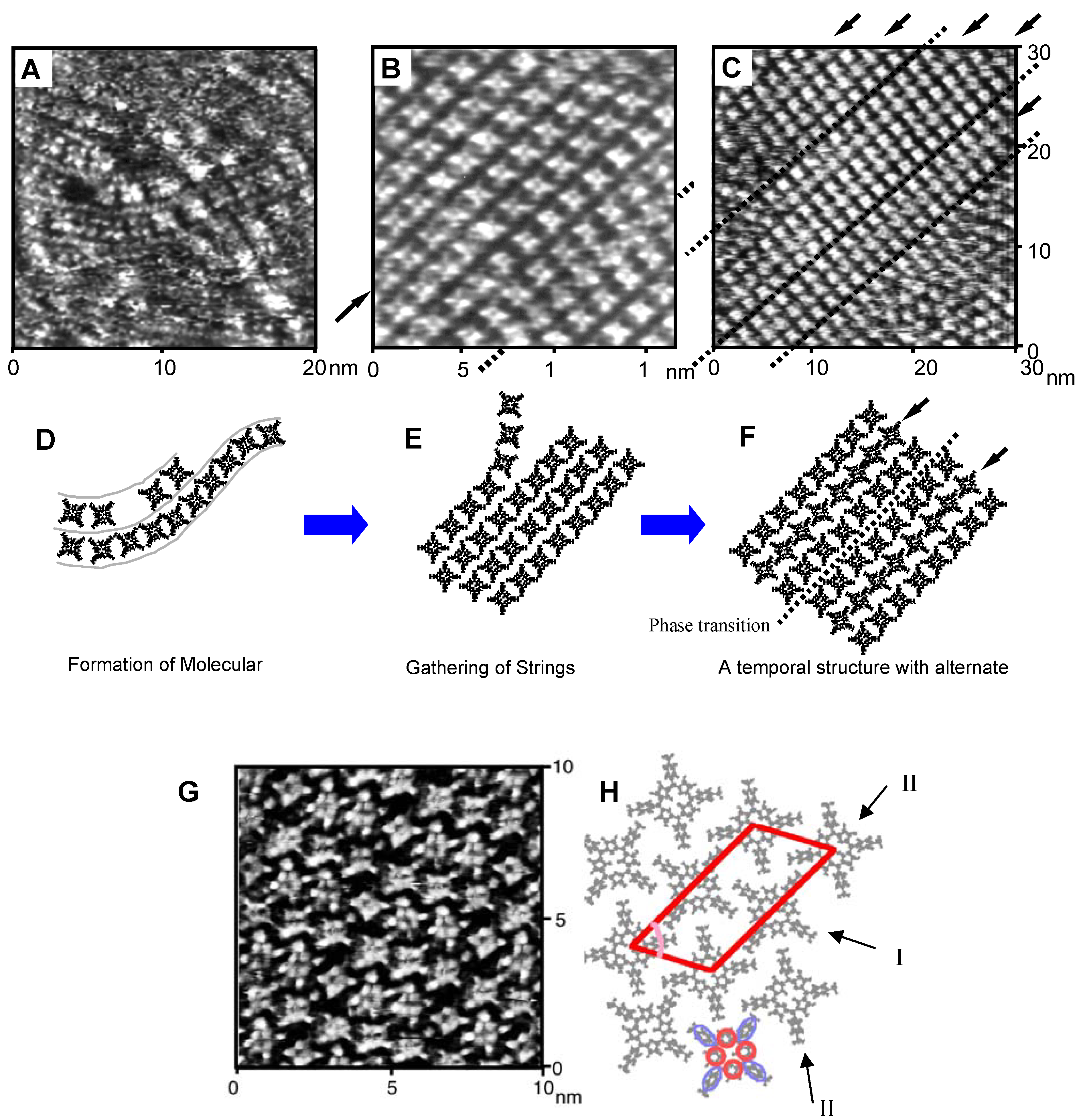
4.2. Ostwald Ripening
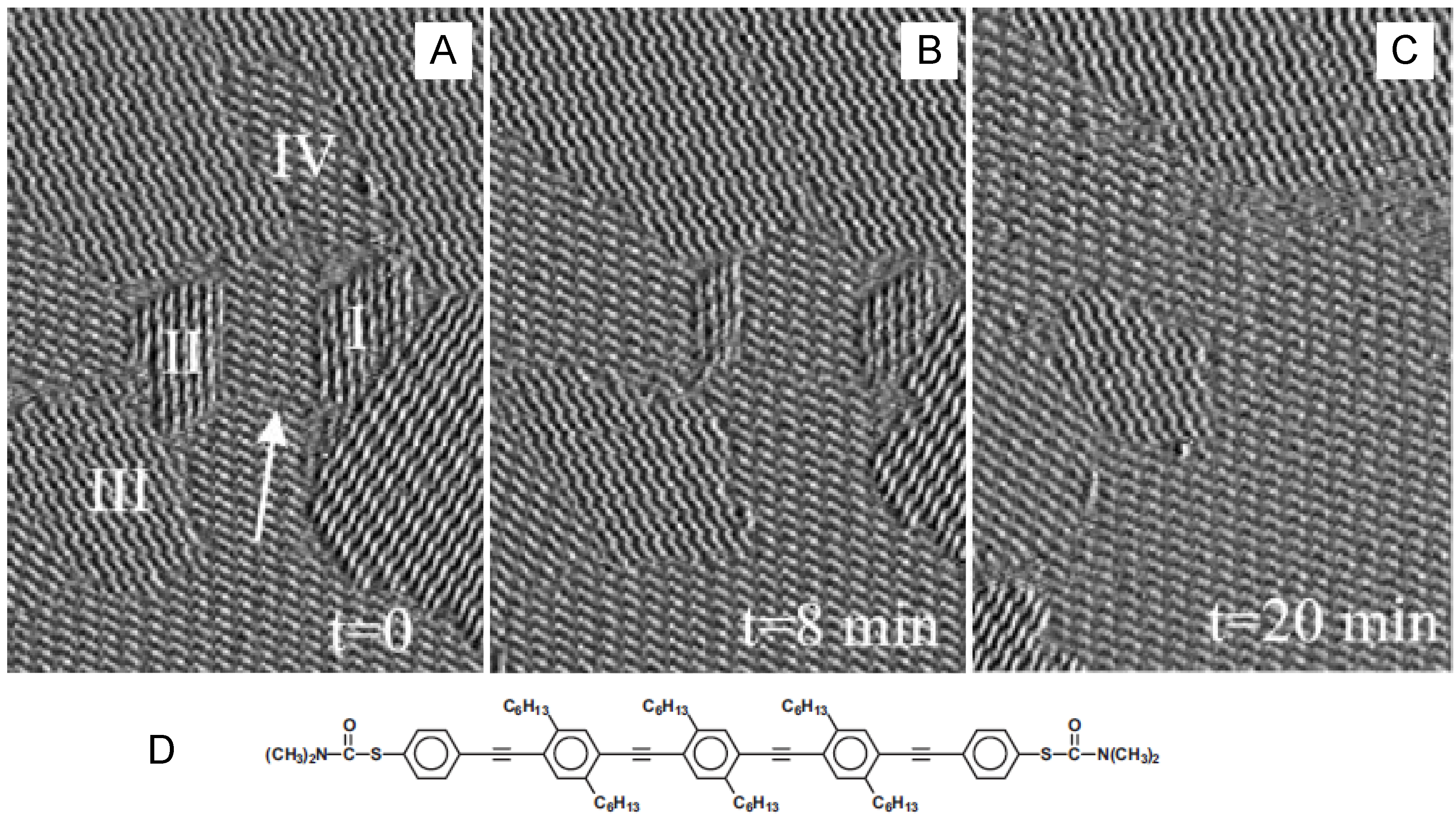
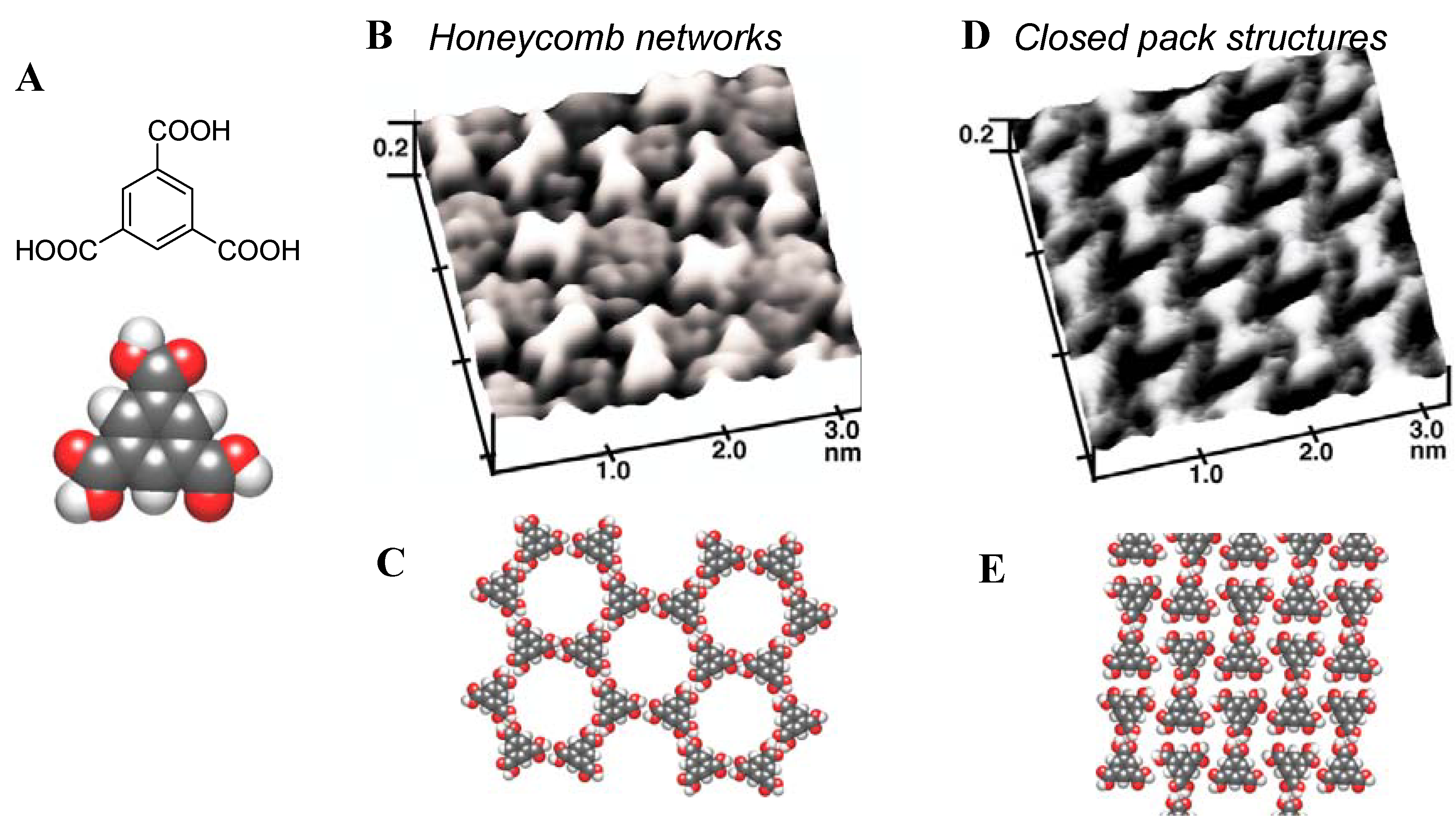
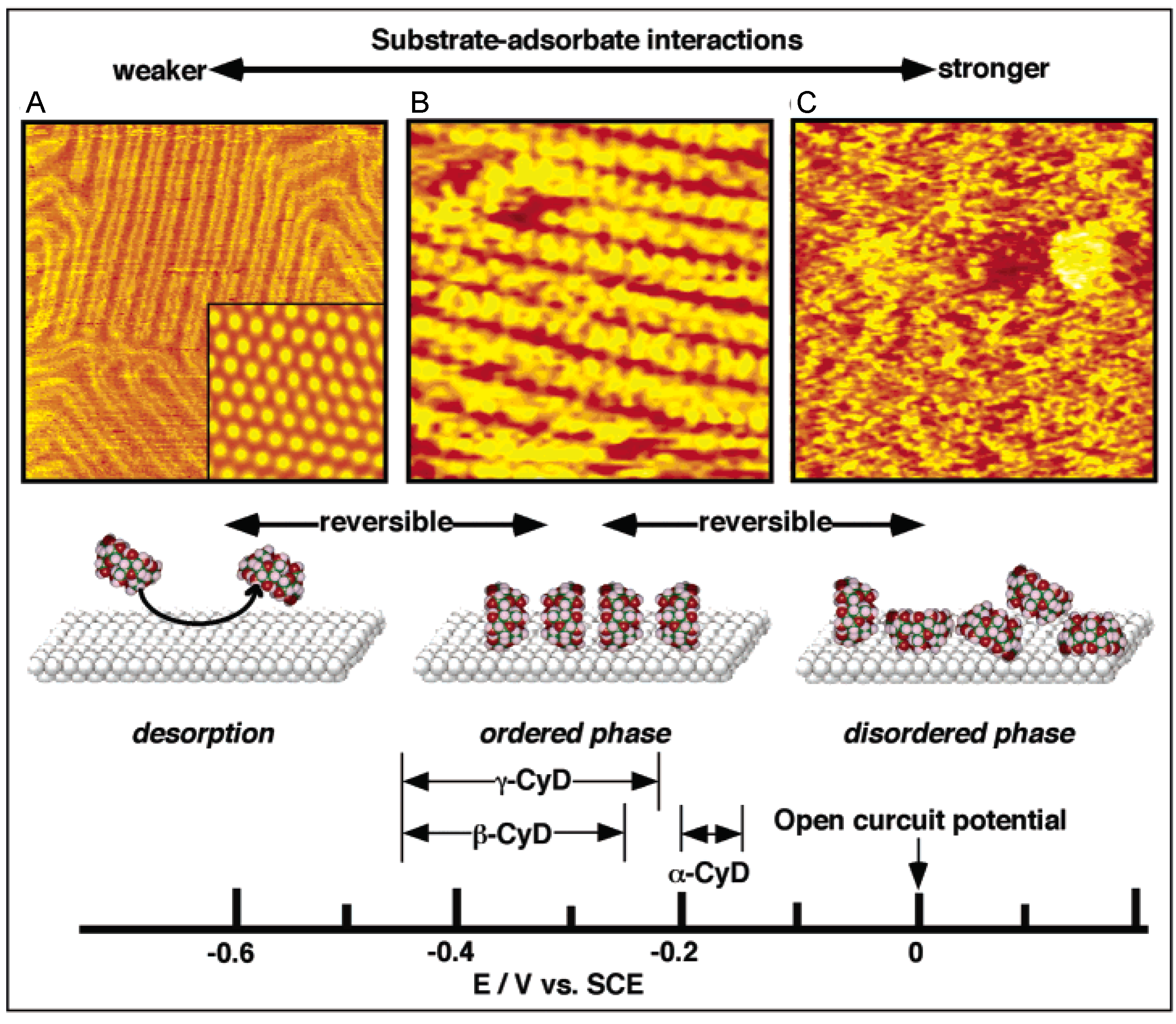
4.3. Two-Dimensional Phase Transition
4.3.1. Single-molecular species

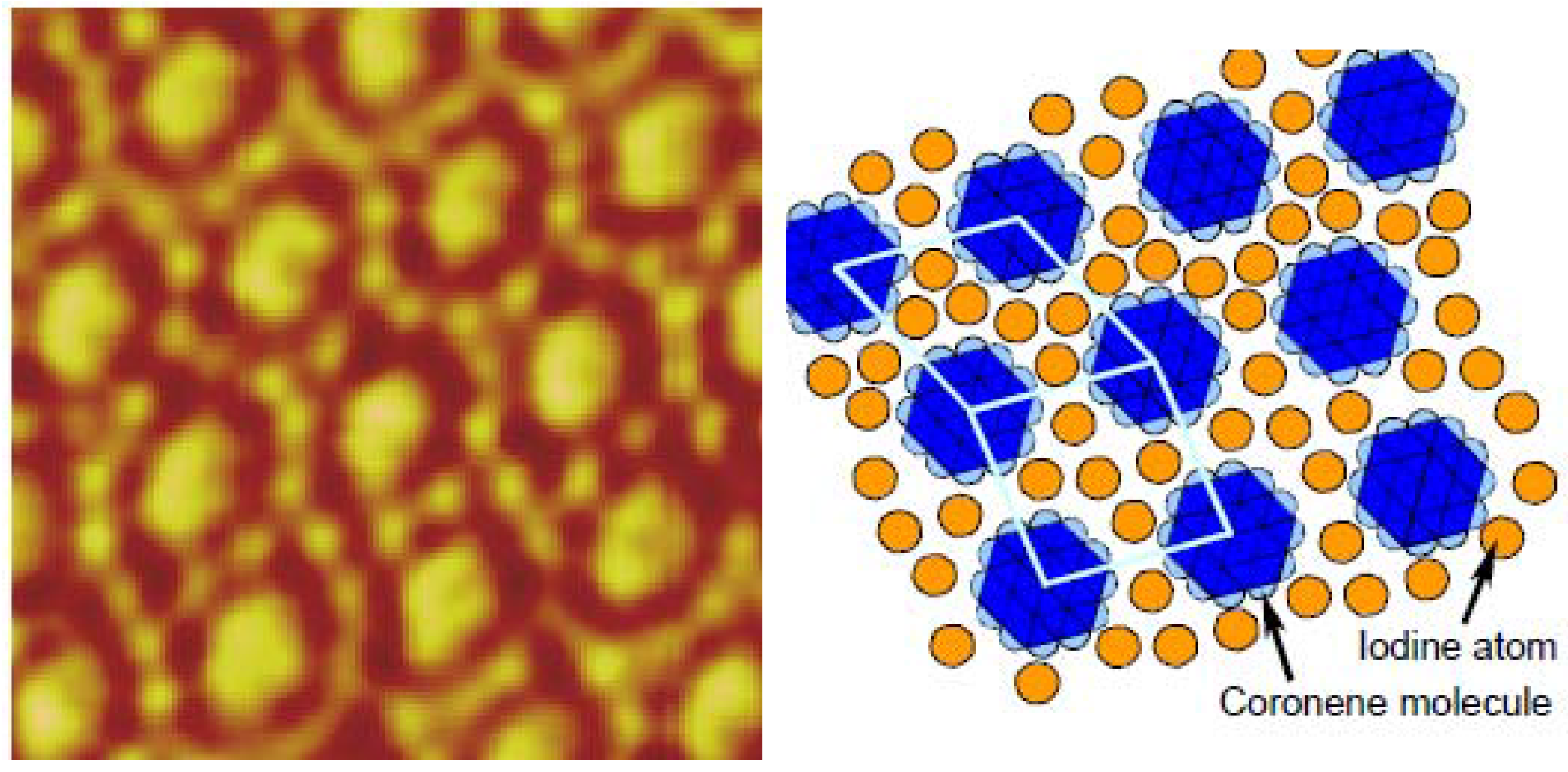
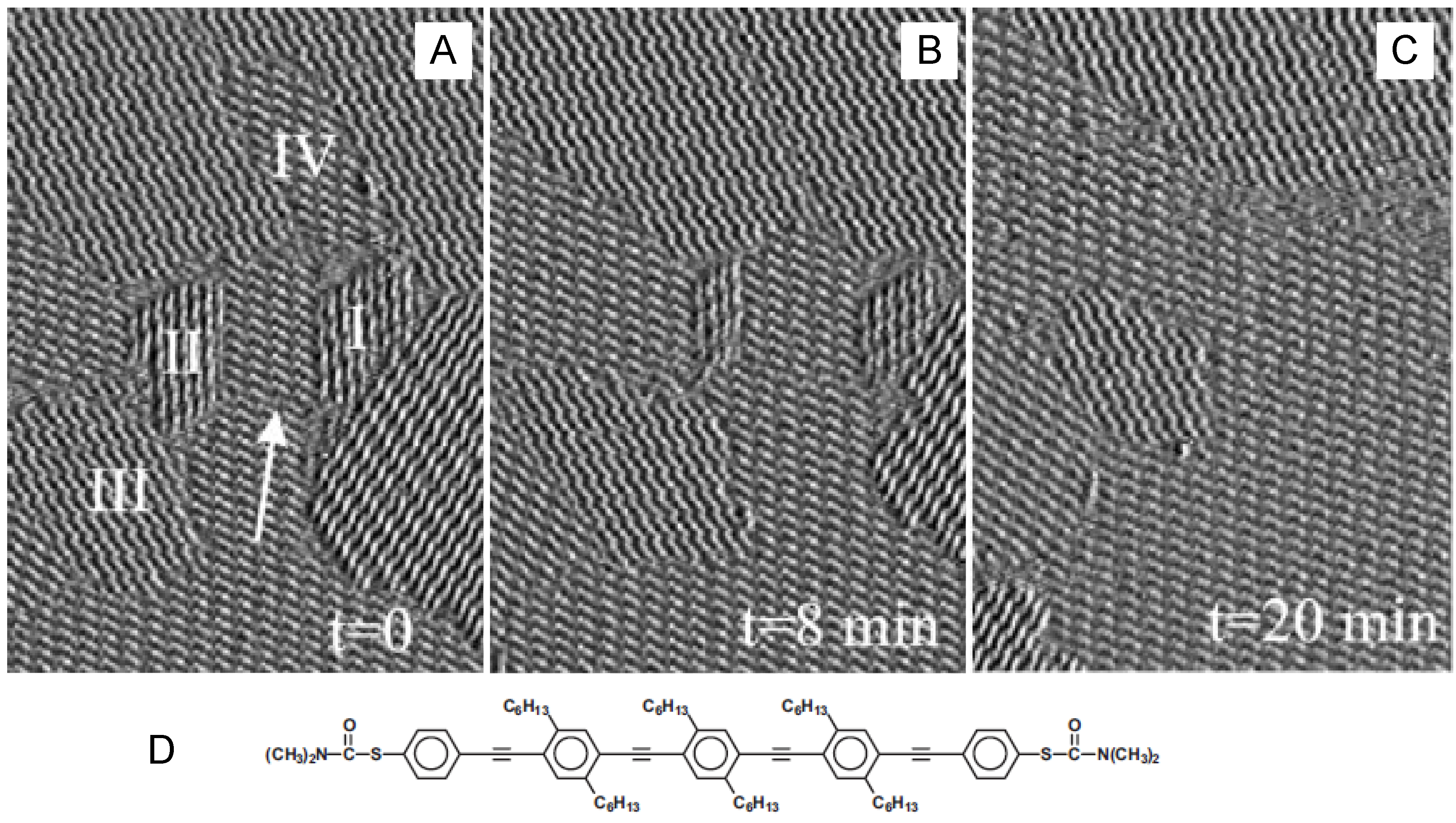
4.3.2. Competitive physisorption dynamics

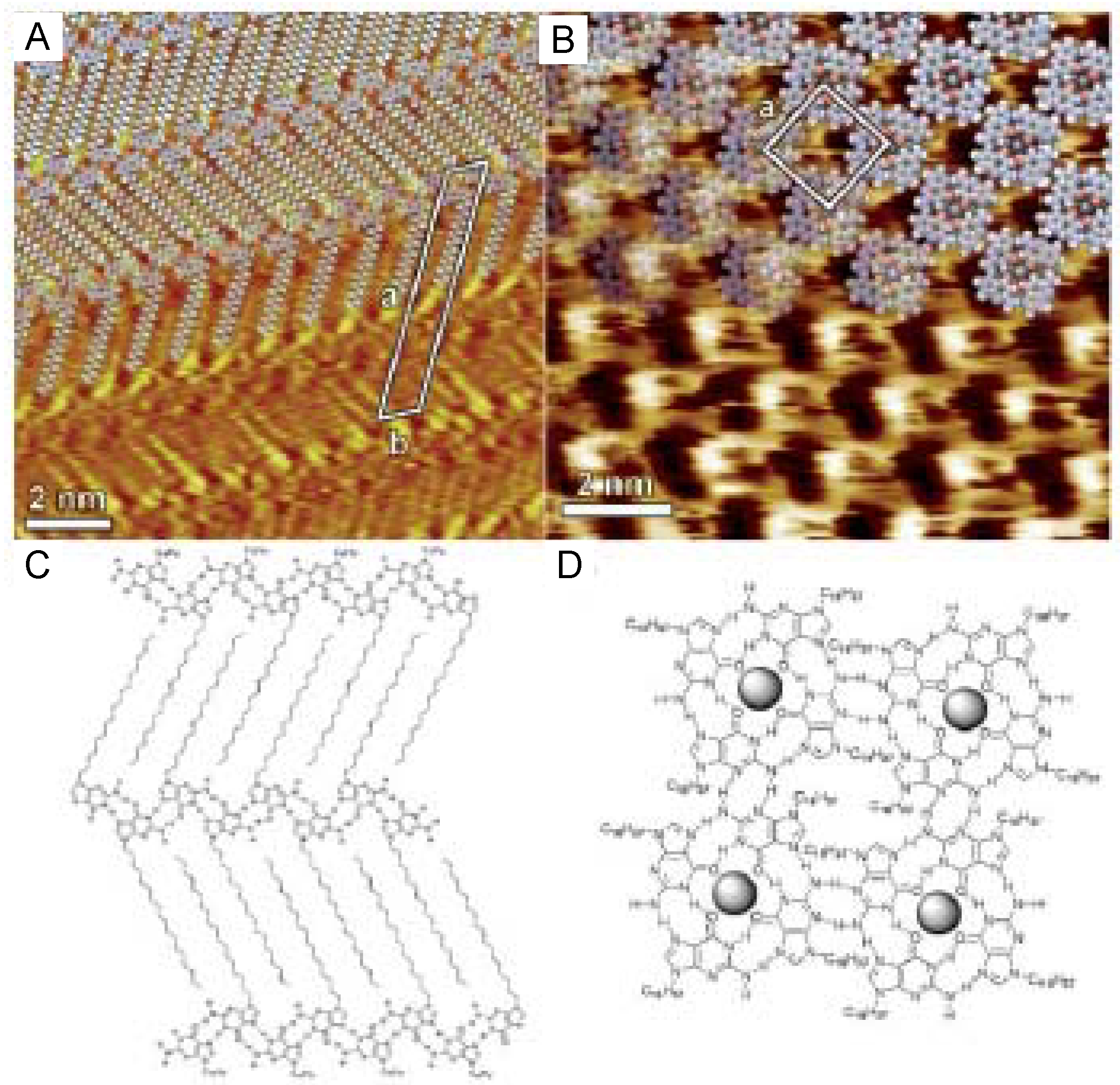
4.4. Conformational Changes


5. Molecular Rotations in Arrays



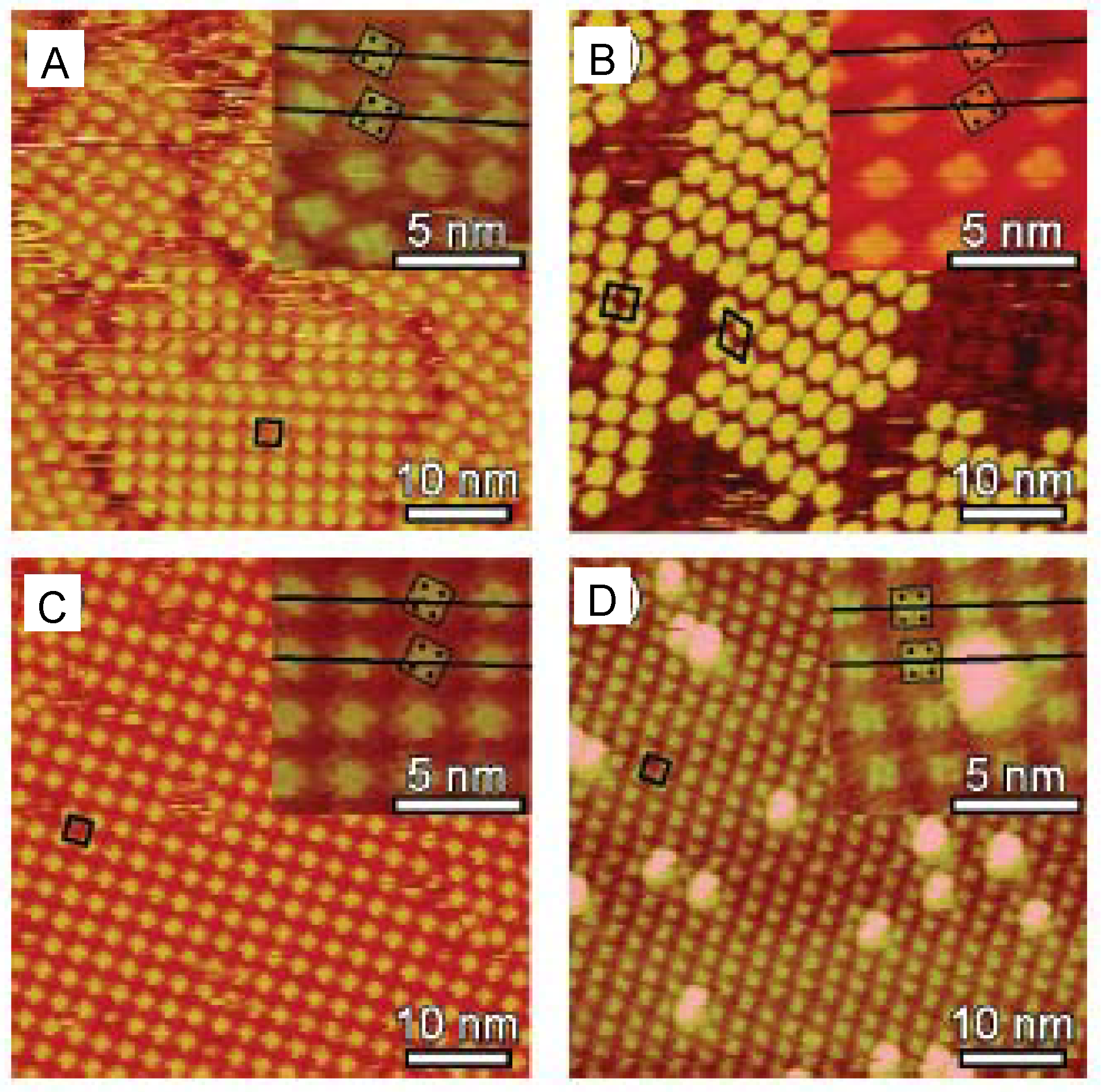
5. Conclusions
References and Notes
- Binnig, G.; Rohrer, H.; Gerber, C.; Weibel, E. Tunneling through a controllable vacuum gap. Appl. Phys. Lett. 1982, 40, 178–180. [Google Scholar] [CrossRef]
- Rabe, J.P. Molecules at interfaces: STM in materials and life sciences. Ultramicroscopy 1992, 42-44, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Weiss, P.S.; Eigler, D.M. Site dependence of the apparent shape of a molecule in scanning tunneling micoscope images: Benzene on Pt{111}. Phys. Rev. Lett. 1993, 71, 3139–3142. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, H.; Wilson, R.J.; Chiang, S.; Mate, C.M. Scanning tunneling microscopy observations of benzene molecules on the Rh(111)-(3 × 3) (C6H6 + 2CO) surface. Phys. Rev. Lett. 1988, 60, 2398–2401. [Google Scholar] [CrossRef] [PubMed]
- Hallmark, V.M.; Chiang, S.; Brown, J.K.; Wöll, Ch. Real-space imaging of the molecular organization of naphthalene on Pt(111). Phys. Rev. Lett. 1991, 66, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Lippel, P.H.; Wilson, R.J.; Miller, M D.; Wöll, C.; Chiang, S. High-resolution imaging of copper-phthalocyanine by scanning-tunneling microscopy. Phys. Rev. Lett. 1989, 62, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Somorjai, G.A. Introduction to Surface Chemistry and Catalysis, 1st ed.; John Wiley & Sons Inc: New York, NY, USA, 1994. [Google Scholar]
- Guckenberger, R.; Hartmann, T.; Wiegrabe, W.; Baumeister, W. Scanning Tunneling Microscopy II; Wiesendanger, R., Guntherodt, H.J., Eds.; Springer-Verlag: New York, NY, USA, 1991; Volume 20, Chapter 7. [Google Scholar]
- Palermo, V.; Samorì, P. Molecular self-assembly across multiple length scales. Angew. Chem. Int. Ed. 2007, 46, 4428–4432. [Google Scholar] [CrossRef]
- Elemans, J.A.A.W.; Lei, S.; De Feyter, S. Molecular and supramolecular networks on surfaces: From two-dimenstional crystal engineering to reactivity. Angew. Chem. Int. Ed. 2009, 48, 7298–7333. [Google Scholar] [CrossRef]
- Cunha, F.; Tao, N.J. Surface charge induced ordered-disorder transition in an organic monolayer. Phys. Rev. Lett. 1995, 75, 2376–2379. [Google Scholar] [CrossRef] [PubMed]
- Kunitake, M.; Akiba, U.; Batina, N.; Itaya, K. Structures and dynamic formation processes of porphyrin adlayers on iodine-modified Au(111) in solution: in situ STM study. Langmuir 1997, 13, 1607–1615. [Google Scholar] [CrossRef]
- Koma, A. Van der Waals epitaxy for highly lattice-mismatched systems. J. Cryst. Growth 1999, 201-202, 236–241. [Google Scholar] [CrossRef]
- Hooks, D.E.; Fritz, T.; Ward, M.D. Epitaxy and molecular organization on solid substrates. Adv. Mater. 2001, 13, 227–241. [Google Scholar] [CrossRef]
- Sakurai, T.; Wang, X.D.; Xue, Q.K.; Hasegawa, Y.; Hashizume, T.; Shinohara, H. Scanning tunneling microscopy study of fullerenes. Prog. Surf. Sci. 1996, 51, 263–408. [Google Scholar] [CrossRef]
- Tautz, F.S. Structure and bonding of large aromatic molecules on noble metal surfaces: The example of PTCDA. Prog. Surf. Sci. 2007, 82, 479–520. [Google Scholar] [CrossRef]
- Uemura, S.; Sakata, M.; Taniguchi, I.; Kunitake, M.; Hirayama, C. Novel “wet process” technique based on electrochemical replacement for the preparation of fullerene epitaxial adlayers. Langmuir 2001, 17, 5–7. [Google Scholar] [CrossRef]
- Uemura, S.; Sakata, M.; Hirayama, C.; Kunitake, M. Fullerene adlayers on various single-crystal metal surfaces prepared by transfer from L films. Langmuir 2004, 20, 9198–9201. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Chen, C.Y.; Lee, Y.L. Highly ordered C60 monolayer self-assembled by using an iodine template on an Au(111) surface in solution. Langmuir 2008, 24, 11611–11615. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Hwang, I.S.; Fang, C.K.; Tsong, T.T. Adsorption and motion of C60 molecules on the Pb-covered Si(111) surface. Phys. Rev. B 2008, 77, 155421 1–7. [Google Scholar] [CrossRef]
- Stöhr, M.; Wagner, Th.; Gabriel, M.; Weyers, B.; Möller, R. Direct observation of hindered eccentric rotation of an individual molecule: Cu-phthalocyanine on C60. Phys. Rev. B 2001, 65. [Google Scholar] [CrossRef]
- Fendrich, M.; Wagner, Th.; Stöhr, M.; Möller, R. Hindered rotation of a copper phthalocyanine molecule on C60: Experiments and molecular mechanics calculations. Phys. Rev. B 2006, 73, 1–7. [Google Scholar] [CrossRef]
- Kunitake, M.; Batina, N.; Itaya, K. Self-organized porphyrin array on iodine-modified Au(111) in electrolyte solutions: in situ scanning tunneling microscopy study. Langmuir 1995, 11, 2337–2340. [Google Scholar] [CrossRef]
- Stabel, A.; Heinz, R.; de Schryver, F.C.; Rabe, J.P. Ostwald ripening of two-dimensional crystals at the solid-liquid interface. J. Phys. Chem. 1995, 99, 505–507. [Google Scholar] [CrossRef]
- Samorì, P.; Müllen, K.; Rabe, J.P. Molecular-scale tracking of the self-healing of polycrystalline monolayers at the solid-liquid interface. Adv. Mater. 2004, 16, 1761–1765. [Google Scholar] [CrossRef]
- Lackinger, M.; Griessl, S.; Kampschulte, L.; Jamizky, F.; Heckl, W.M. Dynamics of grain boundaries in two-dimensional hydrogen-bonded molecular networks. Small 2005, 1, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.R.; Lei, S.B.; Pan, G.B.; Wan, L.J.; Fan, Q.H.; Bai, C.L. Monitoring molecular motion and structure near defect with STM. Colloids Surf. A: Phys. Eng. Aspects 2005, 257-258, 9–13. [Google Scholar] [CrossRef]
- Palma, M.; Pace, G.; Roussel, P.; Geerts, Y.; Samorì, P. STM investigation of alkylated thiotriphenylene monolayers at the solid-liquid interface: Structure and dynamics. Aust. J. Chem. 2006, 59, 376–380. [Google Scholar] [CrossRef]
- Askadskaya, L.; Rabe, J.P. Anisotropic molecular dynamics in the vicinity of order-disorder transition in organic monolayers. Phys. Rev. Lett. 1992, 69, 1395–1398. [Google Scholar] [CrossRef]
- Ohira, A.; Ishizaki, T.; Sakata, M.; Kunitake, M.; Taniguchi, I.; Hirayama, C. Self-organization of α-cyclodextrin on Au(111) surfaces induced by potential controlled adsorption. J. Electroanal. Chem. 1999, 472, 163–167. [Google Scholar] [CrossRef]
- Ohira, A.; Ishizaki, T.; Sakata, M.; Taniguchi, I.; Hirayama, C.; Kunitake, M. Formation of the ‘nanotube’ structure of α-cyclodextrin on Au(111) surfaces induced by potential controlled adsorption. Colloids Surf., A: Phys. Eng. Aspects 2000, 169, 27–33. [Google Scholar] [CrossRef]
- Ohira, A.; Sakata, M.; Taniguchi, I.; Hirayama, C.; Kunitake, M. Comparison of nanotube structures constructed from α-, β-, and γ-cyclodextrins by potential-controlled adsorption. J. Am. Chem. Soc. 2003, 125, 5057–5065. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Ohira, A.; Sakata, M.; Hirayama, C.; Kunitake, M. A two-dimensional molecular network structure of trimesic acid prepared by adsorption-induced self-organization. Chem. Commun. 2002, 2652–2653. [Google Scholar] [CrossRef]
- He, Y.; Ye, T.; Borguet, E. Porphyrin Self-assembly at electrochemical interfaces: Role of potential modulated surface mobility. J. Am. Chem. Soc. 2002, 124, 11964–11970. [Google Scholar] [CrossRef] [PubMed]
- Dretschkow, Th.; Wandlowski, Th. An order-disorder-order adlayer transition of 2,2’-bipyridine on Au(111). Electrochim. Acta 1999, 45, 731–740. [Google Scholar] [CrossRef]
- Suto, K.; Yoshimoto, S.; Itaya, K. Electrochemical control of the structure of two-dimensional supramolecular organization consisting of phthalocyanine and porphyrin on a gold single-crystal surface. Langmuir 2006, 22, 10766–10776. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Honda, Y.; Ito, O.; Itaya, K. Supramolecular pattern of fullerene on 2D bimolecular “chessboard” consisting of bottom-up assembly of porphyrin and phthalocyanine molecules. J. Am. Chem. Soc. 2008, 130, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Griessl, S.; Lackinger, M.; Edelwirth, M.; Hietschold, M.; Heckl, W.M. Self-assembled two-dimensional molecular host-guest architectures from trimesic acid. Single Mol. 2002, 3, 25–31. [Google Scholar] [CrossRef]
- Su, G.J.; Li, Z.H.; Aquilar-Sanchez, R. Phase transition of two-dimensional chiral supramolecular nanostructure tuned by electrochemical potential. Anal. Chem. 2009, 81, 8741–8748. [Google Scholar] [CrossRef] [PubMed]
- Kunitake, M.; Ishikawa, Y.; Matsuda, S.; Abe, S.; Miyasato, Y.; Higuchi, R.; Sakata, M. Coronene-iodine coadsorbed adlayers on Au(111) surfaces promoted by electrochemical potential control. ECS Trans. 2007, 3, 147–154. [Google Scholar]
- Gesquière, A.; Abdel-Mottaleb, M.M.; De Feyter, S.; De Schryver, F.C.; Sieffert, M.; Müllen, K.; Cakderone, A.; Lazzaroni, R.; Brédas, J.L. Dynamics in physisorbed monolayers of 5-alkoxy-isophthalic acid derivatives at the liquid/solid interface investigated by scanning tunneling microscopy. Chem. Eur. J. 2000, 6, 3739–3746. [Google Scholar] [CrossRef] [PubMed]
- Bonini, M.; Zalewski, L.; Breiner, T.; Dötz, F.; Kastler, M.; Schädler, V.; Surin, M.; Layyaroni, R.; Samorì, P. Competitive physisorption among alkyl-substituted π-conjugated oligomers at the solid-liquid interface: Towards prediction of self-assembly at surfaces from a multicomponent solution. Small 2009, 5, 1521–1526. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Minoia, A.; Tomović, Ž.; Lazzaroni, R.; Meijer, E.W.; Schenning, A.P.H.J.; De Feyter, S. A multivalent hexapod: Conformational dynamics of six-legged molecules in self-assembled monolayers at a solid–liquid interface. ACS Nano 2009, 3, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Tahara, K.; De Schryver, F.C.; der Auweraer, M.V.; Tobe, Y.; De Feyter, S. Structural transformation of a two-dimensional molecular network in response to selective guest inclusion. Angew. Chem. Int. Ed. 2007, 46, 2831–2834. [Google Scholar] [CrossRef]
- Ciesielski, A.; Lena, S.; Masiero, S.; Spada, G.P.; Samorì, P. Dynamers at the solid-liquid interface: Controlling the reversible assembly/reassembly process between two highly ordered supramolecular guanine motifs. Angew. Chem. Int. Ed. 2010, 49, 1963–1966. [Google Scholar] [CrossRef]
- Piot, L.; Meudtner, R.M.; El Malah, T.; Hecht, S.; Samorì, P. Modulating large-area self-assembly at the solid-liquid interface by pH-mediated conformational switching. Chem. Eur. J. 2009, 15, 4788–4792. [Google Scholar] [CrossRef] [PubMed]
- Lingenfelder, M.A.; Spillmann, H.; Dmitriev, A.; Stepanow, S.; Lin, N.; Barth, J.V.; Kern, K. Towards surface-supported supramolecular architectures: Tailored coorination assembly of 1,4-bensenedicarboxylate and Fe on Cu(100). Chem. Eur. J. 2004, 10, 1913–1919. [Google Scholar] [CrossRef] [PubMed]
- Payer, D.; Rauschenbach, S.; Malinowski, N.; Konuma, M.; Virojanadara, C.; Starke, U.; Dietrich-Buchecker, C.; Collin, J.P.; Sauvage, J.P.; Lin, N.; Kern, K. Toward mechanical switching of surface-adsorbed [2]catenane by in situ copper complexation. J. Am. Chem. Soc. 2007, 129, 15662–15667. [Google Scholar] [CrossRef] [PubMed]
- Vanoppen, P.; Grim, P.C.M.; Rücker, M.; De Feyter, S.; Moessner, G.; Valiyaveettil, S.; Müllen, K.; De Schryver, F.C. Solvent codeposition and cis-trans isomerization of isophthalic acid derivatives studied by STM. J. Phys. Chem. 1996, 100, 19636–19641. [Google Scholar] [CrossRef]
- Katsonis, N.; Kudernac, T.; Walko, M.; van der Molen, S.J.; van Wees, B.J.; Feringa, B.L. Reversible conductance switching of single diarylethenes on a gold surface. Adv. Mater. 2006, 18, 1397–1400. [Google Scholar] [CrossRef]
- Xu, L.P.; Wan, L.J. STM investigation of the photoisomerization of an azobis(benzo-15-crown-5) molecule and its self-assembly on Au(111). J. Phys. Chem. B 2006, 110, 3185–3188. [Google Scholar] [CrossRef] [PubMed]
- Pace, G.; Ferri, V.; Grave, C.; Elbing, M.; von Hänisch, C.; Zharnikov, M.; Mayor, M.; Rampi, M.A.; Samorì, P. Cooperative light-induced molecular movements of highly ordered azobenzene self-assembled monolayers. Proc. Natl. Acad. Sci. USA 2007, 104, 9937–9942. [Google Scholar] [CrossRef] [PubMed]
- Arai, R.; Uemura, S.; Irie, M.; Matsuda, K. Reversible photoinduced two-dimensional molecular ordering change on HOPG/liquid interface. J. Am. Chem. Soc. 2008, 130, 9371–9379. [Google Scholar] [CrossRef] [PubMed]
- Baber, A.E.; Tlerney, H.L.; Sykes, E.C.H. A quantitative single-molecule study of thioether molecular rotors. ACS Nano 2008, 2, 2385–2391. [Google Scholar] [CrossRef] [PubMed]
- Tierney, H.L.; Baber, A.E.; Jewell, A.D.; Iski, E.V.; Boucher, M.B.; Sykes, E.C.H. Mode-selective electrical excitation of a molecular rotor. Chem. Eur. J. 2009, 15, 9678–9680. [Google Scholar] [CrossRef] [PubMed]
- Tlerney, H.L.; Baber, A.E.; Sykes, E.C.H.; Aklmov, A.; Kolomelsky, A.B. Dynamics of thioether molecular rotors: Effects of surface interactions and chain flexibility. J. Phys. Chem. C 2009, 113, 10913–10920. [Google Scholar] [CrossRef]
- Wilson, R.J.; Meijer, G.; Bethune, D.S.; Johnson, R.D.; Chambliss, D.D.; de Vries, M.S.; Hunziker, H.E.; Wendt, H.R. Imaging C60 clusters on a surface using a scanning tunnelling microscope. Nature 1990, 348, 621–622. [Google Scholar] [CrossRef]
- Behler, S.; Lang, H.P.; Pan, S.H.; Thommen-Geiser, V.; Guntherodt, H.J. Imaging C60 fullerite at 4.5 K by scanning tunneling microscopy. Z. Phys. B 1993, 91, 1–2. [Google Scholar] [CrossRef]
- Uemura, S.; Ohira, A.; Ishizaki, T.; Sakata, M.; Kunitake, M.; Taniguchi, I.; Hirayama, C. In situ STM visualization of fullerene epitaxial adlayers on Au(111) surfaces prepared by the transfer of Langmuir films. Chem. Lett. 1999, 279–280, Correction 1999, 535–537. [Google Scholar]
- Marchenko, A.; Cousty, J. C60 self-organization at the interface between a liquid C60 solution and a Au(111) surface. Surf. Sci. 2002, 513, 233–237. [Google Scholar] [CrossRef]
- Uemura, S.; Samorì, P.; Kunitake, M.; Hirayama, C.; Rabe, J.P. Crystalline C60 monolayers at the solid–organic solution interface. J. Mater. Chem. 2002, 12, 3366–3367. [Google Scholar] [CrossRef]
- Gaisch, R.; Berndt, R.; Gimzewski, J.K.; Reihl, B.; Schlittler, R.R.; Schneider, W.D.; Tschudy, M. Internal structure of C60 fullerene molecules as revealed by low-temperature STM. Appl. Phys. A 1993, 57, 207–210. [Google Scholar] [CrossRef]
- Gimzewski, J.K.; Joachim, C.; Schlittler, R.R.; Langlais, V.; Tang, H.; Johannsen, I. Rotation of a single molecule within a supramolecular bearing. Science 1998, 281, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Wintjes, N.; Bonifazi, D.; Cheng, F.; Kiebele, A.; Stöhr, M.; Jung, T.; Spillmann, H.; Diederich, F. A supramolecular multiposition rotary device. Angew. Chem. Int. Ed. 2007, 46, 4089–4092. [Google Scholar] [CrossRef]
- Binnemans, K.; Sleven, J.; De Feyter, S.; De Schryver, F.C.; Donnio, B.; Guillon, D. Structure and mesomorphic behavior of alkoxy-substituted bis(phthalocyaninato)lanthanide(III) complexes. Chem. Mater. 2003, 15, 3930–3938. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Gan, L.H.; Lei, S.B.; Wan, L.J.; Wang, C.; Jiang, J.Z. Self-assembly of PcOC8 and its sandwich lanthanide complex Pr(PcOC8)2 with oligo(phenylene-ethynylene) molecules. J. Phys. Chem. B 2005, 109, 19859–19865. [Google Scholar] [CrossRef] [PubMed]
- Klymchenko, A.S.; Sleven, J.; Binnemans, K.; De Feyter, S. Two-dimensional self-assembly and phase behavior of an alkoxylated sandwich-type bisphthalocyanine and its phthalocyanine analogues at the liquid–solid interface. Langmuir 2006, 22, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Takami, T.; Arnold, D.P.; Fuchs, A.V.; Will, G.D.; Goh, R.; Waclawik, E.R.; Bell, J.M.; Weiss, P.S.; Sugiura, K.; Liu, W.; Jiang, J. Two-dimensional crystal growth and stacking of bis(phthalocyaninato) rare earth sandwich complexes at the 1-phenyloctane/graphite interface. J. Phys. Chem. B 2006, 110, 1661–1664. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, J.; Kawaguchi, S.; Yamakawa, T.; Asakawa, M.; Miyake, K. Arrays of double-decker porphyrins on highly oriented pyrolytic graphite. Langmuir 2006, 22, 5708–5715. [Google Scholar] [CrossRef] [PubMed]
- Takami, T.; Ye, T.; Arnold, D.P.; Sugiura, K.; Wang, R.; Jiang, J.; Weiss, P.S. Controlled adsorption orientation for doulbe-decker complexes. J. Phys. Chem. C 2007, 111, 2077–2080. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Sawaguchi, T.; Su, W.; Jiang, J.; Kobayashi, N. Suprastructure formation and rearrangement in the adlayer of a rare-earth-metal triple-decker sandwich complex at the electrochemical interface. Angew. Chem. Int. Ed. 2007, 46, 1071–1074. [Google Scholar] [CrossRef]
- Lei, S.B.; Deng, K.; Yang, Y.L.; Zeng, Q.D.; Wang, C.; Jiang, J.Z. Electric driven molecular switching of asymmetric tris(phthalocyaninato)lutetium triple-decker complex at the liquid/solid interface. Nano Lett. 2008, 8, 1836–1843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; Isshiki, H.; Katoh, K.; Yoshida, Y.; Yamashita, M.; Miyasaki, H.; Breedlove, B.K.; Kajiwara, T.; Takaishi, S.; Komeda, T. Low-temperature scanning tunneling microscopy investigation of bis(phthalocyaninato)yttrium growth on Au(111): from individual molecules to two-dimensional domains. J. Phys. Chem. C 2009, 113, 9826–9830. [Google Scholar] [CrossRef]
- Miyake, K.; Fukuta, M.; Asakawa, M.; Hori, Y.; Ikeda, T.; Shimizu, T. Molecular motion of surface-immobilized double-decker phthalocyanine complexes. J. Am. Chem. Soc. 2009, 131, 17808–17813. [Google Scholar] [CrossRef]
- Tanaka, H.; Ikeda, T.; Yamashita, K.; Takeuchi, M.; Shinkai, S.; Kawai, T. Network of tris(porphyrinato)cerium(III) arranged on the herringbone structure of an Au(111) surface. Langmuir 2010, 26, 210–214. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Uemura, S.; Tanoue, R.; Yilmaz, N.; Ohira, A.; Kunitake, M. Molecular Dynamics in Two-Dimensional Supramolecular Systems Observed by STM. Materials 2010, 3, 4252-4276. https://doi.org/10.3390/ma3084252
Uemura S, Tanoue R, Yilmaz N, Ohira A, Kunitake M. Molecular Dynamics in Two-Dimensional Supramolecular Systems Observed by STM. Materials. 2010; 3(8):4252-4276. https://doi.org/10.3390/ma3084252
Chicago/Turabian StyleUemura, Shinobu, Ryota Tanoue, Neval Yilmaz, Akihiro Ohira, and Masashi Kunitake. 2010. "Molecular Dynamics in Two-Dimensional Supramolecular Systems Observed by STM" Materials 3, no. 8: 4252-4276. https://doi.org/10.3390/ma3084252