Thermodynamic Origin of the Vitreous Transition

Centre National de la Recherche Scientifique, Université Joseph Fourier, Consortium de Recherches pour l’Emergence de Technologies Avancées, B.P. 166, 38042 Grenoble Cedex 09, France
Materials 2011, 4(5), 869-892; https://doi.org/10.3390/ma4050869
Submission received: 22 March 2011
/
Revised: 19 April 2011
/
Accepted: 5 May 2011
/
Published: 9 May 2011
(This article belongs to the Special Issue Advances in Bulk Metallic Glasses)
Abstract
:The vitreous transition is characterized by a freezing of atomic degrees of freedom at a temperature Tg depending on the heating and cooling rates. A kinetic origin is generally attributed to this phenomenon instead of a thermodynamic one which we develop here. Completed homogeneous nucleation laws reflecting the energy saving due to Fermi energy equalization of nascent crystals and their melt are used. They are applied to bulk metallic glasses and extended to inorganic glasses and polymers. A transition T*g among various Tg corresponds to a crystal homogeneous nucleation temperature, leading to a preliminary formation of a cluster distribution during the relaxation time preceding the long steady-state nucleation time of crystals in small samples. The thermally-activated energy barrier ΔG*2ls/kBT at T*g for homogeneous nucleation is nearly the same in all glass-forming melts and determined by similar values of viscosity and a thermally-activated diffusion barrier from melt to cluster. The glass transition T*g is a material constant and a linear function of the energy saving associated with charge transfers from nascent clusters to the melt. The vitreous transition and the melting temperatures alone are used to predict the free-volume disappearance temperature equal to the Vogel-Fulcher-Tammann temperature of fragile glass-forming melts, in agreement with many viscosity measurements. The reversible thermodynamic vitreous transition is determined by the disappearance temperature T*g of the fully-relaxed enthalpy Hr that is not time dependent; the observed specific heat jump at T*g is equal to the proportionality coefficient of Hr with (T*g − Ta) for T ≤ T*g as expected from the enthalpy excess stored by a quenched undercooled melt at the annealing temperature Ta and relaxed towards an equilibrium vitreous state. However, the heat flux measurements found in literature over the last 50 years only gave an out-of-equilibrium Tg since the enthalpy is continuous at T*g without visible heat jump.
1. Introduction
The vitreous state is described, up to now, as a freezing of liquid-state below a temperature Tg called vitreous or glass transition, below which the viscosity becomes time dependent with values above 1012–1013 Pa.s. This transformation at Tg is also observed in the heat flow, measured with a technique of differential scanning calorimetry (DSC); endothermic and exothermic heats respectively, depending on the heating and the cooling rates, characterize glass-melt out-of-equilibrium transformations. The glass-forming melt viscosity follows a Vogel-Fulcher-Tammann (VFT) law diverging when the temperature tends to T0; T0 is much smaller than Tg and called the ideal glass transition temperature [1,2,3]. Recent work has shown that the size of heterogeneous regions simultaneously moving to allow a viscous flow grows in the vicinity of the glass transition [4]. The heterogeneous dynamics could also be the result of critical-like fluctuations of static structural order, characterized by a static correlation length diverging towards the ideal glass-transition point T0 in the absence of a thermodynamic transition at Tg. Two glass transition temperatures T0 and Tg could exist without any connection between the two [5]. In addition, the residual entropy available in undercooled melt, as compared to the crystal one at the glass transition, varies strongly among glasses. The Kauzmann temperature Tk has been defined as the temperature at which the crystal fusion entropy would be consumed upon cooling. It could also lead, at thermodynamic equilibrium, to a hidden phase transition following several speculations found in literature [1,2,6].
The high temperature viscosity of some polymer melts, including measurements above the melting point, follows a VFT scaling law giving T0 = T01 = 0.77 × Tg [7]. In the vicinity of the glass transition, the enthalpy relaxation times or the viscosity gives a value T0 = T02 smaller than T0l. Therefore, if T0 increases above Tg within a narrow range of temperature, it explains why the viscosity values measured at high temperatures do not determine the ideal glass transition. The change of T0 occurs around the temperature Ts where a break is seen in some volume-versus-temperature plots [8]. Many experimental results tend to prove that the ideal glass transition temperature is equal to T02 with a viscosity close to Tg following a VFT law with T0 = T02 [3]. The viscosity is an exponential function of B/(T − T0) with B nearly proportional to (Tg − T0) [9].
Following Doolittle’s and Ramachandrarao and Dubey’s works, the free-volume of glass-forming melts would disappear at the ideal glass transition temperature T0 [10,11]; then, the two free-volume disappearance temperatures T0g and T0m correspond respectively to our two VFT temperatures T02 and T01 respectively. Recent numerical simulations of critical-like behavior of glass-forming melts down to T0 (assuming that the glass transition only corresponds to a slowing-down of dynamics) suggests that the melt entropy excess at equilibrium must tend to zero for T = T02 = T0g instead of T = Tk, Tk being the Kauzmann temperature [5,6,12]. New universal and coherent relations between Tg, T0g = T02 and T0m = T01 are proposed in this paper and checked for a series of data in real systems. The associated predictions still need to be clarified.
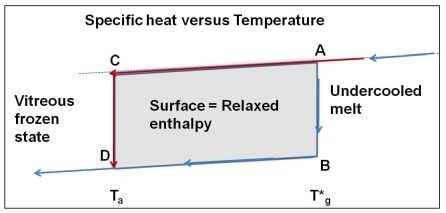
The vitreous transition, observed by DSC techniques in undercooled melts, is generally time dependent and not strictly reversible, because using the same cooling and heating rates do not lead to the same transformation temperature [13]. These properties underline the kinetic aspects of the transition. A rapid cooling at temperatures far below Tg followed by an annealing as a function of time at a temperature Ta < Tg induces an enthalpy relaxation, saturating to a value Hr; Hr itself tends to zero for the transition temperature T*g. From published values of Hr, it is shown that the reversible transition occurs at T*g, and that the transformation into the vitreous state is postponed by quenching the undercooled melt. It is achieved after a time lag equal to the relaxation time by annealing at T = Ta.
Some DSC techniques are able to separate the specific heat at T*g in two parts; the temperature dependent one is reversible and attached to the thermodynamic aspect of the transition, and the time dependent “irreversible” one is attached to the kinetic aspect. The reversible specific heat jump temperature measured by this technique does not depend on cooling and heating rates and is only a function of the chemical composition [13].
In this paper, we show that crystal homogeneous nucleation occurs at the glass transition T*g of fragile glass-forming melts without any adjustable parameter. This nucleation only depends on the melt composition, and the obtained frozen state is a preliminary step on the long way leading to crystallization. The free energy change associated with a crystal formation in a melt has been accepted for many years, and, regardless of its radius, as if it had the same state equation as a solid outside the melt, which does not reflect the fact that the Fermi energy of a nascent crystal in a metallic melt becomes equal to that of the melt. In order to determine why the vitreous state replaces the crystallized state, a “volume energy saving” εv has been added to the Gibbs free-energy change associated with crystal homogeneous nucleation in melt, to obtain the equalization of Fermi energies transferring free electrons from the crystal to the melt [14,15,16]. The energy saving εv is equal to εlpsΔHm/Vm, ΔHm, Vm, and εlps respectively being the molar fusion heat, the molar volume, and the energy saving coefficient, where εlps is a numerical coefficient depending on the reduced temperature θ = (T − Tm)/Tm [16]. The indexes s and l are related to solid and liquid states. The index p is suppressed for unmelted crystals acting as growth nuclei and replaced by the index g for crystals resulting from homogeneous nucleation in glass-forming melts. The value of εlps can be predicted using the VFT temperatures T01 and T02 viewed as the disappearance temperatures Tog and T0m of fragile-glass-forming melt free-volume and of Fermi energy difference between crystal and melt [14,15]. The vitreous transition T*g only depends on the energy saving coefficient. The experimental values of T0g can also be used to predict the vitreous transition temperature T*g.
In this model, the crystal growth starts at the crystal nucleation temperature with a cluster preliminary formation on the long way leading to crystallization. It is locked at the vitreous transition by a freezing without any change of enthalpy and entropy. The same analysis is successfully applied to some polymers and non-metallic glass-formers. The presence of a similar “volume energy saving”, governing the vitreous transition, is determined. This phenomenon is probably due to a free energy which depends on the number of molecules or atoms in a small crystal having a noncritical radius or to an electrostatic interaction between uncompensated average charges proportional to n1/2 carried by nascent crystals built from a random distribution of ions on various sub-lattices and screened by ionic charges of the melt.
This paper is built as follows: Section 2. Model; 2.1. New equations governing the crystal nucleation; 2.2. The ideal glass transition T0 and the energy saving associated with crystal formation; Section 3. Review of experimental results and discussion; 3.1 Presentation of Table 1 and Figure 1; 3.2. Homogeneous nucleation time of crystals and relaxation time; 3.3. The thermodynamic vitreous transition T*g at the disappearance temperature of the fully-relaxed enthalpy; 3.4. The crystal homogeneous nucleation temperature at T*g; 3.5. Volume energy saving associated with nascent crystals in non-metallic glass-forming melts; 3.6. Thermodynamic origin of the relaxed enthalpy and out-of-equilibrium transition temperatures Tg; Section 4. Summary and complementary information on the two crystal nucleation temperatures; Section 5. Conclusions.
2. Model
2.1. New Equations Governing the Crystal Nucleation
The classical equation describing the Gibbs free-energy change associated with a crystal formation, predicts the absence of surviving crystals above the melting temperature Tm [17]. On the contrary, their existence is predicted far above Tm if an energy saving per volume unit εv = εlps × ΔHm/Vm is added. The maximum undercooling ratio θ1 = (T1 − Tm)/Tm is observed as being of the order of −0.2 in liquid elements using droplet sizes of 50–10,000 micrometers instead of −2/3 [18]. This pseudo-maximum has until now been considered to be the maximum of the homogeneous nucleation rate in contradiction with a detailed study of crystallization temperature of gallium droplets as a function of their diameter. Two undercooling temperature dwells, instead of one, corresponding to θ1= −0.28 and θ1= −0.5 have been observed for diameters varying from 1 to 1,000 micrometers [19]. This phenomenon is induced by a distribution of surviving crystal radii between two boundary radii after overheating. A large overheating rate has to be applied in order to melt a part of them and obtain an undercooling rate dwell of about −0.2 [20]. The second dwell corresponding to stronger undercooling rates, is due to numerous surviving crystals having the smallest radius [21,22,23]. The sample radius has to be strongly reduced to observe it. The energy saving εv depends on the Fermi energy difference between solid and liquid and is an even function of the reduced temperature θ = (T − Tm)/Tm as shown in (1) [14,16]:
![Materials 04 00869 i001]()

Consequently, the fusion heat of unmelted crystals remains equal to ΔHm regardless of their radius, because dεls/dθ = 0 and d(ΔG*2ls)/dθ = ΔHm × 4πR3/3/Vm at θ = 0, with ΔG*2ls being the completed Gibbs free energy change for a crystal formation in a melt given by (2):
![Materials 04 00869 i002]() kB being the Boltzmann constant, ΔSm the fusion entropy per mole and lnKlps given by (12). The volume energy saving εv is added to the classical Gibbs free energy θ × ΔHm/Vm and the surface energy is modified by the factor (1 + εls). The classical equation ΔG1ls (θ) is deduced from (2) with εls = 0 [17,18,19]. The experimental values of surface energy and lowest undercooling temperatures of many liquid elements have been used to determine the new surface energy and εls in (1,2) [17]. This Equation (2) allows us to calculate the unique homogeneous nucleation temperature θ2ls = −2/3 and the crystallization temperatures of liquid elements from intrinsic growth nuclei without any adjustable parameter [21,22,23].
kB being the Boltzmann constant, ΔSm the fusion entropy per mole and lnKlps given by (12). The volume energy saving εv is added to the classical Gibbs free energy θ × ΔHm/Vm and the surface energy is modified by the factor (1 + εls). The classical equation ΔG1ls (θ) is deduced from (2) with εls = 0 [17,18,19]. The experimental values of surface energy and lowest undercooling temperatures of many liquid elements have been used to determine the new surface energy and εls in (1,2) [17]. This Equation (2) allows us to calculate the unique homogeneous nucleation temperature θ2ls = −2/3 and the crystallization temperatures of liquid elements from intrinsic growth nuclei without any adjustable parameter [21,22,23].

The energy saving becomes equal to zero at Tm above a radius a little larger than the critical radius for crystal growth nucleation to obey, at equilibrium, the classical J. W. Gibbs’s phase coexistence rule and because a free electron cannot be transferred from the crystal to the melt with an energy saving larger than the Fermi energy . The crystal homogeneous nucleation maximum-rate temperature T2lps (or θ2lps) defined by (3) in the undercooled melt is respectively called θ2lgs or θ2ls with values of εlps0 equal to εlgs0 or εls0:
![Materials 04 00869 i003]()

The θ2lps only depends on the coefficient εlps defined by (1) and (3) and does not depend on other material properties. It is equal to (Tolps − Tm)/Tm where Tolps is equal to T0m or T0g. The critical energy barrier, the critical radius and θ2lps given by (3) have been calculated assuming that εlps is not radius dependent. This assumption works because the influence of dεlps/dR is negligible on the critical parameters of a lot of melts. Equations (1–3) have already been used to predict the time-temperature-transformation diagrams of Mg65Y10Cu25, Zr41.2Ti13.8Cu12.5Ni10Be22.5 and Pd43Cu27Ni10P20 melts [14,15] and the undercooling temperature dwells of liquid elements, in agreement with the experiments without using any adjustable parameter [21,22,23]. Unequal coefficients εls ≠ εlgs would lead to θ2ls ≠ θ2lgs. Equal coefficients εlps = εls = εlgs would lead to the same homogeneous nucleation temperature; the glass transition would be equal to the crystallization temperature.
2.2. The Ideal Glass Transition Temperature T0 and The Energy Saving Associated with Crystal Formation
Many experiments show the presence of numerous intrinsic growth nuclei in melts. Glasses can give rise to about 1025 nanocrystals per m3 within a few hours when they are annealed above the vitreous transition. This number is much too large to be compared with the classical homogeneous nucleation rate. High resolution microscopy reveals the existence of “mean range order” clusters called MRO with a radius of about one nanometer in amorphous Fe83B17 [24]. These entities are not viewed as surviving crystals because they do not exist in the literature. They are as numerous as the nanocrystals and could be growth nuclei. A recent observation of an irreversible viscosity of Fe85B15 far above the liquidus temperature could also be a sign of the existence of surviving crystals up to temperatures as high as 1.3 Tm [25].
A liquid-solid transition is accompanied by Fermi energy and volume changes in metallic glass-forming melts. The free volume change ΔV and the Fermi energy difference ΔEF are expected to disappear at the same reduced temperature θ0lps > −2/3 and to be maximum at the melting temperature (θ = 0) in agreement with the thermal variation given by (1) of the energy saving coefficient εlps of liquid elements between θ0lps = θ2lps ≅ −2/3 and θ = 0 [14,15,16].
The glass-forming melt viscosity is represented by a VFT relation given by (4) depending on three parameters η0, B and T0, in the region of the glass transition Tg [1,2,3]. Its variation by several orders of magnitude above and close to Tg is used to determine T0 [3,26] because including viscosity values measured far above the melting temperature increases the T0 value [27,28]; it is important to fix η0 ≅ NA × h/Vm with NA Avogadro’s number, h Planck’s constant and Vm the molar volume to evaluate T0 and B in (4) [28]:
![Materials 04 00869 i004]() The relaxation time dependence in the temperature range between the onset and the end of the endothermic transition is observed by DSC. At a constant heating rate, the relaxation time τ is also described by the VFT-type relation (5):
The relaxation time dependence in the temperature range between the onset and the end of the endothermic transition is observed by DSC. At a constant heating rate, the relaxation time τ is also described by the VFT-type relation (5):
![Materials 04 00869 i005]() T0 and B are determined using a pre-exponential coefficient τ0 of about 10−14 s [3,27,28].
T0 and B are determined using a pre-exponential coefficient τ0 of about 10−14 s [3,27,28].


The free-volume of glass-forming melts is a linear function of (T−T0). Doolittle’s relation introduces the free volume in the exponential of (4,5) [10]; ΔV would be equal to zero for T = T0 in the absence of vitreous transition. The values of T0 would correspond to the extrapolated free-volume disappearance temperature. Some measurements exist [29,30]. For example, the Pd43Cu27Ni10P20 volume in the liquid and solid states are known down to an extrapolated value ΔV = 0 occurring for T02 ≅ 452 K. In Pd40Cu30Ni10P20, the VFT law leads to T02 = 447 K [31].
The minimum value of εlps0 in undercooled melt can be calculated only knowing T01 (or θ01) and T02 (or θ02) from VFT equations [14,15]. The quadratic equation (6) is obtained applying (1) and (3) for θ = θ2lps:
![Materials 04 00869 i006]()

There are two solutions for θ2lps when εlps0 is larger than a minimum value as already described [14,15]. The relations (7) and (8) between εlps0, θ2lps and ![Materials 04 00869 i007]() are respected when the double solution corresponds to a minimum value of εlps0 larger than 1. It is given by (7,8) and occurs when 9 − 4 × (2 − εlps0) × εlps0/θ20lps = 0:
are respected when the double solution corresponds to a minimum value of εlps0 larger than 1. It is given by (7,8) and occurs when 9 − 4 × (2 − εlps0) × εlps0/θ20lps = 0:
![Materials 04 00869 i008]()
![Materials 04 00869 i009]()
 are respected when the double solution corresponds to a minimum value of εlps0 larger than 1. It is given by (7,8) and occurs when 9 − 4 × (2 − εlps0) × εlps0/θ20lps = 0:
are respected when the double solution corresponds to a minimum value of εlps0 larger than 1. It is given by (7,8) and occurs when 9 − 4 × (2 − εlps0) × εlps0/θ20lps = 0:


The knowledge of θ02 and θ01 from VFT equations chosen respectively equal to θ0lgs and θ0ls and the use of (7) or (8) determine εlps0 and θ2lps and the minimum value of εlps0. The existence of two glass-former classes and their boundaries are predicted completing Angel’s description, if we assume that θ2lgs = θ*g = (T*g − Tm)/Tm [3].
Fragile bulk glasses correspond to θ0lgs = θ0g > −2/3 and εlgs0 > 1, and fragile and quenched glasses to εlgs0 < 1 and θ2lgs = θ0g = −2/3. The undercooled liquid state can be recovered by heating the glass above T2lgs (or θ2lgs) because the condition εlgs0 > 1 stabilizes it. It is not recovered when εlgs0 < 1 and θ0g = −2/3 because there is no minimum value of εlgs0. All predictions of (7,8) are related to the free-volume disappearance temperature of fragile undercooled melts which is equal to the ideal glass transition temperature. Angel’s classification does not fix a quantified boundary between strong and fragile ones.
The strong glass-forming melts correspond to θ0lgs ≤ −2/3, εlgs0 < 2, and θ2lgs > −2/3. They also have a viscosity larger than fragile melts with temperature dependence close to Arrhenius law. Their vitreous transition temperature can be a very large fraction of the melting temperature. The largest value of εlps0 is deduced from the experimental values of θ0lps and θ2lps using (6). Strong glass-forming melts have a homogeneous nucleation temperature always larger than Tm/3 without metastable values of εlps0 regardless of the energy saving.
3. Review of Experimental Results and Discussion
3.1. Presentation of Table 1 and Figure 1
The melting temperatures Tm, the experimental glass transition temperatures Tg or θg = (Tg − Tm)/Tm, the VFT temperatures T01 determined up to temperatures much higher than Tg, the VFT temperatures T02 determined by viscosity or relaxation time measurements in the vicinity of Tm, the free-volume disappearance temperatures T0g = T0lgs calculated using (7,8) and (18) considering that Tg is, in a first approximation, nearly equal to the thermodynamic glass nucleation temperature T*g, the saving energy coefficients εls0(θ = 0) of crystals surviving in the melt far above Tg calculated using (19), the saving energy coefficients εlgs0 (θ = 0) of nascent crystals homogeneously nucleated in the melt near Tg calculated using (18), the free-volume disappearance temperatures T0m = T01 calculated using (7,8,19,20), Tg, and the references [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63] are given in Table 1.
Properties of 20 non-metallic glasses and polymers are numbered with references. B2O3 is numbered 11 and 12. Two values of Tg are used. This glass is not easily crystallized. It gives rise to two crystallographic structures and its highest melting temperature is 783 K. SiO2 N°3 is a strong glass (θ0l < −0.666). Hevea rubber N°50 is just at the limit separating strong glasses from fragile ones because Tog is a little larger than Tm/3.
Properties of 28 bulk metallic glasses are used and numbered with references. The difference of liquidus and solidus temperatures is sometimes too large. A homogeneous melt has a well-defined Fermi energy. The melting temperature has been chosen between these two limits looking at the DSC profile. Two melting temperatures Tm = 728 K and Tl = 925 K are used for La55Al25Ni20 N° 21 and 22. The first one corresponds to the largest endothermic peak and the second one to the liquidus. We find εls0 = 1.65 and about 1.51 respectively. Two melting temperatures are also used for Pd40Ni40P20 N°24 and 25, Tm = 987 K and Tm = 884 K leading to εls0 = 1.63 and 1.56 respectively. The melting temperatures of La55Al25Ni5Cu15 N°28, La55Al25Ni15Cu5 N°32 and La55Al25Ni5Cu10Co5 N°41 are respectively chosen equal to 700 K instead of 878 K, 729 K instead of 899 K and 754 K instead of 822.5 K.
Experimentalists call all glasses characterized by a crystallization temperature Tx occurring near Tg “conventional”. Among them, glasses have a value of Tg close to Tm/2. The glass transitions of Al87Co4Ce9, Al87Co6Ce7, Al87Co8Ce5, Al85Co10Ce5 and Al90Co5Ce5 are not reported because they cannot be distinguished from the crystallization temperature Tx, in the absence of endothermic heat before crystallization in a DSC run [62]. The values of θx = (Tx − Tm)/Tm) are respectively equal to −0.481, −0.523, −0.533, −0.543, −0.567. Au0.77Ge0.136Si0.094 N°51 is characterized by a crystallization temperature Tx occurring above and very close to Tg with θg = −0.539 [63]. All these alloys have an energy saving coefficient larger than 1. They could belong to the fragile glass class because they have a VFT temperature T0 larger than Tm/3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Some properties of 46 glass-forming melts are presented: Tm the melting temperature; Tg the vitreous transition temperature, θg = (Tg − Tm)/Tm, T01 and T02 the Vogel-Fulcher-Tammann temperatures, as found in various references; T0g the free-volume disappearance temperature calculated from Tg and not from T*g; εls0 the energy saving coefficient of tiny crystals surviving in the melt and acting as growth nuclei; εlgs0 the energy saving coefficient of homogeneously-nucleated crystals in the melt; εls0 and εlgs0 being used to calculate T0m and T0g; T0m the free-volume disappearance temperature also calculated from Tg and not from T*g; and references.
| Glass | Tm | Tg | θg | T01 | T02 | T0g | εls0 | εlgs0 | T0m | References | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | As2S3 | 585 | 481 | −0.178 | 270 | 319 | 1.822 | 1.732 | 363 | [32,33] | |
| 2 | Propylene glycol | 214 | 167 | −0.220 | 117 | 108 | 1.780 | 1.671 | 125 | [26,34,35] | |
| 3 | SiO2 | 1,993 | 1,473 | −0.261 | 300 | 1.36 | [2,36] | ||||
| 4 | Propylene carbonate | 217 | 160 | −0.263 | 130 | 102 | 1.737 | 1.606 | 119 | [35] | |
| 5 | polystyrene | 513 | 375 | −0.269 | 323 | 239 | 1.731 | 1.596 | 280 | [26,37] | |
| 6 | Pd43Ni10Cu27P20 | 802 | 585 | −0.271 | 452 | 372 | 1.729 | 1.594 | 436 | [29,38,39] | |
| 7 | O-Terphenil | 329 | 240 | −0.271 | 208 | 153 | 1.729 | 1.593 | 179 | [3,26,40] | |
| 8 | Pd40Cu30Ni10P20 | 823 | 578 | −0.298 | 447 | 366 | 1.702 | 1.553 | 432 | [29,31,39] | |
| 9 | Salol | 315 | 220 | −0.302 | 183 | 139 | 1.698 | 1.548 | 165 | [3,26,35] | |
| 10 | As2Se3 | 645 | 450 | −0.302 | 335 | 285 | 1.698 | 1.547 | 337 | [41] | |
| 11 | B2O3 | 783 | 545 | −0.304 | 402 | 345 | 1.696 | 1.544 | 408 | [3,11,32] | |
| 12 | B2O3 | 783 | 521 | −0.335 | 263 | 330 | 1.665 | 1.498 | 393 | [3,11,32] | |
| 13 | Bromopentane | 158 | 107 | −0.323 | 74 | 68 | 1.677 | 1.516 | 80 | [26] | |
| 14 | ZnCl2 | 565 | 380 | −0.327 | 274 | 241 | 1.673 | 1.509 | 286 | [2,26] | |
| 15 | Butene 1 | 88 | 59 | −0.330 | 37 | 1.670 | 1.506 | 44 | [2] | ||
| 16 | Zr41.2Ti13.8Cu12.5Ni10Be22.5 | 937 | 625 | −0.333 | 413 | 396 | 1.667 | 1.501 | 472 | [39,42] | |
| 17 | La55Al25Ni10Cu10 | 662 | 441 | −0.334 | 255 | 280 | 1.666 | 1.499 | 333 | [43,44,45] | |
| 18 | 2 Methylpentane | 120 | 80 | −0.338 | 58 | 50 | 1.663 | 1.494 | 60 | [2,26,46] | |
| 19 | Toluene | 178 | 117 | −0.343 | 104 | 74 | 1.657 | 1.485 | 89 | [47] | |
| 20 | Glycerol | 293 | 190 | −0.352 | 128 | 121 | 1.648 | 1.473 | 144 | [26,34,35] | |
| 21 | La55Al25Ni20 | 728 | 470 | −0.354 | 307 | 299 | 1.646 | 1.469 | 358 | [43,44,45] | |
| 22 | La55Al25Ni20 | 925 | 470 | −0.492 | 309 | 330 | 1.508 | 1.262 | 394 | [43,44,45] | |
| 23 | PET = (C10H8O4)n | 542 | 342 | −0.369 | 219 | 1.631 | 1.446 | 262 | [2] | ||
| 24 | Pd40Ni40P20 | 884 | 554 | −0.373 | 356 | 355 | 1.627 | 1.440 | 425 | [48,49] | |
| 25 | Pd40Ni40P20 | 987 | 554 | −0.439 | 356 | 369 | 1.561 | 1.342 | 442 | [48,49] | |
| 26 | Pt57.5Cu14.7Ni5.3P22.5 | 813 | 509 | −0.374 | 336 | 326 | 1.626 | 1.439 | 390 | [50] | |
| 27 | Pd0.775Cu0.06Si0.165 | 1,015 | 632 | −0.377 | 515 | 405 | 1.623 | 1.434 | 486 | [51] | |
| 28 | La55Al25Ni5Cu15 | 700 | 436 | −0.378 | 286 | 279 | 1.622 | 1.433 | 335 | [43,44,45] | |
| 29 | Zr52.5Cu17.9Ni14.6Al10Ti5 | 1,090 | 675 | −0.381 | 521 | 434 | 1.619 | 1.429 | 519 | [52] | |
| 30 | Pt57.3Cu14.6Ni5.3P22.8 | 788 | 487 | −0.382 | 336 | 313 | 1.618 | 1.427 | 375 | [50] | |
| 31 | Se | 491 | 303 | −0.383 | 220 | 195 | 1.617 | 1.426 | 233 | [2,53] | |
| 32 | La55Al25Ni15Cu5 | 729 | 449 | −0.384 | 273 | 289 | 1.616 | 1.424 | 346 | [43,44,45] | |
| 33 | Au49Ag5.5Pd2.3Cu26.9Si16.3 | 655 | 403 | −0.385 | 250 | 259 | 1.615 | 1.423 | 311 | [39] | |
| 34 | Ethanol | 159 | 97 | −0.390 | 78 | 58 | 63 | 1.610 | 1.415 | 75 | [2,11,32] |
| 35 | Zr58.5Cu15.6Ni12.8Al10.3Nb2.8 | 1,110 | 673 | −0.394 | 437 | 435 | 1.606 | 1.409 | 522 | [54] | |
| 36 | Zr57Cu15.4Ni12.6Al10.3Nb5 | 1,120 | 678 | −0.395 | 525 | 438 | 1.605 | 1.408 | 526 | [52] | |
| 37 | Pr55Ni25Al20 | 820 | 494 | −0.398 | 296 | 320 | 1.602 | 1.404 | 384 | [55] | |
| 38 | Ti41.5Cu37.5Ni7.5Zr2.5Hf5Sn5Si1 | 1,176 | 693 | −0.411 | 452 | 1.589 | 1.384 | 543 | [56] | ||
| 39 | Cu47Ti34Zr11Ni8 | 1,144 | 673 | −0.412 | 500 | 439 | 1.588 | 1.382 | 527 | [57] | |
| 40 | Y56Al24Co20 | 1,085 | 636 | −0.414 | 614 | 416 | 1.586 | 1.379 | 499 | [58] | |
| 41 | La55Al25Ni5Cu10Co5 | 754 | 439 | −0.418 | 241 | 288 | 1.582 | 1.374 | 345 | [43,44,45] | |
| 42 | Mg59.5Cu22.9Ag6.6Gd11 | 734 | 425 | −0.421 | 249 | 279 | 1.579 | 1.369 | 335 | [59] | |
| 43 | Mg61Cu28Gd11 | 737 | 422 | −0.427 | 256 | 278 | 1.573 | 1.359 | 334 | [59] | |
| 44 | Mg65Cu25Y10 | 739 | 400 | −0.459 | 363 | 260 | 271 | 1.541 | 1.312 | 325 | [27] |
| 45 | Zr46.75Ti8.25Cu7.5Ni10Be27.5 | 1,070 | 595 | −0.444 | 372 | 398 | 1.556 | 1.334 | 477 | [60] | |
| 46 | Cyclo-octanol | 298 | 165 | −0.446 | 92 | 110 | 1.554 | 1.331 | 133 | [26] | |
| 47 | Zr65Al10Ni10Cu15 | 1,161 | 641 | −0.448 | 437 | 430 | 1.552 | 1.328 | 516 | [39] | |
| 48 | Ce60Al10Ni10Cu20 | 677 | 373 | −0.449 | 331 | 250 | 1.551 | 1.326 | 300 | [61] | |
| 49 | Al87Co4Ce9 | 1,104 | 573 | −0.481 | 397 | 1.519 | 1.279 | 475 | [62] | ||
| 50 | Hevea rubber | 421 | 200 | −0.525 | 136 | 147 | 1.475 | 1.213 | 174 | [32] | |
| 51 | Au0.77Ge0.136Si0.094 | 629 | 290 | −0.539 | 241 | 217 | 1.461 | 1.192 | 257 | [63] |
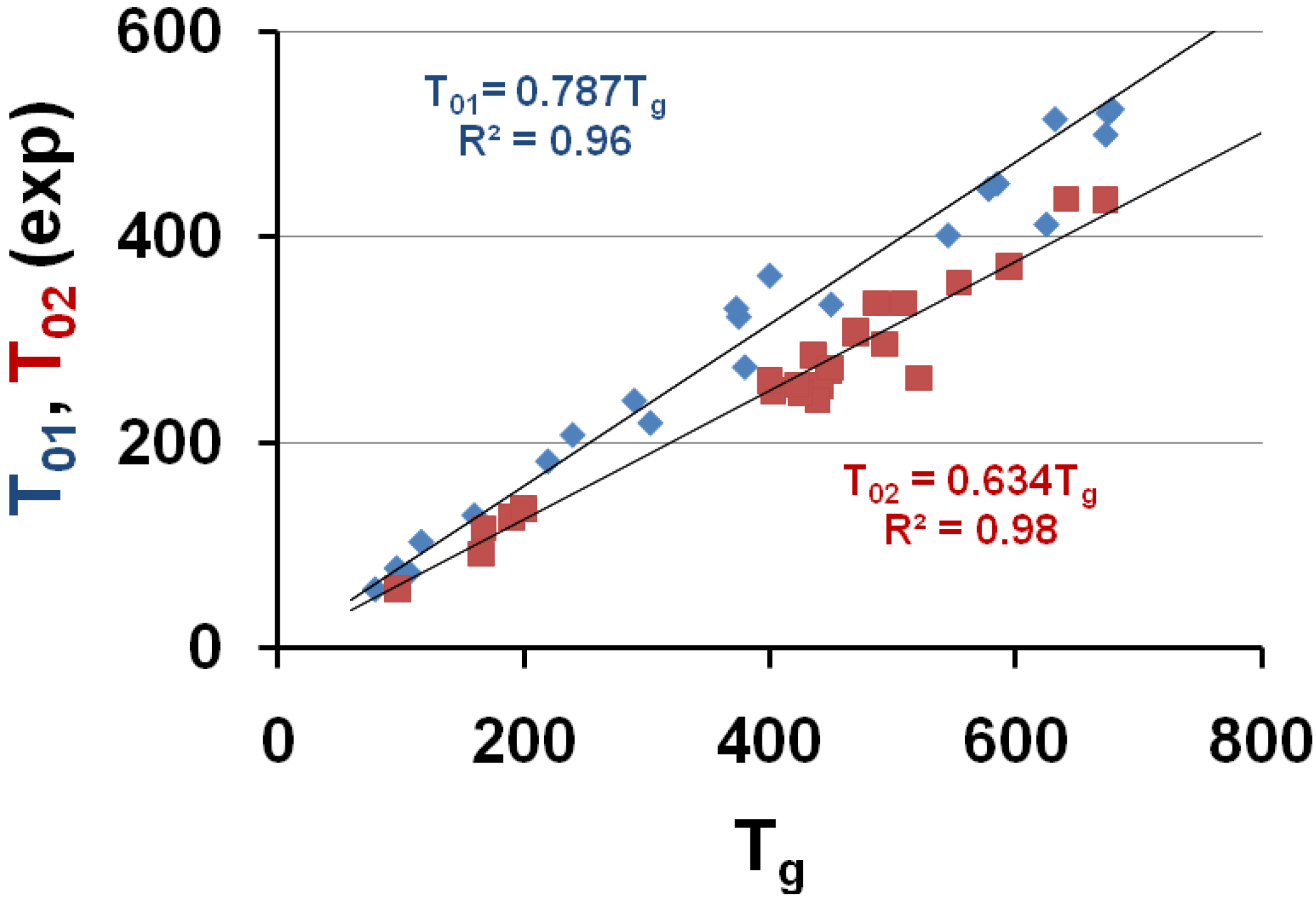
Figure 1.
The VFT temperatures T01 and T02 given in Table 1 are plotted versus Tg; T02 corresponds to measurements in the vicinity of Tg and T01 includes viscosity measurements at higher temperatures. T01 ≅ 0.787 Tg and T02 ≅ 0.634 Tg.
Figure 1.
The VFT temperatures T01 and T02 given in Table 1 are plotted versus Tg; T02 corresponds to measurements in the vicinity of Tg and T01 includes viscosity measurements at higher temperatures. T01 ≅ 0.787 Tg and T02 ≅ 0.634 Tg.
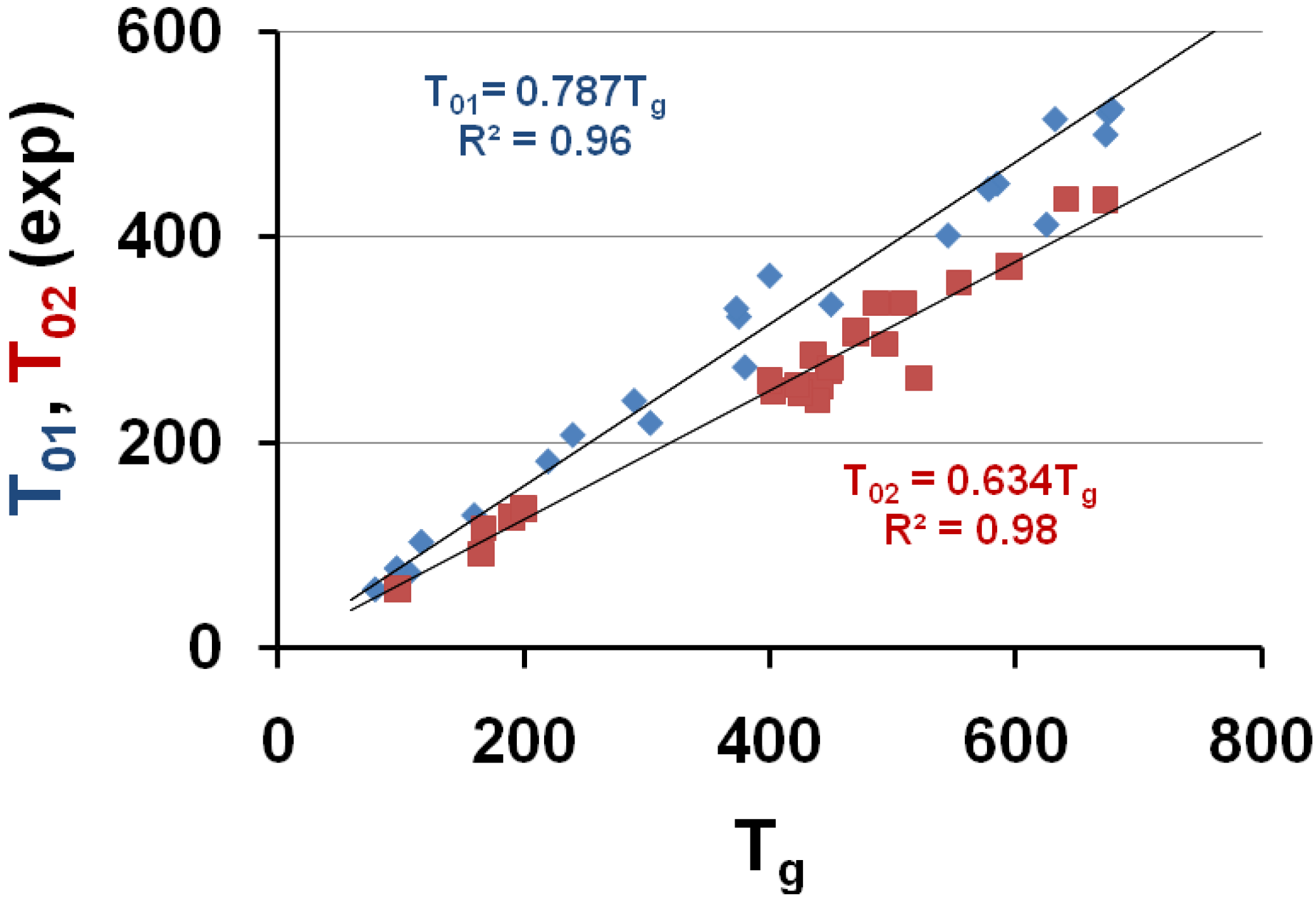
The temperatures T01 and T02 are plotted as a function of the vitreous transition temperature Tg in Figure 1; the upper straight line uses the equation T01 = 0.787 × Tg and corresponds to a similar law T01 = 0.77 × Tg already observed for 7 other polymers [7,64]; the lower straight line uses the equation T02 = 0.634 × Tg.
3.2.Homogeneous Nucleation Temperature of Vitreous Phase and Relaxation Time
The calculation of the crystal nucleation full time t includes not only the steady-state nucleation time tsn defined by v × tsn = 1 in (10) at the nucleation temperature, but also the time lag τns in crystal transient nucleation defined by (9) [2] with Klgs defined by (12):
![Materials 04 00869 i010]()
![Materials 04 00869 i011]()
![Materials 04 00869 i012]()
![Materials 04 00869 i013]()




The steady-state nucleation rate is J, the volume sample v, Zeldovitch’s factor Γ, the critical energy barrier ΔG*2lgs/kBT, a*0 = π2/6, the atom or molecule number per volume unit N. The quantity lnKls is equal to lnA ≅ 90 ± 2 for liquid elements in a broad temperature scale; lnA is a little smaller for crsystallization of glass-forming melts. In the vicinity of T*g, and assuming that T*g (or θ*g) is equal to a crystal homogeneous nucleation temperature T2lgs (or θ2lgs), when J = 1, the crystal transient nucleation time τns viewed as the relaxation time, can be calculated with (15) using the critical parameters (13) and (14):
![Materials 04 00869 i014]()
![Materials 04 00869 i015]()
![Materials 04 00869 i016]()



The coefficient of lnKlgs in (13) is equal to 1 for θ = θ2lgs = (εls − 2)/3 when the crystal-steady-state nucleation time is minimum. The Ag is the A value defined by (12) at the glass transition, which is used for the homogeneously-nucleated cluster formation. Zeldovitch’s factor given by (16) is calculated below, as a function of the number Jc of molecules or atoms in a spherical crystal of critical radius R*2lgs, given by (14) at θ2lgs = θ∗g. The pre-exponential time τ0 given by (17) depends on Ag and ln(τns/τ0) is equal to B/(T*g − T0g) ≅ 36.5 − 39 at T*g [3,9]:
![Materials 04 00869 i017]()
![Materials 04 00869 i018]() where ΔSm is the fusion entropy per mole, NA the Avogadro number, θ2lgs defined by (3), NAkB = 8.32 Joule. The temperature T2lgs (or θ2lgs) is a constant of the material and a unique function of the energy saving εlgs in (3). It does not strictly depend on viscosity. Nevertheless, the viscosity has a strong influence on the occurrence of the maximum nucleation rate temperature because the critical energy barrier ΔG*2lgs/kBT is proportional to lnKlgs and the numerical coefficient of lnKlgs in ΔG*2lgs/kBT is equal to about 1 in a broad window of temperatures above T*g. The critical energy barrier ΔG*2lgs/kBT is nearly the same at T*g for all glass-forming melts and lnKlgs decreases with the increase in viscosity. The nucleation rate is at a maximum when lnKlgs becomes exactly equal to ΔG*2lgs/kBTg. This event occurs at T*g (or θ*g) = T2lgs (or θ2ls) for similar viscosity values because τ0, and the relaxation time
where ΔSm is the fusion entropy per mole, NA the Avogadro number, θ2lgs defined by (3), NAkB = 8.32 Joule. The temperature T2lgs (or θ2lgs) is a constant of the material and a unique function of the energy saving εlgs in (3). It does not strictly depend on viscosity. Nevertheless, the viscosity has a strong influence on the occurrence of the maximum nucleation rate temperature because the critical energy barrier ΔG*2lgs/kBT is proportional to lnKlgs and the numerical coefficient of lnKlgs in ΔG*2lgs/kBT is equal to about 1 in a broad window of temperatures above T*g. The critical energy barrier ΔG*2lgs/kBT is nearly the same at T*g for all glass-forming melts and lnKlgs decreases with the increase in viscosity. The nucleation rate is at a maximum when lnKlgs becomes exactly equal to ΔG*2lgs/kBTg. This event occurs at T*g (or θ*g) = T2lgs (or θ2ls) for similar viscosity values because τ0, and the relaxation time ![Materials 04 00869 i019]() are nearly the same in all glass-forming melts at Tg.
are nearly the same in all glass-forming melts at Tg.


 are nearly the same in all glass-forming melts at Tg.
are nearly the same in all glass-forming melts at Tg.The relaxation time ![Materials 04 00869 i019]() is generally of the order of 100 s at
Tg. A value τ0 = 1.4 × 10−14 s is deduced with B/(Tg − T0g) = 36.5 and τ0 = 3.14 × 10−15 s with B/(Tg − T0g) = 38 in all glasses [3,9,27]. Equation (17) giving τ0 is used to determine lnAg only, depending on {ln(1 + θ2lgs] − ln[Vm × ΔSm].
is generally of the order of 100 s at
Tg. A value τ0 = 1.4 × 10−14 s is deduced with B/(Tg − T0g) = 36.5 and τ0 = 3.14 × 10−15 s with B/(Tg − T0g) = 38 in all glasses [3,9,27]. Equation (17) giving τ0 is used to determine lnAg only, depending on {ln(1 + θ2lgs] − ln[Vm × ΔSm].
is generally of the order of 100 s at
Tg. A value τ0 = 1.4 × 10−14 s is deduced with B/(Tg − T0g) = 36.5 and τ0 = 3.14 × 10−15 s with B/(Tg − T0g) = 38 in all glasses [3,9,27]. Equation (17) giving τ0 is used to determine lnAg only, depending on {ln(1 + θ2lgs] − ln[Vm × ΔSm].With τ0 = 1.4 × 10−14 s, the lnAg is equal to 100.7 in N°6 Pd43Ni10Cu27P20 (Vm = 8 × 10−6 m3, ΔSm = 8.74 J/K, θ2lgs = −0.271) and to 96.4 in N°20 Glycerol (Vm =73.07 × 10−6 m3, ΔSm = 62.42 J/K, θ2lgs = −0.352).
With τ0 = 3.14 × 10−15 s, the lnAg is respectively equal to 102.2 and 97.9 for the same liquids with an increase of the product Vm × ΔSm by a factor 66.
With τ0 = 1.4 × 10−14 s, the average value of lnAg is 98.5 ± 2 and with τ0 = 3.14 × 10−15 s, lnAg = 100 ± 2. The lnKlgs and the thermally-activated energy barrier ΔG*2lgs/kBT2 are always equal to 62 ± 2 in all glass-forming melts at T = Tg.
Equation (15) shows that the assumption of a value of τ0 being the same in all melts can be replaced by a nearly-constant value of Ag; lnAg is about 15% larger than the one found for crystal nucleation from surviving crystals in several glass-forming melts at higher temperatures [14]. Then, the time lag of a transient nucleation to produce a crystal nuclei distribution, built from surviving crystals and ready for steady-state nucleation, is about 106 times larger than the time lag required for a homogeneously-nucleated cluster distribution formation. The steady-state nucleation time tsn is, in addition, equal to 109 s for v = 10−9 m3. A nucleus distribution with cluster size close to the critical value is created just near Tg when the relaxation time is minimum. In Turnbull and Fisher’s model, lnKls is nearly equal to ln(NAkBT*g/Vmh) − Δf*/kBT*g where h is Planck’s constant and Δf*/kBT*g is a thermally-activated energy barrier for atom diffusion from the melt to the homogeneously-nucleated cluster which is smaller than the one from the melt to a surviving crystal [65]. This weakening could be due to formation of clusters containing many vacancies on their various sub-lattices during homogeneous nucleation. Surviving crystals are expected to be well-crystallized because they are part of previously-crystallized materials. They did not melt above Tm and they are very stable with their fusion heat equal to the bulk one [21,22,23].
3.3. The Thermodynamic Vitreous Transition T*g at the Disappearance Temperature of the Fully-Relaxed Enthalpy
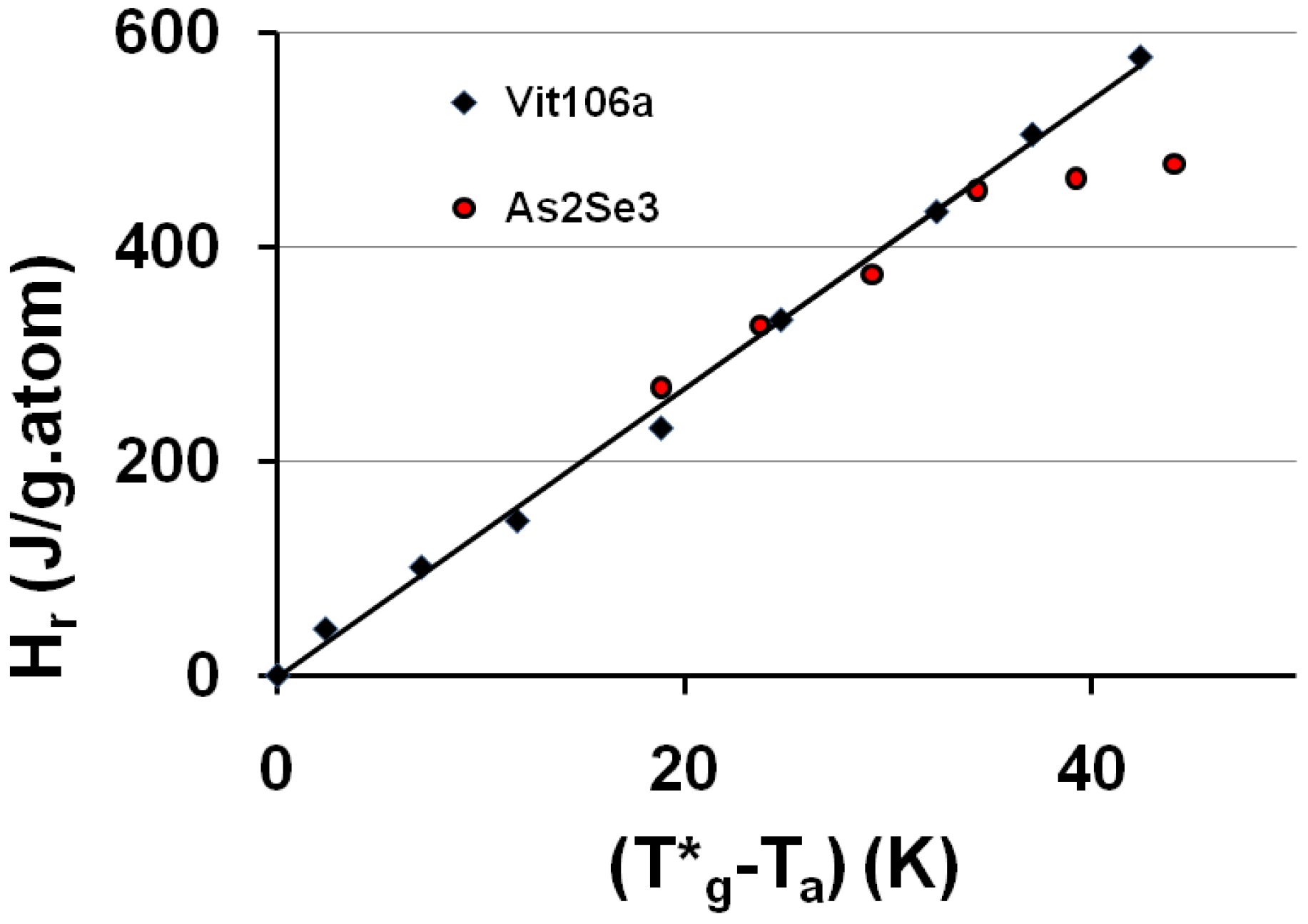
A relaxed enthalpy is measured by DSC, after quenching the undercooled liquid to much lower temperatures than Tg and annealing it at a temperature Ta smaller than Tg, during the relaxation time necessary to obtain its maximum value Hr.. The structural relaxation is viewed as a transformation of the quenched undercooled liquid state in a fully-frozen state. This exothermic heat varies linearly with (T*g − Ta) as shown in Figure 2 and Figure 3. The fully-relaxed enthalpies Hr of As2Se3 and Zr58.5Cu15.6Ni12.8Al10.3Nb2.8 (vit106a) are plotted in Figure 2 using values of T*g equal to 462 and 690 K instead of Tg = 450 and 673 K respectively [54,66,67]. The calculated temperatures T0g are respectively 293 K and 437 K as compared to T02 = 335 and 437 K. The vitreous transition T*g of As2Se3 exactly corresponds to the mid-point of the reversible specific heat jump [67]. In Figure 3 we describe the relaxed enthalpy variation of propylene glycol and glycerol extracted from two different publications with T*g = 171 and 189.8 K instead of Tg = 167 and 190 K respectively [68,69]. The calculated temperatures T0g are respectively 112 K and 121 K as compared to T02 = 117 and 128 K.
The specific heat excess of the undercooled melt ΔCpgl can be directly calculated from dHr/dTa = ΔCpgl because Hr is a linear function of Ta and ![Materials 04 00869 i020]() . The following calculated values of ΔCpgl are in good agreement with the specific heat difference ΔCpls between solid and liquid; for propylene glycol, ΔCpgl = 52.3 and ΔCpls = 67.3 J/mole/K [68,69]; for glycerol, ΔCpgl = 69.9 and ΔCpls ≅ 79.4 J/mole/K [32,68,69]; for vit106a, ΔCpgl = 13.5 and ΔCpls ≅ 15.5 J/mole/K [54]; for As2Se3, ΔCpgl = 67 J/mole/K from the relaxed enthalpy and 67 J/mole/K from the reversible specific heat [67]. The specific heat jump ΔCpls is a little too large at Tg when it is measured at a too low heating rate because it still contains an endothermic contribution except when stepscan techniques are used [13].
. The following calculated values of ΔCpgl are in good agreement with the specific heat difference ΔCpls between solid and liquid; for propylene glycol, ΔCpgl = 52.3 and ΔCpls = 67.3 J/mole/K [68,69]; for glycerol, ΔCpgl = 69.9 and ΔCpls ≅ 79.4 J/mole/K [32,68,69]; for vit106a, ΔCpgl = 13.5 and ΔCpls ≅ 15.5 J/mole/K [54]; for As2Se3, ΔCpgl = 67 J/mole/K from the relaxed enthalpy and 67 J/mole/K from the reversible specific heat [67]. The specific heat jump ΔCpls is a little too large at Tg when it is measured at a too low heating rate because it still contains an endothermic contribution except when stepscan techniques are used [13].
 . The following calculated values of ΔCpgl are in good agreement with the specific heat difference ΔCpls between solid and liquid; for propylene glycol, ΔCpgl = 52.3 and ΔCpls = 67.3 J/mole/K [68,69]; for glycerol, ΔCpgl = 69.9 and ΔCpls ≅ 79.4 J/mole/K [32,68,69]; for vit106a, ΔCpgl = 13.5 and ΔCpls ≅ 15.5 J/mole/K [54]; for As2Se3, ΔCpgl = 67 J/mole/K from the relaxed enthalpy and 67 J/mole/K from the reversible specific heat [67]. The specific heat jump ΔCpls is a little too large at Tg when it is measured at a too low heating rate because it still contains an endothermic contribution except when stepscan techniques are used [13].
. The following calculated values of ΔCpgl are in good agreement with the specific heat difference ΔCpls between solid and liquid; for propylene glycol, ΔCpgl = 52.3 and ΔCpls = 67.3 J/mole/K [68,69]; for glycerol, ΔCpgl = 69.9 and ΔCpls ≅ 79.4 J/mole/K [32,68,69]; for vit106a, ΔCpgl = 13.5 and ΔCpls ≅ 15.5 J/mole/K [54]; for As2Se3, ΔCpgl = 67 J/mole/K from the relaxed enthalpy and 67 J/mole/K from the reversible specific heat [67]. The specific heat jump ΔCpls is a little too large at Tg when it is measured at a too low heating rate because it still contains an endothermic contribution except when stepscan techniques are used [13].
Figure 2.
The saturated value of the relaxed enthalpy Hr is plotted versus (T*g − Ta), T*g being the thermodynamic vitreous transition and Ta the annealing temperature; for N°35 vit 106a (Zr58.5Cu15.6Ni12.8Al10.3Nb2.8), T*g = 690 K and the slope of the straight line ΔCpgl = 13.5 J/g.atom and for N° 10 As2Se3, T*g = 462 K and ΔCpgl = 13.5 J/g.atom [54,66,67]. The corresponding values of T0g calculated from T*g are 443 K and 293 K. The deviation from the straight line is due to the approach of the Kauzmann temperature of As2Se3 [67].
Figure 2.
The saturated value of the relaxed enthalpy Hr is plotted versus (T*g − Ta), T*g being the thermodynamic vitreous transition and Ta the annealing temperature; for N°35 vit 106a (Zr58.5Cu15.6Ni12.8Al10.3Nb2.8), T*g = 690 K and the slope of the straight line ΔCpgl = 13.5 J/g.atom and for N° 10 As2Se3, T*g = 462 K and ΔCpgl = 13.5 J/g.atom [54,66,67]. The corresponding values of T0g calculated from T*g are 443 K and 293 K. The deviation from the straight line is due to the approach of the Kauzmann temperature of As2Se3 [67].
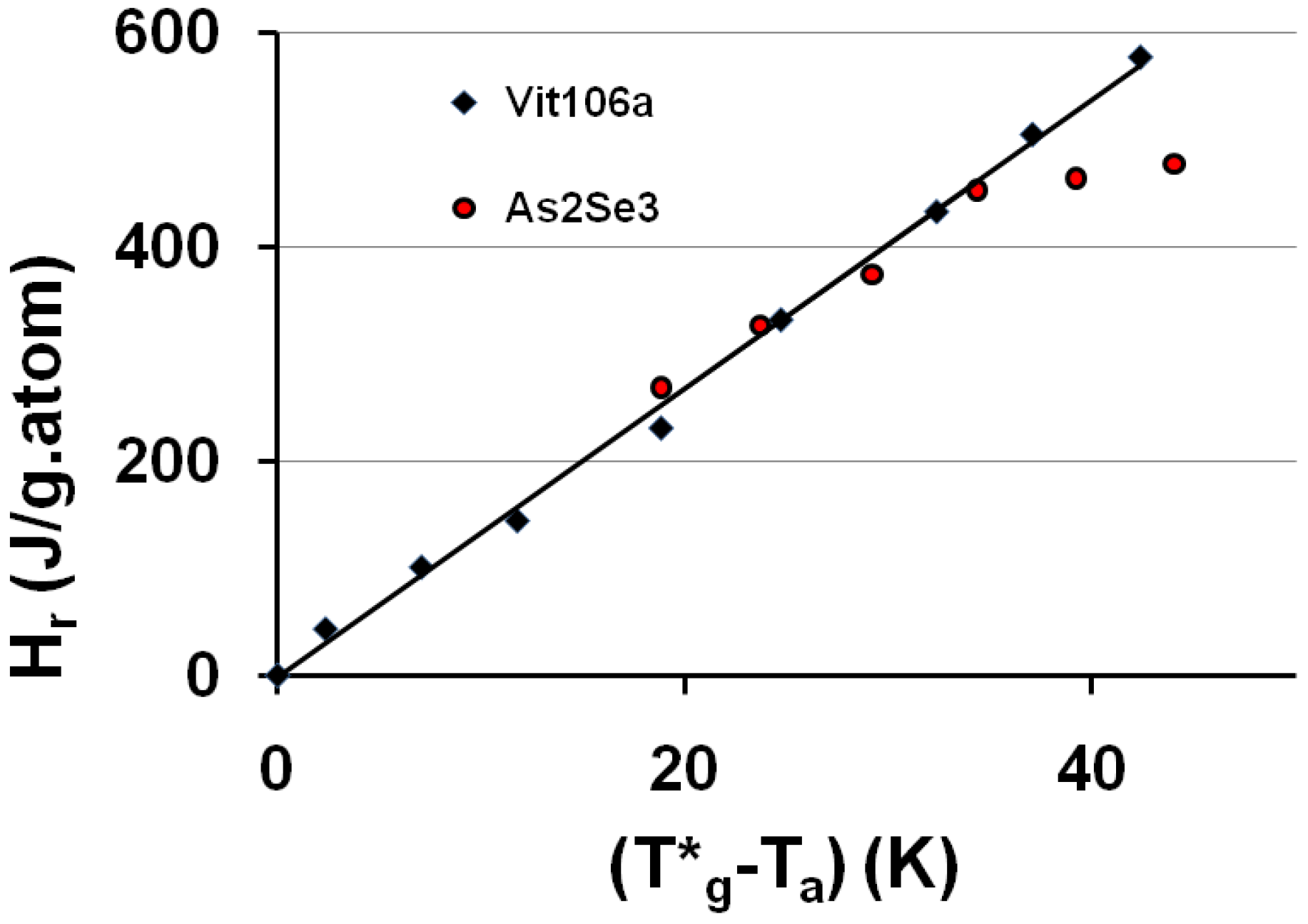
Figure 3.
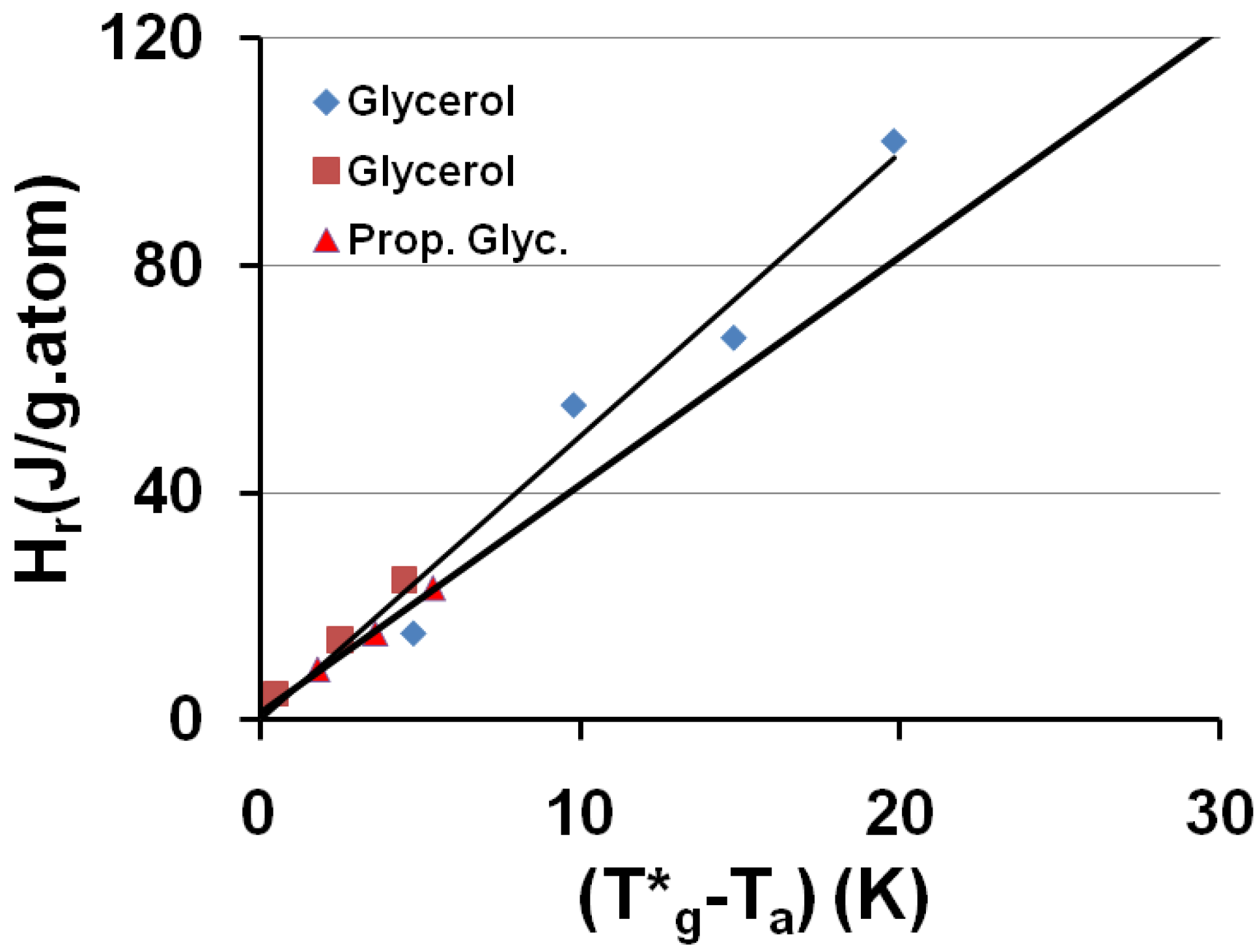
The saturated value of the relaxed enthalpy Hr is plotted versus (T*g − Ta), T*g being the thermodynamic vitreous transition and Ta the annealing temperature; for N° 20 glycerol, T*g = 189.8 K and the slope of the straight line ΔCpgl = 4.99 J/g.atom and for N° 2 propylene glycol T*g = 171 K and ΔCpgl = 4.02 J/g.atom. The corresponding values of T0g calculated from T*g are 121 and 112 K respectively. The largest values of Hr are due to [68] and the smallest ones to [69].
Figure 3.
The saturated value of the relaxed enthalpy Hr is plotted versus (T*g − Ta), T*g being the thermodynamic vitreous transition and Ta the annealing temperature; for N° 20 glycerol, T*g = 189.8 K and the slope of the straight line ΔCpgl = 4.99 J/g.atom and for N° 2 propylene glycol T*g = 171 K and ΔCpgl = 4.02 J/g.atom. The corresponding values of T0g calculated from T*g are 121 and 112 K respectively. The largest values of Hr are due to [68] and the smallest ones to [69].

The specific heat excess of an undercooled melt tends to zero at the Kauzmann temperature as shown by the fact that the derivative dΔHr/dT of As2Se3 tends to zero at this temperature as reproduced in Figure 2 [66,67]; the Kauzmann temperature Tk is an actual temperature of undercooled melts instead of a virtual one [67].
3.4. The Crystal Homogeneous Nucleation Temperature at T*g
The vitreous transition T*g (or θ*g) is viewed as occurring at the crystal steady–state nucleation maximum-rate temperature T2lgs (or θ2lgs). In this case, the glass transition being a material constant has to obey (6). The energy saving approximate coefficients εlgs0 in Table 1 are given by (18) using Tg which is not known instead of T*g:
![Materials 04 00869 i021]()

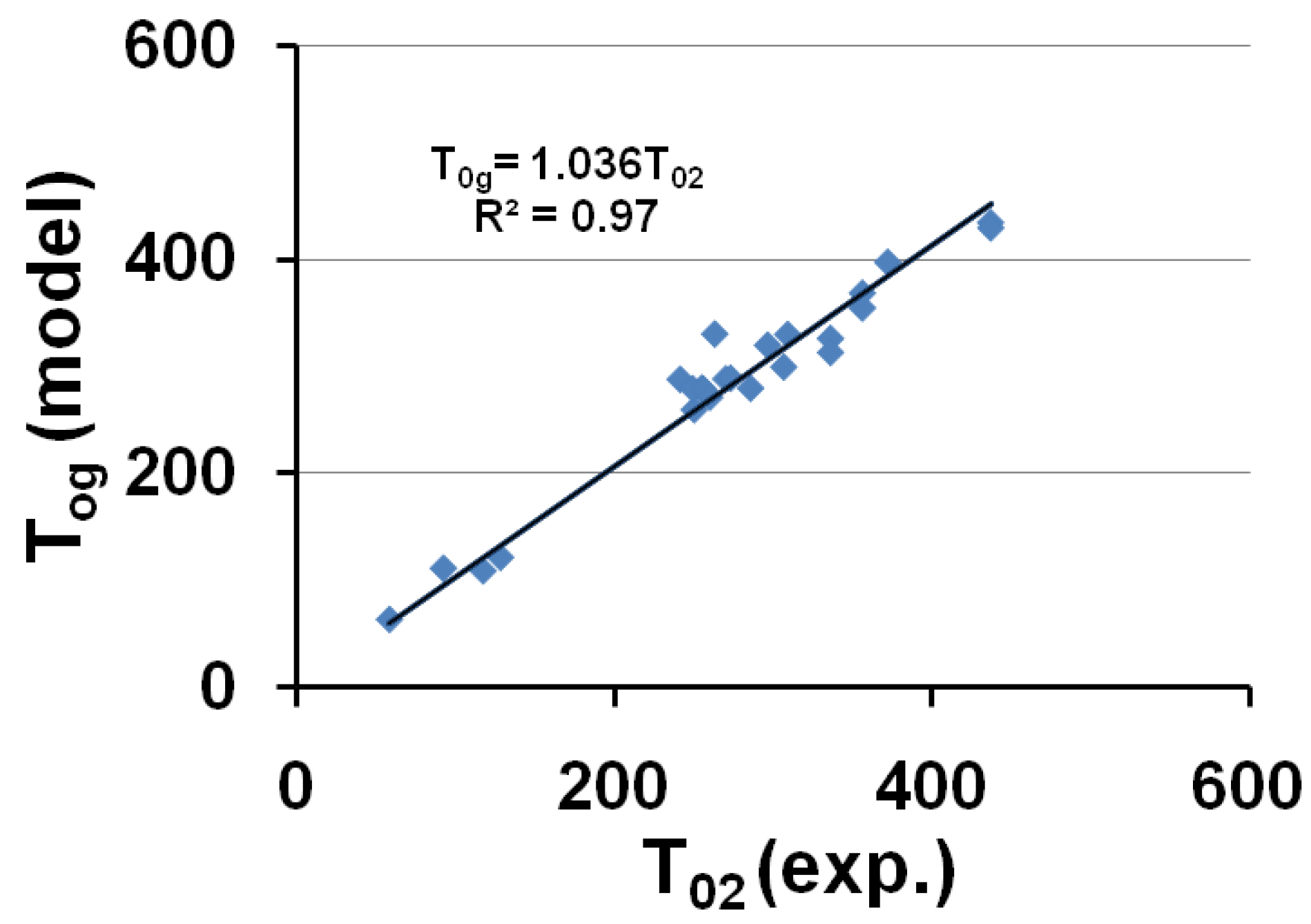
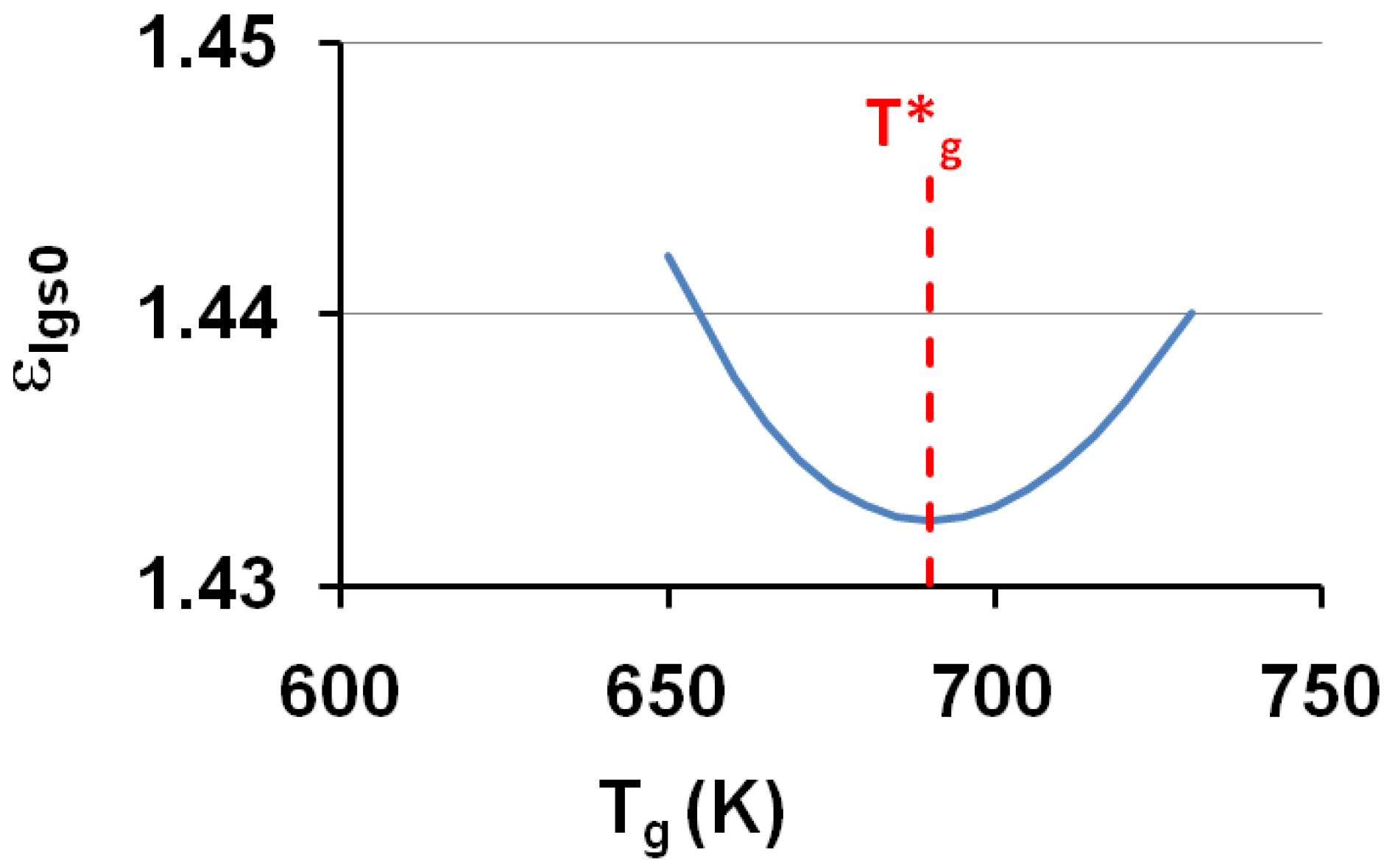
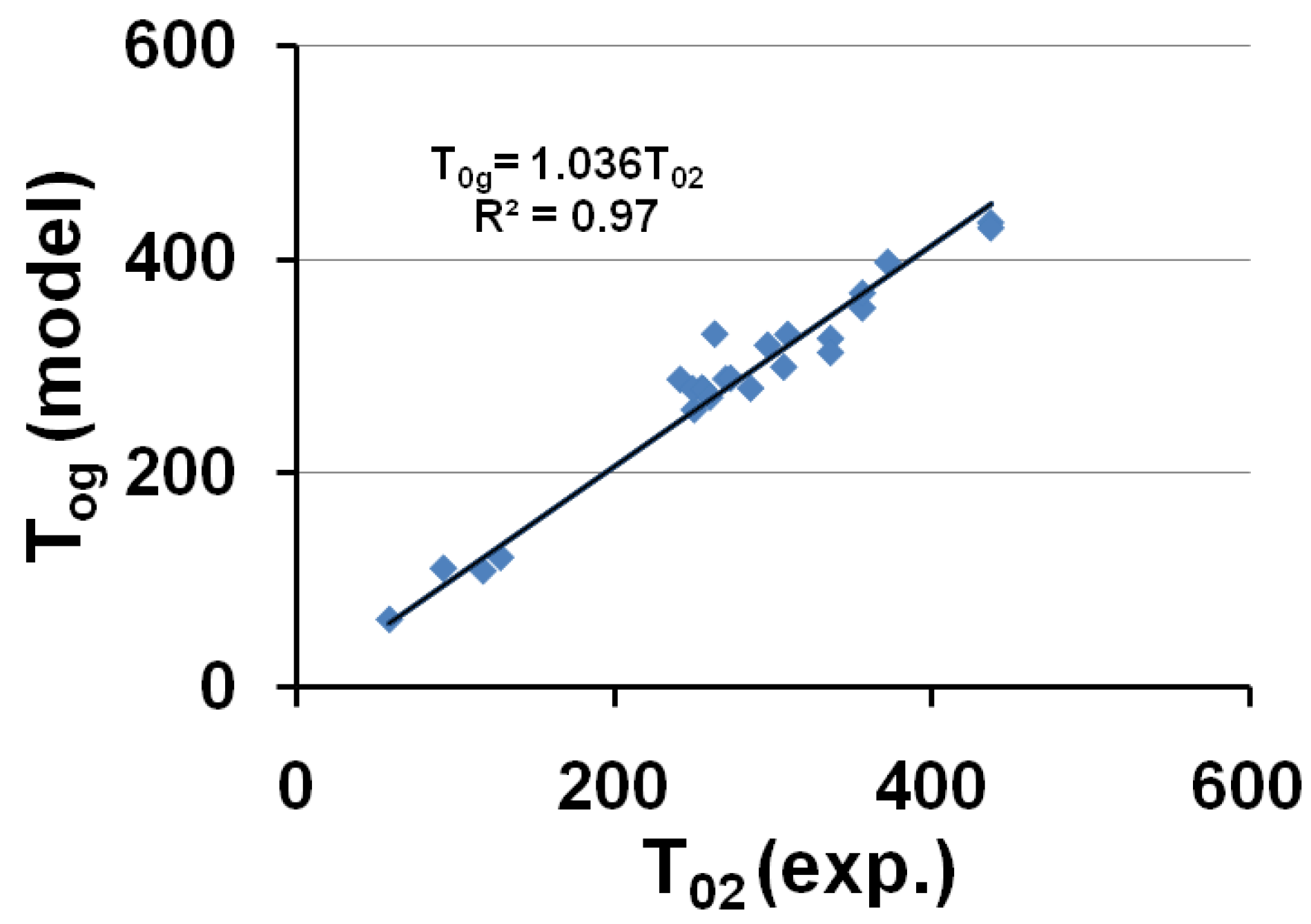
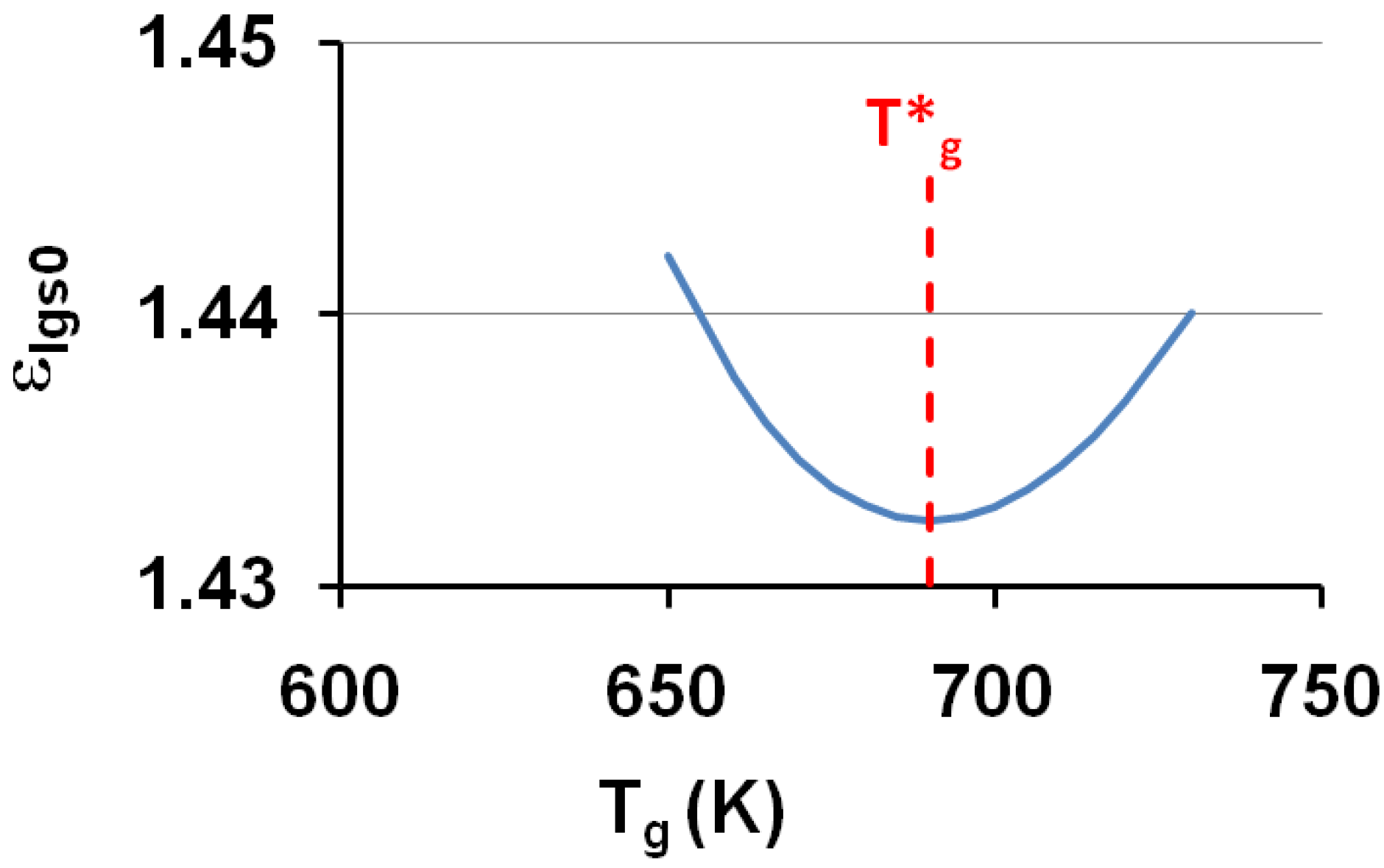
The corresponding temperatures T0g = T0lgs (or θ0g = θ0lgs) listed in Table 1, are calculated from εlgs0 determined by (18) and (7,8). The calculated temperature T0g of the free volume disappearance is plotted as a function of the VFT temperature T02 in Figure 4. The average of T0g is 3.6% larger than that of T02. These quantities are nearly equal if we consider that Tg is an out-of-equilibrium temperature which is not exactly equal to the thermodynamic transition T*g. The model works and is able to predict the VFT temperature of fragile glass-forming melts when T*g is known.
Figure 4.
The calculated values of the free-volume disappearance temperature T0g of fragile glass-forming melts are plotted versus the VFT temperatures T02 determined by measurements in the vicinity of Tg; T0g ≅ 1.036 T02.
Figure 4.
The calculated values of the free-volume disappearance temperature T0g of fragile glass-forming melts are plotted versus the VFT temperatures T02 determined by measurements in the vicinity of Tg; T0g ≅ 1.036 T02.
The energy saving coefficients εlgs0 and εls0 are not equal and correspond to the two reduced temperatures θ0g and θ0m given by (6). The coefficient εls0 is calculated using T0m and Tg of N° 29 vit105 (Zr52.5Cu17.9Ni14.6Al10Ti5) because the scaling law T0m = 0.77 × Tg [7,64] is obeyed by this material. Δε (θg) = (εls0 − εlg0) is equal to 0 for θg = 0 and 0.19 for θg = −0.381. These two particular values are used to determine a possible scaling law followed by Δε. The crystal nucleation maximum-rate temperature θ2ls is given by (7,8) as a function of εls0; then, εls0 and εls0 − εlg0 = Δε have to be linear functions of θg given by (11):
![Materials 04 00869 i022]()
![Materials 04 00869 i023]()


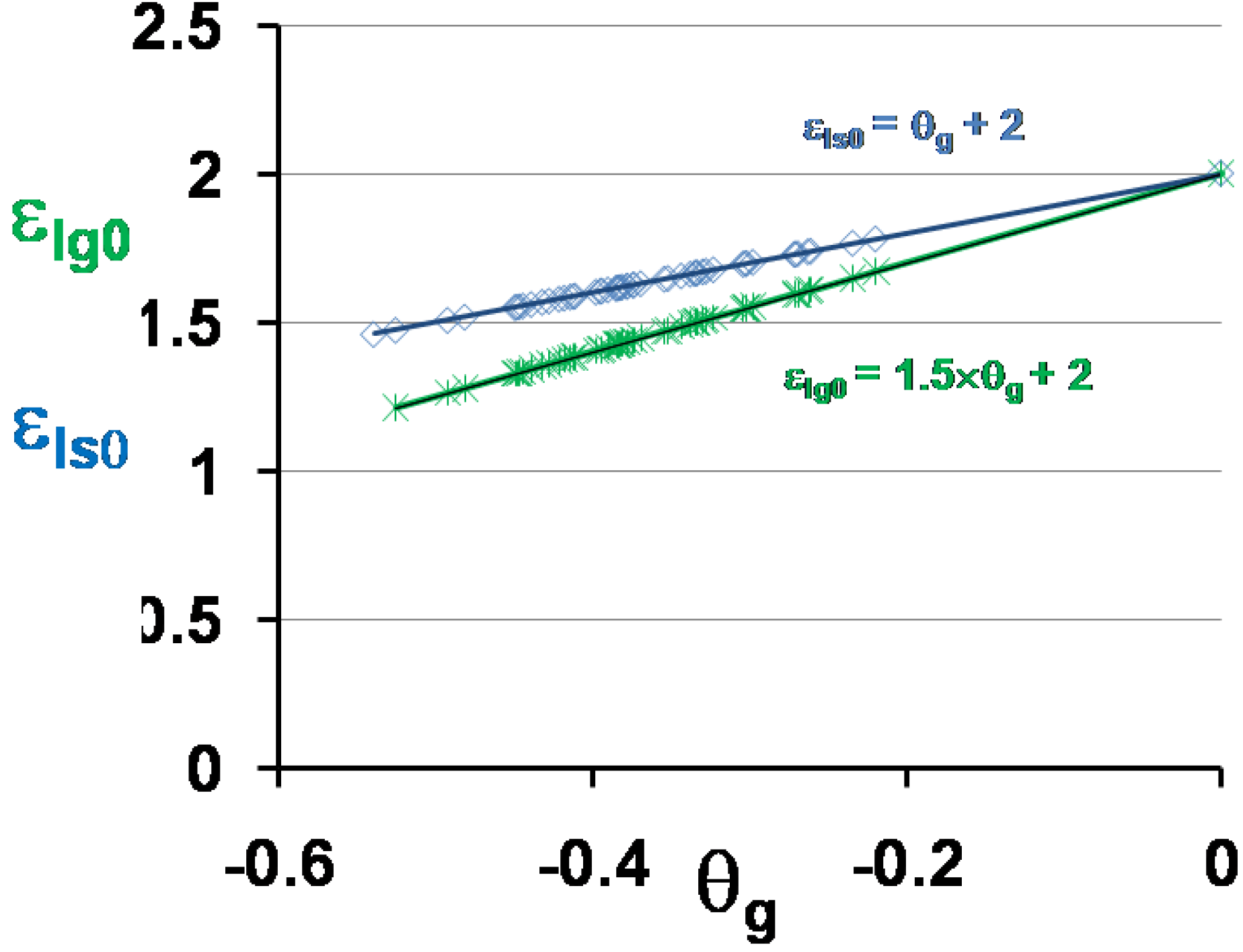
The two energy saving coefficients εls0 and εlg0 given in Table 1 are plotted as a function of θg in Figure 5. Equations (7) and (8) are used to predict the temperatures T0g and T0m also given in Table 1 and plotted as a function of Tg in Figure 6. These values are the free-volume disappearance temperatures of glass-forming melts having a thermodynamic glass transition occurring at T*g = Tg.
Figure 5.
The energy saving coefficients εls0 and εlg0 are calculated respectively using the scaling laws (18) and (19,20) and are plotted versus θg = (Tg − Tm)/Tm.
Figure 5.
The energy saving coefficients εls0 and εlg0 are calculated respectively using the scaling laws (18) and (19,20) and are plotted versus θg = (Tg − Tm)/Tm.

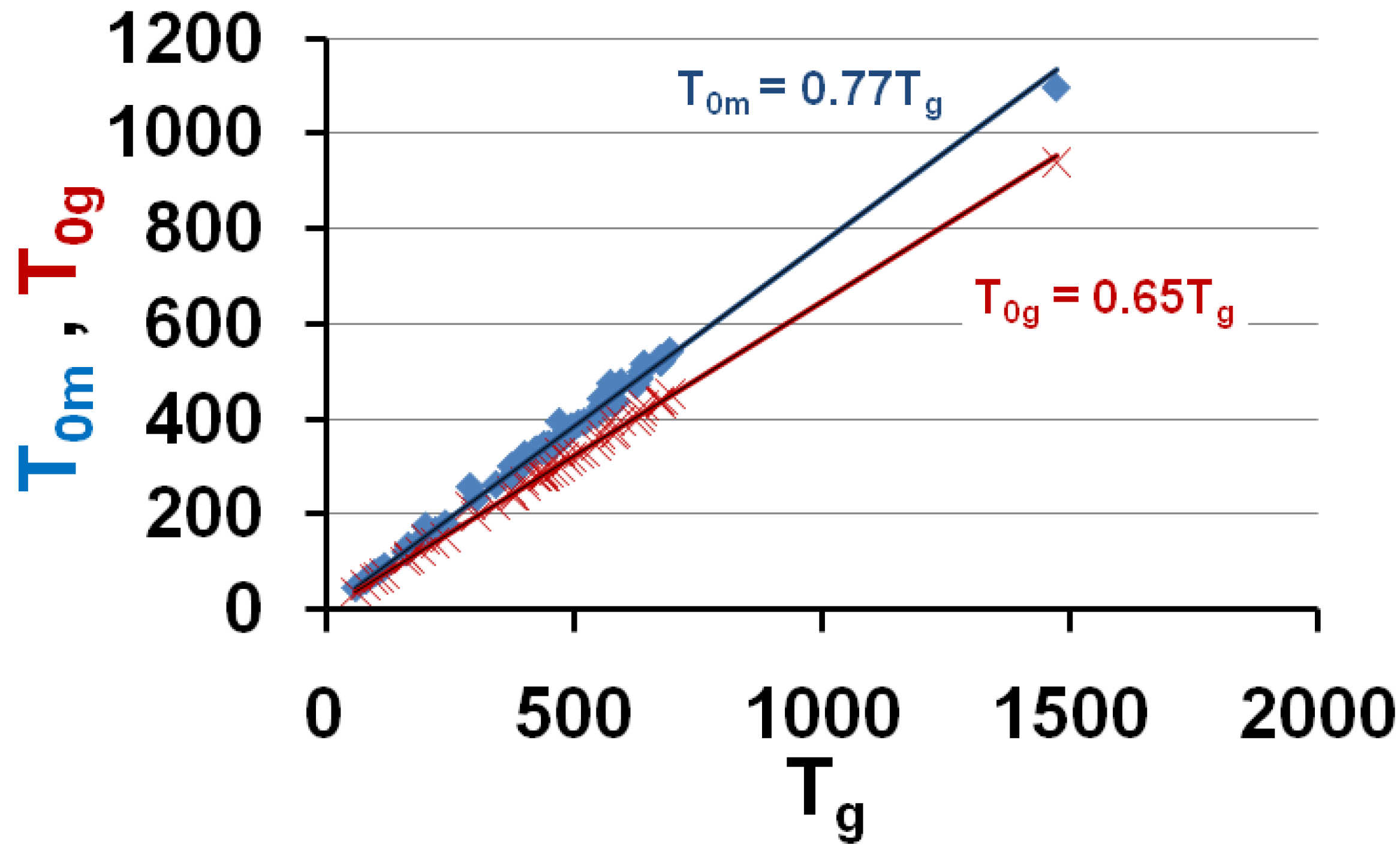
Figure 6.
The calculated values of the free-volume disappearance temperatures T0m and T0g are plotted versus Tg; they are equal to the VFT temperatures T01 and T02 represented in Figure 1. T0m = 0.77 Tg and T0g = 0.65 Tg.
Figure 6.
The calculated values of the free-volume disappearance temperatures T0m and T0g are plotted versus Tg; they are equal to the VFT temperatures T01 and T02 represented in Figure 1. T0m = 0.77 Tg and T0g = 0.65 Tg.
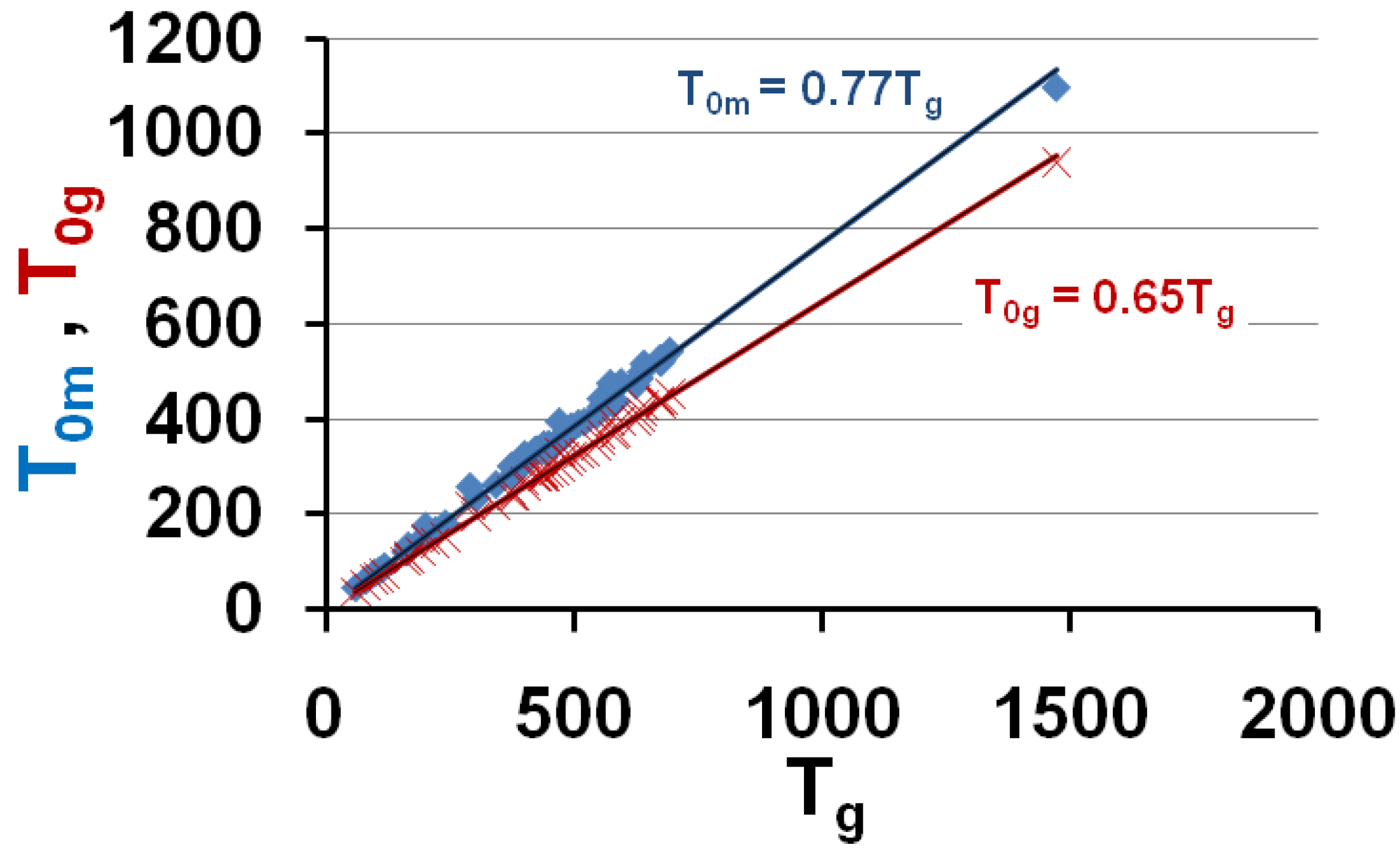
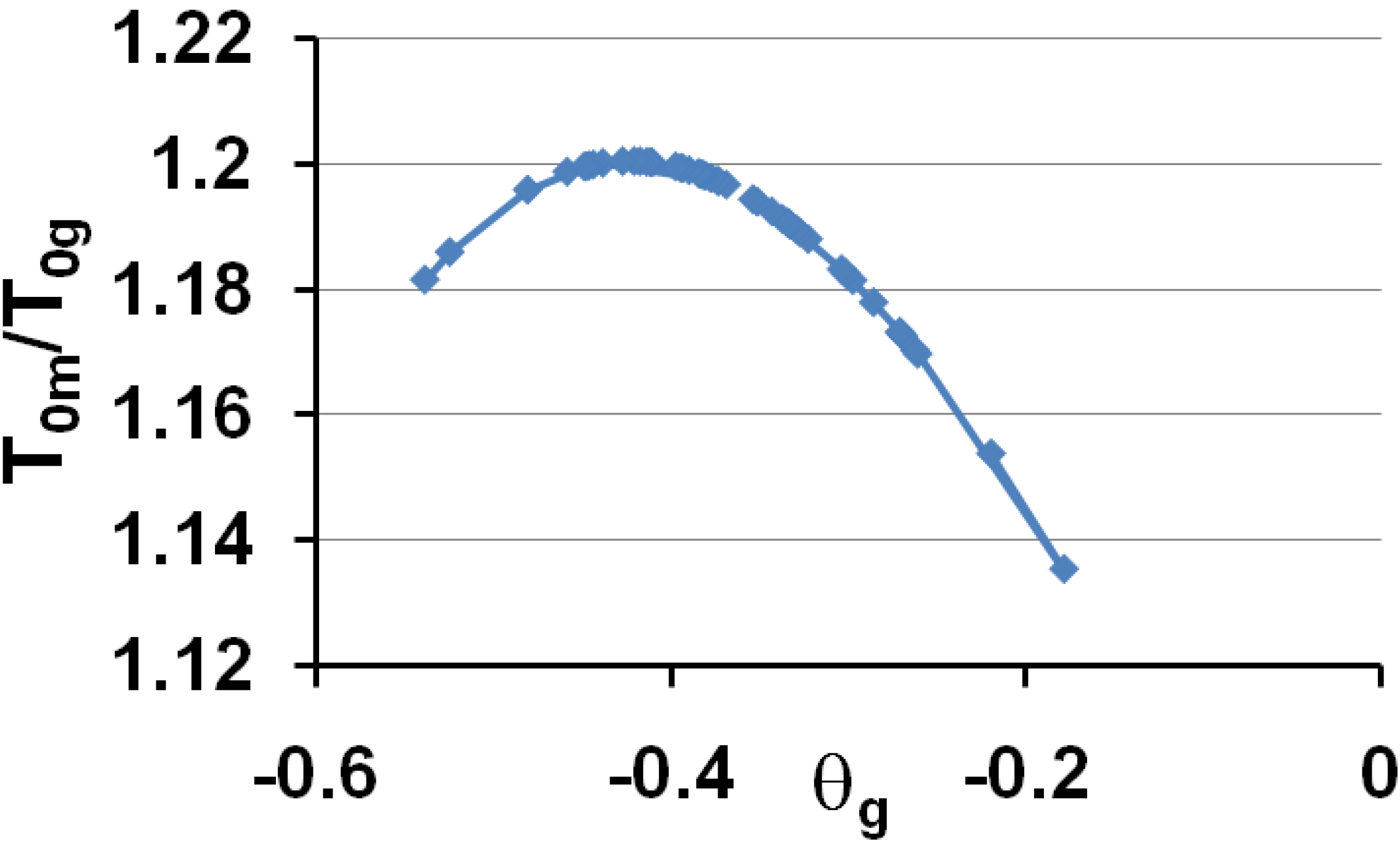
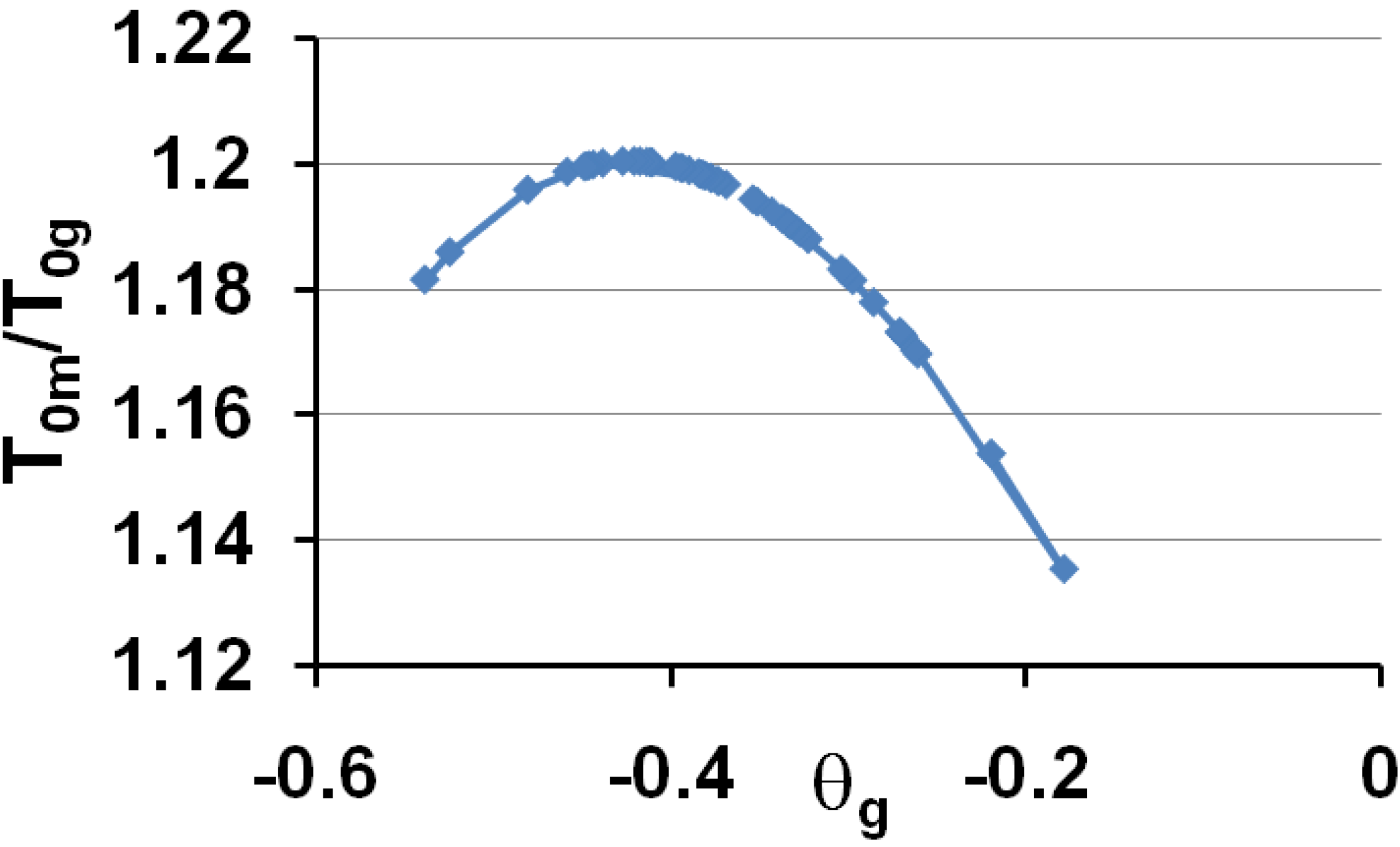
These predictions are in very good agreement with experiments when we compare Figure 6 to Figure 1 and Figure 4. Then, the vitreous transition corresponds to a crystal homogeneous nucleation temperature. A distribution of homogeneously-nucleated clusters is created when the temperature decreases down to T*g. The scaling laws (18) and (19) are obeyed and reflect intrinsic properties of glass-forming melts. The two VFT temperatures corresponding to two free-volume disappearance temperatures follow intrinsic scaling laws related to a change of the energy saving in all melts. These predictions can be more precise as shown in Figure 7 and Figure 8. In fact, the ratios Tom/Tg and T0g/Tg are weakly varying with the glass transition; the proportionality coefficients 0.77 and 0.65 are mean values for a lot of glasses and polymers having θg values larger than −0.45 and smaller than −0.2. The ratio T0m/T0g is nearly constant in the same interval of θg values as shown in Figure 6; it tends to 1 when θg tends to 0 and −2/3.
Figure 7.
The ratios Tog/Tg and T0m/Tg of the free-volume disappearance temperatures to the glass transition temperature Tg are plotted versus θg = (Tg − Tm)/Tm.
Figure 7.
The ratios Tog/Tg and T0m/Tg of the free-volume disappearance temperatures to the glass transition temperature Tg are plotted versus θg = (Tg − Tm)/Tm.
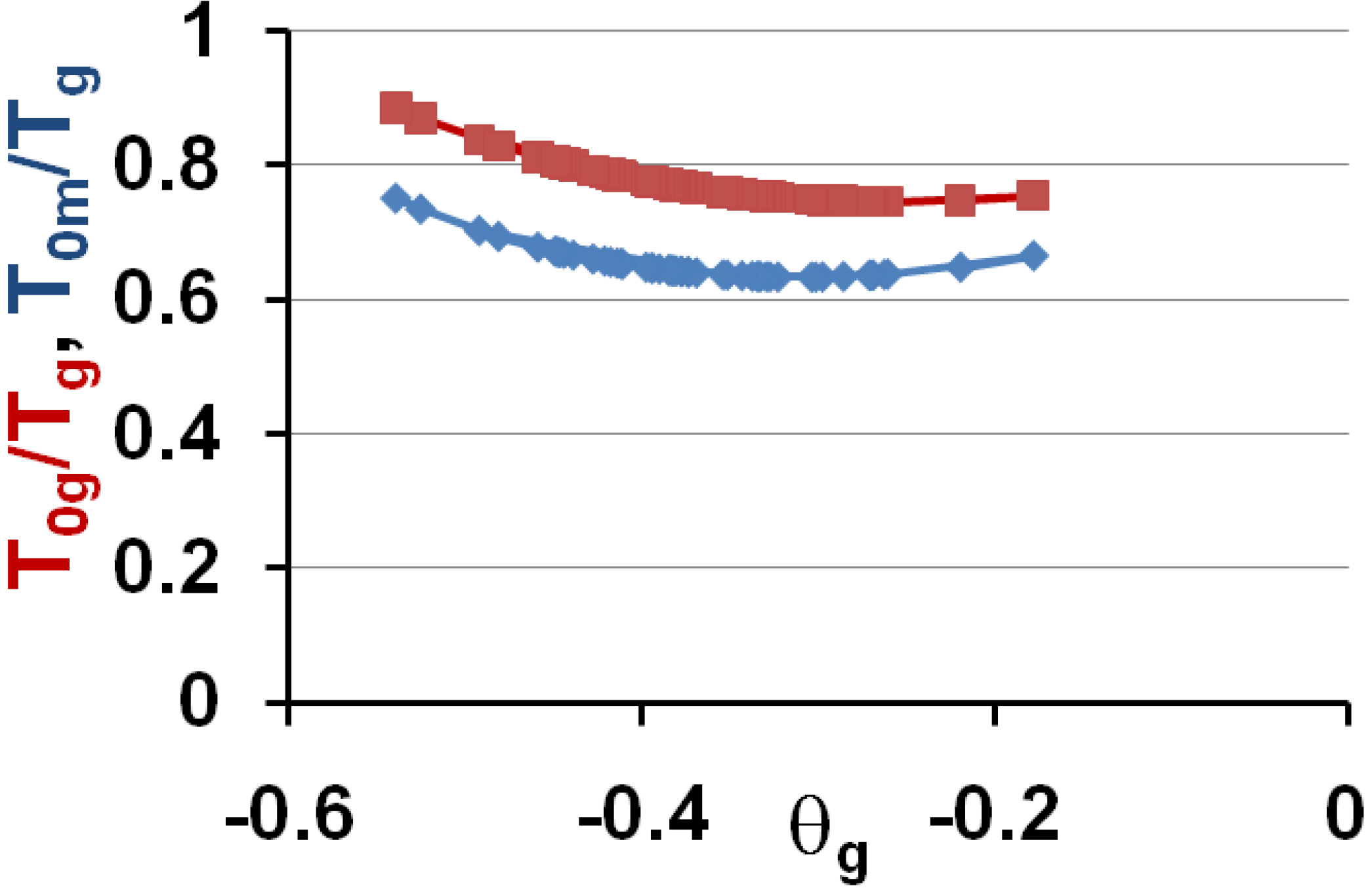
Figure 8.
The ratios of the free-volume disappearance temperatures Tom/Tog are plotted versus θg = (Tg − Tm)/Tm.
Figure 8.
The ratios of the free-volume disappearance temperatures Tom/Tog are plotted versus θg = (Tg − Tm)/Tm.
3.5. Volume Energy Saving Associated with Nascent Crystals in Non-Metallic Glass-Forming Melts
Volume energy saving associated with homogeneously-nucleated crystal formation also exists in non-metallic liquids. It could be due to an electrostatic interaction between a screen of ionic charges present in the melt and charges carried by homogeneously-nucleated crystals containing unoccupied ionic sites. Homogeneous nucleation gives rise, in a first step, to ultra-fine crystals among density fluctuations. Ions of opposite charges could be randomly distributed inside their own sub-lattices in a crystal. The mean charge carried by such crystals would be proportional to the square root of their atom number n. Counter-ions of opposite charge would screen the grain charge and induce an attractive interaction proportional to the square of the grain charge and then to the atom number n. Neel already made a similar assumption to explain the superparamagnetic (ferrimagnetic) properties of antiferromagnetic ultra-fine grains. The superparamagnetic Curie constant of ultra-fine grains is equal to the Curie constant of n paramagnetic atoms because the magnetic moments carried by different ions are randomly distributed in their own sub-lattices and the grain uncompensated magnetic moment is proportional to n1/2 [70].
The presence of volume energy saving in nonmetallic glass-forming melts is also due to a more general phenomenon associated with the formation of noncritical clusters in melts containing n atoms. The chemical potential of a small cluster is expected to differ from the bulk value. A new contribution −(p − p0)Vm ought to be added to the classical Gibbs free energy change. It depends on the Laplace pressure p applied to the cluster when a cluster is formed, p0 being the classical pressure of the melt on the solid particle, regardless of its size. This complementary contribution is not involved in the classical Gibbs free energy change because the pressure p is not homogeneous in the melt [71]. The Laplace pressure p increases with a decreasing atom number n. In addition, the energy saving is quantified when the critical cluster radius and the number of transferred electrons are very small in metallic glass-forming melts [14,15]. The temperature dependence of εls given by (1) is a general law for nascent clusters in all melts.
3.6. Thermodynamic Origin of Relaxed Enthalpy and of Out-of-Equilibrium Nucleation Temperatures Tg
Enthalpy is relaxed at the annealing temperature Ta < T*g applied after quenching the undercooled melt down to a much lower temperature. The fully-relaxed enthalpy is equal to Hr = ΔCplg (T*g) × [T*g − Ta] for Tk < Ta < T*g instead of being related to the enthalpy excess stored by an undercooled melt quenched from Ta down to the Kauzmann temperature Tk and to the entropy available below Ta. So this relaxed enthalpy correlated to the thermodynamic transition.
The annealing temperature Ta is an out-of-equilibrium homogeneous nucleation temperature of a fragile glass-forming melt and a solution of (6) corresponding, for the same value of θ0lgs, to an energy saving coefficient of nascent crystals at Tm being a little larger than the equilibrium value at a nucleation temperature equal to T*g [15]. The annealing temperature Ta is a temporary nucleation temperature during the time lag of the transient nucleation. The undercooled melt progressively relaxes enthalpy and entropy excesses stored between Ta and T*g towards their equilibrium values at T*g. The existence of this relaxed enthalpy is a strong argument in favor of a thermodynamic equilibrium at T*g. The time-dependent vitreous transition Tg is due to this endothermic heat appearing at a temperature varying with the heating rate in a DSC run. A nucleation temperature T2ls = Tg could also exist above T*g when high heating rates are used because the homogeneous nucleation temperature T2ls only depends on the energy saving coefficient εlgs0 for a well-defined ideal glass transition temperature T0g (or θ0g) as shown by (6) and Figure 9.
Figure 9.
The out-of-equilibrium homogeneous nucleation temperatures T2ls = Tg equal to homogeneous nucleation temperatures depend on energy saving coefficients εlgs0 through (6). The equilibrium transition of vit 106a (Zr58.5Cu15.6Ni12.8Al10.3Nb2.8) at T*g = 690 K has been previously determined as shown in Figure 2. The temperature T0g = 437 K is equal to T02.
Figure 9.
The out-of-equilibrium homogeneous nucleation temperatures T2ls = Tg equal to homogeneous nucleation temperatures depend on energy saving coefficients εlgs0 through (6). The equilibrium transition of vit 106a (Zr58.5Cu15.6Ni12.8Al10.3Nb2.8) at T*g = 690 K has been previously determined as shown in Figure 2. The temperature T0g = 437 K is equal to T02.
A spin-glass transition is also characterized in zero field by the presence at once of a time-dependent susceptibility cusp temperature, a phase transition temperature and, in the magnetic field, by two lines of transition Hc(T) and Hm(T) [72]. The phase diagram (H,T) of Cu–Mn has been investigated by measuring magnetocaloric effects showing the importance of the entropy S(T,H) in understanding the physics of spin glass transition. The Hc(T) is the boundary line spin-glass/non-Curie paramagnet. The Hm(T) is a cross-over line Curie/Non-Curie paramagnet corresponding to an irreversibility line and to a freezing of rigid clusters of spins [72,73,74]. The spin-glass phase transition can be separated from the irreversibility line when the magnetic field frequency increases. The existence of this type of phenomenon in glasses remains an open question because the reversible specific heat jump always occurs in As2Se3 at the same vitreous transition regardless of the heating and cooling rates [67].
4. Summary and Complementary Information on the Two Crystal Nucleation Temperatures
New equations governing the crystal nucleation, reflecting the energy saving associated with Fermi energies equalization of nascent crystals and melt, have been used and applied to all glass-forming melts. The vitreous transition is characterized by freezing at a crystal homogeneous nucleation temperature only determined by thermodynamics considerations. We have shown, for the first time, that an energy scale governs the vitreous transition. This material constant does not strictly depend on the viscosity, even if the viscosity is high and nearly the same at T*g, because the energy barrier for crystal growth nucleation ΔG*2ls divided by kBTg is nearly the same in all glass-forming melts. The energy barrier Δf* to transfer an atom from the melt to a nascent crystal divided by kBT*g is also nearly the same and is a little smaller than the one from transport across the melt-crystal interfaces at the first crystallization temperature which is induced by surviving intrinsic crystals.
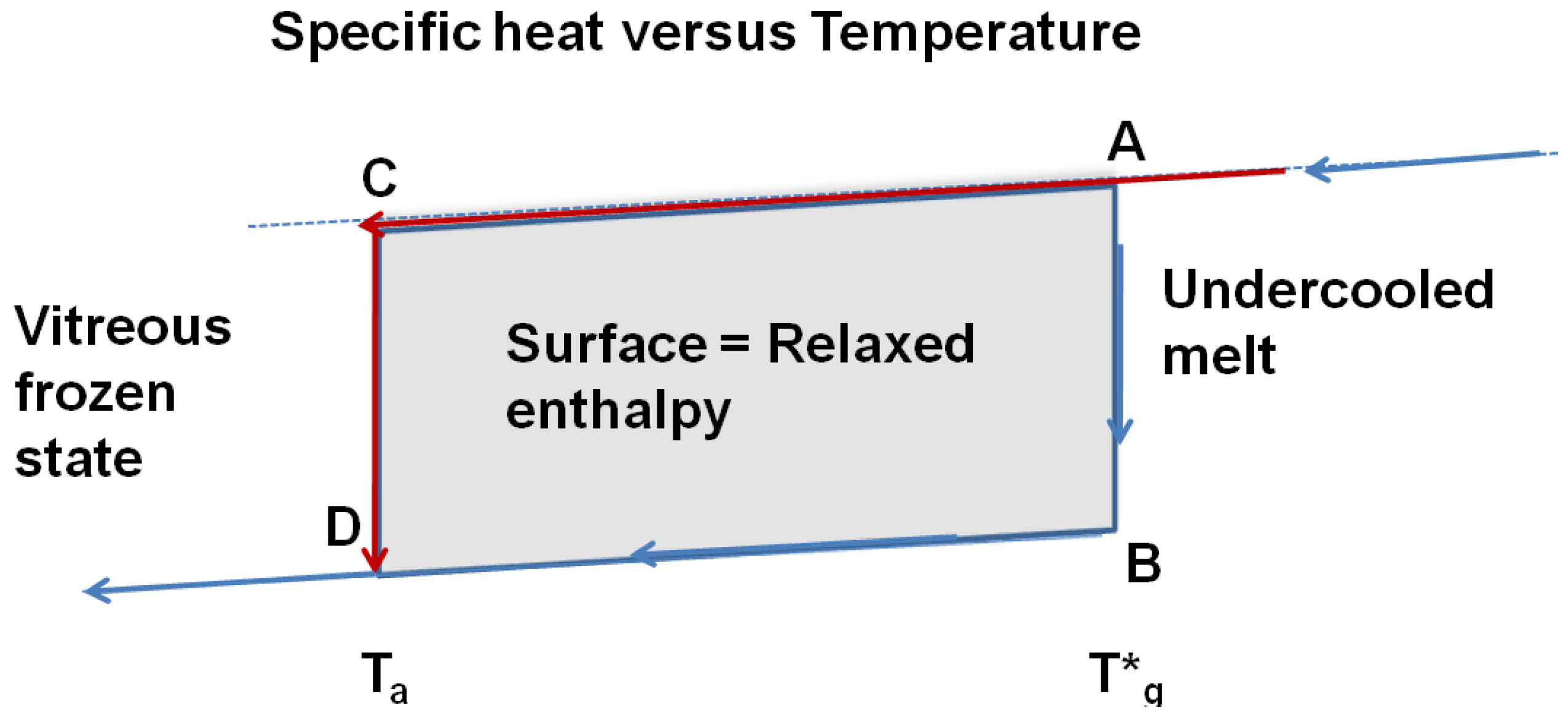
These findings are in agreement with published works having shown that the reversible specific heat jump at T*g does not depend on time and on sample thermal history. In addition, the relaxed enthalpy disappears at the thermodynamic transition T*g and its maximum value obtained at each annealing temperature Ta after quenching the undercooled liquid to lower temperatures, is given by ![Materials 04 00869 i020]() , ΔCpgl being the specific heat jump at T*g. The apparent specific heat jump at Tg calculated from the heat flux measurement is equal to the reversible one. The specific heat jump deduced from heat flux measurement occurs at a temperature Tg depending on the heating rate. There is no visible anomaly at T*g in a DSC run. This phenomenon is schematized in Figure 10. The glass transition temperatures Tg determined by DSC correspond to out-of-equilibrium crystal homogeneous nucleation temperatures and to out-of-equilibrium values of the energy saving coefficient εlg0.
, ΔCpgl being the specific heat jump at T*g. The apparent specific heat jump at Tg calculated from the heat flux measurement is equal to the reversible one. The specific heat jump deduced from heat flux measurement occurs at a temperature Tg depending on the heating rate. There is no visible anomaly at T*g in a DSC run. This phenomenon is schematized in Figure 10. The glass transition temperatures Tg determined by DSC correspond to out-of-equilibrium crystal homogeneous nucleation temperatures and to out-of-equilibrium values of the energy saving coefficient εlg0.
, ΔCpgl being the specific heat jump at T*g. The apparent specific heat jump at Tg calculated from the heat flux measurement is equal to the reversible one. The specific heat jump deduced from heat flux measurement occurs at a temperature Tg depending on the heating rate. There is no visible anomaly at T*g in a DSC run. This phenomenon is schematized in Figure 10. The glass transition temperatures Tg determined by DSC correspond to out-of-equilibrium crystal homogeneous nucleation temperatures and to out-of-equilibrium values of the energy saving coefficient εlg0.
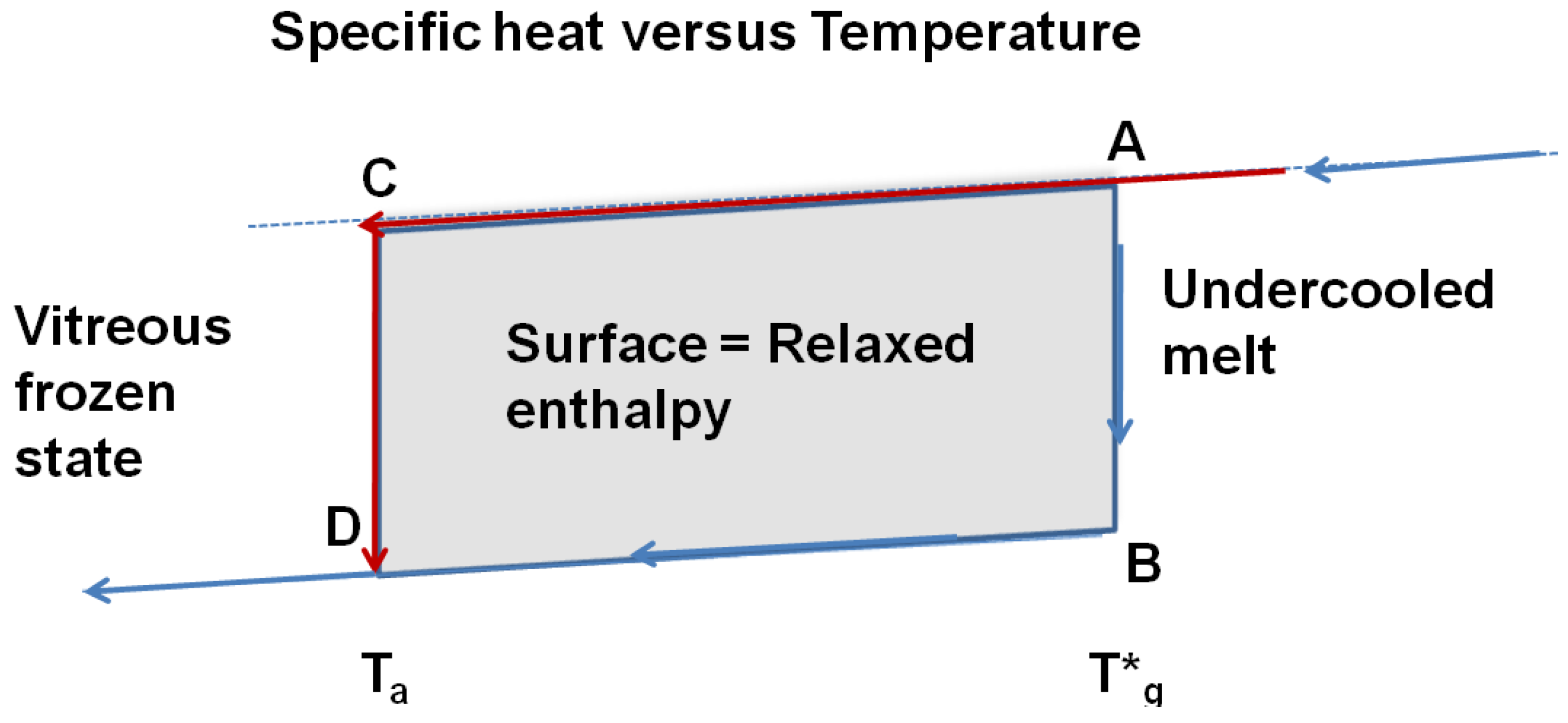
Figure 10.
The reversible thermodynamic vitreous transition occurs at T = T*g; the specific heat decreases along AB; the specific heat of the vitreous fully-frozen state along BD is equal to the crystallized state. The undercooled melt is quenched at low temperatures and annealed at the temperature Ta. The transformation from C to D relaxes an enthalpy equal to the surface ABDC when the time-lag necessary for cluster formation has evolved.
Figure 10.
The reversible thermodynamic vitreous transition occurs at T = T*g; the specific heat decreases along AB; the specific heat of the vitreous fully-frozen state along BD is equal to the crystallized state. The undercooled melt is quenched at low temperatures and annealed at the temperature Ta. The transformation from C to D relaxes an enthalpy equal to the surface ABDC when the time-lag necessary for cluster formation has evolved.
The thermodynamic transition T*g is a linear function of the energy saving divided by the fusion heat associated, in a nascent crystal formation, with the equalization of Fermi energy or chemical potential of nascent crystals and glass-forming melts. It is possible to predict, only using T*g and the melting temperature Tm, a free-volume disappearance temperature equal to the VFT temperature of fragile-glass-forming melts deduced from viscosity and relaxation time measurements above and near T*g. There are two crystal homogeneous nucleation temperatures which follow scaling laws linearly dependent on two energy saving coefficients εls0 in the crystal formation as there are two VFT temperatures.
Experimentally, the first-crystallization temperature occurs when cooling the glass-forming melt at a lesser rate than the critical one, down to a temperature that is higher than the homogeneous nucleation temperature and is generally induced by intrinsic heterogeneous crystals which reduce the energy barrier for crystal growth. The isothermal crystallization time depends on overheating and undercooling temperatures and leads to a time-temperature-transformation diagram induced by intrinsic nuclei. The nose temperature of this diagram depends on the overheating temperature, the surviving crystal size and the energy saving εls.
The second nucleation temperature is lower and gives rise by homogeneous nucleation to imperfect crystals having an energy barrier for diffusion from the melt to the crystal slightly smaller than the first. The free-volume disappearance temperature of the undercooled melt decreases from T0m to T0g. A glass state is obtained by quenching the melt using a cooling rate larger than its critical value. The vitreous transition temperature T*g occurs at a homogeneous nucleation maximum-rate temperature determined by a smaller value εlgs(θg) of the energy saving associated with a smaller VFT temperature. The relaxation time leading to vitreous state is the time lag for initial formation of a homogeneously-nucleated-cluster distribution during the transient nucleation. These entities could be imperfect crystals. Their formation is a preliminary step during the long time leading to crystallization. The time dependence of various properties depends on the time lag τns associated to the transient nucleation and to the steady-state nucleation time tsn depending on the energy barrier for crystal growth.
The model used in this paper is also based on previous publications related to the classical Gibbs free energy change for a crystal formation in an undercooled melt that has been completed by an energy saving associated with the equalization of Fermi energies or chemical potentials of melt and nascent crystal. This analysis only works for nascent crystals in an out-of-equilibrium state having a radius smaller than the critical radius for crystal growth because J. W. Gibbs’s phase coexistence rule predicts the absence of energy saving for radii larger than the critical one when solid and liquid phases are at equilibrium at the melting temperature.
5. Conclusions
The vitreous transition is a new type of phase transition from undercooled melt to frozen state, without entropy and enthalpy change occurring at a temperature T*g, which corresponds to the maximum nucleation rate temperature of homogeneously-nucleated crystals in bulk metallic and non-metallic glass-forming melts. Because of the melt freezing, the steady-state nucleation time is too long to ever reach the divergence of the correlation length in critical phenomenon and the crystal’s growth.
These nascent crystals would be formed with a free energy change which differs from the classical Gibbs free energy change used in many nucleation models. A complementary energy saving exists which depends on the atom or molecule number n involved in these crystals. We have shown the existence of two homogeneous nucleation temperatures associated with two energy savings, which follow scaling laws as a function of the vitreous transition temperature. The nascent crystals acting at the vitreous transition could contain a lot of unoccupied ionic sites as compared with crystals surviving in the melt and acting as growth nuclei at higher temperatures.
The glass freezing occurs without entropy and enthalpy changes; these changes can only appear at unattainable times when crystallization occurs. This analysis shows that the frozen and the solid states have the same equilibrium specific heat below the glass transition, eliminating all speculations about other configurational contributions and phase transitions.
The disappearance temperature of the fully-relaxed enthalpy, as described in previous publications, does not depend on time and is equal to the thermodynamic temperature T*g. The specific heat jump accompanying this phase transition is deduced from the linear variation of the relaxed enthalpy with temperature. The DSC runs are not able to detect T*g because the enthalpy is continuous at this temperature.
The existence of a vitreous transition viewed as a constant of material was initially established by experiments eight years ago and published by the University of Pardubice.
Acknowledgements
This work was also sponsored by the sino-french Laboratory for the Application of Superconductors and Magnetic Materials involving the Northwest Institute for Non-ferrous Metal Research (NIN), the Northwestern Polytechnical University (NPU) in Xi’an (P.R. China). Thanks are due to Lian Zhou for NIN support as, Jean Etourneau, Philippe Odier, Jean-Louis Soubeyroux, Jean-Louis Tholence for their suggestions or their criticisms, Dr Jinna Mei from NPU and J.L. Soubeyroux for fully-relaxed enthalpy measurements of some glass-forming melts [75].
References
- Zarzycki, J. Les Verres et l’Etat Vitreux; Masson: Paris, France, 1982. [Google Scholar]
- Gutzov, I.; Schmeltzer, J. The Vitreous State; Springer-Verlag: Berlin, Germany, 1995. [Google Scholar]
- Angell, C.A. Formation of glasses from liquids and biopolymers. Science 1995, 267, 1924–1935. [Google Scholar] [CrossRef]
- Berthier, L.; Biroli, G.; Bouchaud, J.; Cipelletti, L.; Masri, D.E.; L’Hôte, D.; Ladieu, F.; Pierno, M. Direct experimental evidence of a growing length scale accompanying the glass transition. Science 2005, 310, 1797–1800. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Awasaki, T.; Shintani, H.; Watanabe, K. Critical-like behavior of glass-forming liquids. Nature Mater. 2010, 9, 324–331. [Google Scholar] [CrossRef]
- Kauzmann, W. The nature of the glassy state and the behavior of liquids at low temperatures. Chem. Rev. 1948, 43, 219–256. [Google Scholar] [CrossRef]
- Liu, C.-Y.; He, J.; Keunings, R.; Bailly, C. New linearized relation for the universal viscosity-temperature behavior of polymer melts. Macromolecules 2006, 39, 8867–8869. [Google Scholar] [CrossRef]
- Williams, M.L.; Landel, R.F.; Ferry, J.D. Temperature dependence of relaxation mechanisms in amorphous polymers and other glass-forming melts. J. Am. Chem. Soc. 1955, 77, 3701–3707. [Google Scholar] [CrossRef]
- Takeuchi, A.; Kato, H.; Inoue, A. Vogel-Fulcher-Tammann plot for viscosity scaled with temperature interval between actual and ideal glass transitions for metallic glasses in liquid and supercooled liquid states. Intermetallics 2010, 18, 406–411. [Google Scholar]
- Doolittle, A.K. Studies in Newtonian Flow. II. The dependence of the viscosity of liquids on free-space. J. Appl. Phys. 1951, 22, 1471–1475. [Google Scholar] [CrossRef]
- Ramachandrarao, P.; Dubey, K.S. A thermodynamic approach to the viscous behaviour of glass-forming liquids. Mat. Sc. Eng. B 1995, 32, 285–293. [Google Scholar] [CrossRef]
- Gibbs, J.H.; DiMarzio, E.A. Nature of the glass transition and the glassy state. J. Chem. Phys. 1958, 28, 373–383. [Google Scholar] [CrossRef]
- Holubovà, J.; Cernozek, Z.; Cernoskovà, E. The study of the glass transition by the stepscan DSC technique. J. Optoelectron. Adv. Mat. 2005, 7, 2671–2676. [Google Scholar]
- Tournier, R.F. Crystal growth nucleation and Fermi energy equalization of intrinsic spherical nuclei in glass-forming melts. Sci. Technol. Adv. Mater. 2009, 10, 014607:1–014607:12. [Google Scholar]
- Tournier, R.F. Crystal nucleation and equalization of Fermi energies of intrinsic nuclei and glass-forming melts. J. Phys. Confer. Ser. 2009, 144, 012116:1–012116:4. [Google Scholar] [CrossRef]
- Tournier, R.F. Presence of intrinsic growth nuclei in overheated and undercooled liquid elements. Phys. B 2007, 392, 79–93. [Google Scholar] [CrossRef]
- Vinet, B.; Magnusson, L.; Fredriksson, H.; Desré, P.J. Correlations between surface and interface energies with respect to crystal nucleation. J. Coll. Interf. Sci. 2002, 255, 363–374. [Google Scholar] [CrossRef]
- Turnbull, D.; Cech, R.E. Microscopic observation of the solidification of small metal droplets. J. Appl. Phys. 1950, 21, 804–810. [Google Scholar] [CrossRef]
- Bosio, L.; Defrain, A.; Epelboin, I. Changements de phase du gallium à la pression atmosphérique. J. Phys. France 1966, 27, 61–71. [Google Scholar] [CrossRef]
- Hays, C.C.; Johnson, W.L. Undercooling of bulk metallic glasses processed by electrostatic levitation. J. Non-Cryst. Solids 1999, 250-252, 596–600. [Google Scholar] [CrossRef]
- Tournier, R.F. Expected properties of gold melt containing intrinsic nuclei. In Proceedings of the 6th International Conference on Electromagnetic Processing of Materials, EPM 2009, Dresden, Germany, 19–23 October 2009; pp. 304–307.
- Tournier, R.F. Undercooling versus Overheating of Liquid Elements Containing Intrinsic Nuclei: Application to Magnetic Texturing. In Presented at the 4th International Workshop on Materials Analysis and Processing in Magnetic Fields Organized by Hamid Garmestani, Atlanta, GA, USA, 10–12 May 2010.
- Tournier, R.F. Nucleation of crystallization in titanium and vitreous state in glass-forming melt. In Proceedings of the 12th World Conference on Titanium, Beijing, China, 19–24 June 2011.
- Hirata, A.; Hirotsu, Y.; Ohkubo, T.; Hanada, T.; Bengus, V.Z. Compositional dependence of local atomic structures in amorphous Fe100−xBx alloys studied by electron diffraction and high-resolution electron microscopy. Phys. Rev. B 2006, 74, 214206:1–214206:9. [Google Scholar]
- Sidorov, V.; Popel, P.; Calvo-Dahlborg, M.; Dahlborg, U.; Manov, V. Heat treatment of iron-based melts before quenching. Mater. Sc. Eng. A 2001, 304-306, 480–486. [Google Scholar] [CrossRef]
- Kokshenev, V.B. Characteristic temperatures of liquid-glass transition. Phys. A 1999, 262, 88–97. [Google Scholar] [CrossRef]
- Busch, R.; Liu, W.; Johnson, W.L. Thermodynamics and kinetics of the Mg65Cu25Y10 bulk metallic glass forming liquid. J. Appl. Phys. 1998, 83, 4134–4441. [Google Scholar] [CrossRef]
- Shadowspeaker, L.; Busch, R. On the fragility of Nb-Ni-based and Zr-based bulk metallic glasses. Appl. Phys. Lett. 2004, 85, 2508–2510. [Google Scholar] [CrossRef]
- Lu, I.-R.; Görler, G.P.; Willnecker, R. Specific volume of glass-forming liquid Pd43Cu27Ni10P20 and related thermodynamic aspects of the glass transition. Appl. Phys. Lett. 2002, 80, 4534–4536. [Google Scholar] [CrossRef]
- Yavari, A.R.; Le Moulec, A.; Nishiyama, N.; Inoue, A.; Vaughan, G.; Kvick, A.; Botta, W.J. Glass Transition Tg and quenched-in free volume in bulk metallic glasses measured by X-ray diffraction. J. Metast. Nanocryst. Mater. 2004, 20-21, 23–28. [Google Scholar] [CrossRef]
- Nishiyama, N.; Inoue, A. Glass transition behavior and viscous flow working of Pd40Cu30Ni10P20 amorphous alloy. Mater. Trans. JIM 1999, 40, 64–71. [Google Scholar] [CrossRef]
- Dubey, K.S.; Ramachandrarao, P.; Lele, S. Thermodynamic and viscous behavior of undercooled liquids. Thermochim. Acta 1996, 280/281, 25–62. [Google Scholar] [CrossRef]
- Kouamé, N.; Sei, J.; Houphouët-Boigny, D.; Kra, G.; Jumas, J.C.; Olivier-Fourcade, J. Propriétés thermiques et optiques des verres du système Sb2S3–As2S3. C. R. Chimie 2007, 10, 498–501. [Google Scholar] [CrossRef]
- Birge, N.O. Specific heat spectroscopy of glycerol and propylene glycol near the glass transition. Phys. Rev. B. 1986, 34, 1631–1642. [Google Scholar] [CrossRef]
- Schönhals, A.; Kremer, F.; Hofmann, A.; Fischer, E.W. Anomalies in the scaling of the α-relaxation studied by dielectric spectroscopy. Phys. A 1993, 201, 263–269. [Google Scholar] [CrossRef]
- Mishra, R.K.; Dubey, K.S. Glass-forming ability of materials: A thermodynamic approach. J. Non-Cryst. Sol. 2009, 355, 2199–2204. [Google Scholar] [CrossRef]
- Yang, Z.; Lam, C.H.; Dimasi, E.; Bouet, N.; Jordan-Sweet, J.; Tsui, O.K.C. Method to measure the viscosity of nanometer liquid films from the surface fluctuations. Appl. Phys. Lett. 2009, 94, 251906:1–251906:3. [Google Scholar]
- Lu, I.-R.; Wilde, G.; Görler, G.P.; Willnecker, R. Thermodynamic properties of Pd-based glass-forming alloys. J. Non-Cryst. Sol. 1999, 250-252, 577–581. [Google Scholar] [CrossRef]
- Schroers, J. On the formability of bulk metallic glass in its supercooled liquid state. Acta Mater. 2008, 56, 471–478. [Google Scholar] [CrossRef]
- Johari, G.P. Heat capacity and entropy of an equilibrium liquid from Tg to 0 K, and examining the conjectures of an underlying thermodynamic transition. Chem. Phys. 2001, 265, 217–231. [Google Scholar] [CrossRef]
- Henderson, D.W.; Ast, D.G. Viscosity and crystallization kinetics of As2Se3. J. Non-Cryst. Sol. 1984, 64, 43–70. [Google Scholar] [CrossRef]
- Waniuk, T.A.; Busch, R.; Masuhr, A.; Johnson, W.L. Equilibrium viscosity of the Zr41.2Ti13.8Cu12.5Ni10Be22.5 bulk metallic glass-forming liquid and viscous flow during relaxation, phase separation, and primary crystallization. Acta Mater. 1998, 46, 5229–5236. [Google Scholar] [CrossRef]
- Lu, Z.P.; Li, Y.; Liu, C.T. Glass-forming tendency of bulk La–Al–Ni–Cu–(Co) metallic glass-forming melts. J. Appl. Phys. 2003, 93, 286–290. [Google Scholar] [CrossRef]
- Lu, Z.P.; Liu, C.T. A new glass-forming ability criterion for bulk metallic glasses. Acta Mater. 2002, 50, 3501–3512. [Google Scholar] [CrossRef]
- Lu, Z.P.; Li, Y.; Ng, S.C. Reduced glass transition temperature and glass forming ability of bulk glass forming alloys. J. Non-Cryst. Sol. 2000, 270, 103–114. [Google Scholar] [CrossRef]
- Angell, C.A.; Rao, K.J. Configurational excitations in condensed matter, and the “bond lattice” model for liquid-glass transition. J. Chem. Phys. 1971, 57, 470–481. [Google Scholar] [CrossRef]
- Hatase, M.; Hanaya, M.P.; Hikima, T.; Oguni, M. Discovery of homogeneous-nucleation-based crystallization in simple glass-forming liquid of toluene below its glass-transition temperature. J. Non-Cryst. Sol. 2003, 307-310, 257–263. [Google Scholar]
- Kato, H.; Wada, T.; Hasegawa, M.; Saida, J.; Inoue, A. Fragility and thermal stability of Pt- and Pd-based bulk glass forming liquids and their correlation with deformability. Scripta Materialia 2006, 54, 2023–2027. [Google Scholar] [CrossRef]
- Kui, H.-W.; Turnbull, D. The heat capacity of Ni40Pd40P20 in the liquid, glass and crystallized states. J. Non-Cryst. Sol. 1987, 94, 62–69. [Google Scholar] [CrossRef]
- Legg, B.A.; Schroers, J.; Busch, R. Thermodynamics, kinetics, and crystallization of Pt57.3 Cu14.6 Ni5.3 P22.8 bulk metallic glass. Acta Mater. 2007, 55, 1109–1116. [Google Scholar] [CrossRef]
- Chen, H.S.; Goldstein, M. Anomalous visco-elastic behavior of metallic-glasses of Pd–Si-based alloys. J. Appl. Phys. 1972, 43, 1642–1648. [Google Scholar] [CrossRef]
- Mukherjee, S.; Schroers, J.U.; Zhou, Z.; Johnson, W.L.; Rhim, W.-K. Viscosity and specific volume of bulk metallic glass-forming alloys and their correlation with glass-forming stability. Acta Mater. 2004, 52, 3689–3695. [Google Scholar] [CrossRef]
- Màlek, J.; Svoboda, R.; Pustkova, P.; Cicmanec, P. Volume and enthalpy relaxation of a-Se in the glass transition region. J. Non-Cryst. Sol. 2009, 355, 264–272. [Google Scholar] [CrossRef]
- Gallino, I.; Shah, M.B.; Busch, R. Enthalpy relaxation and its relation to the thermodynamics and crystallization of the Zr58.5Cu15.6Ni12.8Al10.3Nb2.8 bulk metallic glass-forming alloy. Acta Mater. 2007, 55, 1367–1376. [Google Scholar] [CrossRef]
- Meng, Q.G; Zhang, S.G.; Li, J.G.; Bian, X.F. Strong liquid behavior of Pr55Ni25Al20 bulk metallic glass. J. Alloys Compounds 2007, 431, 191–196. [Google Scholar] [CrossRef]
- Huang, H.J.; Shen, J.; Sun, X.B.; Yu, J.F. A new Ti–Zr–Hf–Cu–Ni–Si–Sn bulk amorphous alloy with high glass-forming ability. J. Alloys Compounds 2007, 427, 171–175. [Google Scholar] [CrossRef]
- Glade, S.C.; Johnson, W.L. Viscous flow of the Cu47Ti34Zr11Ni8 glass forming alloy. J. Appl. Phys. 2000, 87, 7249–7251. [Google Scholar] [CrossRef]
- He, S.; Liu, Y.; Huang, B.; Li, Z.; Wu, H. Effect of Zr on glass-forming ability and crystallization kinetics of Y56Al24Co20 metallic glass. J. Mater. Proc. Techn. 2008, 204, 179–183. [Google Scholar] [CrossRef]
- Zheng, Q.; Xu, J.; Ma, E. High glass-forming ability correlated with fragility of Mg–Cu(Ag)–Gd alloys. J. Appl. Phys. 2007, 102, 113519:1–113519:5. [Google Scholar]
- Busch, R.; Bakke, E.; Johnson, W.L. Viscosity of the supercooled liquid and relaxation at the glass transition of the Zr46.75Ti8.25Cu7.5Ni10Be27.5 bulk metallic glass forming alloy. Acta Metall. 1998, 46, 4725–4732. [Google Scholar]
- Zhang, B.; Wang, R.J.; Zhao, D.Q.; Pan, M.X.; Wang, W.H. Properties of Ce-based bulk metallic glass-forming alloys. Phys. Rev. B 2004, 70, 224208:1–224208:7. [Google Scholar]
- Bian, X.F.; Sun, B.A.; Hu, L.N.; Jia, Y.B. Fragility of superheated melts and glass-forming ability in Al-based alloys. Phys. Lett. A 2005, 335, 61–67. [Google Scholar] [CrossRef]
- Chen, H.; Turnbull, D.J. Evidence of a glass-liquid transition in gold-germanium-silicon. J. Chem. Phys. 1968, 48, 2560–2571. [Google Scholar] [CrossRef]
- Adam, G.; Gibbs, J.H. On the temperature dependence of cooperative relaxation properties in glass-forming liquids. J. Chem. Phys. 1965, 43, 139–146. [Google Scholar] [CrossRef]
- Turnbull, D.; Fisher, J.C. Rate of nucleation in condensed systems. J. Chem. Phys. 1949, 17, 71–73. [Google Scholar] [CrossRef]
- Cernosek, Z.; Holubova, J.; Cernoskova, E. Kauzmann temperature and the glass transition. J. Optoelectron. Adv. Mater. 2005, 7, 2941. [Google Scholar]
- Cernosek, Z.; Holubova, J.; Cernoskova, E.; Liska, M. Enthalpic relaxation and the glass transition. J. Optoel. Adv. Mater. 2002, 4, 489–503. [Google Scholar]
- Mehl, P.M. Determination of enthalpy relaxation times using traditional differential scanning calorimetry for glycerol and for propylene glycol. Thermochem. Acta 1996, 272, 201–209. [Google Scholar] [CrossRef]
- Fransson, A.; Bäckström, G. Isothermal enthalpy relaxation of glycerol. Int. J. Thermophys. 1987, 8, 351–362. [Google Scholar] [CrossRef]
- Néel, L. Superparamagnétisme de grains très fins antiferromagnétiques. C. R. Am. Sci. 1961, 252, 4075–4080. [Google Scholar]
- Wu, D.T.; Granasy, L.; Spaepen, F. Nucleation and the solid-liquid interfacial free energy. MRS Bulletin. Available online: http://www.mrs.org/publications/bulletin (accessed on December 2004).
- Berton, A.; Chaussy, J.; Odin, J.; Rammal, R.; Tournier, R. Magnetocaloric investigation of (H,T) phase diagram of Cu-Mn spin glass. J. Physique-Lett. 1982, 43, L153–L158. [Google Scholar] [CrossRef]
- Tholence, J.L; Tournier, R. Susceptibility and remanent magnetization of a spin glass. J. Phys. Suppl. Coll. 1974, 35, C4:229–C4:235. [Google Scholar]
- Mydosh, J.A. Spin Glasses: An Experimental Introduction; Taylor & Francis: London, UK, 1993. [Google Scholar]
- Mei, J.N.; Soubeyroux, J.L.; Blandin, J.J.; Li, J.S.; Kou, H.C.; Fu, H.Z.; Zhou, L. Structural relaxation of Ti40Zr25Ni8Cu9Be18 bulk metallic glass. J. Non-Cryst. Sol. 2011, 357, 110–115. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Tournier F., R. Thermodynamic Origin of the Vitreous Transition. Materials 2011, 4, 869-892. https://doi.org/10.3390/ma4050869
AMA Style
Tournier F. R. Thermodynamic Origin of the Vitreous Transition. Materials. 2011; 4(5):869-892. https://doi.org/10.3390/ma4050869
Chicago/Turabian StyleTournier F., Robert. 2011. "Thermodynamic Origin of the Vitreous Transition" Materials 4, no. 5: 869-892. https://doi.org/10.3390/ma4050869