Hysteretic Tricolor Electrochromic Systems Based on the Dynamic Redox Properties of Unsymmetrically Substituted Dihydrophenanthrenes and Biphenyl-2,2'-Diyl Dications: Efficient Precursor Synthesis by a Flow Microreactor Method

Abstract
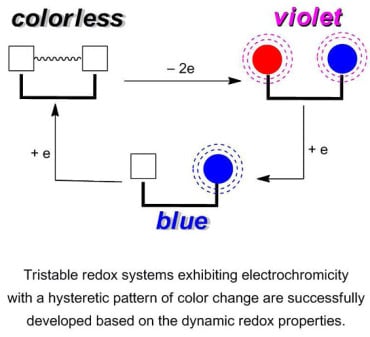
:
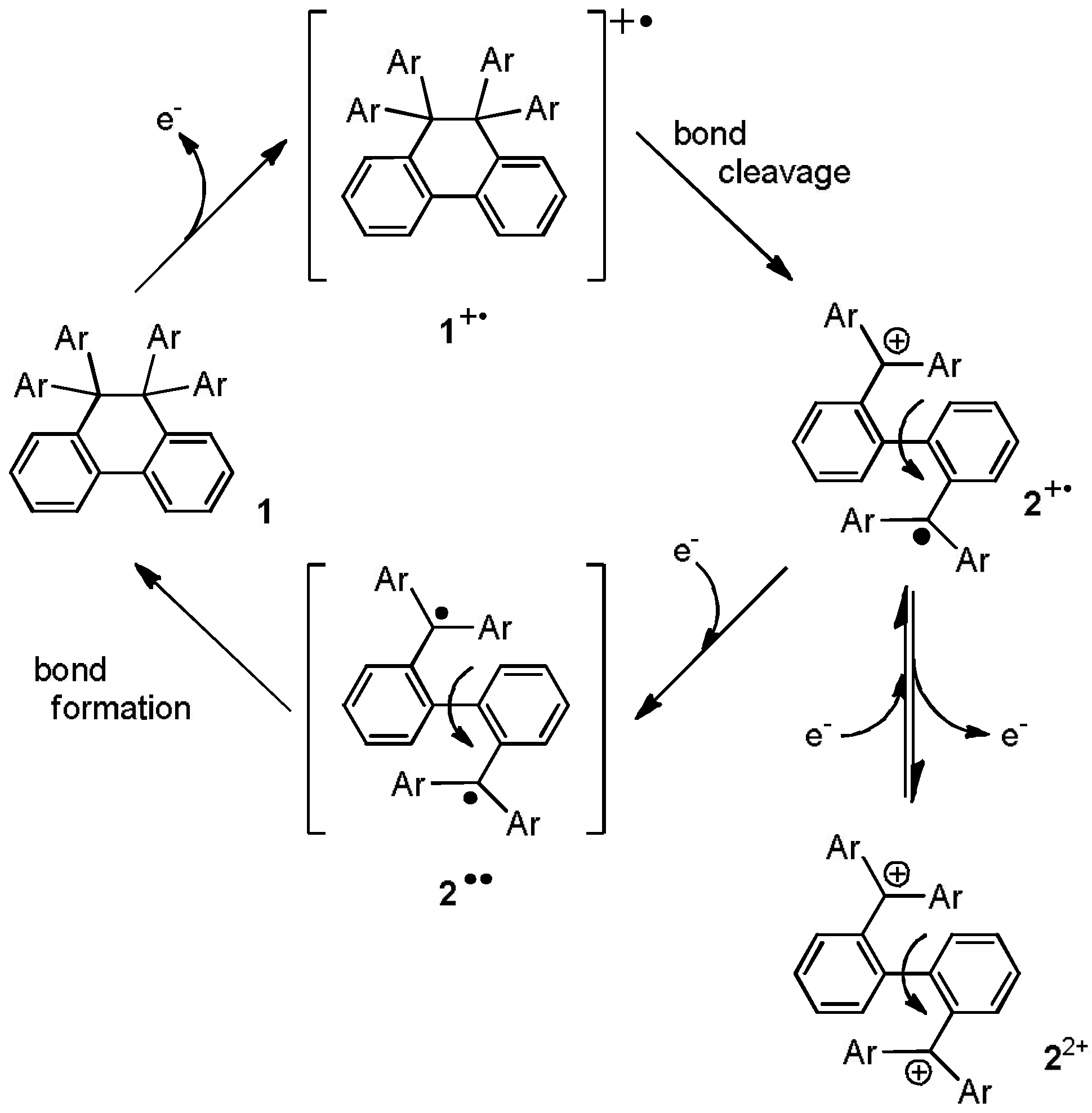
1. Introduction
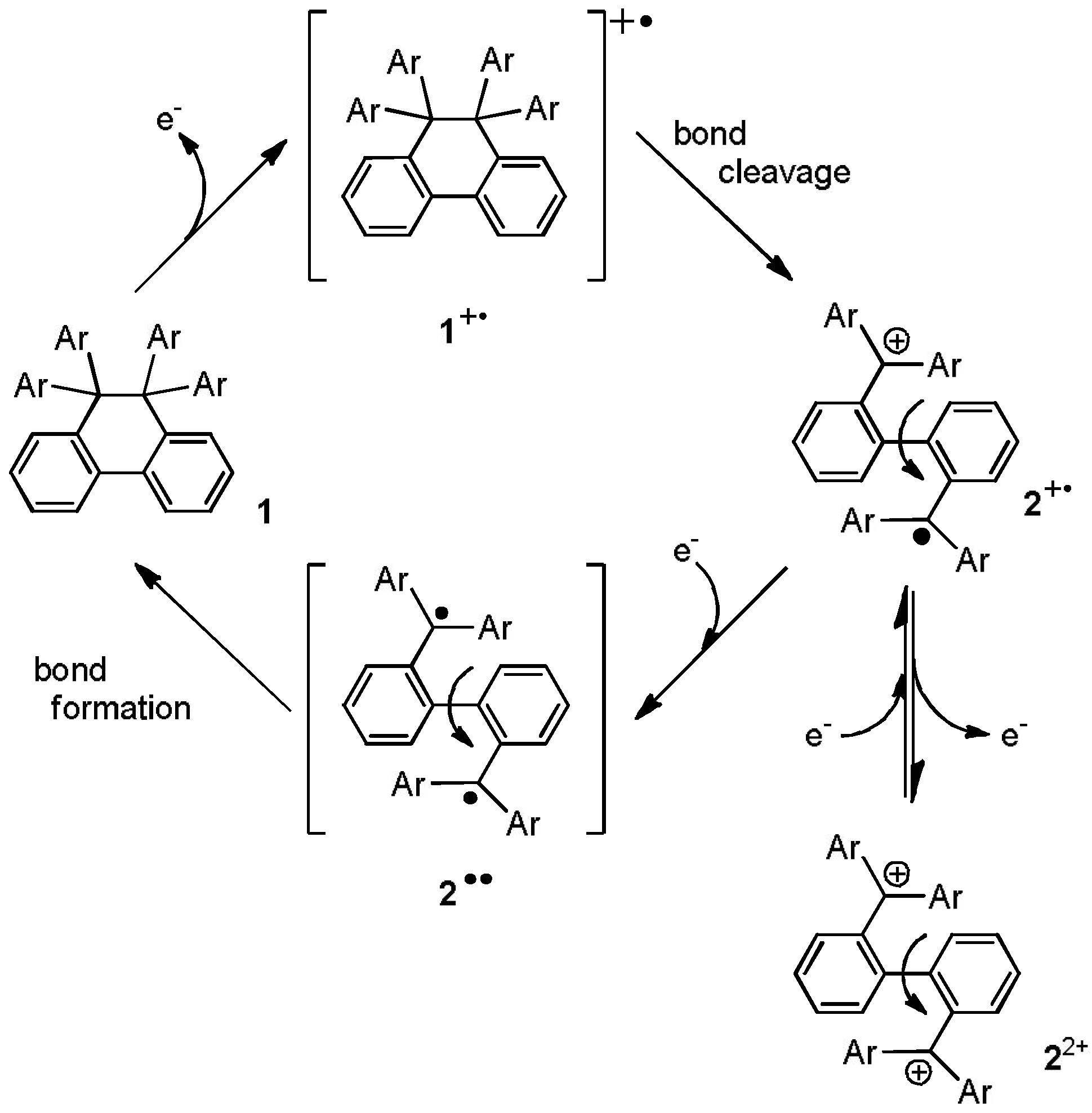
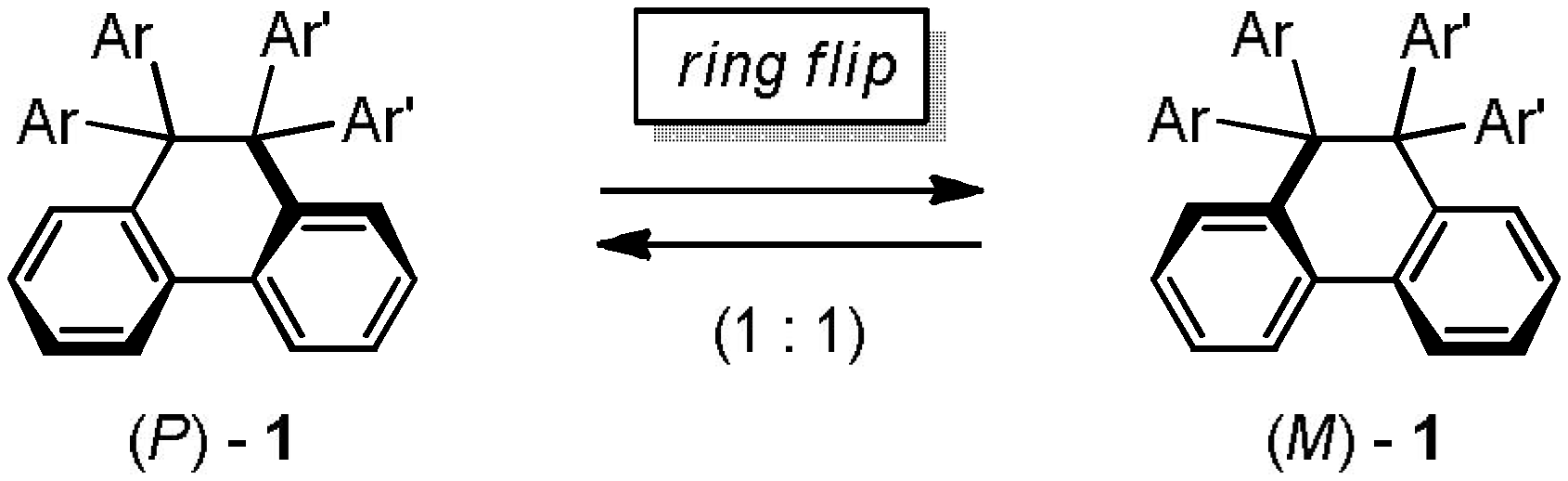
2. Results and Discussion
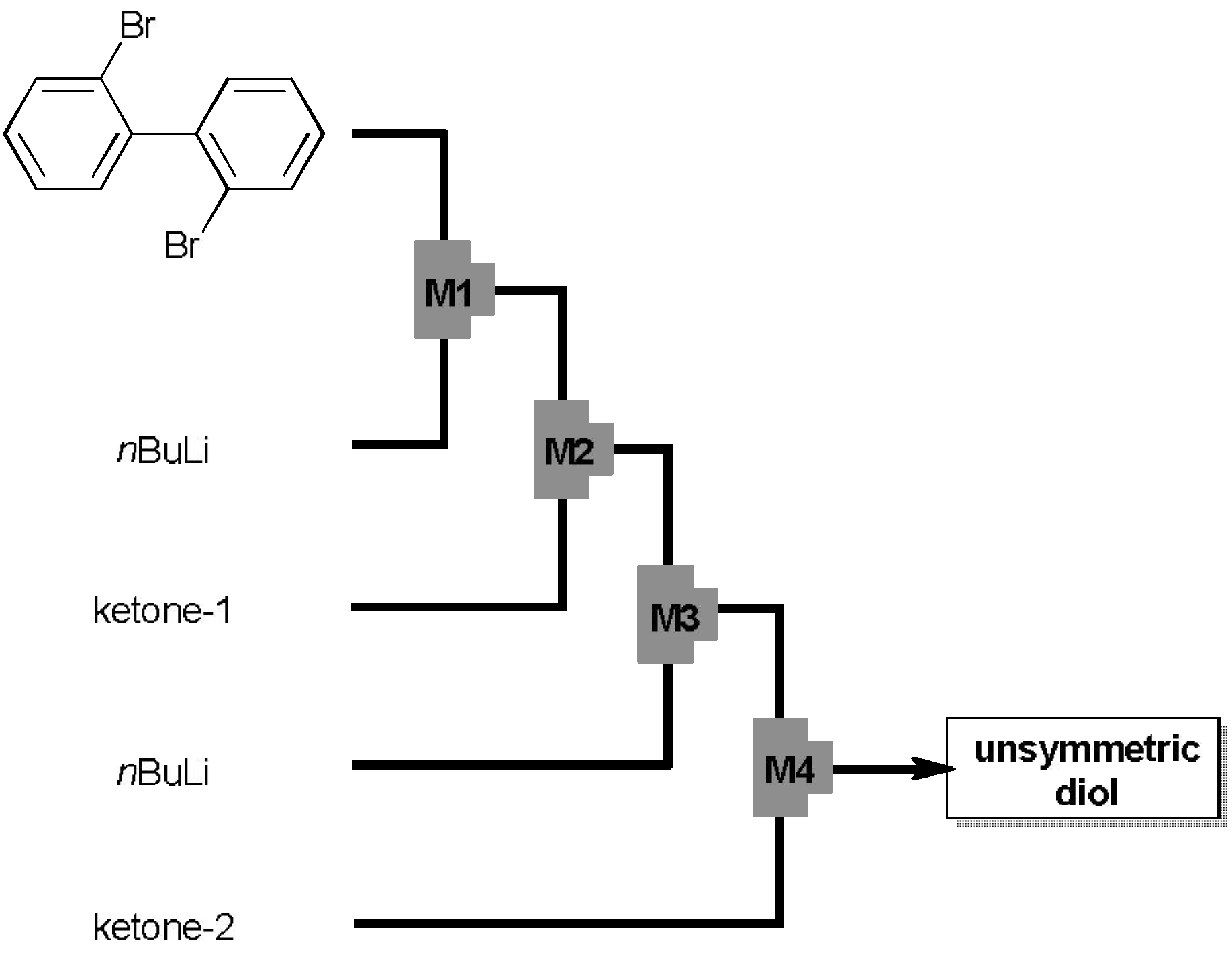
2.1. Preparation of Unsymmetrically Substituted Diol-Precursors
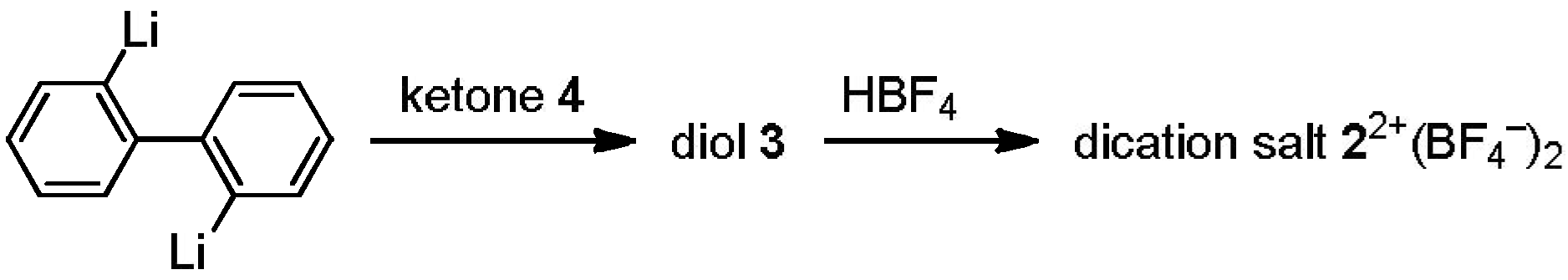
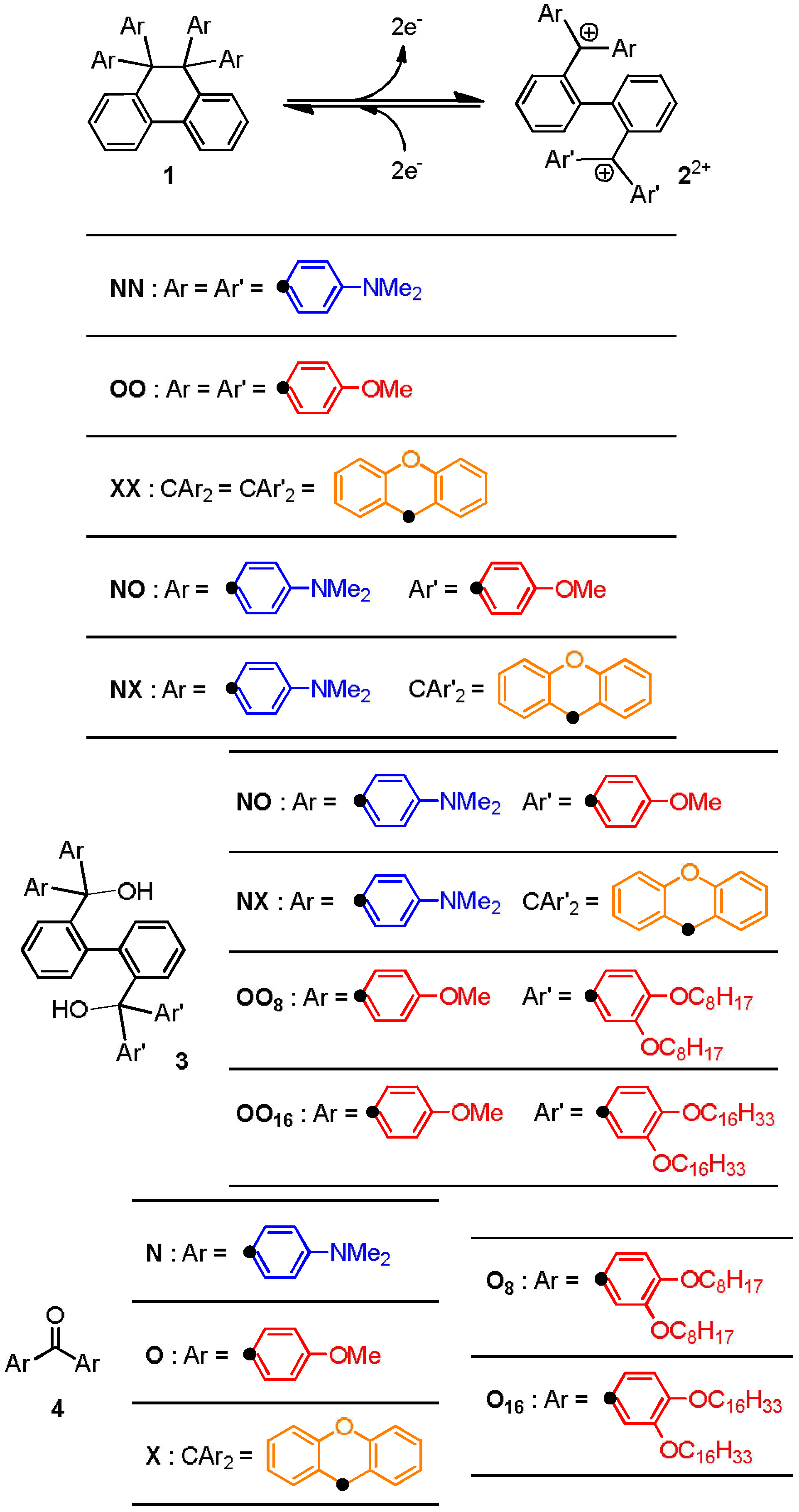
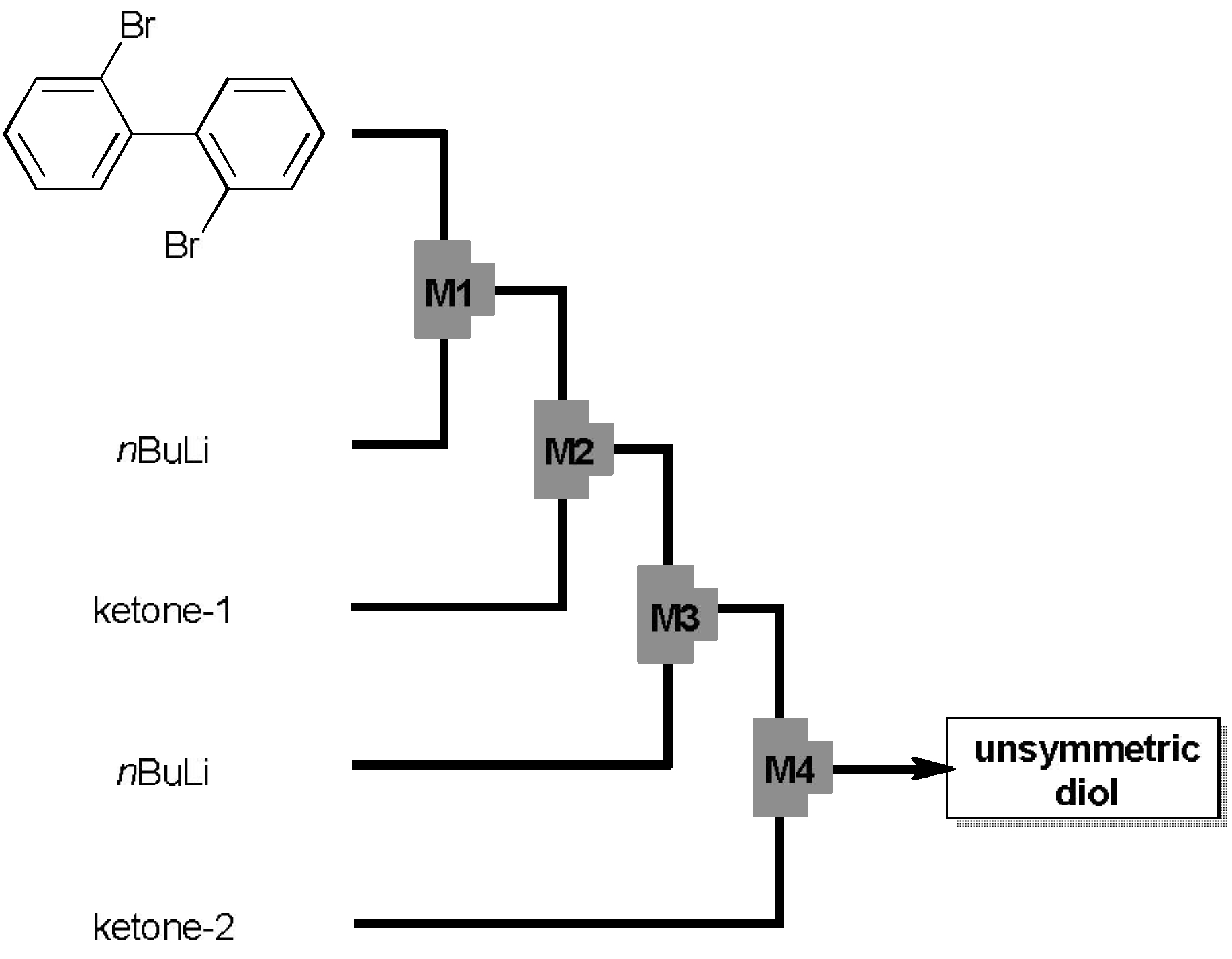
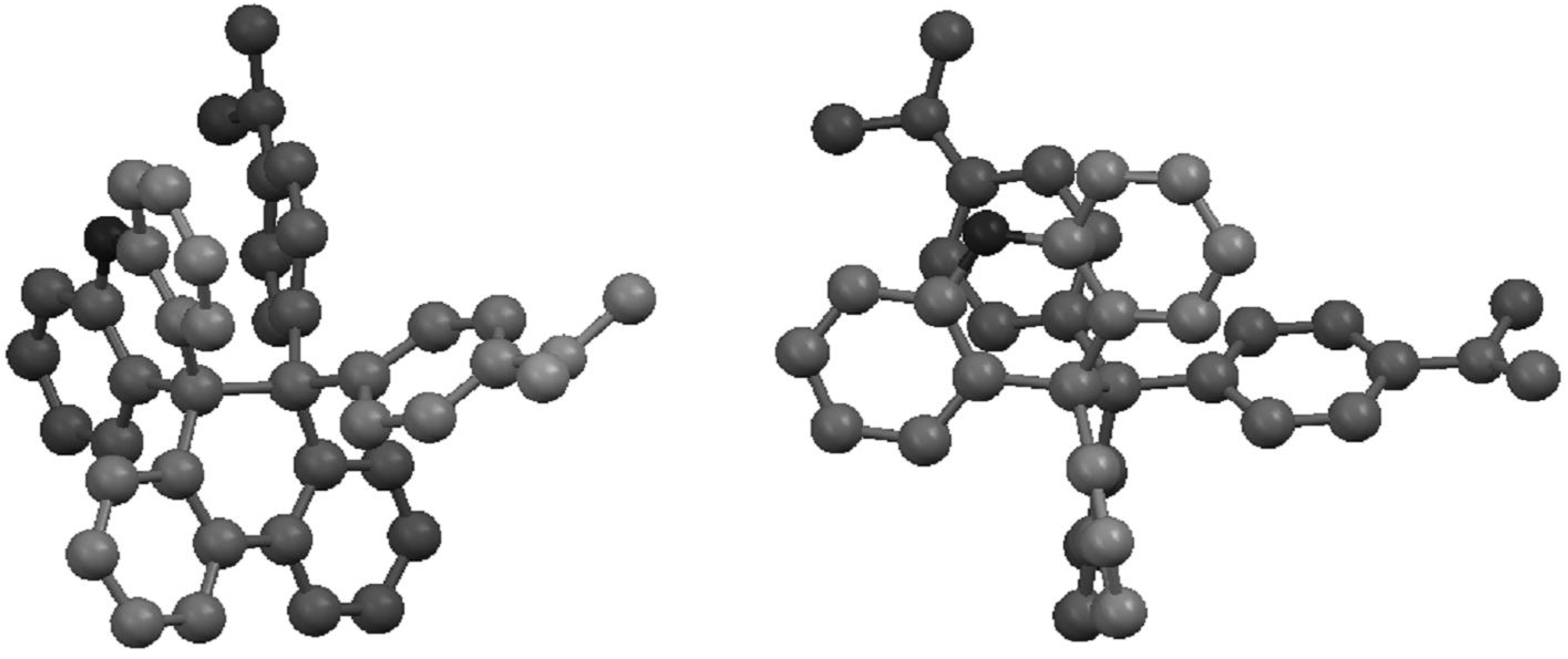
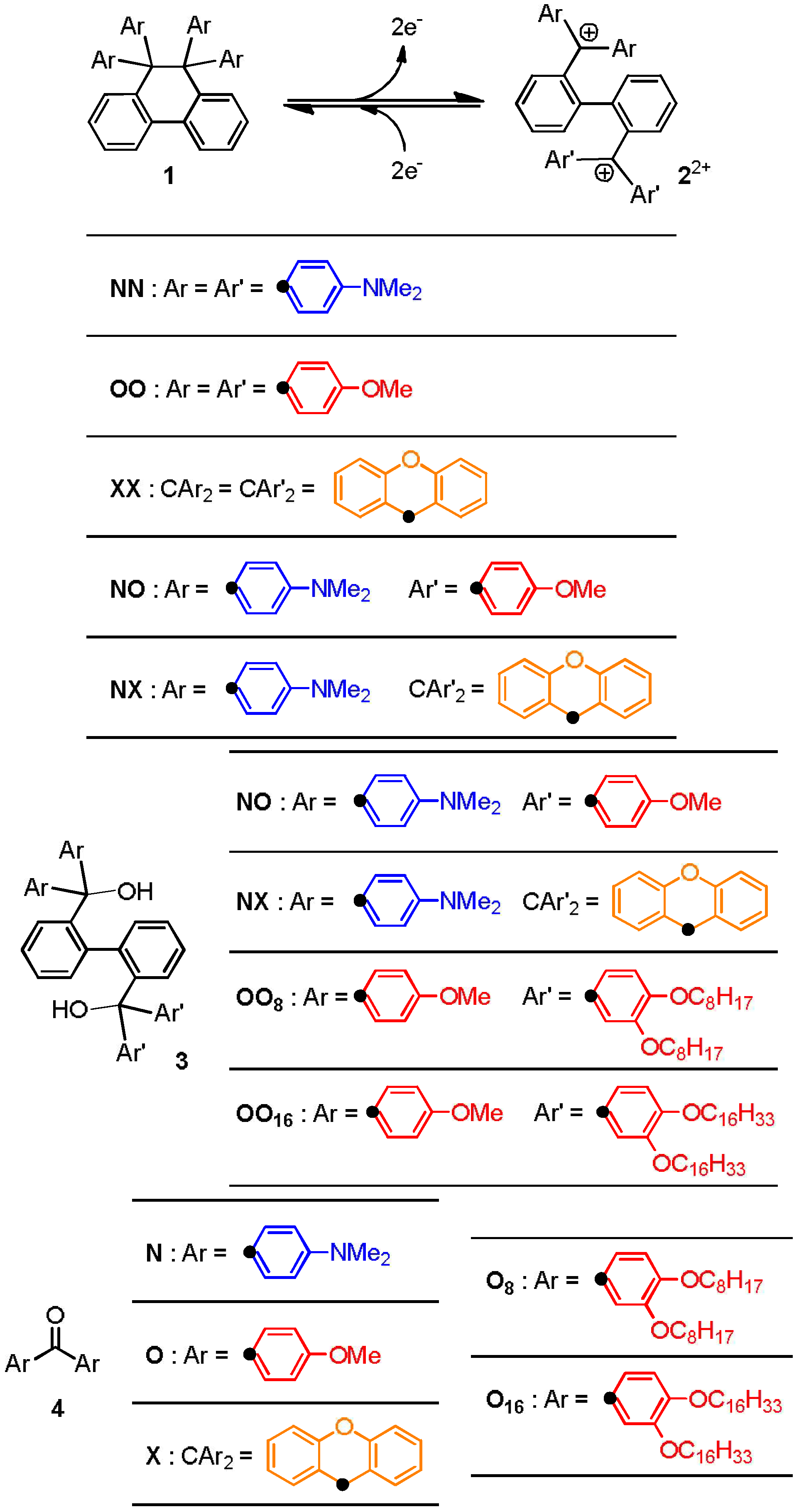
2.2. Preparation and Two-Electron Interconversion of Unsymmetrically Substituted Redox Pairs
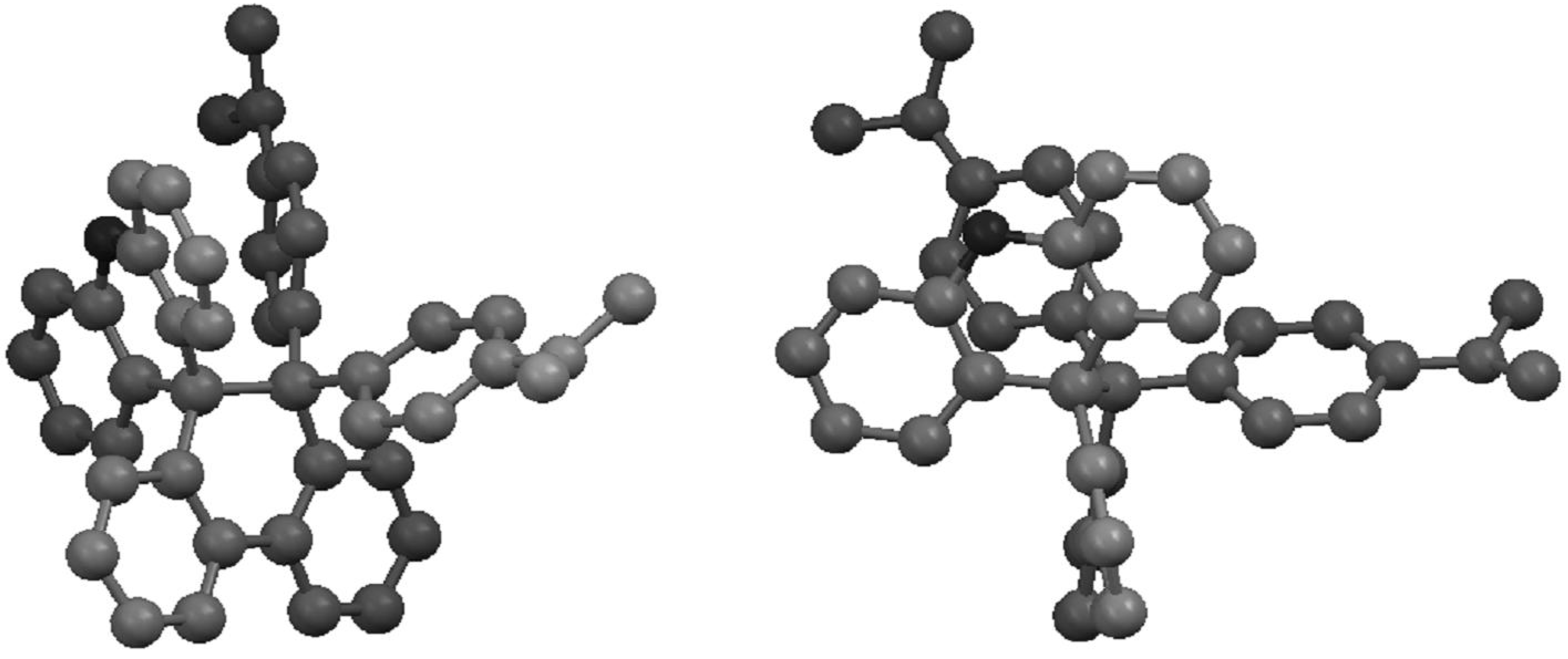
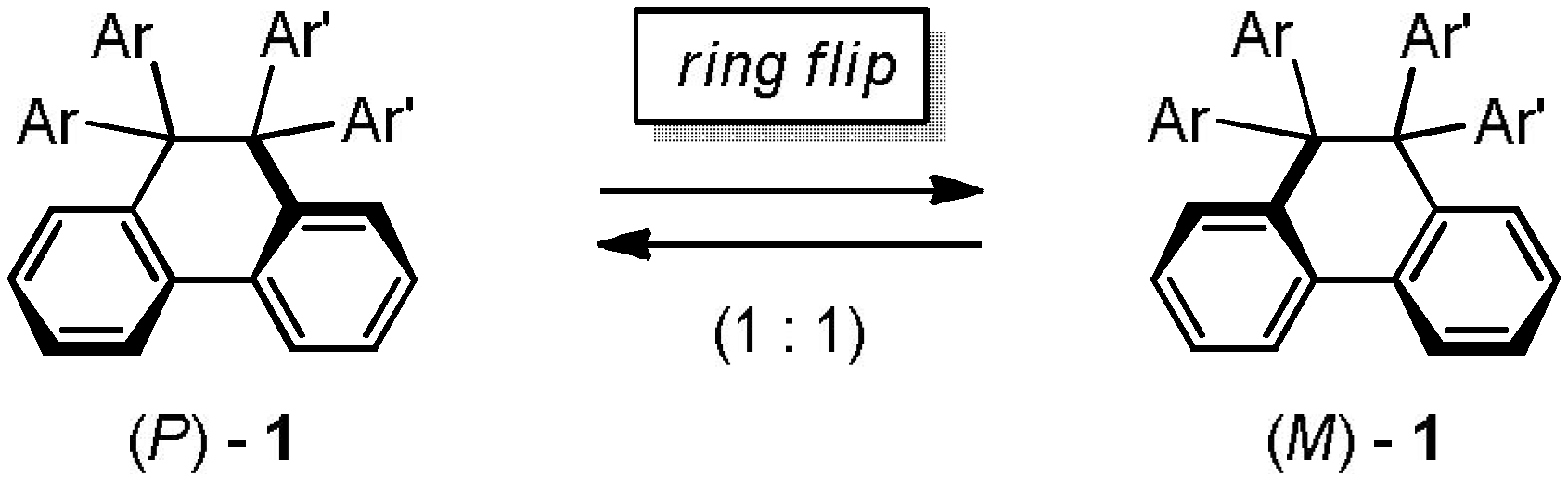
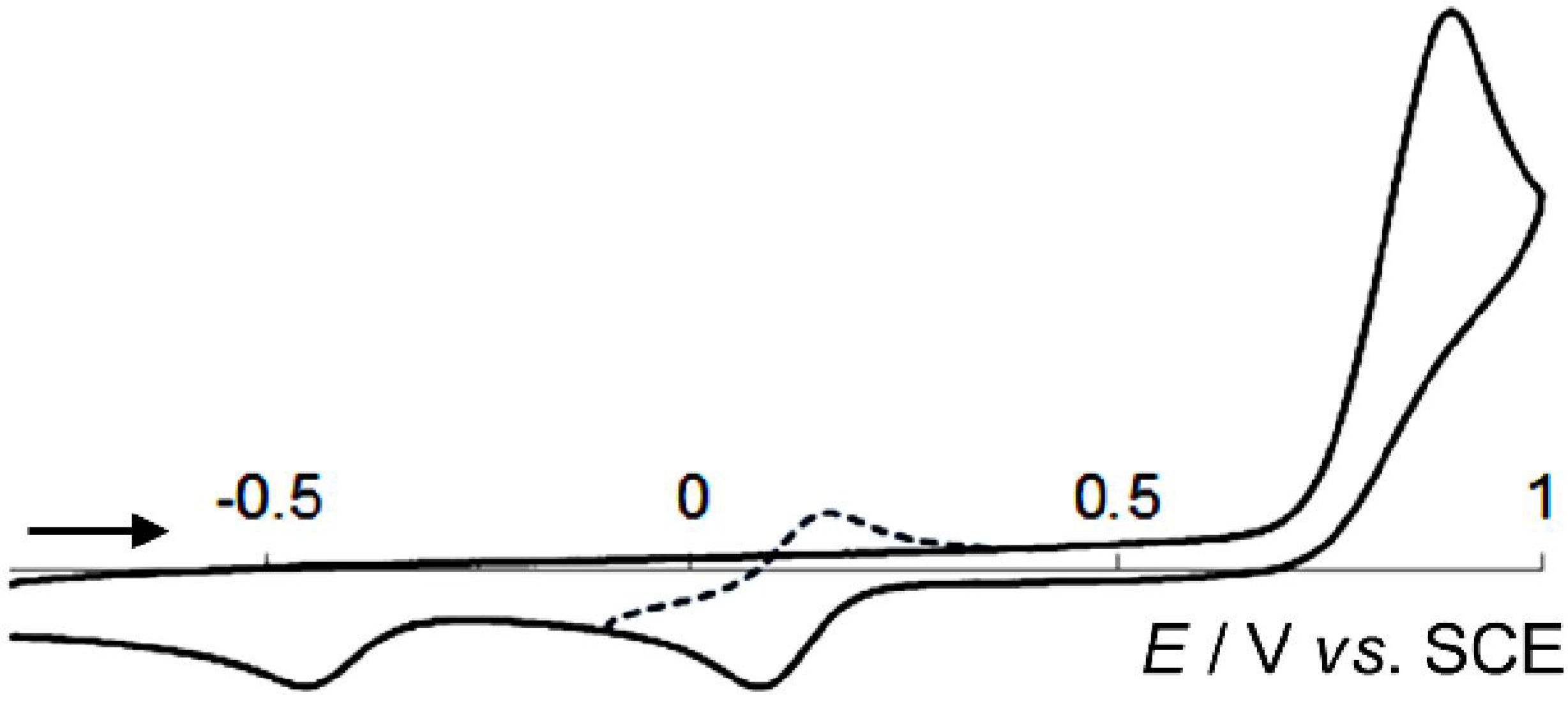
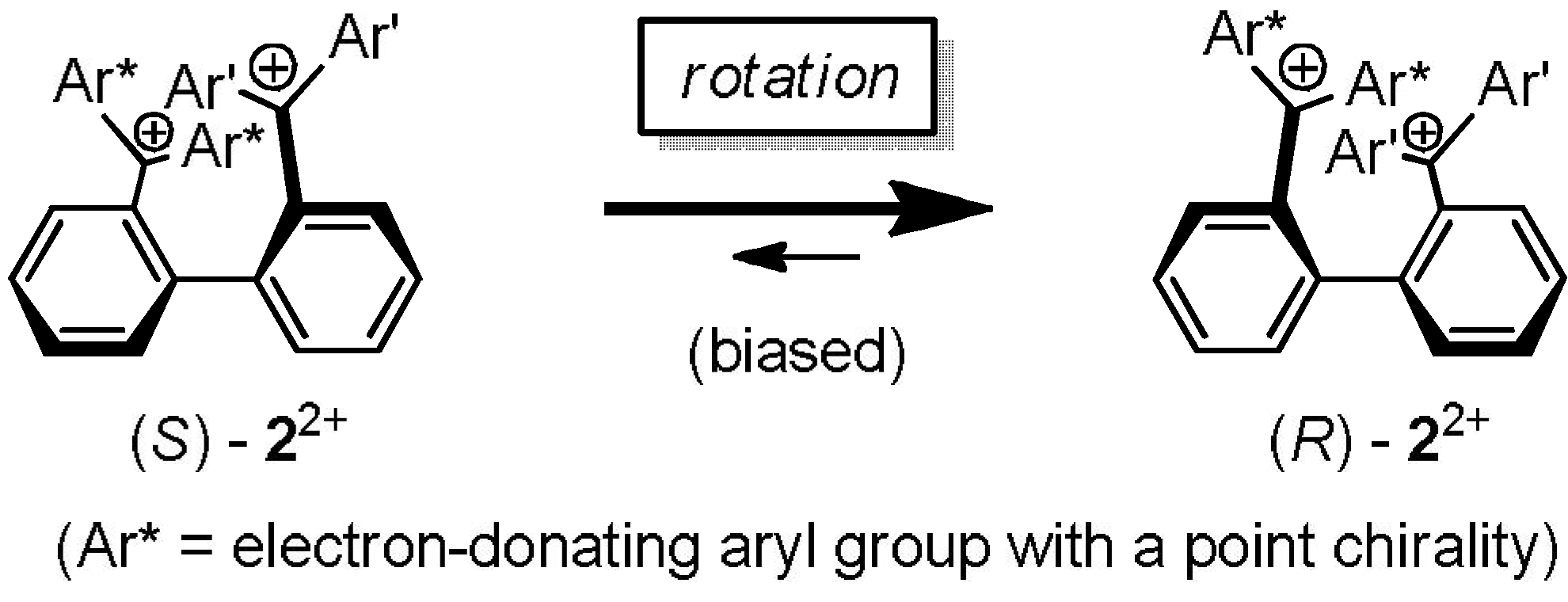
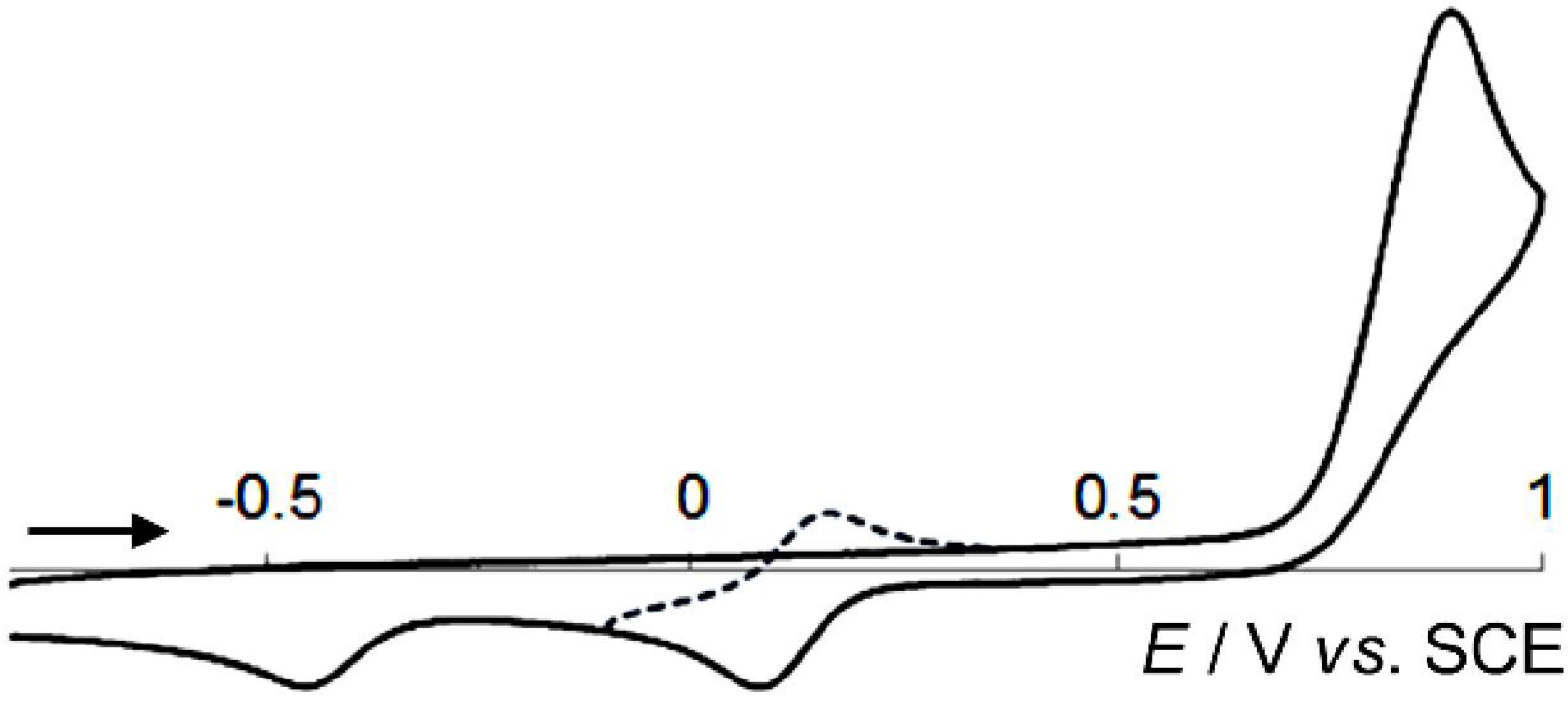
2.3. Hysteretic Redox Behavior of Unsymmetrically Substituted Redox Pairs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
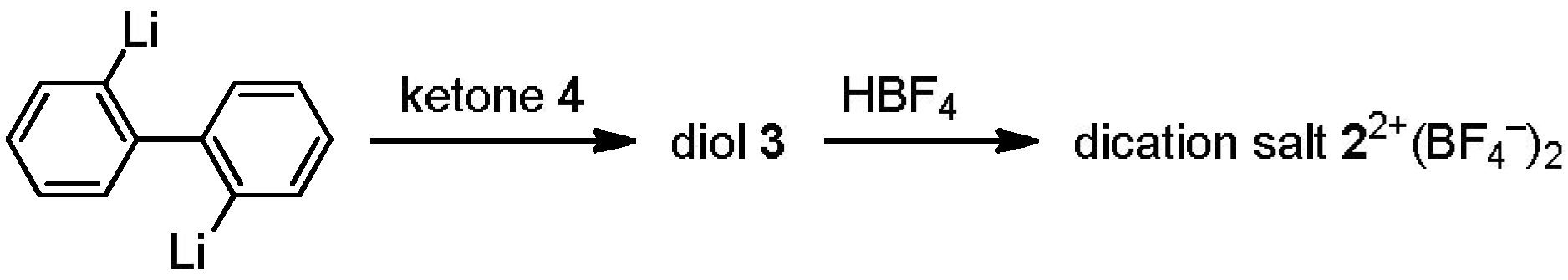{kind=link}
{kind=link}
{kind=link}
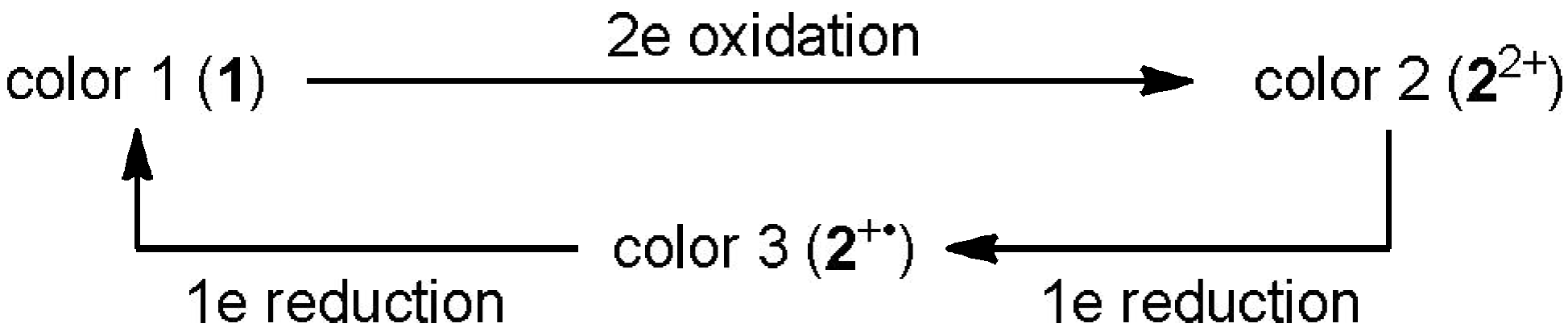{kind=link}
{kind=link}
{kind=link}
| compd. | Eox (1) | E1red (22+) | E2red (2+•) |
|---|---|---|---|
| NO | +0.83b,c | +0.10c | −0.45c |
| NX | +0.76b,c | +0.24c | −0.19c |
| NN | +0.74b,c | −0.42b,c | |
| OO | +1.44b,c | +0.21b,c | |
| XX | +1.39b,c | +0.53b,c |
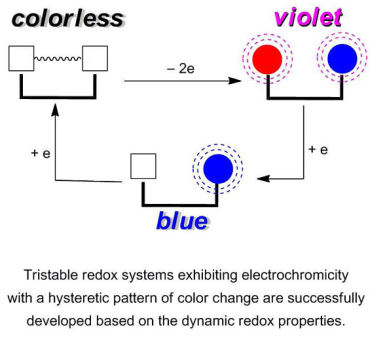
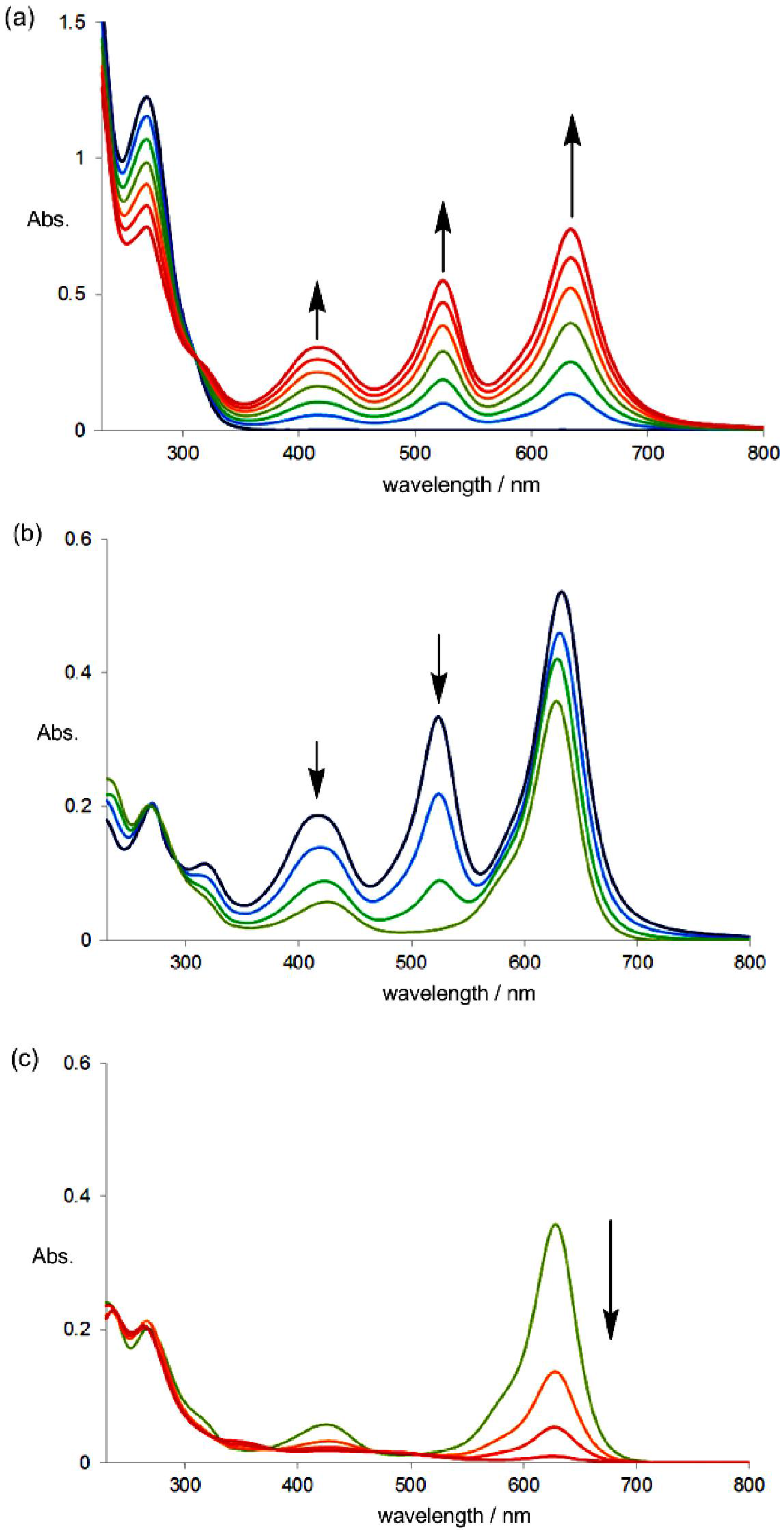
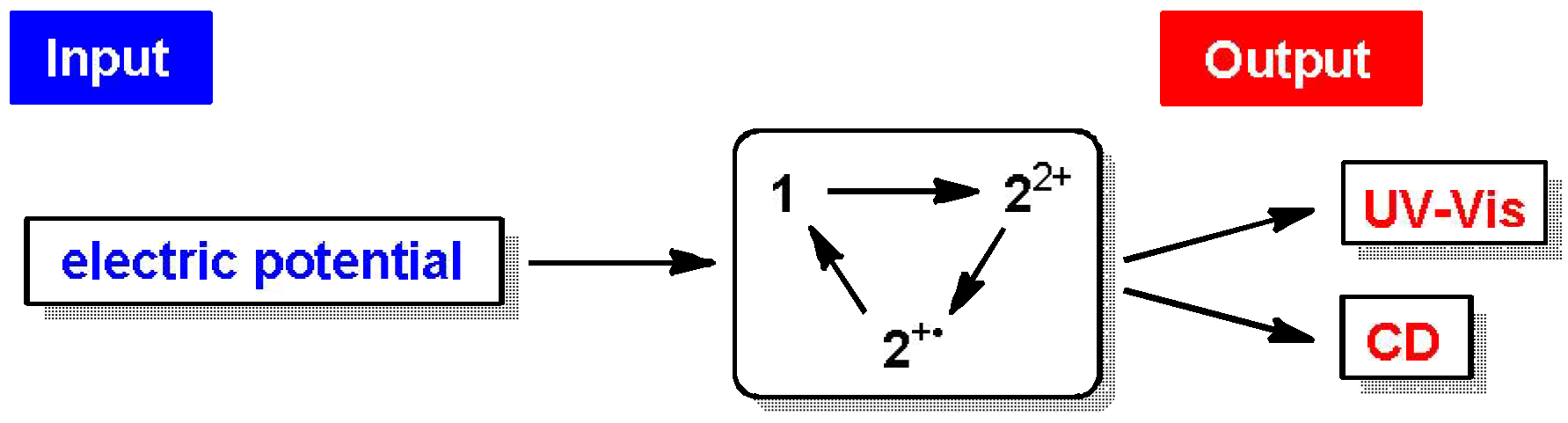
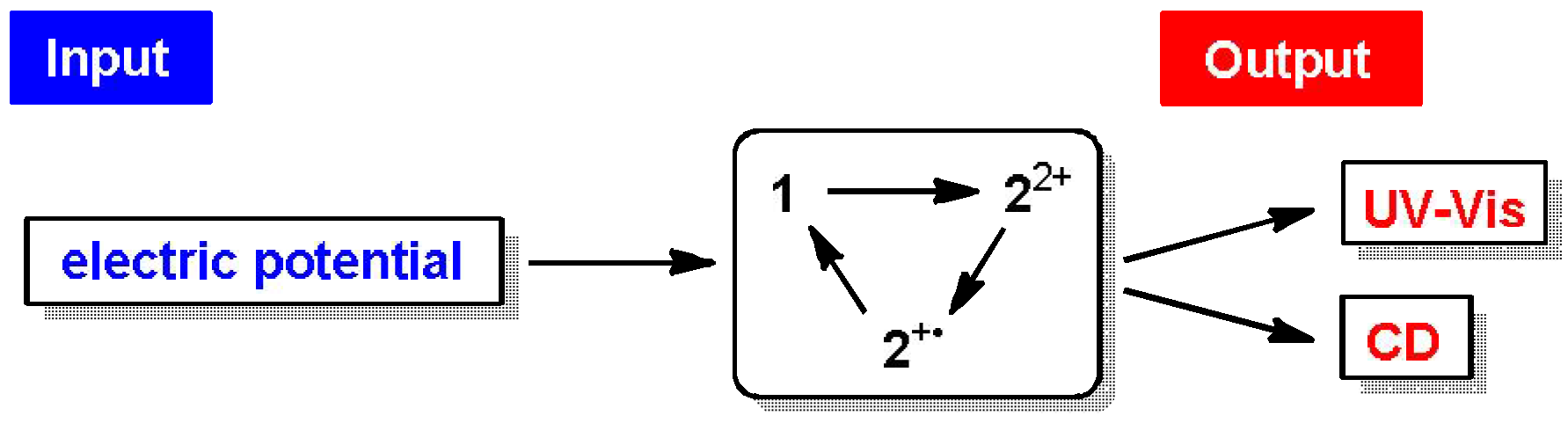
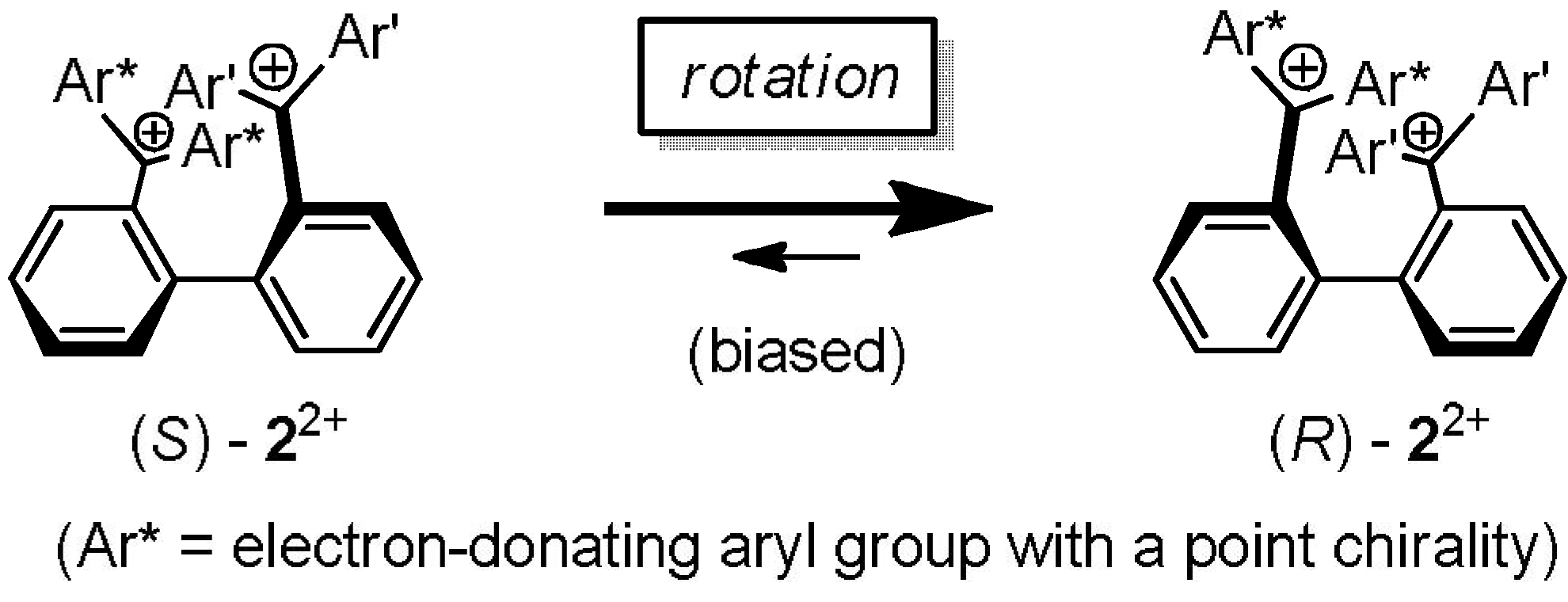
2.4. Hysteretic Tricolor Electrochromicity of Unsymmetrically Substituted Redox Pairs
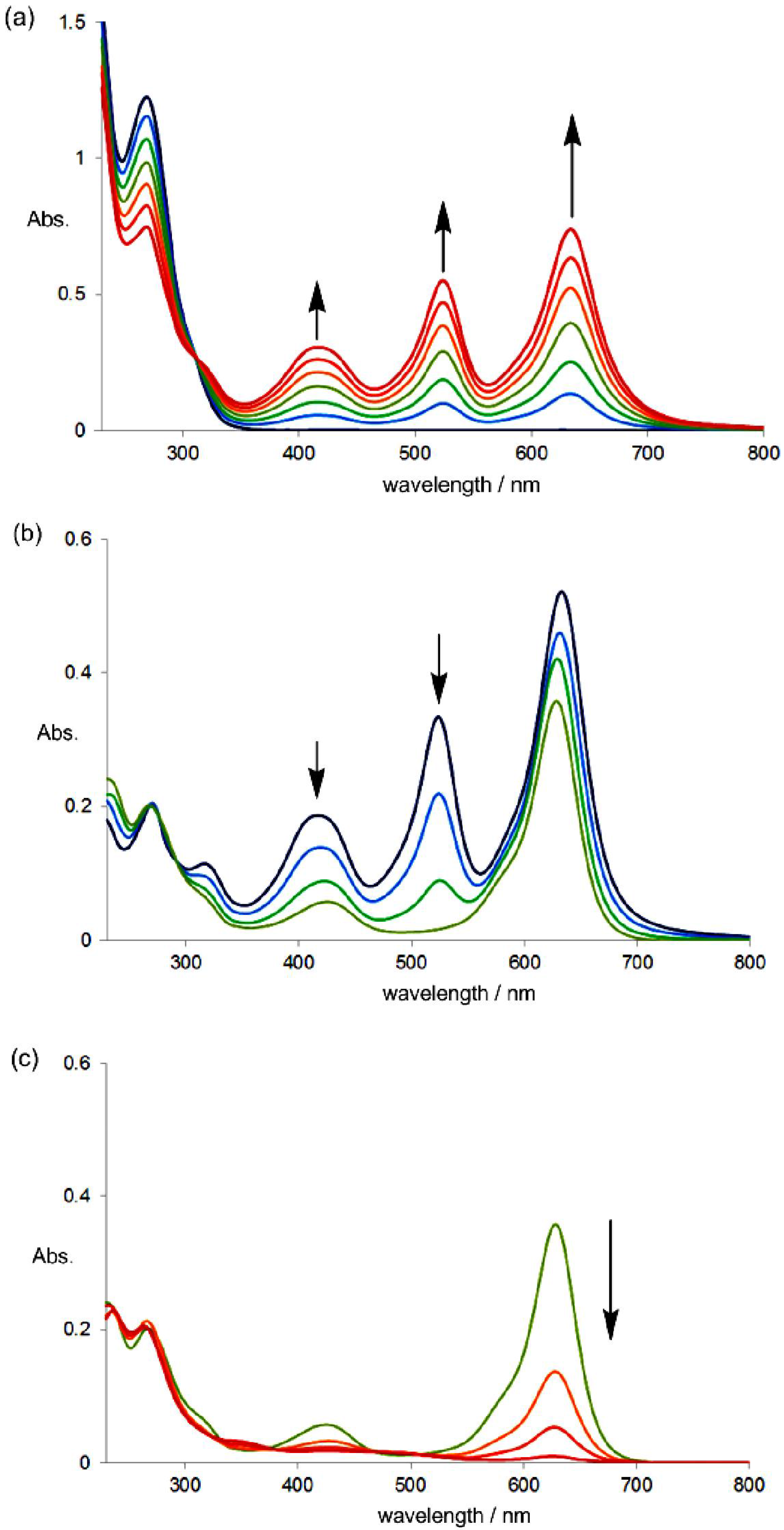
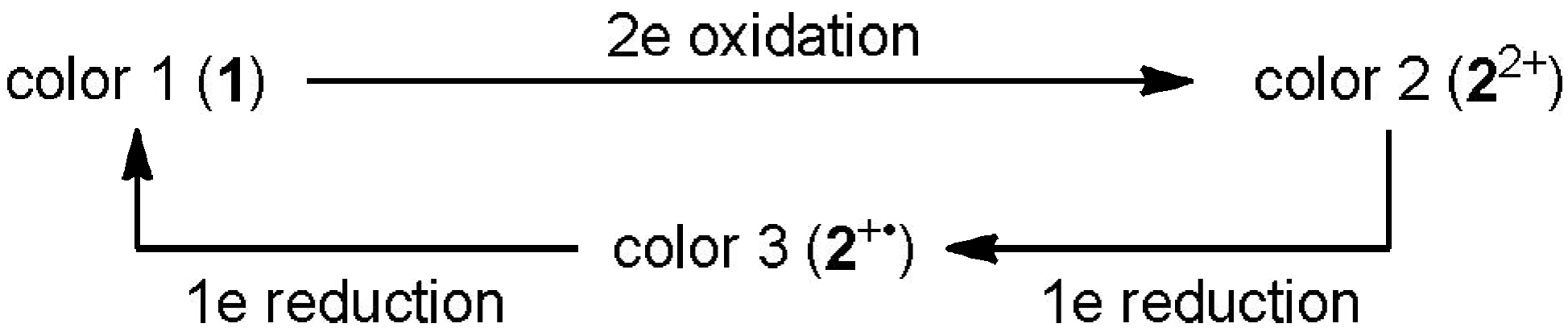
3. Conclusions
4. Experimental Section
4.1. Preparation of 9,9-Bis(4-Dimethylaminophenyl)-10,10-Bis(4-Methoxyphenyl)-9,10-Dihydrophenanthrene 1NO
4.2. Preparation of Spiro[10,10-Bis(4-Dimethylaminophenyl)-9,10-Dihydrophenanthrene-9,9'-[9H]Xanthene] 1NX
4.3. Preparation of Biphenyl-2-yl[Bis(4-Dimethylaminophenyl)Methylium]-2'-yl[Bis(4'-Methoxyphenyl)Methylium] Bis(Tetrafluoroborate) 2NO2+(BF4−)2
4.4. Preparation of Biphenyl-2-yl[Bis(4-Dimethylaminophenyl)Methylium]-2'-yl[9-Xanthenylium] Bis(Tetrafluoroborate) 2NX2+(BF4−)2
4.5. Oxidation to Biphenyl-2-yl[Bis(4-Dimethylaminophenyl)Methylium]-2'-yl[Bis(4'-Methoxyphenyl)Methylium] Bis(Hexachloroantimonate) 2NO2+(SbCl6−)2
4.6. Oxidation to Biphenyl-2-yl[Bis(4-Dimethylaminophenyl)Methylium]-2'-yl[9-Xanthenylium] Bis(Hexachloroantimonate) 2NX2+(SbCl6−)2
4.7. Preparation of 2-[Bis(4-Dimethylaminophenyl)Hydroxymethyl]-2'-[Bis(4'-Methoxylphenyl)Hydroxymethyl]Biphenyl 3NO via Flow Microreactor Method
4.8. Preparation of 2-[Bis(4-Dimethylaminophenyl)Hydroxymethyl]-2'-[Bis(4'-Methoxylphenyl)Hydroxymethyl]Biphenyl 3NO via Macro Batch Method
4.9. Preparation of 9-[2'-Bis(4-Dimethylaminophenyl)Hydoroxymethylbiphenyl-2-yl]-9-Hydroxyxanthene 3NX via Flow Microreactor Method
4.9. Preparation of 9-[2'-Bis(4-Dimethylaminophenyl)Hydoroxymethylbiphenyl-2-yl]-9-Hydroxyxanthene 3NX via Macro Batch Method
4.10. Preparation of 2-{Bis[3,4-Bis(Octyloxy)Phenyl]Hydroxymethyl}-2'-[Bis(4'-Methoxylphenyl)Hydroxymethyl]Biphenyl 3OO8
4.11. Preparation of 2-{Bis[3,4-Bis(Hexadecyloxy)Phenyl]Hydroxymethyl}-2'-[Bis(4'-Methoxylphenyl)Hydroxymethyl]Biphenyl 3OO16
4.12. Preparation of 3,3',4,4'-Tetrakis(Octyloxy)Benzophenone 4O8
4.13. Preparation of 3,3',4,4'-Tetrakis(Hexadecyloxy)Benzophenone 4O16
4.14. Preparation of 4-Bromo-1,2-Bis(Hexadecyloxy)Benzene
4.15. Measurement of Redox Potentials
4.16. X-Ray Structural Analyses
Acknowledgments
References
- Monk, P.M.S.; Mortimer, R.J.; Rosseinsky, D.R. Electrochromism and Electrochromic Devices; Cambridge University Press: Cambridge,MA, USA and New York, NY, USA, 2007. [Google Scholar]
- Muthyala, R. Chemistry and Application of Leuco Dyes; Plenum Press: New York, NY, USA and London, UK, 1997. [Google Scholar]
- Suzuki, T.; Higuchi, H.; Tsuji, T.; Nishida, J.; Yamashita, Y.; Miyashi, T. Chemistry of Nanomolecular Systems. Chapter 1: Dynamic Redox Systems; Nakamura, T., Matsumoto, T., Tada, T., Sugiura, K., Eds.; Springer: Heidelberg, Germany, 2003; pp. 3–24. [Google Scholar]
- Suzuki, T.; Ohta, E.; Kawai, H.; Fujiwara, K.; Fukushima, T. Dynamic redox systems as electrochromic materials: Bistability and advanced response. Synlett (Account) 2007, 38, 851–869. [Google Scholar] [CrossRef]
- Suzuki, T.; Nishida, J.; Tsuji, T. Hexaphenylethane derivatives exhibiting novel electrochromic behavior. Angew. Chem. Int. Ed. Engl. 1997, 36, 1329–1331. [Google Scholar] [CrossRef]
- Suzuki, T.; Nishida, J.; Tsuji, T. A new type of tricolor electrochromic system based on the dynamic redox properties of hexaarylethane derivatives. Chem. Commun. 1998, 20, 2193–2194. [Google Scholar] [CrossRef]
- Suzuki, T.; Ishigaki, Y.; Iwai, T.; Kawai, H.; Fujiwara, K.; Ikeda, H.; Kano, Y.; Mizuno, K. Multi-input/multi-output molecular response system based on the dynamic redox behavior of 3,3,4,4-tetraaryldihydro[5]helicene derivatives: Reversible formation/destruction of chiral fluorophore and modulation of chiroptical properties by solvent polarity. Chem. Eur. J. 2009, 15, 9434–9441. [Google Scholar] [CrossRef] [PubMed]
- Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Chemistry in microstructured reactors. Angew. Chem. Int. Ed. 2004, 43, 406–446. [Google Scholar] [CrossRef]
- Doku, G.N.; Verboom, W.; Reinhoudt, D.N.; van den Berg, A. On-microchip multiphase chemistry—A review of microreactor design principles and reagent contacting modes. Tetrahedron 2005, 61, 2733–2742. [Google Scholar] [CrossRef]
- Watts, P.; Haswell, S.J. The application of micro reactors for organic synthesis. Chem. Soc. Rev. 2005, 34, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J. Flash chemistry using electrochemical method and microsystems. Chem. Commun. 2005, 36, 4509–4516. [Google Scholar] [CrossRef]
- Geyer, K.; Codee, J.D.C.; Seeberger, P.H. Microreactors as tools for synthetic chemists—The chemists’ round-bottomed flask of the 21st century? Chem. Eur. J. 2006, 12, 8434–8442. [Google Scholar] [CrossRef] [PubMed]
- De Mello, A.J. Control and detection of chemical reactions in microfluidic systems. Nature 2006, 442, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Chen, D.L.; Ismagilov, R.F. Reactions in droplets in microfluidic channels. Angew. Chem. Int. Ed. 2006, 45, 7336–7356. [Google Scholar] [CrossRef]
- Kobayashi, J.; Mori, Y.; Kobayashi, S. Multiphase organic synthesis in microchannel reactors. Chem. Asian. J. 2006, 1, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Mason, B.P.; Price, K.E.; Steinbacher, J.L.; Bogdan, A.R.; McQuade, D.T. Greener approaches to organic synthesis using microreactor technology. Chem. Rev. 2007, 107, 2300–2318. [Google Scholar] [CrossRef] [PubMed]
- Ahmed-Omer, B.; Brandtand, J.C.; Wirth, T. Advanced organic synthesis using microreactor technology. Org. Biomol. Chem. 2007, 5, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, T.; Rahman, M.T.; Sato, M.; Ryu, I. Adventures in inner space: Microflow systems for practical organic synthesis. Synlett 2008, 2, 151–163. [Google Scholar]
- Yoshida, J.; Nagaki, A.; Yamada, T. Flash chemistry: Fast chemical synthesis by using microreactors. Chem. Eur. J. 2008, 14, 7450–7459. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, N.; Kos, A.J.; Schleyer, P.R. Regioselektive dimetallierung von aromaten. Bequemer zugang zu 2,2'-disubstituierten biphenylderivaten. J. Organomet. Chem. 1982, 228, 107–118. [Google Scholar] [CrossRef]
- Leroux, F.; Nicod, N.; Bonnafoux, L.; Quissac, B.; Colobert, F. New vistas in halogen/metal exchange reactions: The discrimination between seemingly equal halogens. Lett. Org. Chem. 2006, 3, 165–169. [Google Scholar]
- Nagaki, A.; Takabayashi, N.; Tomida, Y.; Yoshida, J. Selective monolithiation of dibromobiaryls using microflow systems. Org. Lett. 2008, 10, 3937–3940. [Google Scholar] [CrossRef] [PubMed]
- Nagaki, A.; Takabayashi, N.; Tomida, Y.; Yoshida, J. Synthesis of unsymmetrically substituted biaryls via sequential lithiation of dibromobiaryls using integrated microflow systems. Beilstein J. Org. Chem. 2009, 5. [Google Scholar] [CrossRef]
- Usutani, H.; Tomida, Y.; Nagaki, A.; Okamoto, H.; Nokami, T.; Yoshida, J. Generation and reactions of o-bromophenyllithium without benzyne formation using a microreactor. J. Am. Chem. Soc. 2007, 129, 3046–3047. [Google Scholar] [CrossRef] [PubMed]
- Nagaki, A.; Tomida, Y.; Usutani, H.; Kim, H.; Takabayashi, N.; Nokami, T.; Okamoto, H.; Yoshida, J. Integrated micro flow synthesis based on sequential br–li exchange reactions of p-, m-, and o-dibromobenzenes. Chem. Asian J. 2007, 2, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Suga, S.; Yamada, D.; Yoshida, J. Cationic three-component coupling involving an optically active enamine derivative. From time integration to space integration of reactions. Chem. Lett. 2010, 39, 404–406. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Kahr, B.; Engen, D.V.; Mislow, K. Length of the ethane bond in hexaphenylethane and its derivatives. J. Am. Chem. Soc. 1986, 108, 8305–8307. [Google Scholar] [CrossRef]
- Suzuki, T.; Takeda, T.; Kawai, H.; Suzuki, T. Ultralong C–C bonds in hexaphenylethane derivatives. Pure Appl. Chem. 2008, 80, 547–553. [Google Scholar]
- Kawai, H.; Takeda, T.; Fujiwara, K.; Wakeshima, M.; Hinatsu, Y.; Suzuki, T. Ultralong carbon-carbon bonds in dispirobis(10-methylacridan) derivatives with an acenaphthene, pyracene, or dihydropyracylene skeleton. Chem. Eur. J. 2008, 14, 5780–5793. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Kawai, H.; Herges, R.; Mucke, E.; Sawai, Y.; Fujiwara, K.; Suzuki, T. Negligible diradical character for the ultralong C–C bond in 1,1,2,2-tetraarylpyracene derivatives at room temperature. Tetrahedron Lett. 2009, 50, 3693–3697. [Google Scholar] [CrossRef]
- Dyker, G.; Körning, J.; Bubenitschek, P.; Jones, P.G. Palladium-catalyzed synthesis of propellanes with hexaarylethane structure. Liebigs Ann. Recl. 1997, 1, 203–209. [Google Scholar] [CrossRef]
- Maslak, P.; Chapman, W.H., Jr.; Vallombroso, T.M., Jr.; Watson, B.A. Mesolytic Scission of C–C bonds in radical cations of amino derivatives: Steric and solvent effects. J. Am. Chem. Soc. 1995, 117, 12380–12389. [Google Scholar] [CrossRef]
- Huang, L.-T.; Yen, H.-J.; Chang, C.-W.; Liou, G.-S. Red, green, and blue electrochromism in ambipolar poly(amine-amide-imide)s based on electroactive tetraphenyl-p-phenylenediamine units. J. Polymer Sci. 2010, 48, 4747–4757. [Google Scholar]
- Koyuncu, S.; Zafer, C.; Sefer, E.; Koyuncu, F.B.; Demic, S.; Kaya, I.; Ozdemir, E.; Icli, S. A new conducting polymer of 2,5-bis(2-thienyl)-1H-(pyrrole) (SNS) containing carbazole subunit: Electrochemical, optical and electrochromic properties. Synth. Met. 2009, 159, 2013–2021. [Google Scholar] [CrossRef]
- Zeng, Q.; McNally, A.; Keyes, T.E.; Forster, R.J. Redox induced switching dynamics of a three colour electrochromic metallopolymer film. Electrochim. Acta 2008, 53, 7033–7038. [Google Scholar] [CrossRef]
- Ozyurt, F.; Gunbas, E.G.; Durmus, A.; Toppare, L. Processable and multichromic polymer of bis-3-hexylthiophene substituted 4-tert-butylphenyl quinoxaline. Org. Elect. 2008, 9, 296–302. [Google Scholar] [CrossRef]
- Zeng, Q.; McNally, A.; Keyes, T.E.; Forster, R.J. Three colour electrochromic metallopolymer based on a ruthenium phenolate complex bound to poly(4-vinyl)pyridine. Electrochem. Commun. 2008, 10, 466–470. [Google Scholar] [CrossRef]
- Li, X.-G.; Wang, H.-Y.; Huang, M.-R. Synthesis, film-forming, and electronic properties of o-phenylenediamine copolymers displaying an uncommon tricolor. Macromolecules 2007, 40, 1489–1496. [Google Scholar] [CrossRef]
- Thompson, B.C.; Kim, Y.-G.; McCarley, T.D.; Reynolds, J.R. Soluble narrow band gap and blue propylenedioxythiophene-cyanovinylene polymers as multifunctional materials for photovoltaic and electrochromic applications. J. Am. Chem. Soc. 2006, 128, 12714–12725. [Google Scholar] [CrossRef] [PubMed]
- Sonmez, G.; Wudl, F. Completion of the three primary colours: The final step toward plastic displays. J. Mat. Chem. 2005, 15, 20–22. [Google Scholar] [CrossRef]
- Yano, J.; Yamasaki, S. Three-color electrochromism of an aramid film containing polyaniline and poly(o-phenylenediamine). Synth. Met. 1999, 102, 1157. [Google Scholar] [CrossRef]
- Sotzing, G.A.; Reddinger, J.L.; Katritzky, A.R.; Soloducho, J.; Musgrave, R.; Reynolds, J.R.; Steel, P.J. Multiply colored electrochromic carbazole-based polymers. Chem. Mat. 1997, 9, 1578–1587. [Google Scholar] [CrossRef]
- Yano, J.; Terayama, K.; Yamasaki, S. Electrochemically prepared poly(o-phenylenediamine)—Prussian blue composite film for a three-colour expressible ECD material. J. Mat. Sci. 1996, 31, 4785–4792. [Google Scholar] [CrossRef]
- Yamasaki, S.; Terayama, K.; Yano, J. Poly(p-phenylene terephthalamide) film immobilizing oligomer species as a colored matrix for three-color electrochromic displays. J. Electrochem. Soc. 1996, 143, L212–L214. [Google Scholar] [CrossRef]
- Yang, J.; Kai, S.; Ogura, K. Poly(o-phenylenediamine)-Prussian blue composite film for a three-colour-expressible electrochromic display material. J. Mat. Sci. Lett. 1993, 12, 1791–1792. [Google Scholar] [CrossRef]
- Sugimoto, T.; Nagatomi, T.; Ando, H.; Yoshida, Z. Redox-active thieno[3,2-b]thiophene as a novel, three-color electrochromic system. Angew. Chem. Int. Ed. Engl. 1988, 27, 560–561. [Google Scholar] [CrossRef]
- Li, X.-G.; Wang, H.-Y.; Huang, M.-R. Synthesis, film-forming, and electronic properties of o-phenylenediamine copolymers displaying an uncommon tricolor. Macromolecules 2007, 40, 1489–1496. [Google Scholar] [CrossRef]
- Zeng, Q.; McNally, A.; Forster, R.J. Redox induced switching dynamics of a three colour electrochromic metallopolymer film. Electrochem. Acta 2008, 53, 7033–7038. [Google Scholar]
- Deng, W.; Flood, A.H.; Stoddart, J.F.; Goddard, W.A., III. An electrochemical color-switchable rgb dye: Tristable [2]catenane. J. Am. Chem. Soc. 2005, 127, 15994–15995. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Saha, S.; Aprahamian, I.; Leung, K.C.F.; Williams, A.; Deng, W.; Flood, A.H.; Goddard, W.A., III; Stoddart, J.F. Toward electrochemically controllable tristable three-station [2]catenanes. Chem. Asian J. 2007, 2, 76–93. [Google Scholar] [CrossRef] [PubMed]
- Nishida, J.; Suzuki, T.; Ohkita, M.; Tsuji, T. A redox switch based on dihydro[5]helicene: Drastic chiroptical response induced by reversible C−C bond making/breaking upon electron transfer. Angew. Chem. Int. Ed. 2001, 40, 3251–3254. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, R.; Higuchi, H.; Hirota, E.; Ohkita, M.; Tsuji, T. Electrochiroptical response of a hexaarylethane derivative with a helical π-skeleton: Drastic UV–Vis and CD spectral changes upon electrolysis of 4′,5′-dibromodispiro[xanthene-9,9′(9′H,10′H)-phenanthrene-10′,9″-xanthene]. J. Chem. Soc. Perkin Trans. 2002, 2, 1937–1942. [Google Scholar] [CrossRef]
- Suzuki, T.; Iwai, T.; Ohta, E.; Kawai, H.; Fujiwara, K. Electrochiroptical systems based on biphenyl-2,2′-diyl-type dicationic dyes: Strong chiroptical signals through the transmission of point chirality to axial chirality. Tetrahedron Lett. 2007, 48, 3599–3603. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ishigaki, Y.; Suzuki, T.; Nishida, J.-i.; Nagaki, A.; Takabayashi, N.; Kawai, H.; Fujiwara, K.; Yoshida, J.-i. Hysteretic Tricolor Electrochromic Systems Based on the Dynamic Redox Properties of Unsymmetrically Substituted Dihydrophenanthrenes and Biphenyl-2,2'-Diyl Dications: Efficient Precursor Synthesis by a Flow Microreactor Method. Materials 2011, 4, 1906-1926. https://doi.org/10.3390/ma4111906
Ishigaki Y, Suzuki T, Nishida J-i, Nagaki A, Takabayashi N, Kawai H, Fujiwara K, Yoshida J-i. Hysteretic Tricolor Electrochromic Systems Based on the Dynamic Redox Properties of Unsymmetrically Substituted Dihydrophenanthrenes and Biphenyl-2,2'-Diyl Dications: Efficient Precursor Synthesis by a Flow Microreactor Method. Materials. 2011; 4(11):1906-1926. https://doi.org/10.3390/ma4111906
Chicago/Turabian StyleIshigaki, Yusuke, Takanori Suzuki, Jun-ichi Nishida, Aiichiro Nagaki, Naofumi Takabayashi, Hidetoshi Kawai, Kenshu Fujiwara, and Jun-ichi Yoshida. 2011. "Hysteretic Tricolor Electrochromic Systems Based on the Dynamic Redox Properties of Unsymmetrically Substituted Dihydrophenanthrenes and Biphenyl-2,2'-Diyl Dications: Efficient Precursor Synthesis by a Flow Microreactor Method" Materials 4, no. 11: 1906-1926. https://doi.org/10.3390/ma4111906