Preparation of Polyaminopyridines Using a CuI/l-Proline-Catalyzed C-N Polycoupling Reaction

Abstract
:
1. Introduction
2. Results and Discussion
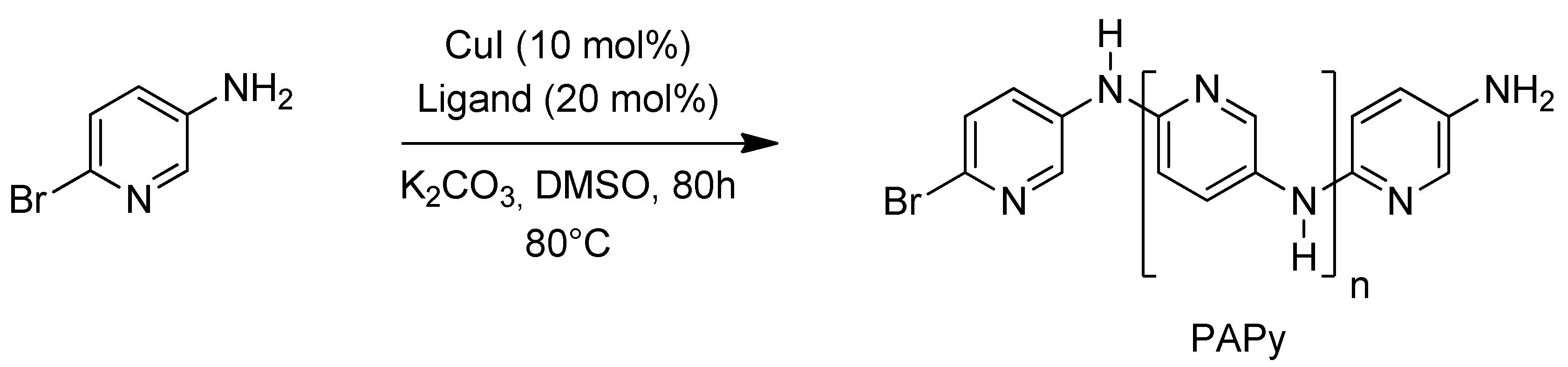
2.1. Solubility and Systematic Study of Polycoupling Reaction
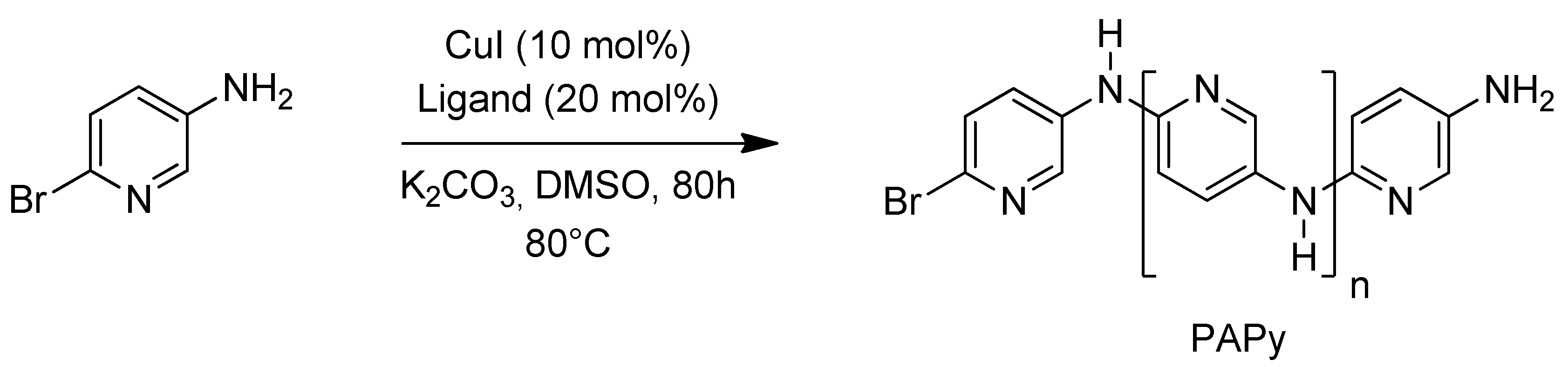
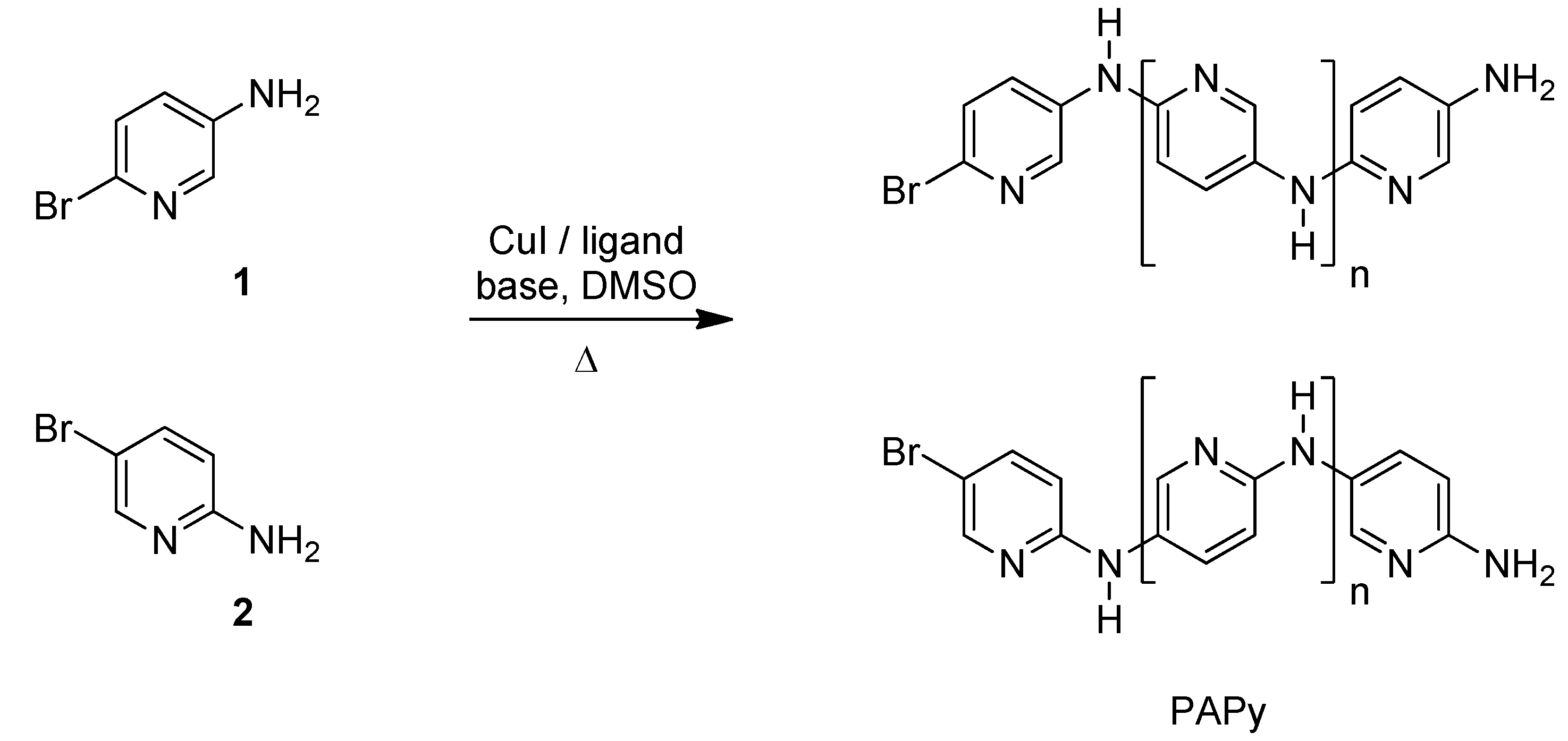

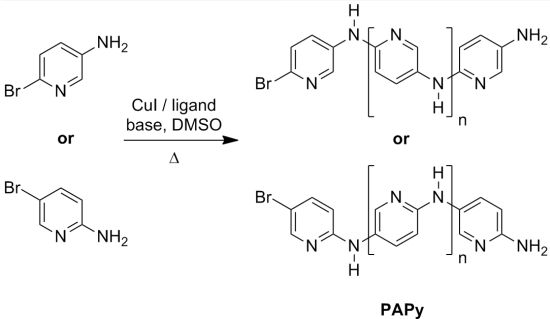{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Monomer | Base | Ligand | Condition (oC/h) | DP/Mn | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 1 | K2CO3 | l-proline | 80/80 | 21/2013 | 44 |
| 2 | 1 | K2CO3 | l-proline | 100/80 | 46/4313 | 61 |
| 3 | 1 | K2CO3 | l-proline | 100/80 | 64/5969 | 73a |
| 4 | 1 | K2CO3 | l-proline | 100/40 | 32/3025 | 62 |
| 5 | 1 | K2CO3 | none | 100/80 | 7/725 | 28b |
| 6 | 1 | K2CO3 | none | 100/80 | 18/1737 | 32b,c |
| 7 | 1 | K2CO3 | o-phenanthroline | 100/80 | 22/2105 | 33 |
| 8 | 1 | K2CO3 | N,N-dimethylglycine | 100/80 | 35/3301 | 48 |
| 9 | 1 | Cs2CO3 | l-proline | 100/80 | 30/2841 | 45 |
| 10 | 1 | K3PO4 | l-proline | 100/80 | 45/4221 | 80 |
| 11 | 1 | K3PO4 | l-proline | 100/80 | 64/5969 | 87a |
| 12 | 2 | K2CO3 | l-proline | 80/80 | - | 0d |
| 13 | 2 | K2CO3 | l-proline | 80/80 | 3/357 | 20a,e |
| 14 | 2 | K2CO3 | l-proline | 100/80 | 6/633 | 23 |
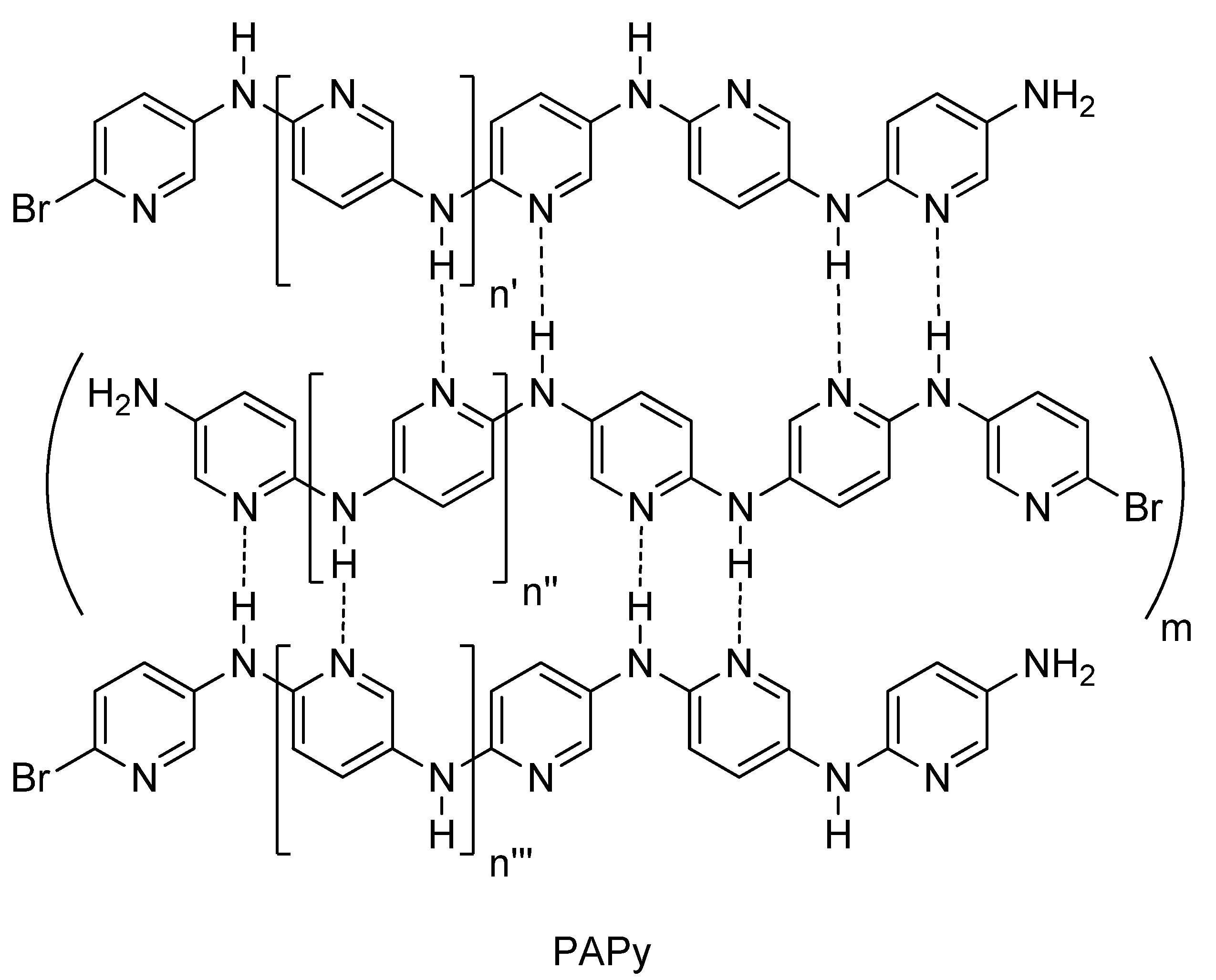
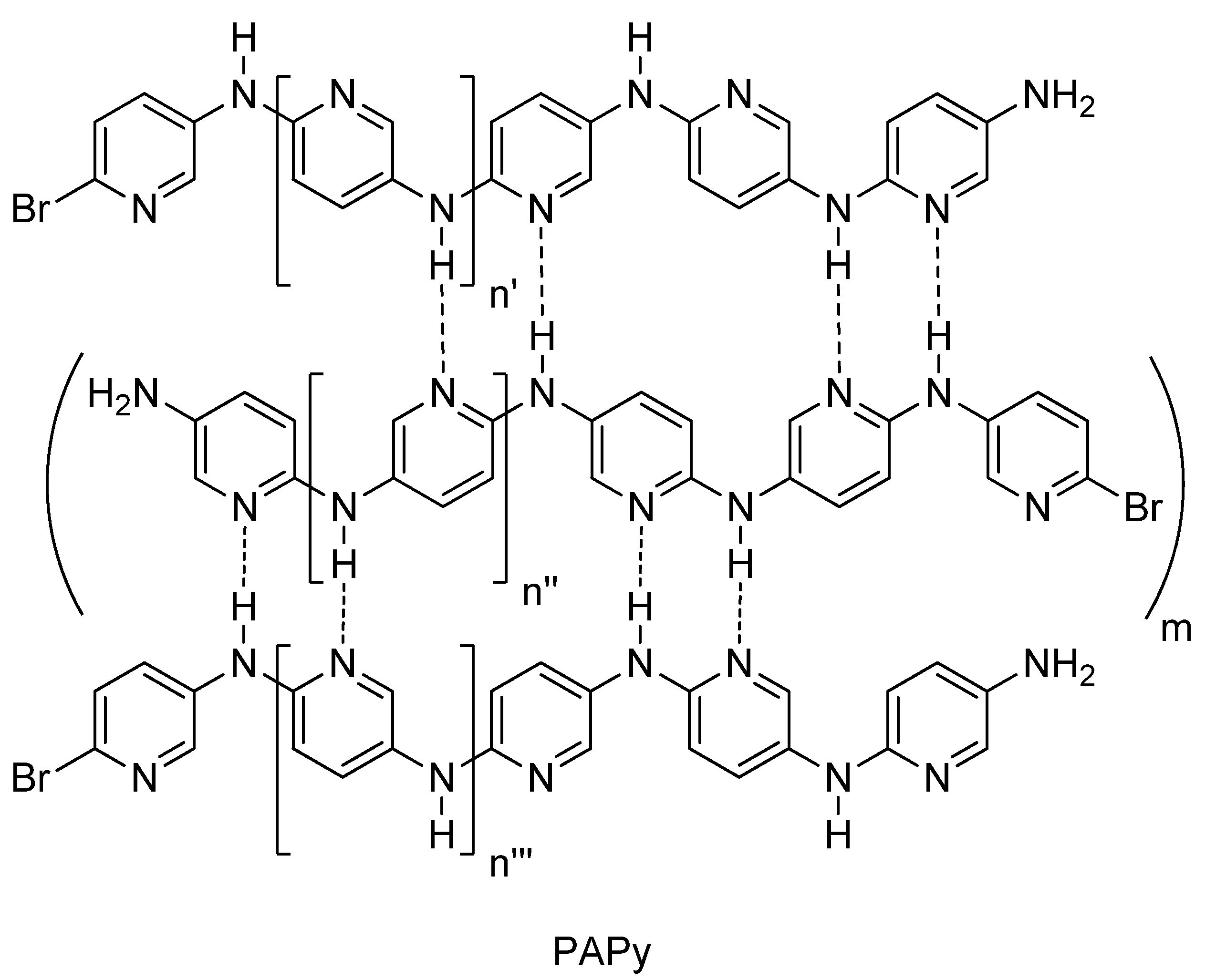
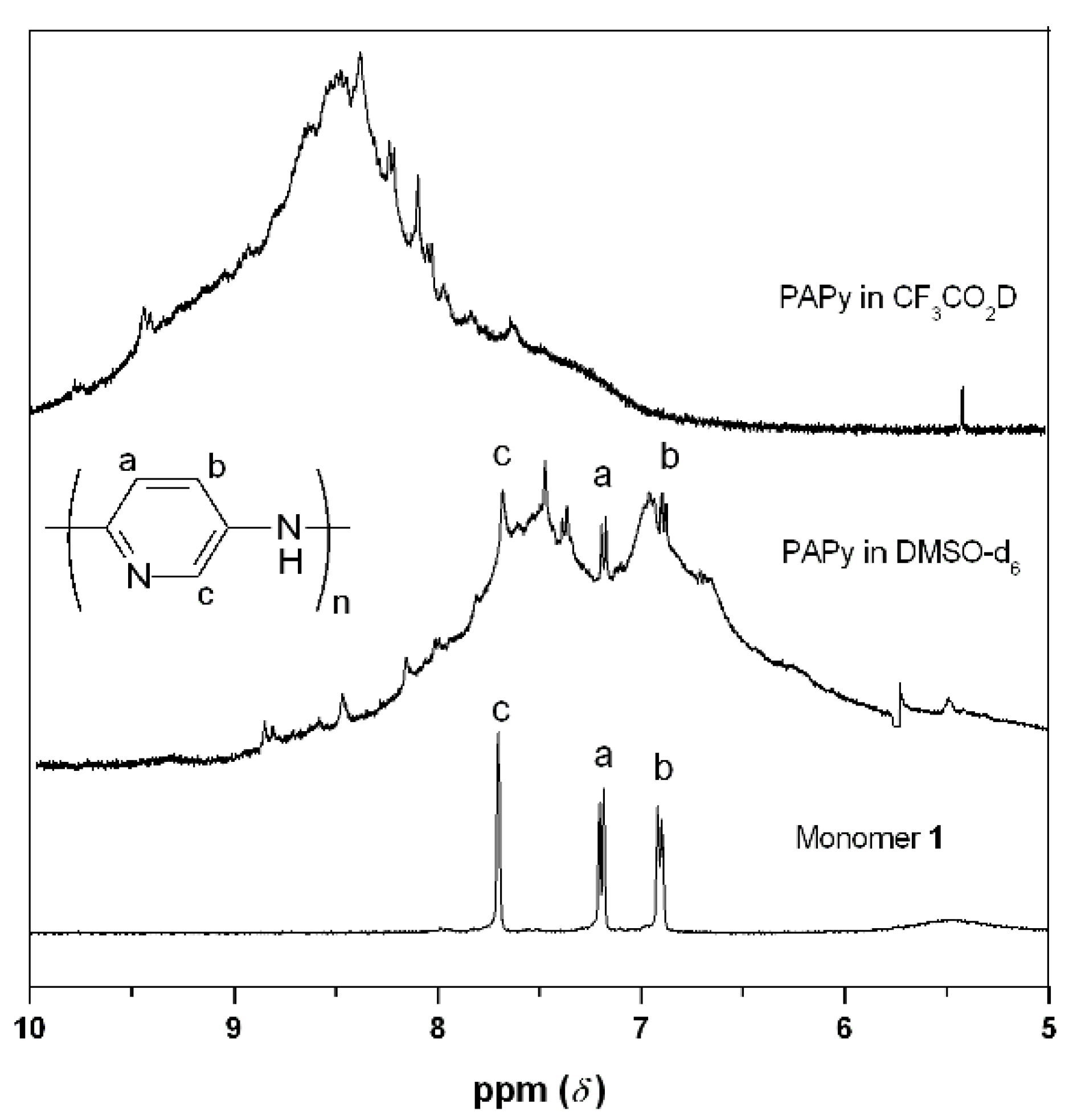
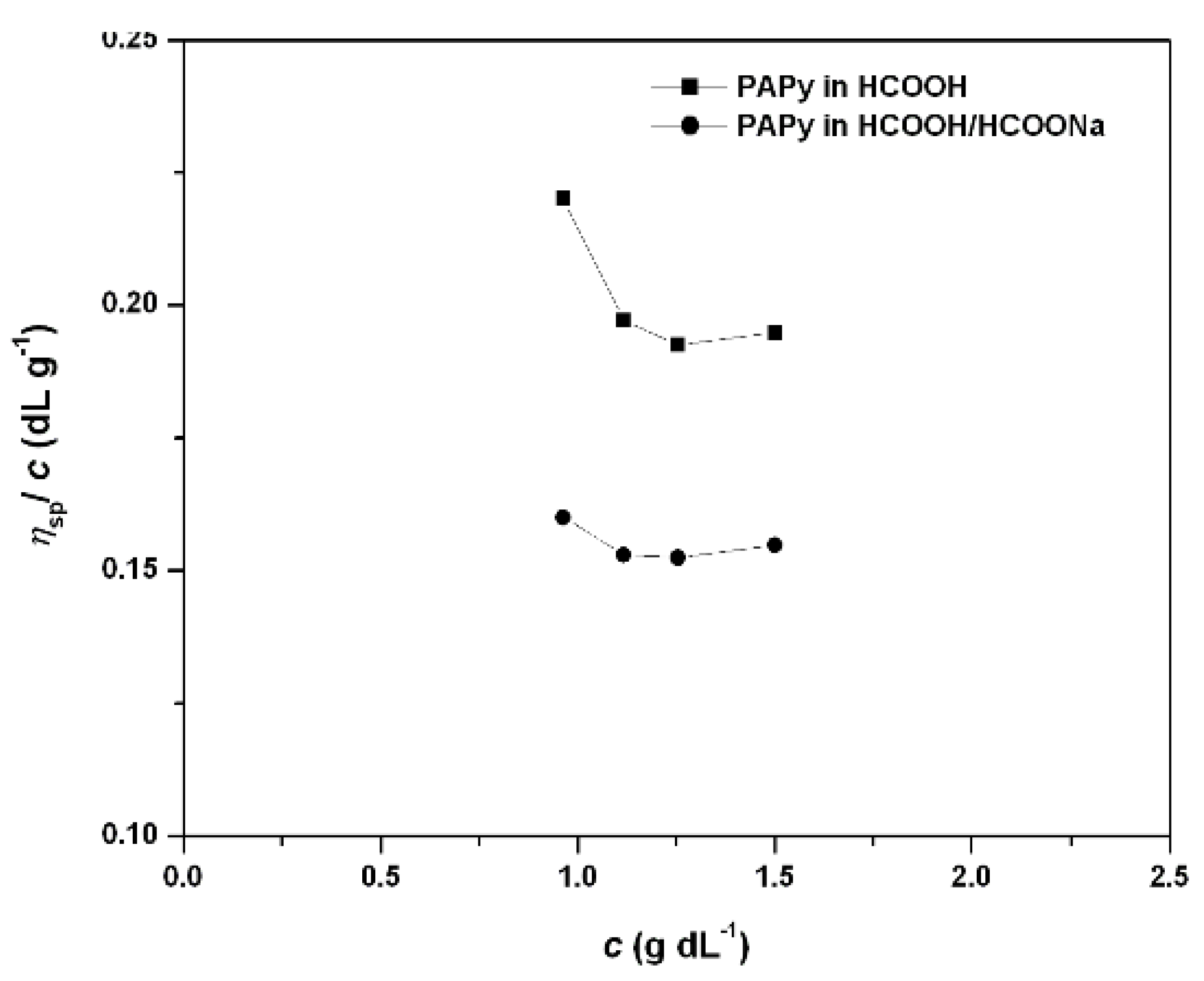
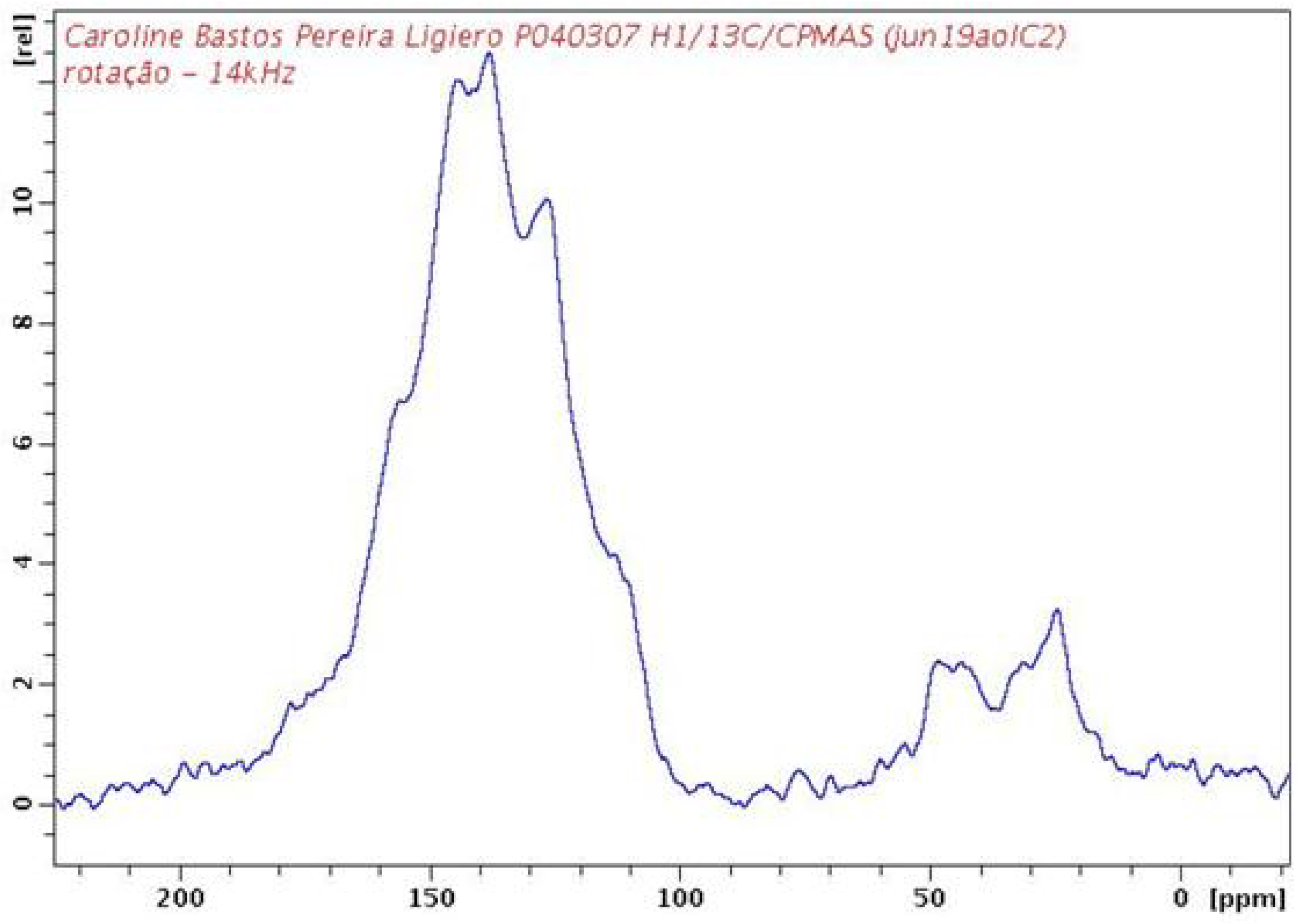
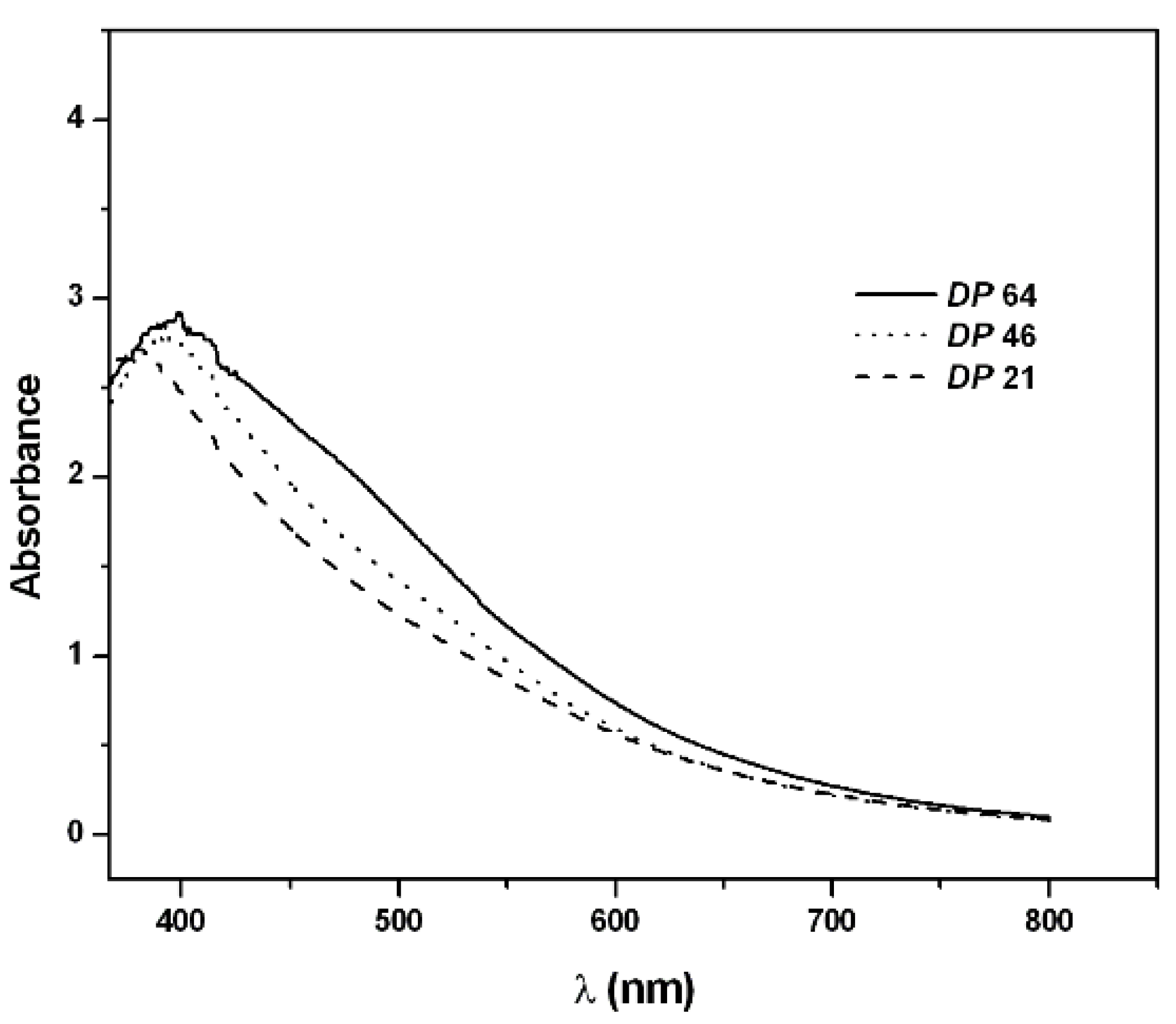
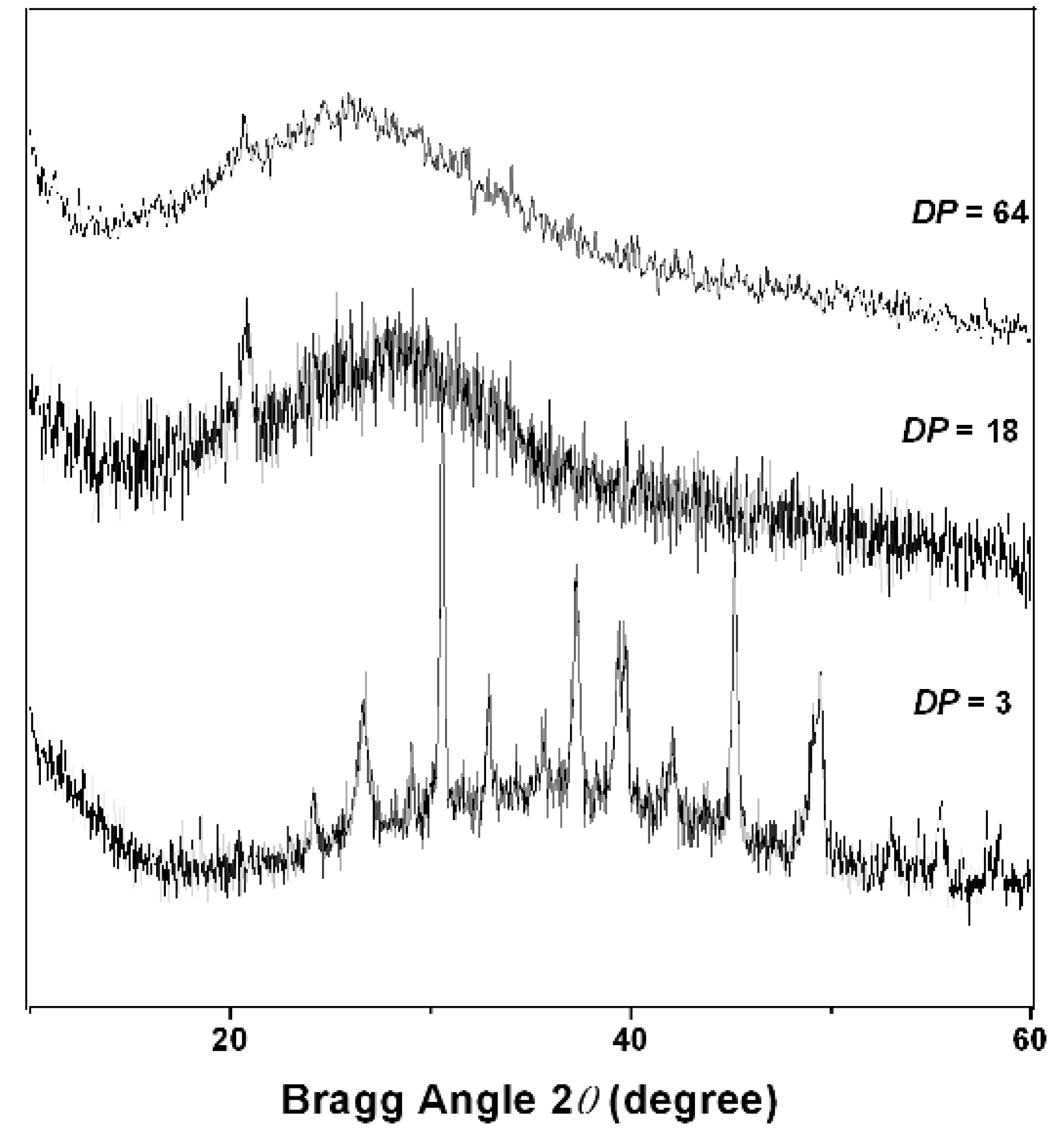
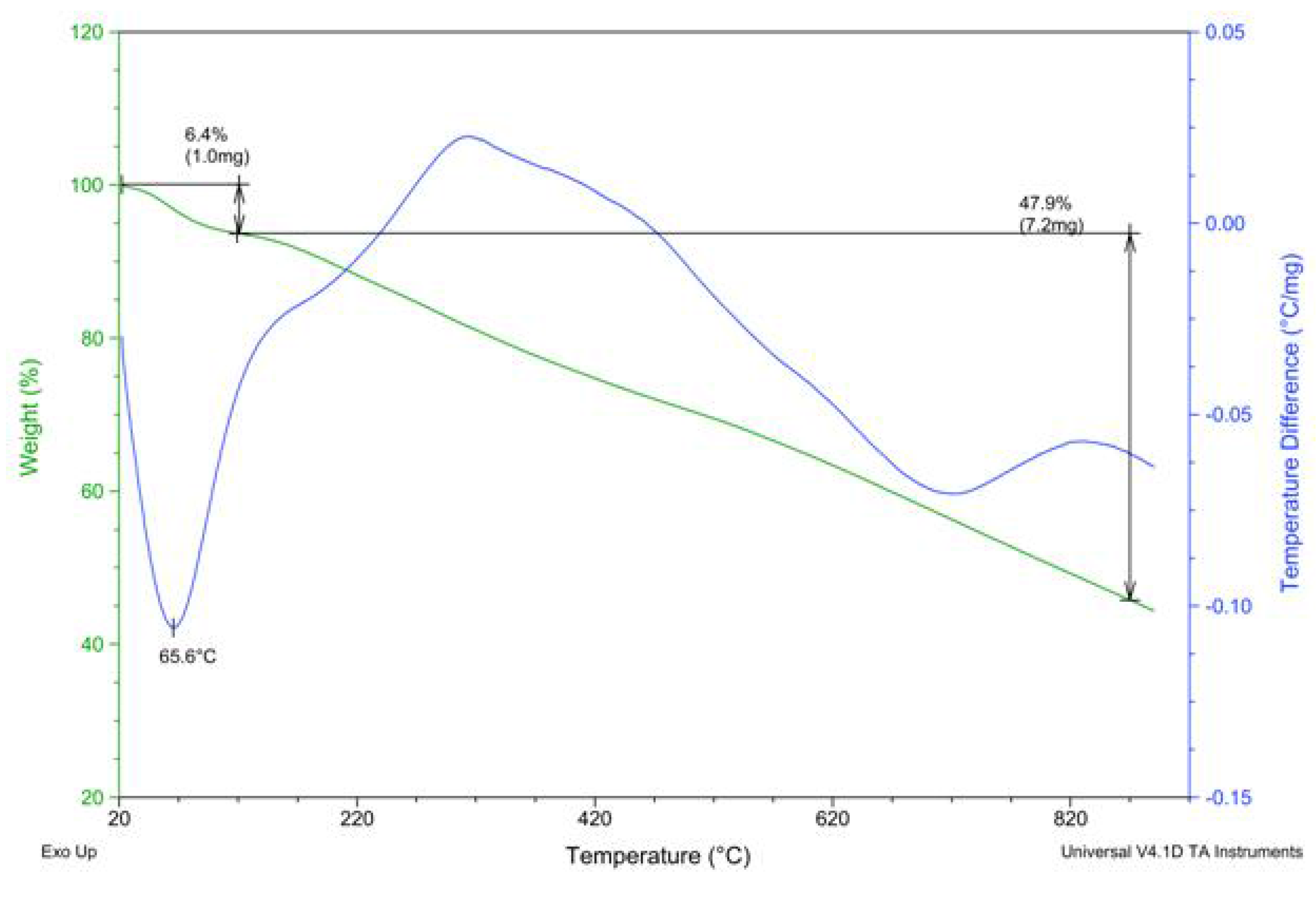
2.2. PAPy Characterization
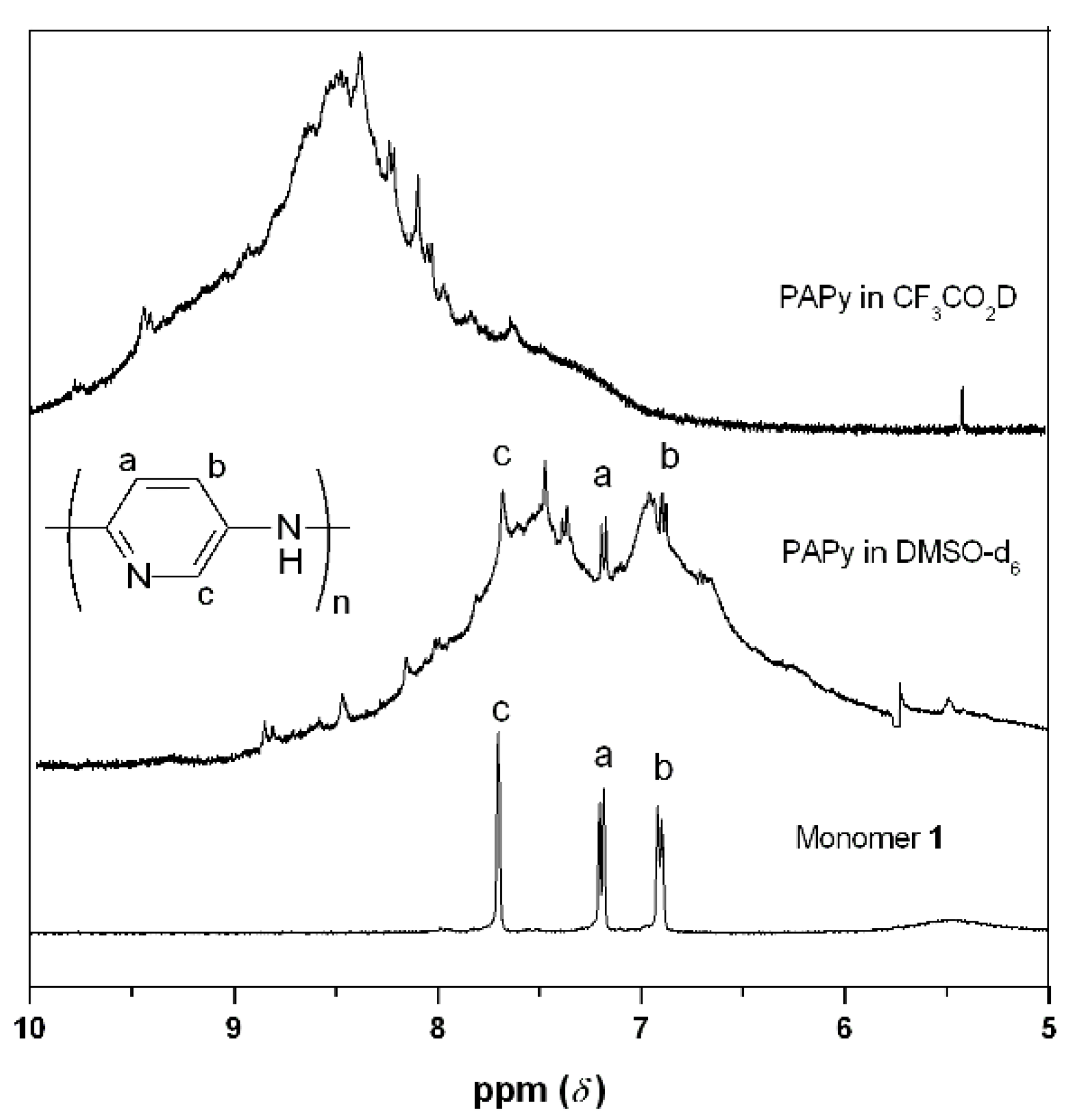
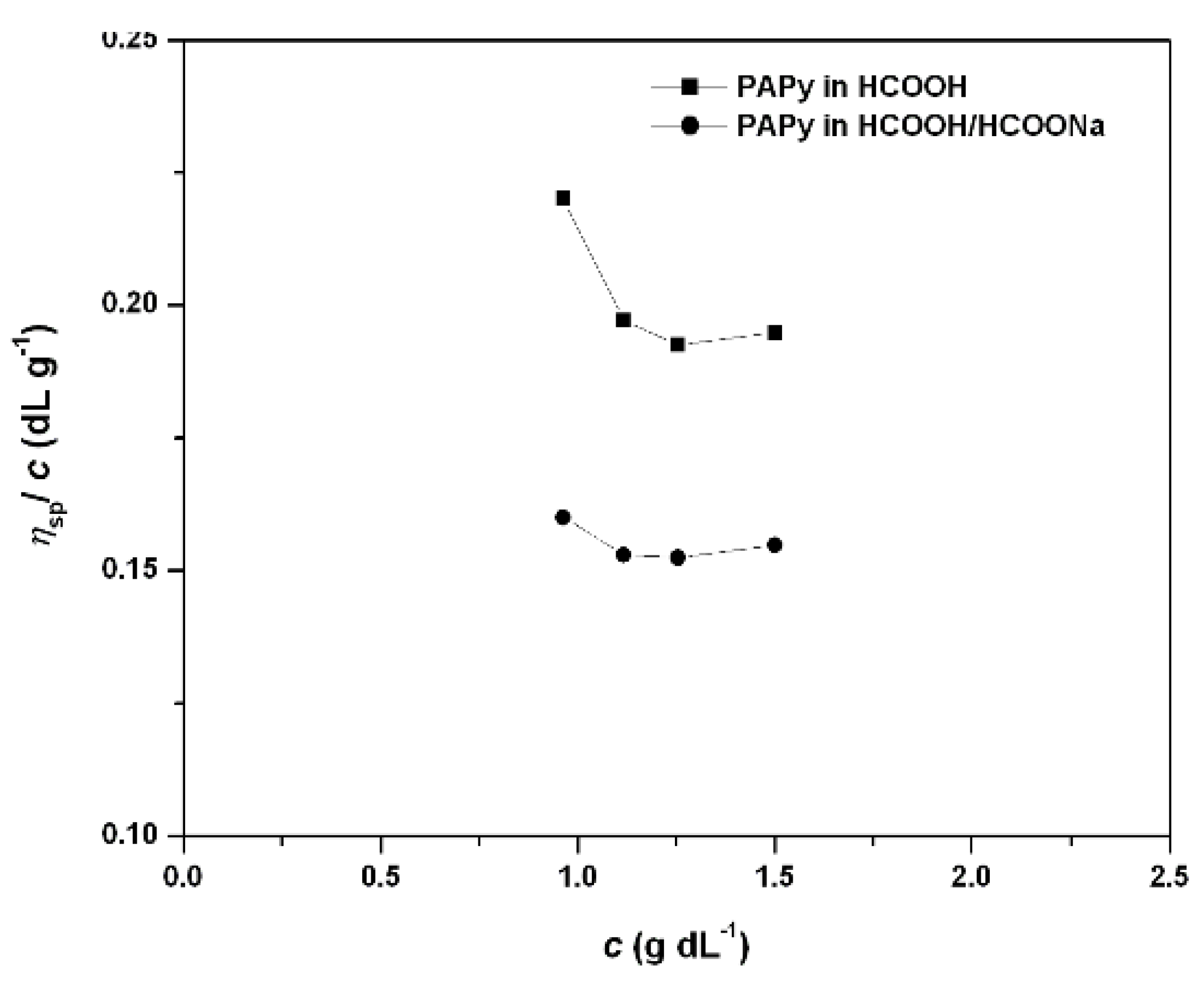
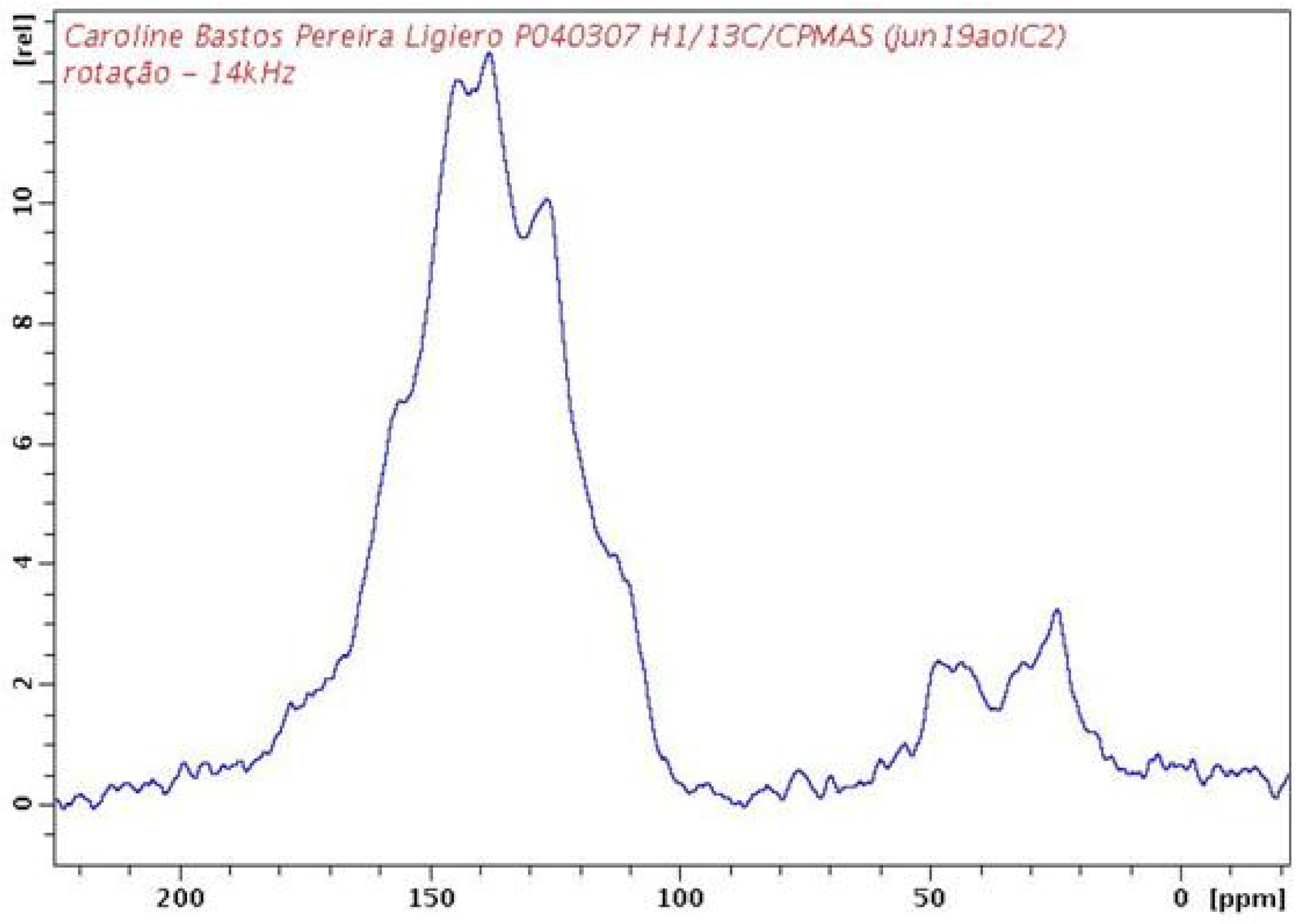

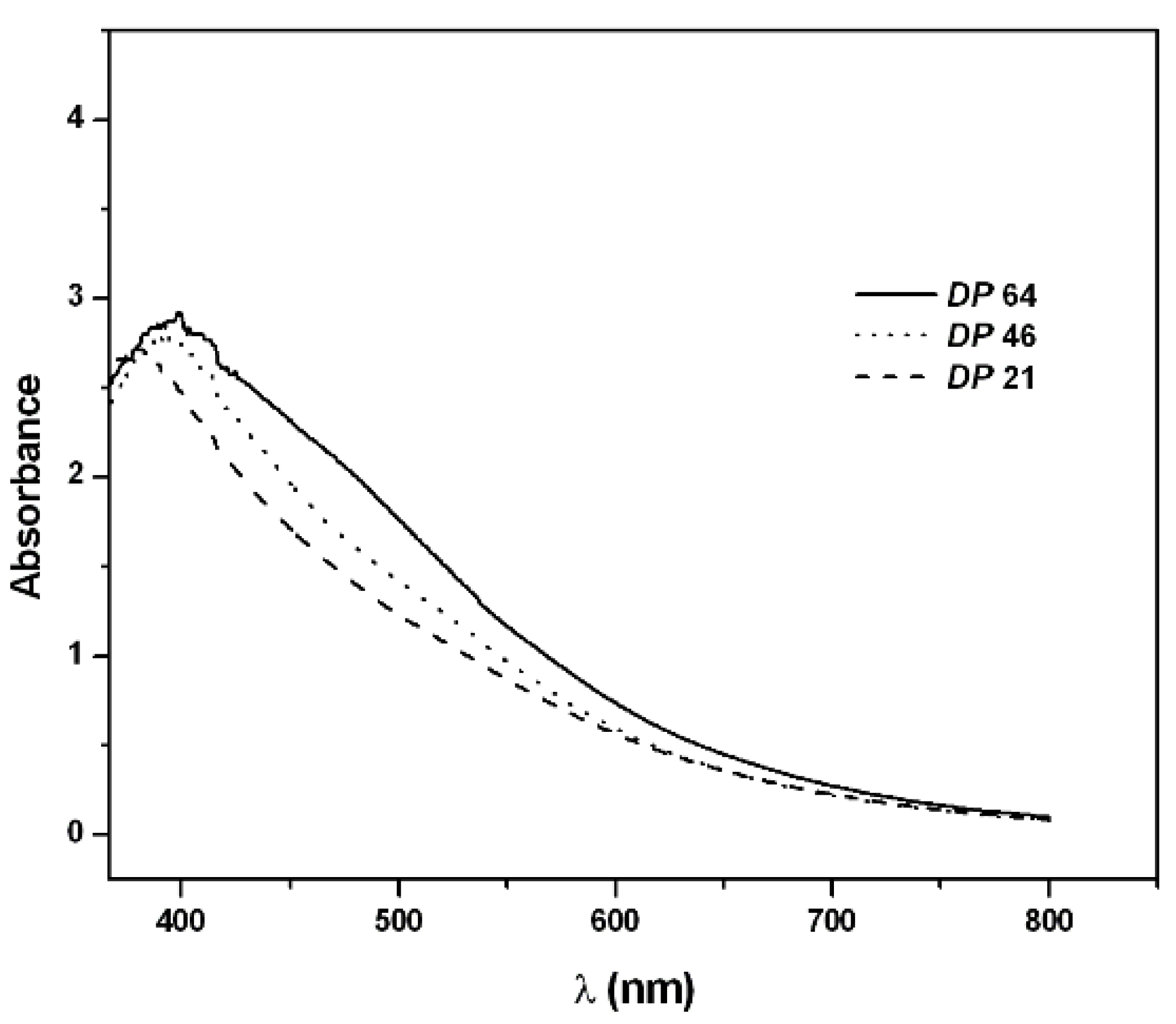
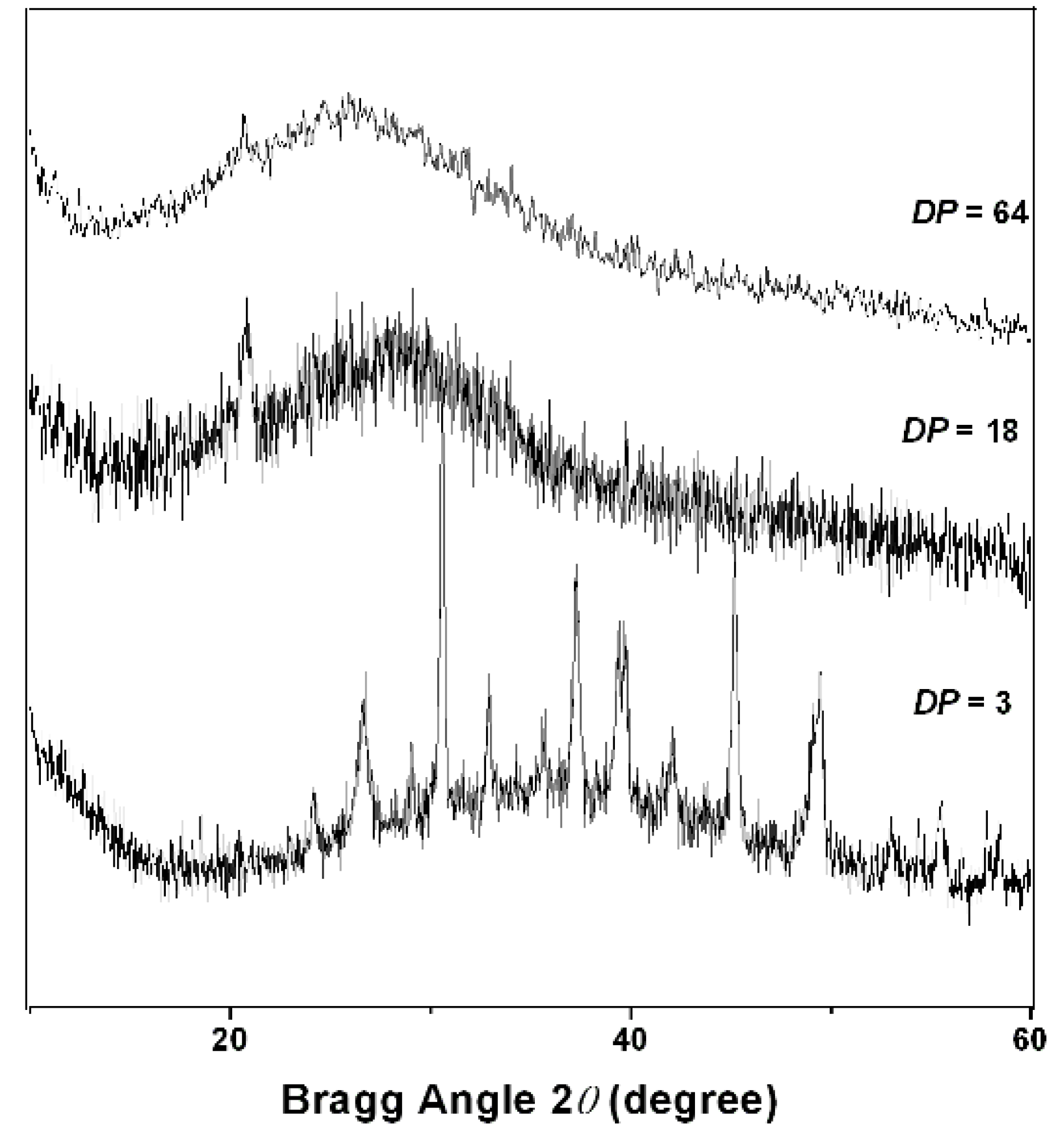
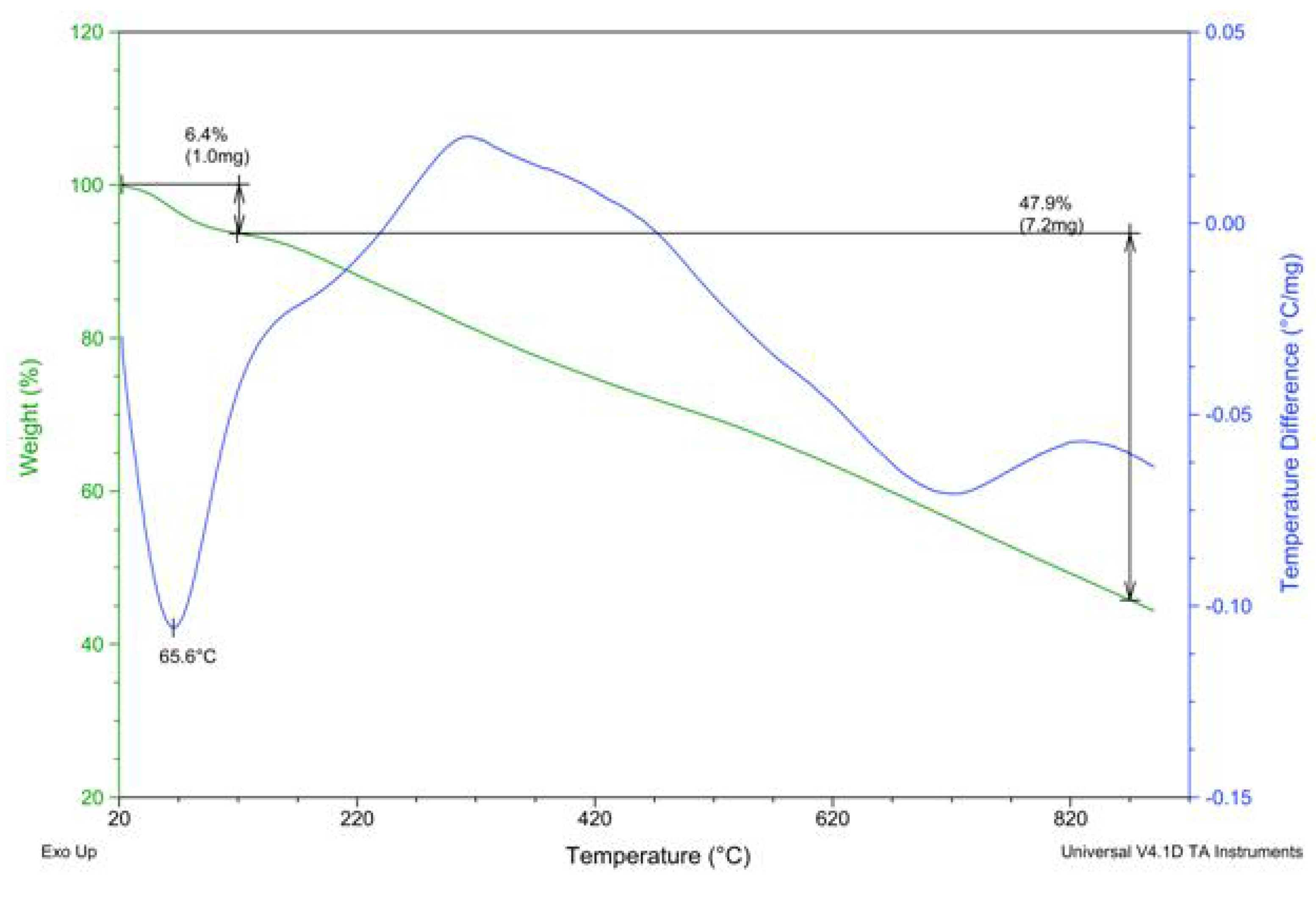
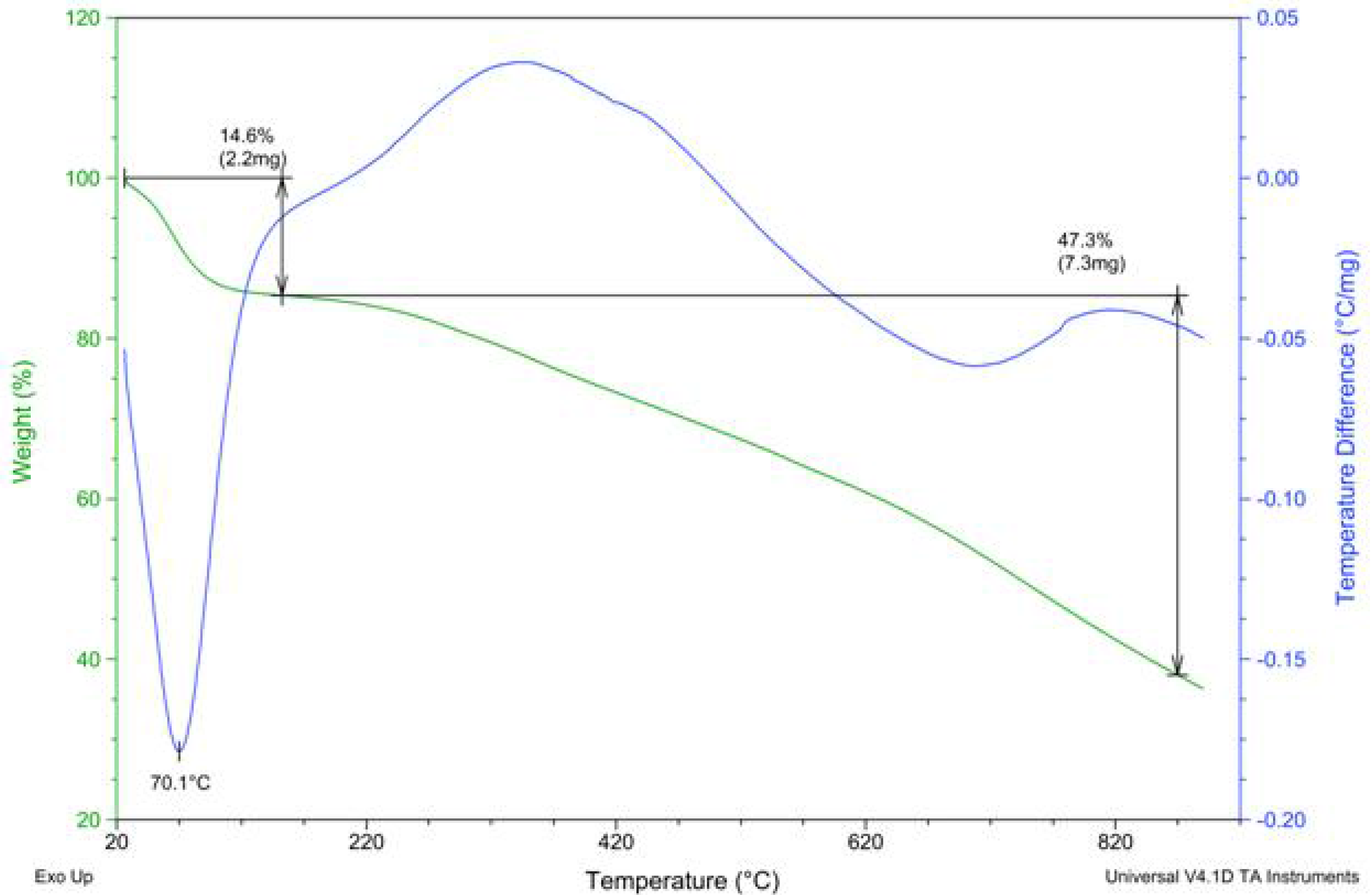
3. Experimental Section
3.1. Measurements
4. Conclusions
Acknowledgments
References
- MacDiarmid, A.G.; Yang, L.S.; Huang, W.S.; Humphrey, B.D. Polyaniline: Electrochemistry and application to rechargeable batteries. Synth. Met. 1987, 18, 393–398. [Google Scholar]
- Gustafsson, G.; Cao, Y.; Treacy, G.M.; Klavetter, F.; Colaneri, N.; Heeger, A.J. Flexible light-emitting diodes made from soluble conducting polymers. Nature 1992, 357, 477–479. [Google Scholar]
- Geniès, E.M.; Boyle, A.; Lapkowski, M.; Tsintavis, C. Polyaniline: A historical survey. Synth. Met. 1990, 36, 139–182. [Google Scholar]
- Sun, Y.; Guo, G.; Yang, B.; Tian, Y.; He, M.; Liu, Y.; Zhao, G. Facile synthesis of polyaniline micro-rods with high yield. Synth. Met. 2011, 161, 2206–2210. [Google Scholar]
- Chen, Z.; Pina, C.D.; Falletta, E.; Faro, M.L.; Pasta, M.; Rossi, M.; Santo, N. Facile synthesis of polyaniline using gold catalyst. J. Catal. 2008, 259, 1–4. [Google Scholar]
- Park, D.S.; Shim, Y.B.; Park, S.M. Characterization of electrochemically prepared polyaminopyridines. Electroanalysis 1996, 8, 44–48. [Google Scholar]
- Kan, J.Q.; Li, X.; Li, Y.F. Synthesis and properties of poly-2-aminopyridine. Acta Phys. Chim. Sin. 2002, 18, 106–111. [Google Scholar]
- Hayat, U.; Bartlett, P.N.; Dodd, G.H. Electrochemical synthesis of poly(2-aminopyridine). Polym. Commun. 1986, 27, 362–363. [Google Scholar]
- Koketsu, J.; Kato, K.; Ando, F.; Fujimura, Y. Electrochemical polymerization of 3-aminopyridine and some characteristics. Synth. Met. 1993, 60, 45–49. [Google Scholar]
- Li, X.G.; Huang, M.R.; Duan, W.; Yang, Y.L. Novel multifunctional polymers from aromatic diamines by oxidative polymerizations. Chem. Rev. 2002, 102, 2925–3030. [Google Scholar]
- Goto, H.; Iino, K.; Akagi, K.; Shirakawa, H. Syntheses and ESR characterizations of conjugated polymers with nitrogen atoms. Synth. Met. 1997, 85, 1683–1684. [Google Scholar]
- Ullmann, F. Ueber eine neue bildungsweise von diphenylaminderivaten. Ber. Dtsch. Chem. Ges. 1903, 36, 2382–2384. [Google Scholar]
- Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl-aryl bond formation one century after the discovery of the Ullmann reaction. Chem. Rev. 2002, 102, 1359–1470. [Google Scholar]
- Hartwig, J.F. Transition metal catalyzed synthesis of arylamines and aryl ethers from aryl halides and triflates: Scope and mechanism. Angew. Chem. Int. Ed. 1998, 37, 2046–2067. [Google Scholar]
- Wolfe, J.P.; Wagaw, S.; Marcoux, J.F.; Buchwald, S.L. The rational development of practical catalysts for aromatic Carbon-Nitrogen bond formation. Acc. Chem. Res. 1998, 31, 805–818. [Google Scholar]
- Kuwabara, J.; Mori, H.; Teratani, T.; Akita, M.; Kanbara, T. Regioregulated syntheses of poly(aminopyridine)s by Pd-catalyzed amination reaction. Macromol. Rapid Commun. 2009, 30, 997–1001. [Google Scholar]
- Vargas, C.; Balu, A.M.; Campelo, J.M.; Arellano, C.G.; Luque, R.; Romero, A.A. Towards greener and more efficient C-C and C-heteroatom couplings: Present and future. Curr. Org. Synth. 2010, 7, 568–586. [Google Scholar]
- Marcoux, J.F.; Doye, S.; Buchwald, S.L. A general Copper-catalyzed synthesis of diaryl ethers. J. Am. Chem. Soc. 1997, 119, 10539–10540. [Google Scholar]
- Kiyomori, A.; Marcoux, J.F.; Buchwald, S.L. An efficient Copper-catalyzed coupling of aryl halides with imidazoles. Tetrahedron Lett. 1999, 40, 2657–2660. [Google Scholar]
- Kunz, K.; Scholz, U.; Ganzer, D. Renaissance of Ullmann and Goldberg reactions: Progress in copper catalyzed C-N, C-O and C-S coupling. Synlett 2003, 15, 2428–2439. [Google Scholar]
- Monnier, F.; Taillefer, M. Catalytic C-C, C-N, and C-O Ullmann-type coupling reactions: Copper makes a difference. Angew. Chem. Int. Ed. 2008, 47, 3096–3099. [Google Scholar]
- Laskoski, M.; Dominguez, D.D.; Keller, T.M. Oligomeric cyanate ester resins: Application of a modified Ullmann synthesis in the preparation of thermosetting polymers. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 4559–4565. [Google Scholar]
- Wang, J.; Gao, Y.; Hlil, A.R.; Hay, A.S. Synthesis of high molecular weight poly(phthalazinone ether)s by Ullmann C-N and C-O condensation reactions. Macromolecules 2008, 41, 298–300. [Google Scholar]
- Zhang, H.; Cai, Q.; Ma, D. Amino acid promoted CuI-catalyzed C-N bond formation between aryl halides and amines or N-containing heterocycles. J. Org. Chem. 2005, 70, 5164–5173. [Google Scholar]
- Dias, H.V.R.; Wang, X.; Rajapakse, R.M.G.; Elsenbaumer, R.L. A mild, copper catalyzed route to conducting polyaniline. Chem. Commun. 2006, 9, 976–978. [Google Scholar]
- Jia, C.; Kitamura, T.; Fujiwara, Y. Catalytic functionalization of arenes and alkanes via C-H bond activation. Acc. Chem. Res. 2001, 34, 633–639. [Google Scholar]
- Daugulis, O.; Do, H.Q.; Shabashov, D. Palladium- and copper-catalyzed arylation of carbon-hydrogen bonds. Acc. Chem. Res. 2009, 42, 1074–1086. [Google Scholar]
- Ferreira, E.M.; Stoltz, B.M. Catalytic C-H bond functionalization with Palladium(II): Aerobic oxidative annulations of indoles. J. Am. Chem. Soc. 2003, 125, 9578–9579. [Google Scholar]
- Liao, Y.H.; Kwei, T.K.; Levon, K. Investigation of the aggregation phenomenon of polyaniline in dilute solutions. Macromol. Chem. Phys. 1995, 196, 3107–3116. [Google Scholar]
- Yang, D.; Adams, P.N.; Goering, R.; Mattes, B.R. New methods for determining the molecular weight of polyaniline by size exclusion chromatography. Synth. Met. 2003, 135–136, 293–294. [Google Scholar]
- Leung, M.K.; Mandal, A.B.; Wang, C.C.; Lee, G.H.; Peng, S.M.; Cheng, H.L.; Her, G.R.; Chao, I.; Lu, H.F.; Sun, Y.C.; Shiao, M.Y.; Chou, P.T. Self-complementarity of oligo-2-aminopyridines: A new class of Hydrogen-bonded ladders. J. Am. Chem. Soc. 2002, 124, 4287–4297. [Google Scholar]
- Strieter, E.R.; Bhayana, B.; Buchwald, S.L. Mechanistic studies on the Copper-catalyzed N-arylation of amides. J. Am. Chem. Soc. 2009, 131, 78–88. [Google Scholar]
- Monnier, F.; Taillefer, M. Catalytic C-C, C-N, and C-O Ullmann-type coupling reactions. Angew. Chem. Int. Ed. 2009, 48, 6954–6971. [Google Scholar]
- Fagan, P.J.; Hauptman, E.; Shapiro, R.; Casalnuovo, A. Using intelligent/random library screening to design focused libraries for the optimization of homogeneous catalysts: Ullmann ether formation. J. Am. Chem. Soc. 2000, 122, 5043–5051. [Google Scholar]
- Moinard, D.; Borsali, R.; Taton, D.; Gnanou, Y. Scattering and viscosimetric behaviors of four- and six-arm star polyelectrolyte solutions. Macromolecules 2005, 38, 7105–7120. [Google Scholar]
- Eckelt, J.; Knopf, A.; Wolf, B.A. Polyelectrolytes: Intrinsic viscosities in the absence and in the presence of salt. Macromolecules 2008, 41, 912–918. [Google Scholar]
- Kolla, H.S.; Surwade, S.P.; Zhang, X.; MacDiarmid, A.G.; Manohar, S.K. Absolute molecular weight of polyaniline. J. Am. Chem. Soc. 2005, 127, 16770–16771. [Google Scholar]
- Gazotti, W.A.; de Paoli, M.A. High yield preparation of a soluble polyaniline derivative. Synth. Met. 1996, 80, 263–269. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Reis, L.A.; Ligiéro, C.B.P.; Andrade, A.A.; Taylor, J.G.; Miranda, P.C.M.L. Preparation of Polyaminopyridines Using a CuI/l-Proline-Catalyzed C-N Polycoupling Reaction. Materials 2012, 5, 2176-2189. https://doi.org/10.3390/ma5112176
Reis LA, Ligiéro CBP, Andrade AA, Taylor JG, Miranda PCML. Preparation of Polyaminopyridines Using a CuI/l-Proline-Catalyzed C-N Polycoupling Reaction. Materials. 2012; 5(11):2176-2189. https://doi.org/10.3390/ma5112176
Chicago/Turabian StyleReis, Lindomar A., Carolina B. P. Ligiéro, Acácio A. Andrade, Jason G. Taylor, and Paulo C. M. L. Miranda. 2012. "Preparation of Polyaminopyridines Using a CuI/l-Proline-Catalyzed C-N Polycoupling Reaction" Materials 5, no. 11: 2176-2189. https://doi.org/10.3390/ma5112176