Difference between Toxicities of Iron Oxide Magnetic Nanoparticles with Various Surface-Functional Groups against Human Normal Fibroblasts and Fibrosarcoma Cells



Abstract
:1. Introduction
2. Results and Discussion
2.1. Physicochemical Characterizations of Fe3O4 MNPs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coating material | Average diameter (nm) | Surface charge (mV) | Functional group |
|---|---|---|---|
| none (bare) | 10 ± 3 | −20 ± 0.5 | –O− |
| TEOS | 100–150 | −30 ± 1.8 | –O− |
| APTMS | 10 ± 4 | 25 ± 1.2 | –NH3+ |
| TEOS/APTMS | 100–150 | 30 ± 2.0 | –NH3+ |
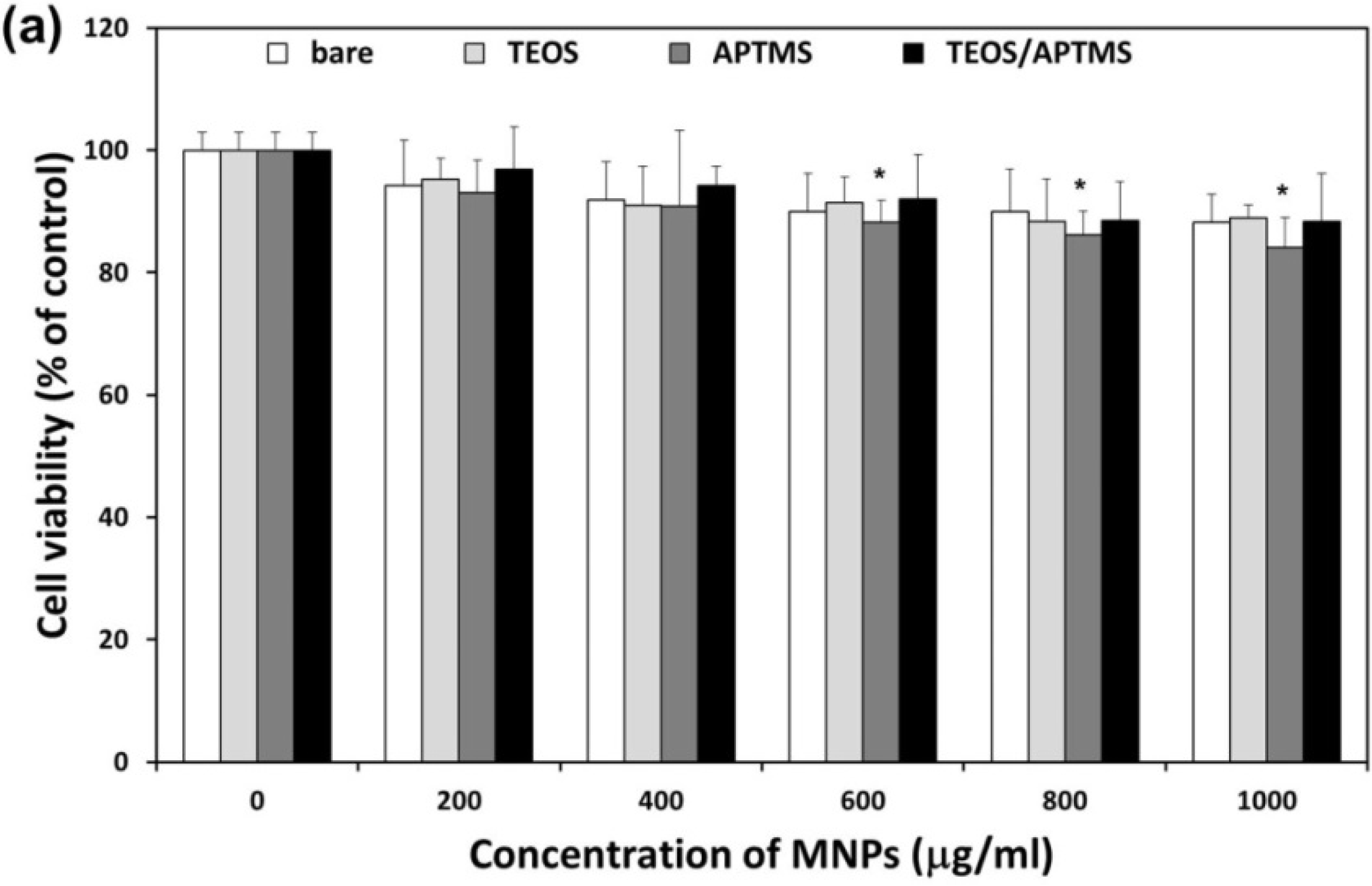
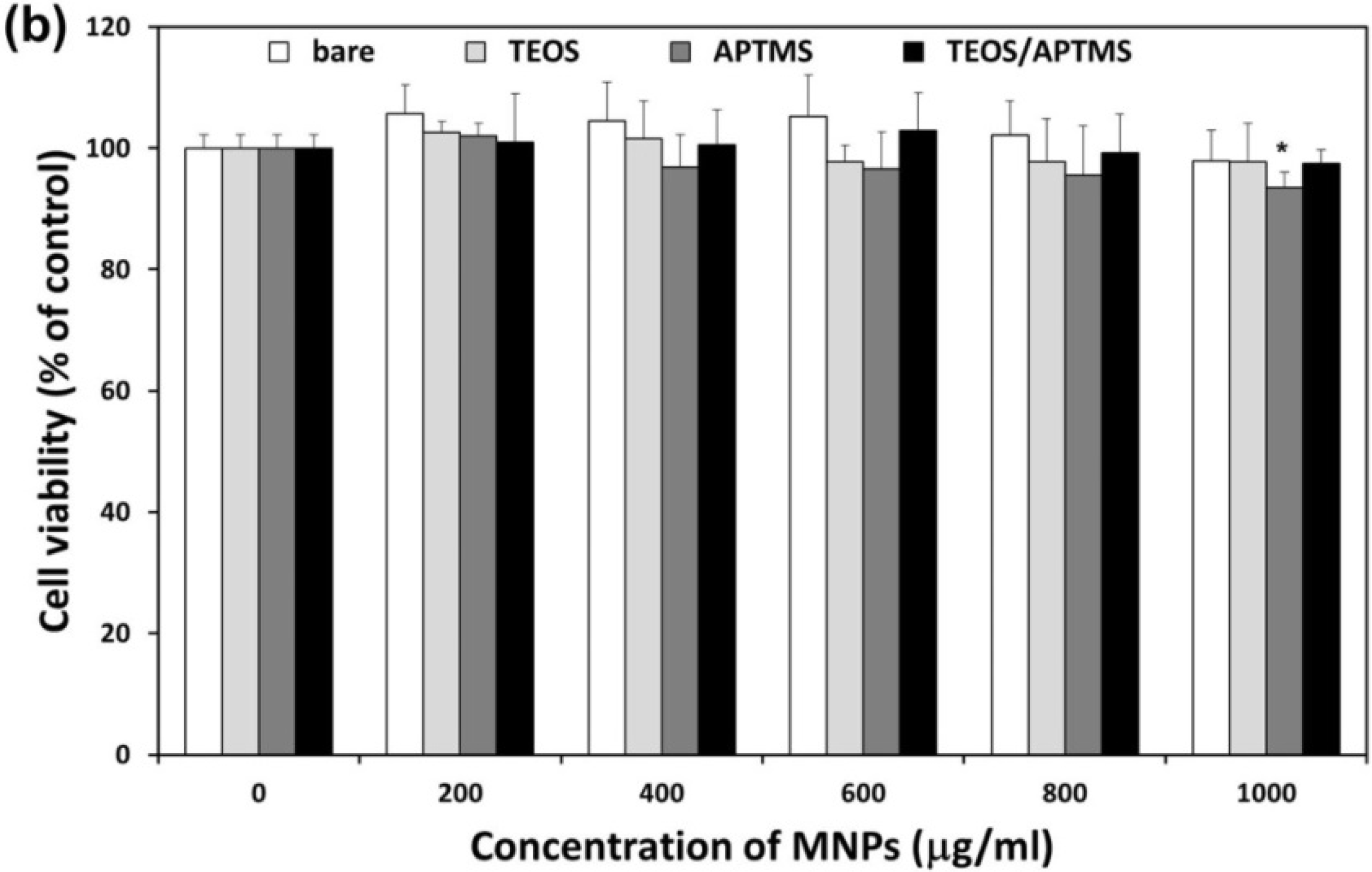
2.2. Effects of MNPs on Metabolic Activity

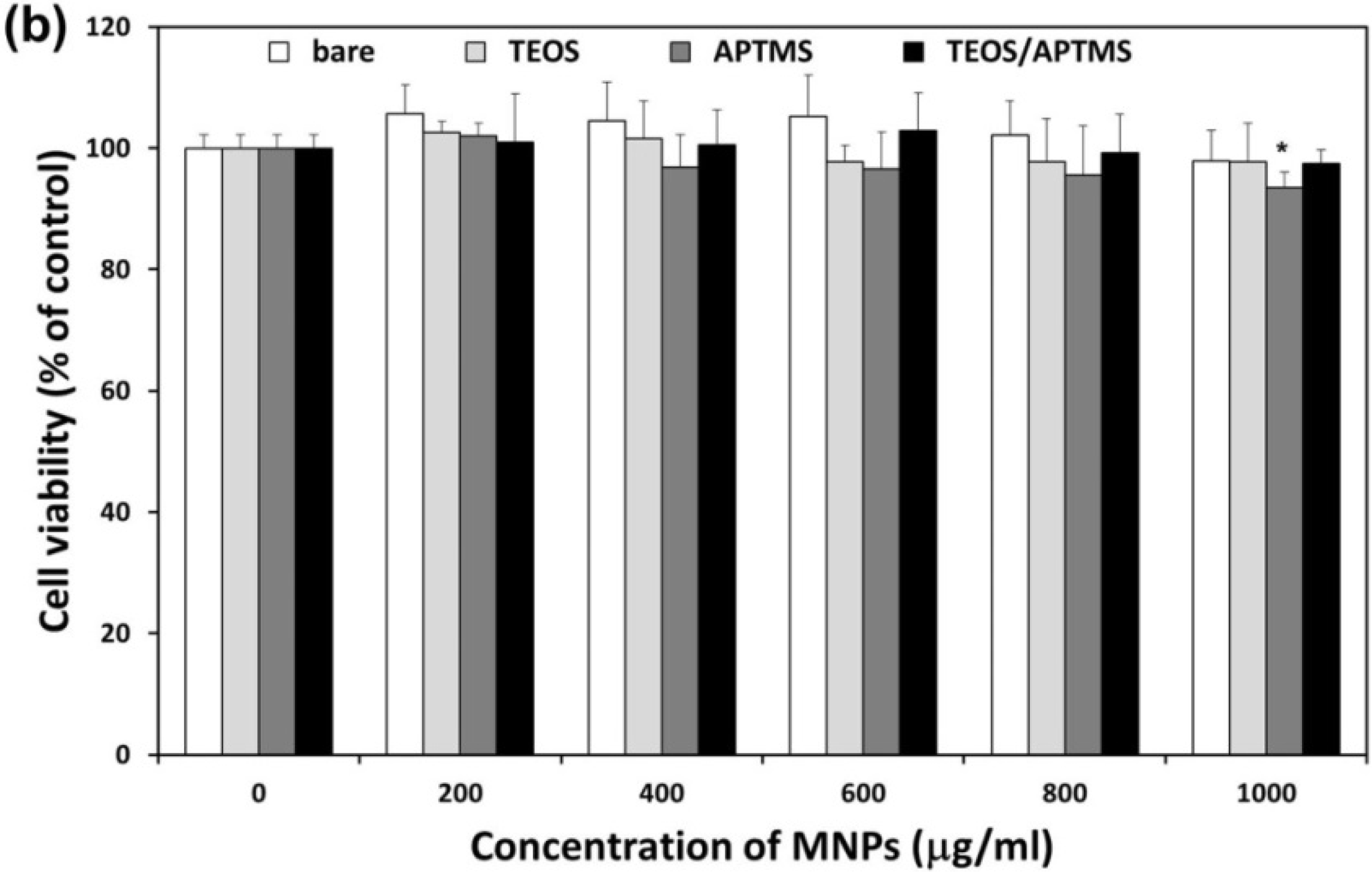
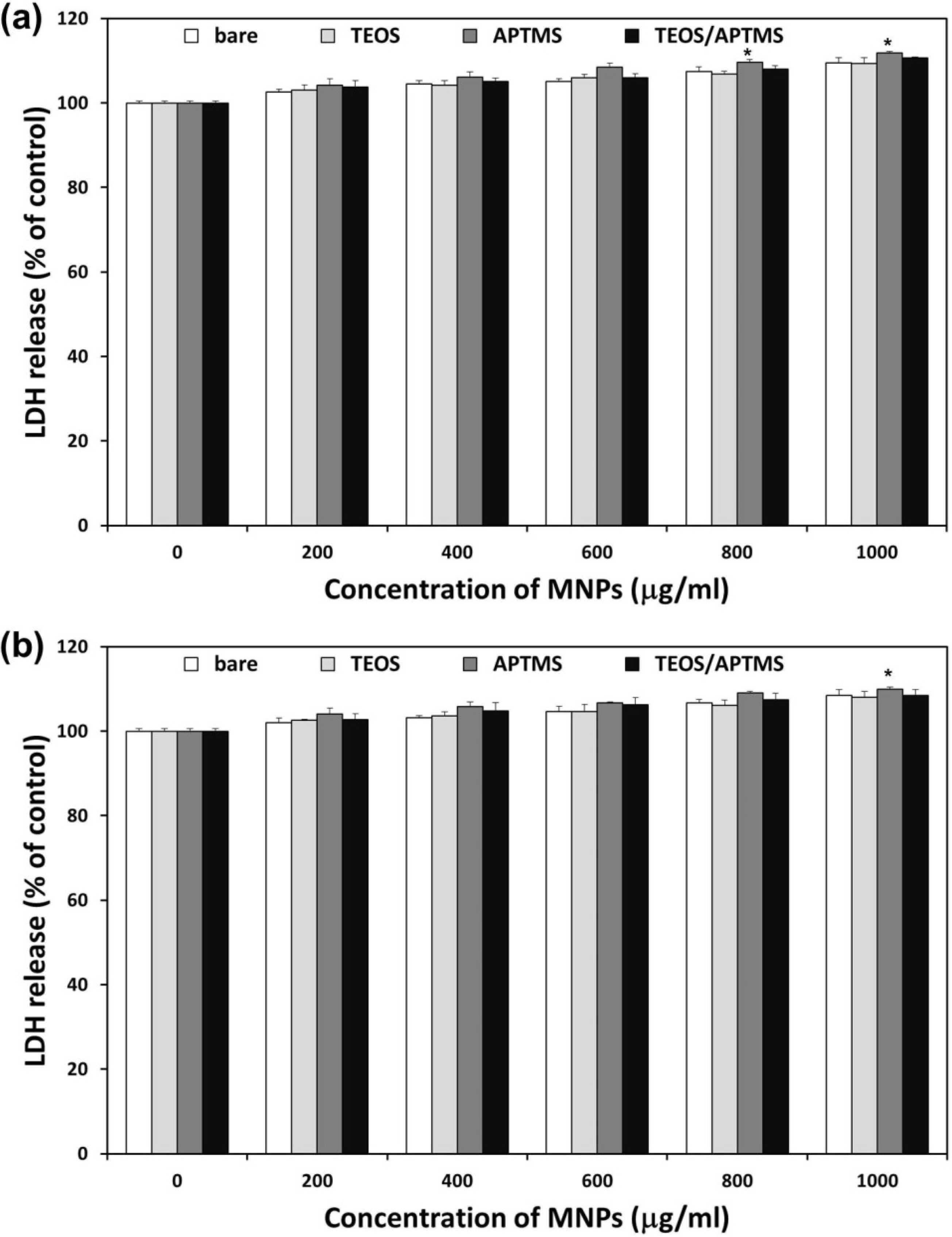
2.3. Effects of MNPs on Cell Membrane Integrity
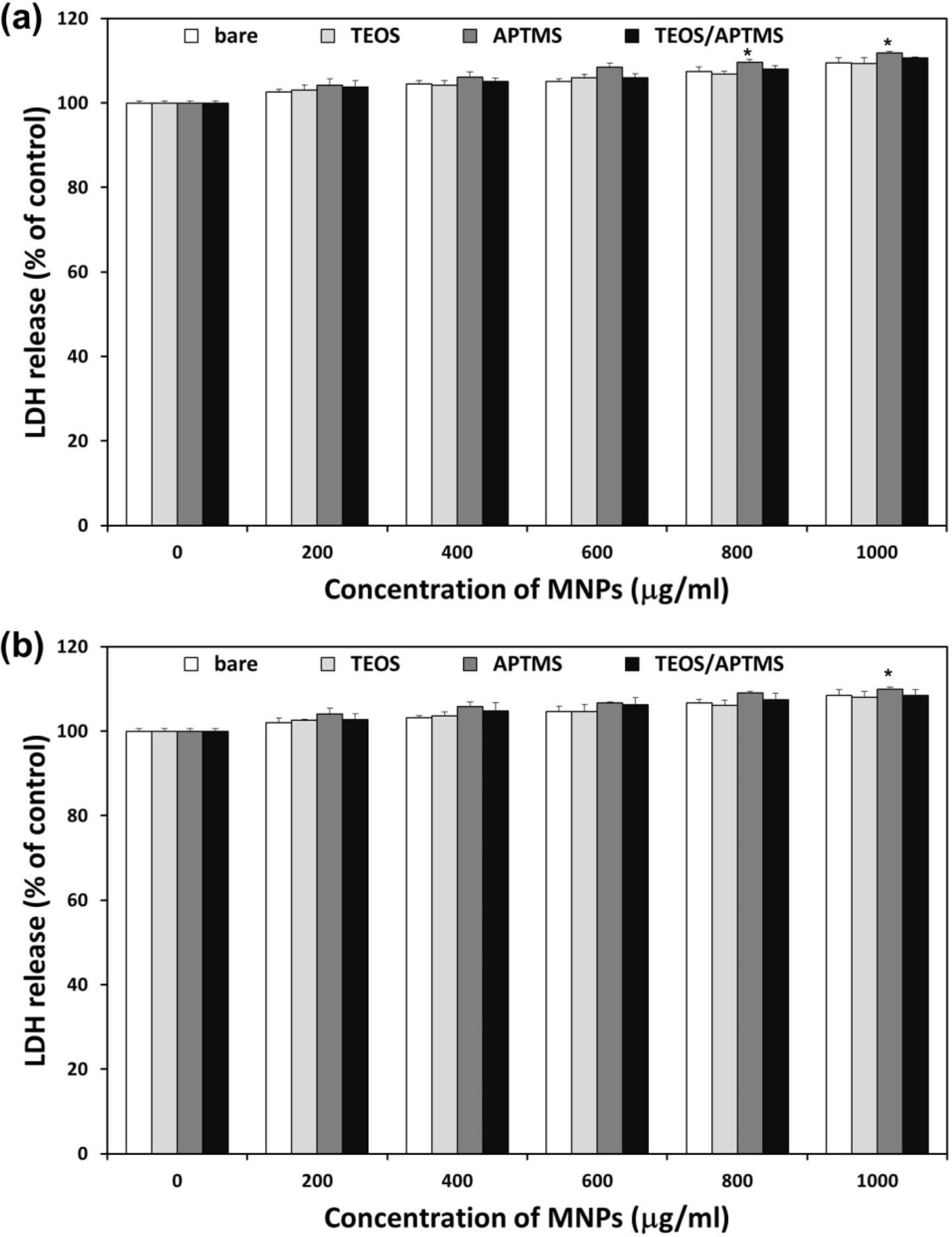
2.4. SEM and TEM Observations


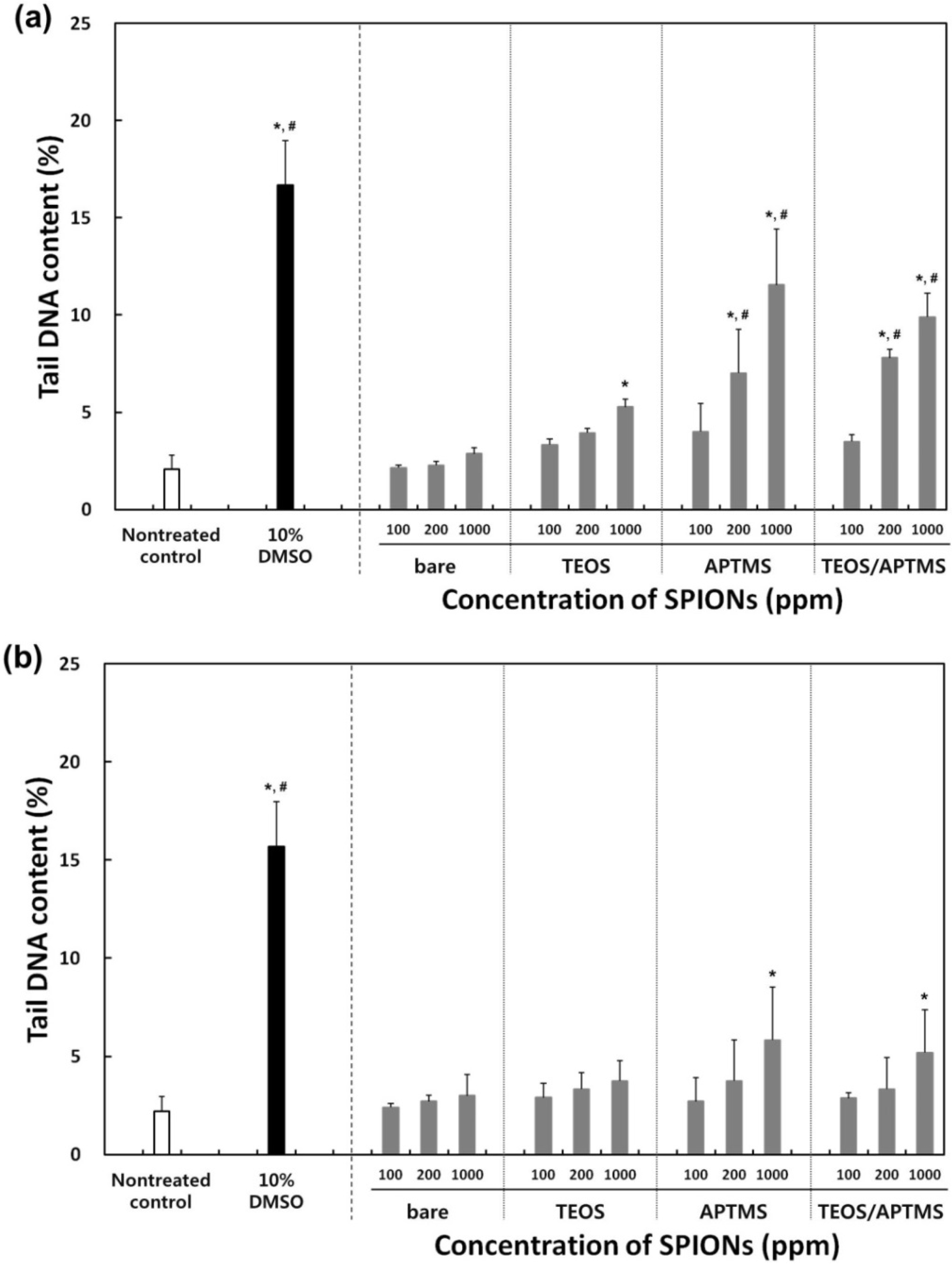
2.5. Effects of MNPs on DNA Stability
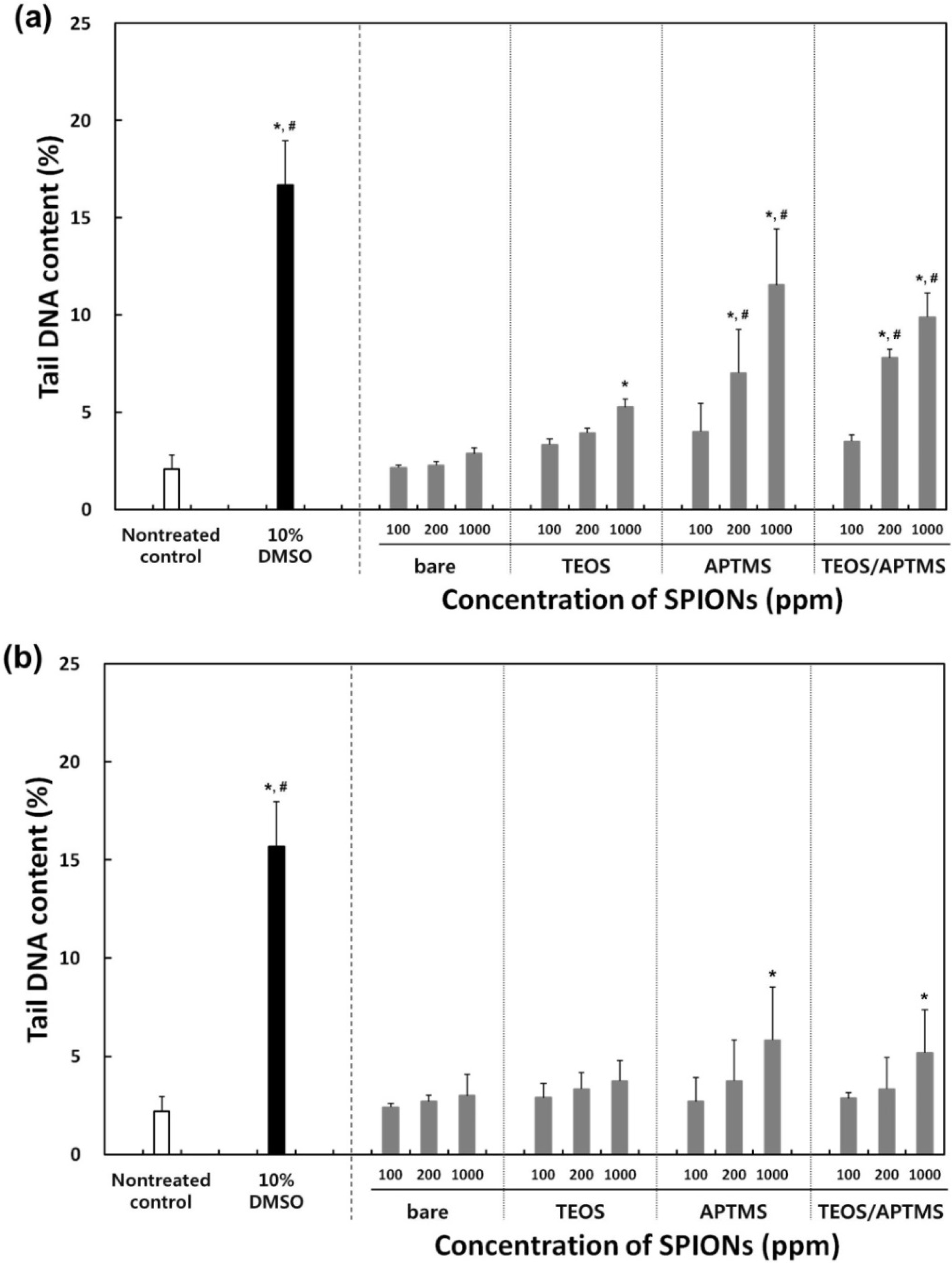
3. Experimental Section
3.1. Synthesis and Characterizations of Surface-Functionalized MNPs
3.1.1. Bare MNPs
3.1.2. TEOS-Coated MNPs
3.1.3. APTMS-Coated MNPs
3.1.4. TEOS/APTMS-Coated MNPs
3.1.5. Physicochemical Characterizations
3.2. Cell Cultures and Conditions
3.3. CCK-8 Assay for Cytotoxicity Determination
3.4. LDH Assay for Cytotoxicity Determination
3.5. Electron Microscopy Observations
3.6. Comet Assay for Genotoxicity Evaluation
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Frey, N.A.; Peng, S.; Cheng, K.; Sun, S. Magnetic nanoparticles: Synthesis, functionalization, and applications in bioimaging and magnetic energy storage. Chem. Soc. Rev. 2009, 38, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Gu, H.; Xu, B. Multifunctional magnetic nanoparticles: Design, synthesis, and biomedical applications. Acc. Chem. Res. 2009, 42, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Frimpong, R.A.; Hilt, J.Z. Magnetic nanoparticles in biomedicine: Synthesis, functionalization and applications. Nanomedicine 2010, 5, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, H.Y.; Zhou, H.; Hwang, S.; Koh, K.; Han, D.-W.; Lee, J. Green synthesis of phytochemical-stabilized Au nanoparticles under ambient conditions and their biocompatibility and antioxidative activity. J. Mater. Chem. 2011, 21, 13316–13326. [Google Scholar] [CrossRef]
- Granitzer, P.; Rumpf, K. Magnetic nanoparticles embedded in a silicon matrix. Materials 2011, 4, 908–928. [Google Scholar] [CrossRef]
- Yu, M.K.; Park, J.; Jon, S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics 2012, 2, 3–44. [Google Scholar] [CrossRef] [PubMed]
- Wahajuddin; Arora, S. Superparamagnetic iron oxide nanoparticles: Magnetic nanoplatforms as drug carriers. Int. J. Nanomedicine 2012, 7, 3445–3471. [Google Scholar]
- Auffan, M.; Rose, J.; Bottero, J.Y.; Lowry, G.V.; Jolivet, J.P.; Wiesner, M.R. Towards a definition of inorganic nanoparticles from an environmental, health and safety perspective. Nat. Nanotechnol. 2009, 4, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.; Xia, T.; Mädler, L.; Li, N. Toxic potential of materials at the nanolevel. Science 2006, 311, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.J.; Gole, A.M.; Stone, J.W.; Sisco, P.N.; Alkilany, A.M.; Goldsmith, E.C.; Baxter, S.C. Gold nanoparticles in biology: Beyond toxicity to cellular imaging. Acc. Chem. Res. 2008, 41, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Boisselier, E.; Astruc, D. Gold nanoparticles in nanomedicine: Preparations, imaging, diagnostics, therapies and toxicity. Chem. Soc. Rev. 2009, 38, 1759–1782. [Google Scholar] [CrossRef] [PubMed]
- Vega-Villa, K.R.; Takemoto, J.K.; Yáñez, J.A.; Remsberg, C.M.; Forrest, M.L.; Davies, N.M. Clinical toxicities of nanocarrier systems. Adv. Drug Deliv. Rev. 2008, 60, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, T.; Schopf, B.; Hofmann, H.; Hofmann, M.; von Rechenberg, B. Superparamagnetic nanoparticles for biomedical applications: possibilities and limitations of a new drug delivery system. J. Magn. Magn. Mater. 2005, 293, 483–496. [Google Scholar] [CrossRef]
- Mbeh, D.A.; França, R.; Merhi, Y.; Zhang, X.F.; Veres, T.; Sacher, E.; Yahia, L. In vitro biocompatibility assessment of functionalized magnetite nanoparticles: Biological and cytotoxicological effects. J. Biomed. Mater. Res. A 2012, 100, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Novotna, B.; Jendelova, P.; Kapcalova, M.; Rossner, P., Jr.; Turnovcova, K.; Bagryantseva, Y.; Babic, M.; Horak, D.; Sykova, E. Oxidative damage to biological macromolecules in human bone marrow mesenchymal stromal cells labeled with various types of iron oxide nanoparticles. Toxicol. Lett. 2012, 210, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Jenkins, G.J.; Nelson, B.C.; Marquis, B.J.; Maffeis, T.G.; Brown, A.P.; Williams, P.M.; Wright, C.J.; Doak, S.H. The role of iron redox state in the genotoxicity of ultrafine superparamagnetic iron oxide nanoparticles. Biomaterials 2012, 33, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Kumar, V.; Yadav, S.K. Therapeutic nanoparticles and associated toxicity. Curr. Nanosci. 2011, 7, 389–395. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Laurent, S.; Shokrgozar, M.A.; Hosseinkhani, M. Toxicity evaluations of superparamagnetic iron oxide nanoparticles: Cell “vision” versus physicochemical properties of nanoparticles. ACS Nano 2011, 5, 7263–7276. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, S.; Behzadi, S.; Laurent, S.; Forrest, M.L.; Stroeve, P.; Mahmoudi, M. Toxicity of nanomaterials. Chem. Soc. Rev. 2012, 41, 2323–2343. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.C.; Lee, J.H.; Lee, J.; Kim, H.Y.; Park, J.Y.; Cho, J.; Lee, J.; Han, D.-W. Subtle cytotoxicity and genotoxicity differences in superparamagnetic iron oxide nanoparticles coated with various functional groups. Int. J. Nanomedicine 2011, 6, 3219–3231. [Google Scholar] [PubMed]
- Zhang, F.; Wang, C.C. Fabrication of one-dimensional iron oxide/silica nanostructures with high magnetic sensitivity by dipole-directed self-assembly. J. Phys. Chem. C 2008, 112, 15151–15156. [Google Scholar] [CrossRef]
- Bucak, S.; Yavuztürk, B.; Sezer, A.D. Magnetic nanoparticles: synthesis, surface modifications and application in drug delivery. In Recent Advances in Novel Drug Carrier Systems, 1st ed.; Ali, D.S., Ed.; InTech: Rijeka, Croatia, 2012; Chapter 7; pp. 165–200. [Google Scholar]
- Karlsson, H.L.; Cronholm, P.; Gustafsson, J.; Möller, L. Copper oxide nanoparticles are highly toxic: a comparison between metal oxide nanoparticles and carbon nanotubes. Chem. Res. Toxicol. 2008, 21, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Rivera, G.P.; Huhn, D.; del Mercato, L.L.; Sasse, D.; Parak, W.J. Nanopharmacy: Inorganic nanoscale devices as vectors and active compounds. Pharmacol. Res. 2010, 62, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; de Haan, L.H.; Evers, N.M.; Jiang, X.; Marcelis, A.T.; Zuilhof, H.; Rietjens, I.M.; Alink, G.M. Role of surface charge and oxidative stress in cytotoxicity of organic monolayer-coated silicon nanoparticles towards macrophage NR8383 cells. Part Fibre Toxicol. 2011, 7. [Google Scholar] [CrossRef]
- Kai, W.; Xiaojun, X.; Ximing, P.; Zhenqing, H.; Qiqing, Z. Cytotoxic effects and the mechanism of three types of magnetic nanoparticles on human hepatoma BEL-7402 cells. Nanoscale Res. Lett. 2011, 6. [Google Scholar] [CrossRef]
- Blank, F.; Gerber, P.; Rothen-Rutishauser, B.; Sakulkhu, U.; Salaklang, J.; de Peyer, K.; Gehr, P.; Nicod, L.P.; Hofmann, H.; Geiser, T.; Petri-Fink, A.; von Garnier, C. Biomedical nanoparticles modulate specific CD4+ T cell stimulation by inhibition of antigen processing in dendritic cells. Nanotoxicology 2011, 5, 606–621. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Huang, S.; Yu, K.J.; Clyne, A.M. Dextran and polymer polyethylene glycol (PEG) coating reduce both 5 and 30 nm iron oxide nanoparticle cytotoxicity in 2D and 3D cell culture. Int. J. Mol. Sci. 2012, 13, 5554–5570. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, N.; Colvin, V.; Drezek, R. Cytotoxicity of nanoparticles. Small 2008, 4, 26–49. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Wells, S. Surface-modified superparamagnetic nanoparticles for drug delivery: Preparation, characterization, and cytotoxicity studies. IEEE Trans. Nanobiosci. 2004, 3, 66–73. [Google Scholar] [CrossRef]
- Hoskins, C.; Cuschieri, A.; Wang, L. The cytotoxicity of polycationic iron oxide nanoparticles: Common endpoint assays and alternative approaches for improved understanding of cellular response mechanism. J. Nanobiotechnol. 2012, 10. [Google Scholar] [CrossRef]
- Singh, N.; Jenkins, G.J.; Asadi, R.; Doak, S.H. Potential toxicity of superparamagnetic iron oxide nanoparticles (SPION). Nano Rev. 2010, 1. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Sahu, S.K.; Banerjee, I.; Das, M.; Mishra, D.; Maiti, T.K.; Pramanik, P. Synthesis, characterization, and in vitro biological evaluation of highly stable diversely functionalized superparamagnetic iron oxide nanoparticles. J. Nanopart. Res. 2011, 13, 4173–4188. [Google Scholar] [CrossRef]
- Vácha, R.; Martinez-Veracoechea, F.J.; Frenkel, D. Receptor-mediated endocytosis of nanoparticles of various shapes. Nano Lett. 2011, 11, 5391–5395. [Google Scholar] [CrossRef] [PubMed]
- Atabaev, T.Sh.; Lee, J.H.; Han, D.-W.; Hwang, Y.-H.; Kim, H.-K. Cytotoxicity and cell imaging potentials of submicron color-tunable yttria particles. J. Biomed. Mater. Res. A 2012, 100, 2287–2294. [Google Scholar] [PubMed]
- Chithrani, B.D.; Chan, W.C. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Kim, B.Y.; Rutka, J.T.; Chan, W.C. Nanoparticle-mediated cellular response is size-dependent. Nat. Nanotechnol. 2008, 3, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Atabaev, T.Sh.; Jin, O.S.; Lee, J.H.; Han, D.-W.; Vu, H.H.T.; Hwang, Y.-H.; Kim, H.-K. Facile synthesis of bifunctional silica-coated core-shell Y2O3:Eu3+, Co2+ composite particles for biomedical applications. RSC Adv. 2012, 2, 9495–9501. [Google Scholar] [CrossRef]
- Sohaebuddin, S.K.; Thevenot, P.T.; Baker, D.; Eaton, J.W.; Tang, L. Nanomaterial cytotoxicity is composition, size, and cell type dependent. Part Fibre Toxicol. 2010, 7. [Google Scholar] [CrossRef]
- Park, K.S.; Lee, H.S.; Kim, Y.S.; Kang, T.M.; Lee, J.H.; Joh, J.W.; Kim, S.J. Improved quantification of islet transplants by magnetic resonance imaging with Resovist. Pancreas 2011, 40, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Fang, J.; Zhang, T.; Wang, B.; Chen, J.; Li, X.; Zhang, S.; Zhang, W. Magnetic resonance imaging targeting of intracranial glioma xenografts by Resovist-labeled endothelial progenitor cells. J. Neurooncol. 2011, 105, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Sant, S.; Wang, B.; Laurent, S.; Sen, T. Superparamagnetic iron oxide nanoparticles (SPIONs): Development, surface modification and applications in chemotherapy. Adv. Drug Deliv. Rev. 2011, 63, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.M.; Kim, D.K.; El Haj, A.J.; Dobson, J. Development of superparamagnetic iron oxide nanoparticles (SPIONS) for translation to clinical applications. IEEE Trans. Nanobiosci. 2008, 7, 298–305. [Google Scholar] [CrossRef]
- Bae, J.E.; Huh, M.I.; Ryu, B.K.; Do, J.Y.; Jin, S.U.; Moon, M.J.; Jung, J.C.; Chang, Y.; Kim, E.; Chi, S.G.; et al. The effect of static magnetic fields on the aggregation and cytotoxicity of magnetic nanoparticles. Biomaterials 2011, 32, 9401–9414. [Google Scholar] [CrossRef] [PubMed]
- Rossi, L.M.; Quach, A.D.; Rosenzweig, Z. Glucose oxidase—magnetite nanoparticle bioconjugate for glucose sensing. Anal. Bioanal. Chem. 2004, 380, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Blazer-Yost, B.L.; Banga, A.; Amos, A.; Chernoff, E.; Lai, X.; Li, C.; Mitra, S.; Witzmann, F.A. Effect of carbon nanoparticles on renal epithelial cell structure, barrier function, and protein expression. Nanotoxicology 2011, 5, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Cemeli, E.; Anderson, D. Mechanistic investigation of ROS-induced DNA damage by oestrogenic compounds in lymphocytes and sperm using the comet assay. Int. J. Mol. Sci. 2011, 12, 2783–2796. [Google Scholar] [CrossRef] [PubMed]
- Klarić, M.S.; Darabos, D.; Rozgaj, R.; Kasuba, V.; Pepeljnjak, S. Beauvericin and ochratoxin A genotoxicity evaluated using the alkaline comet assay: Single and combined genotoxic action. Arch. Toxicol. 2010, 84, 641–650. [Google Scholar] [CrossRef] [PubMed]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, W.J.; Lee, J.H.; Hong, S.C.; Lee, J.; Lee, J.; Han, D.-W. Difference between Toxicities of Iron Oxide Magnetic Nanoparticles with Various Surface-Functional Groups against Human Normal Fibroblasts and Fibrosarcoma Cells. Materials 2013, 6, 4689-4706. https://doi.org/10.3390/ma6104689
Yang WJ, Lee JH, Hong SC, Lee J, Lee J, Han D-W. Difference between Toxicities of Iron Oxide Magnetic Nanoparticles with Various Surface-Functional Groups against Human Normal Fibroblasts and Fibrosarcoma Cells. Materials. 2013; 6(10):4689-4706. https://doi.org/10.3390/ma6104689
Chicago/Turabian StyleYang, Won Jun, Jong Ho Lee, Seong Cheol Hong, Jaewook Lee, Jaebeom Lee, and Dong-Wook Han. 2013. "Difference between Toxicities of Iron Oxide Magnetic Nanoparticles with Various Surface-Functional Groups against Human Normal Fibroblasts and Fibrosarcoma Cells" Materials 6, no. 10: 4689-4706. https://doi.org/10.3390/ma6104689