Homogenized Elastic Properties of Graphene for Small Deformations

Abstract
:1. Introduction
2. Graphene Model Properties and Homogenization Procedure
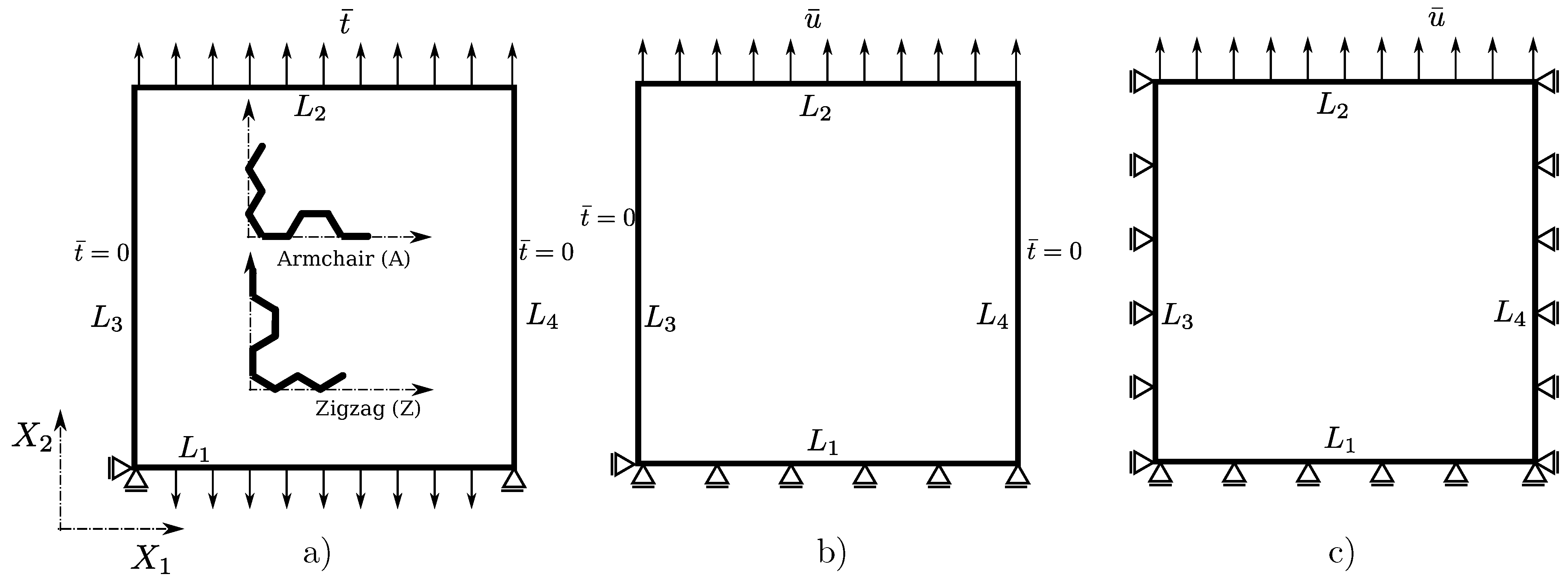
2.1. Problem Statement
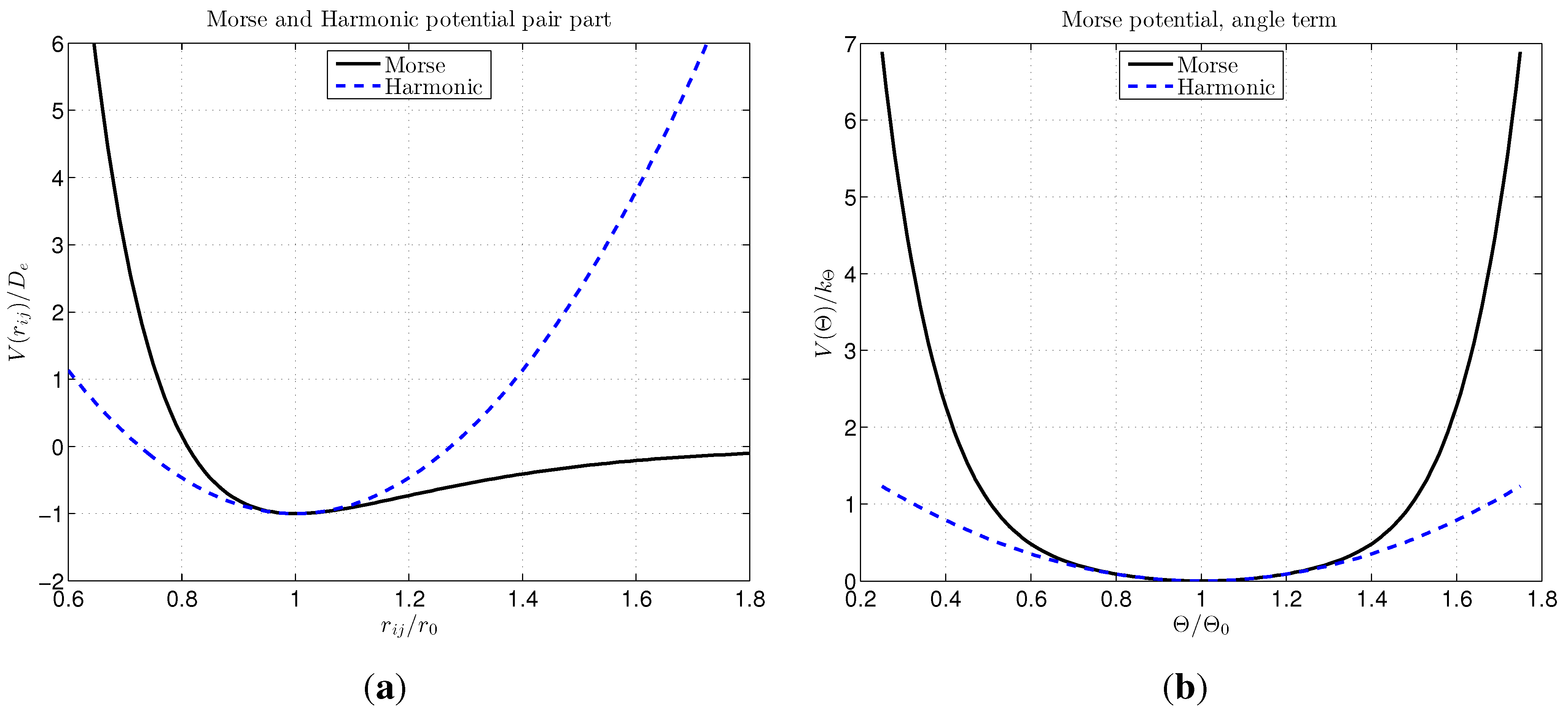
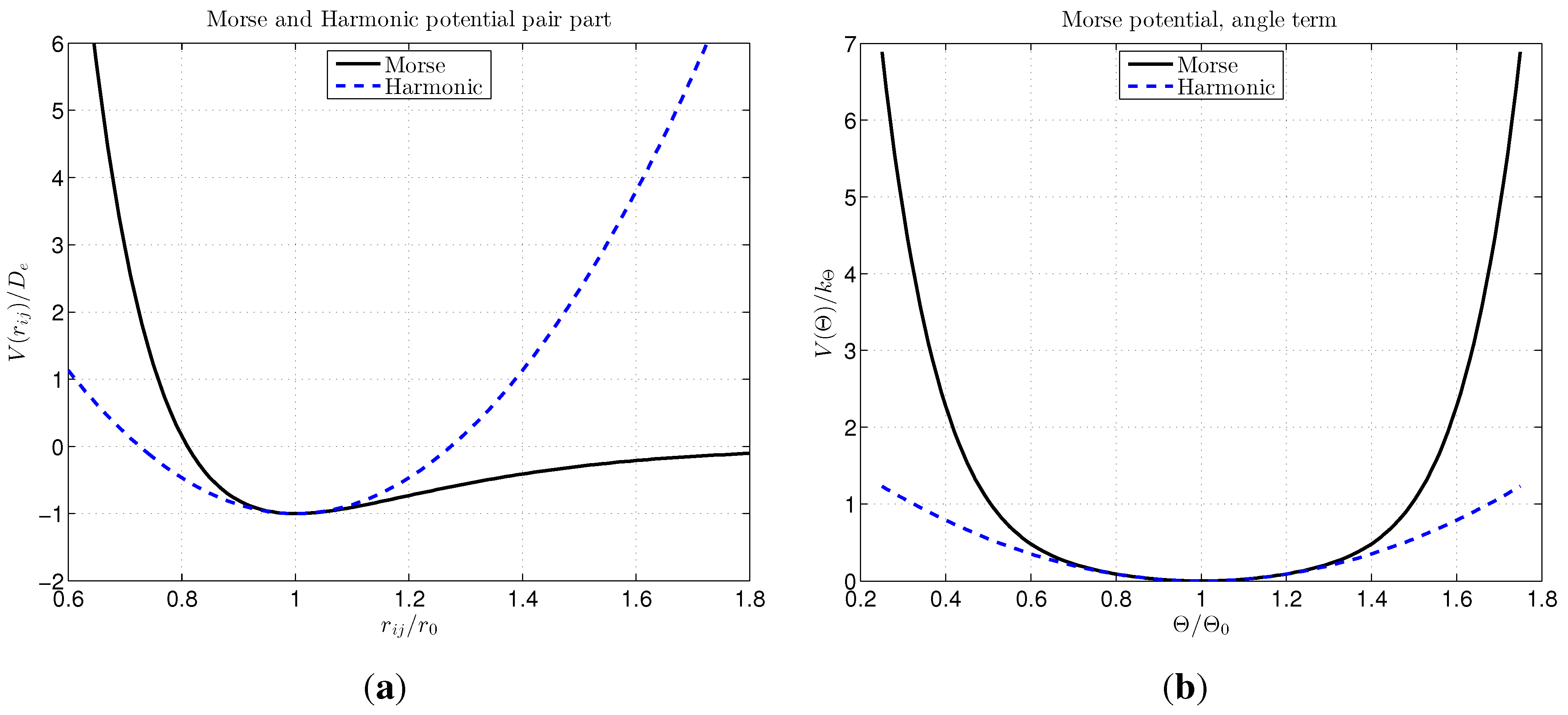
2.2. Choice of Interatomic Potential
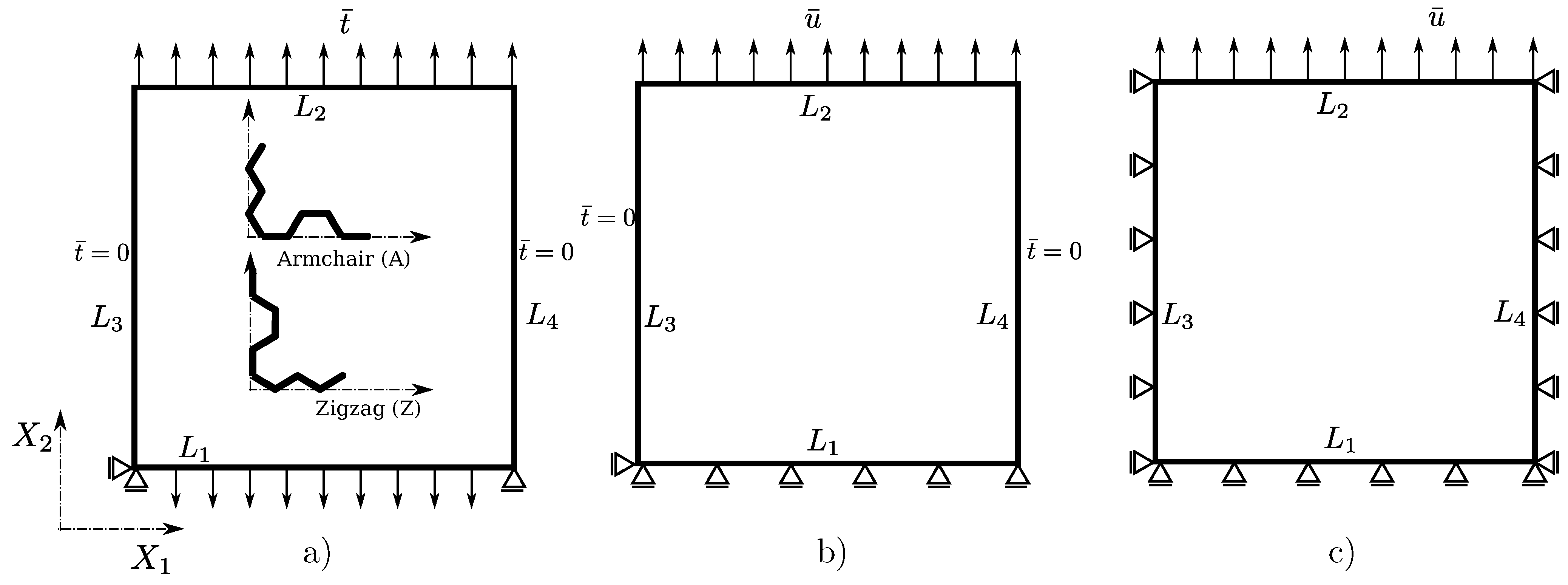
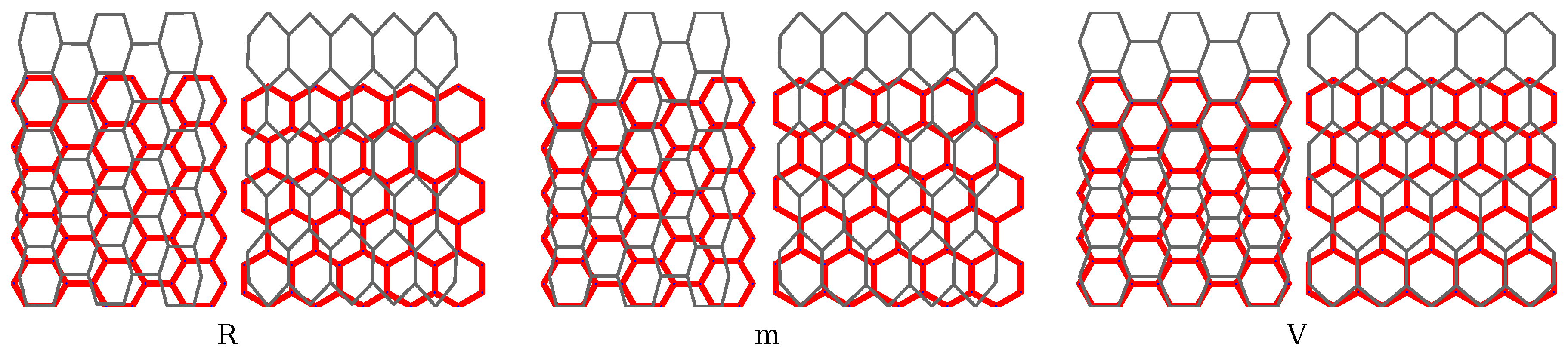
2.3. Choice of Boundary Conditions and Computational Procedure
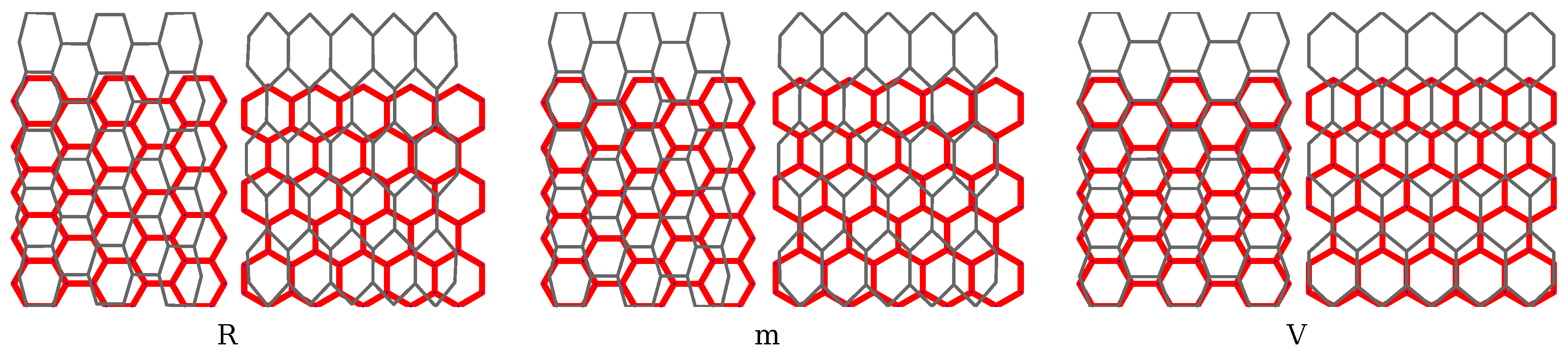
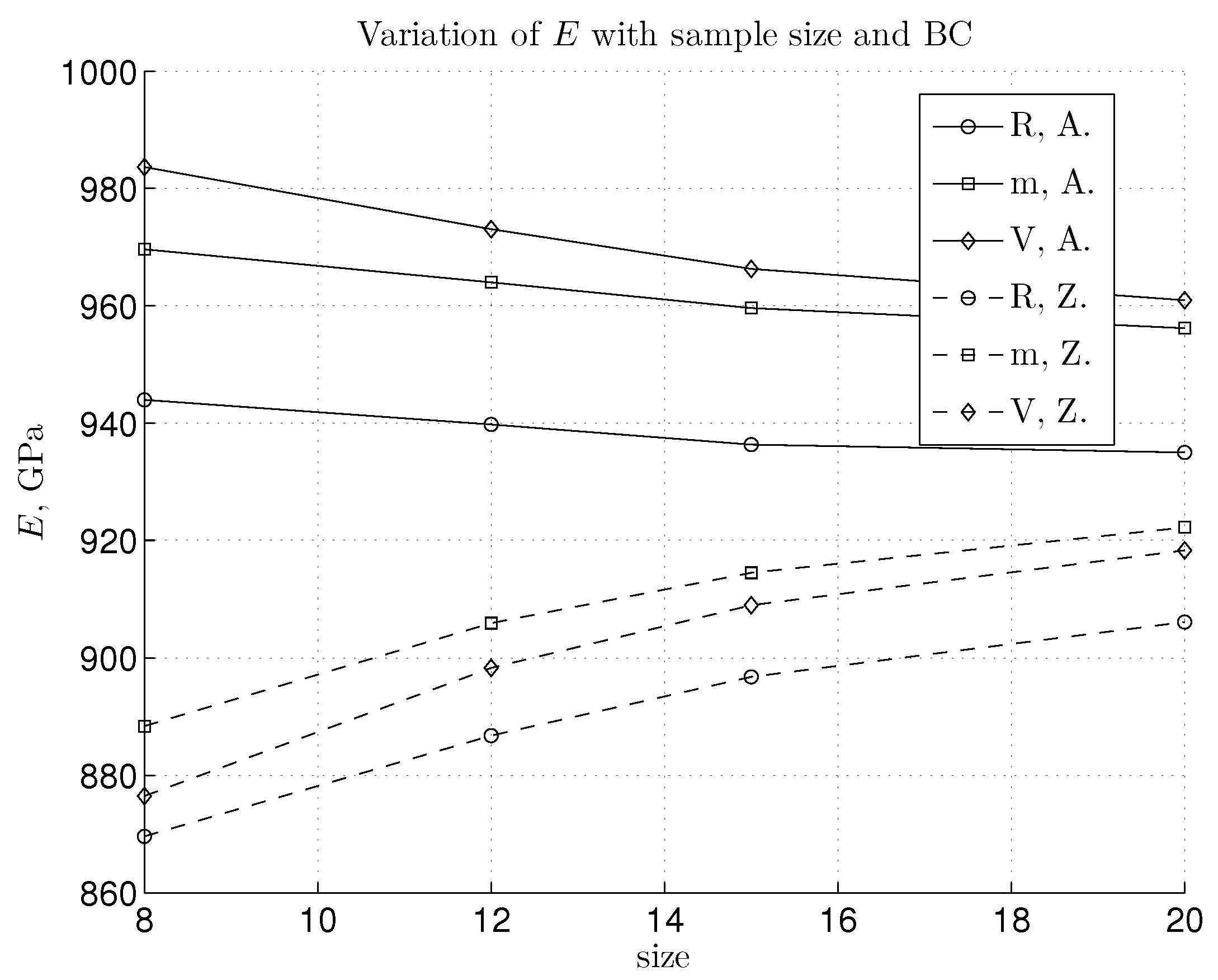
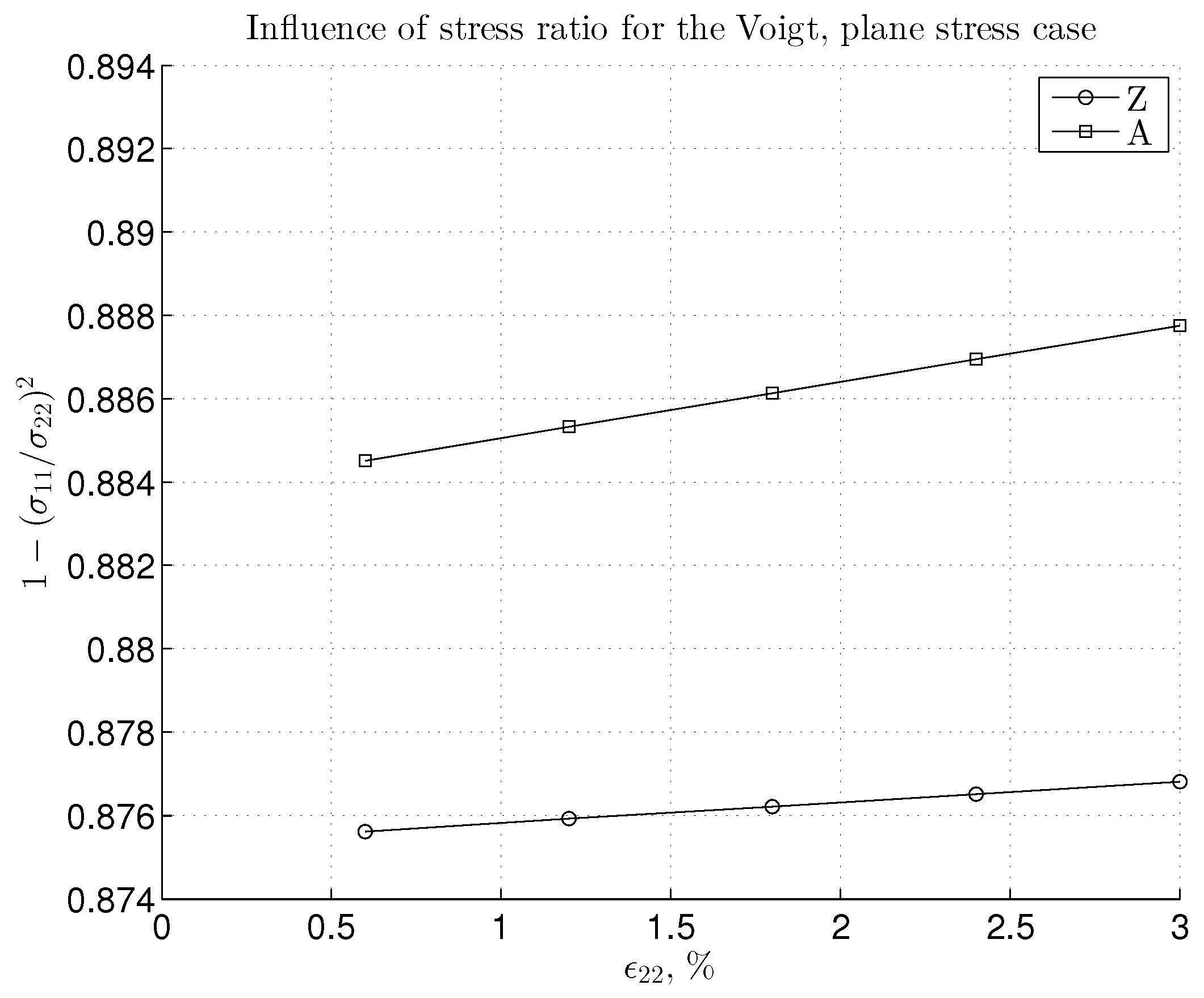
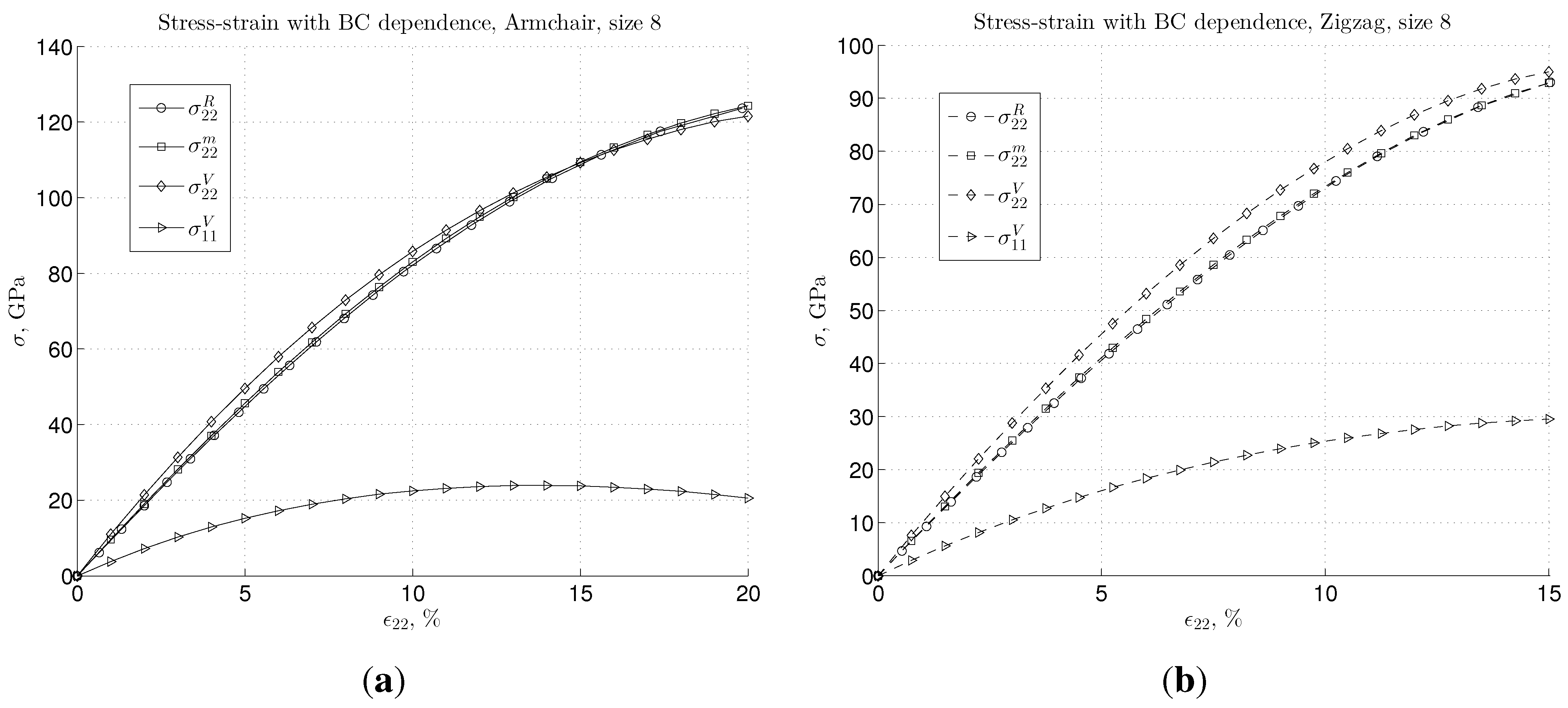
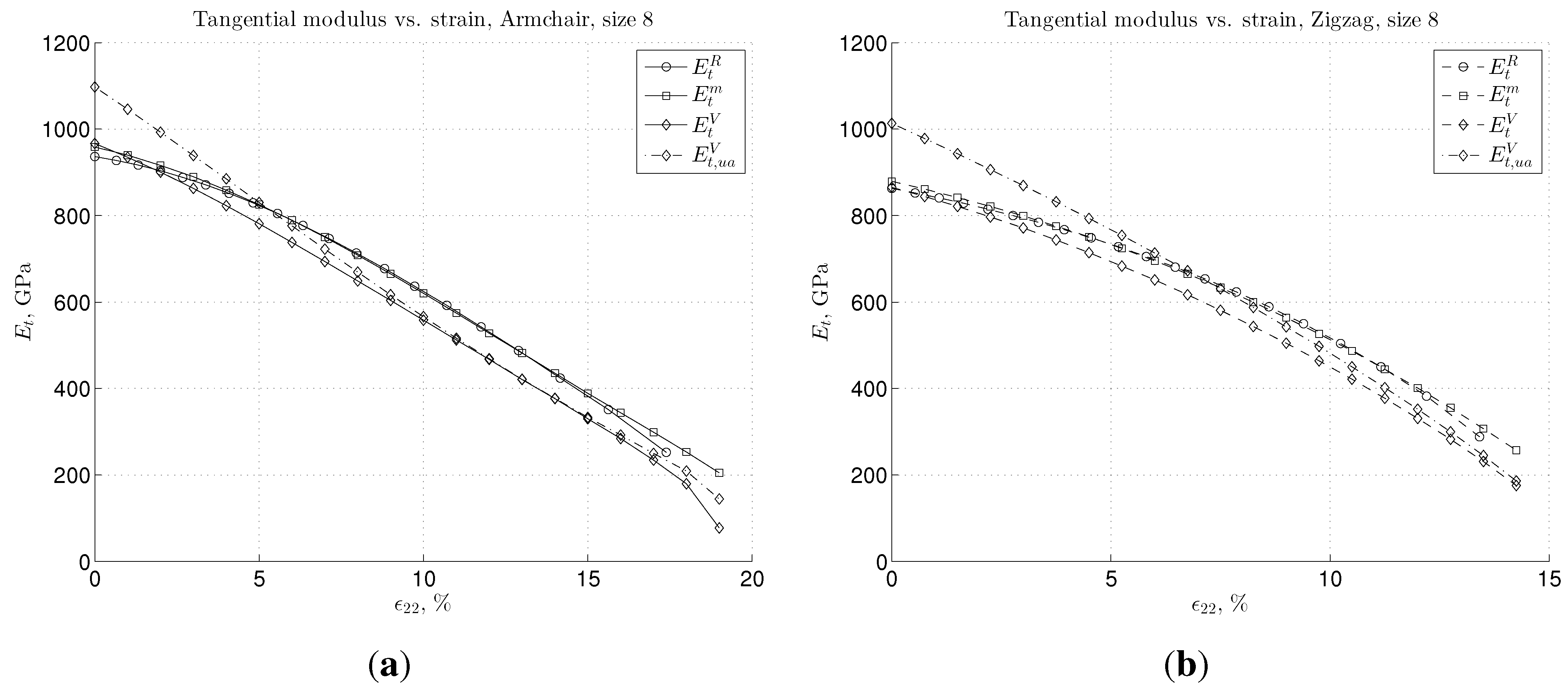
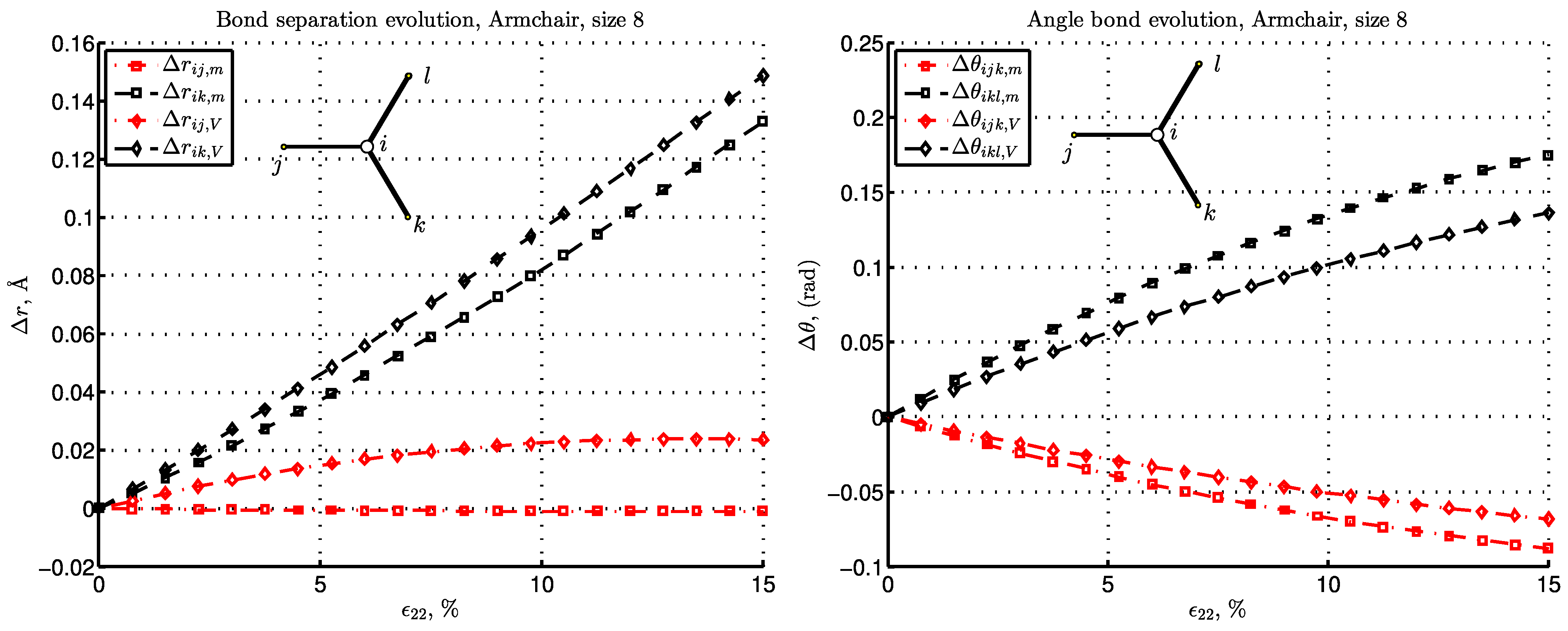
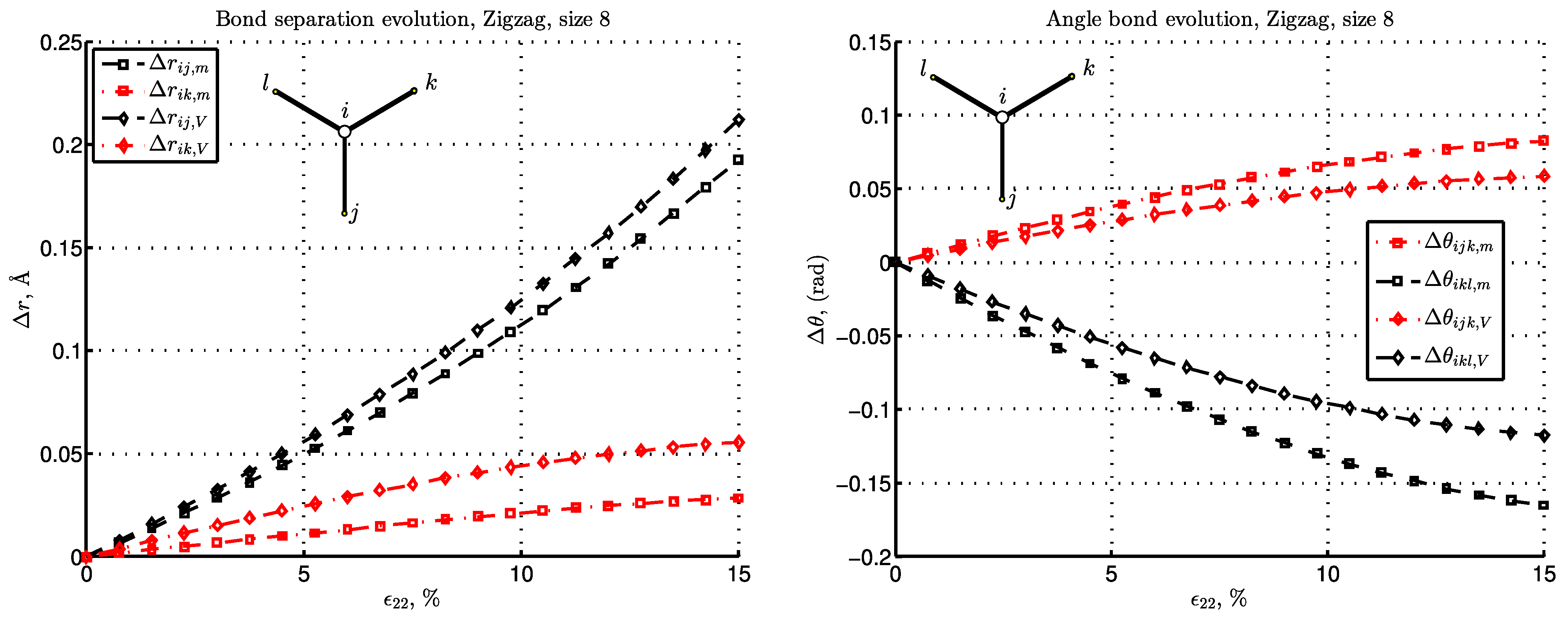
3. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| size parameter | 5 | 8 | 12 | 16 | 20 | 24 | 28 |
|---|---|---|---|---|---|---|---|
| , Å | 12.03 | 19.26 | 28.89 | 38.52 | 48.15 | 57.78 | 67.41 |
| number of atoms | 66 | 170 | 350 | 660 | 984 | 1372 | 1824 |
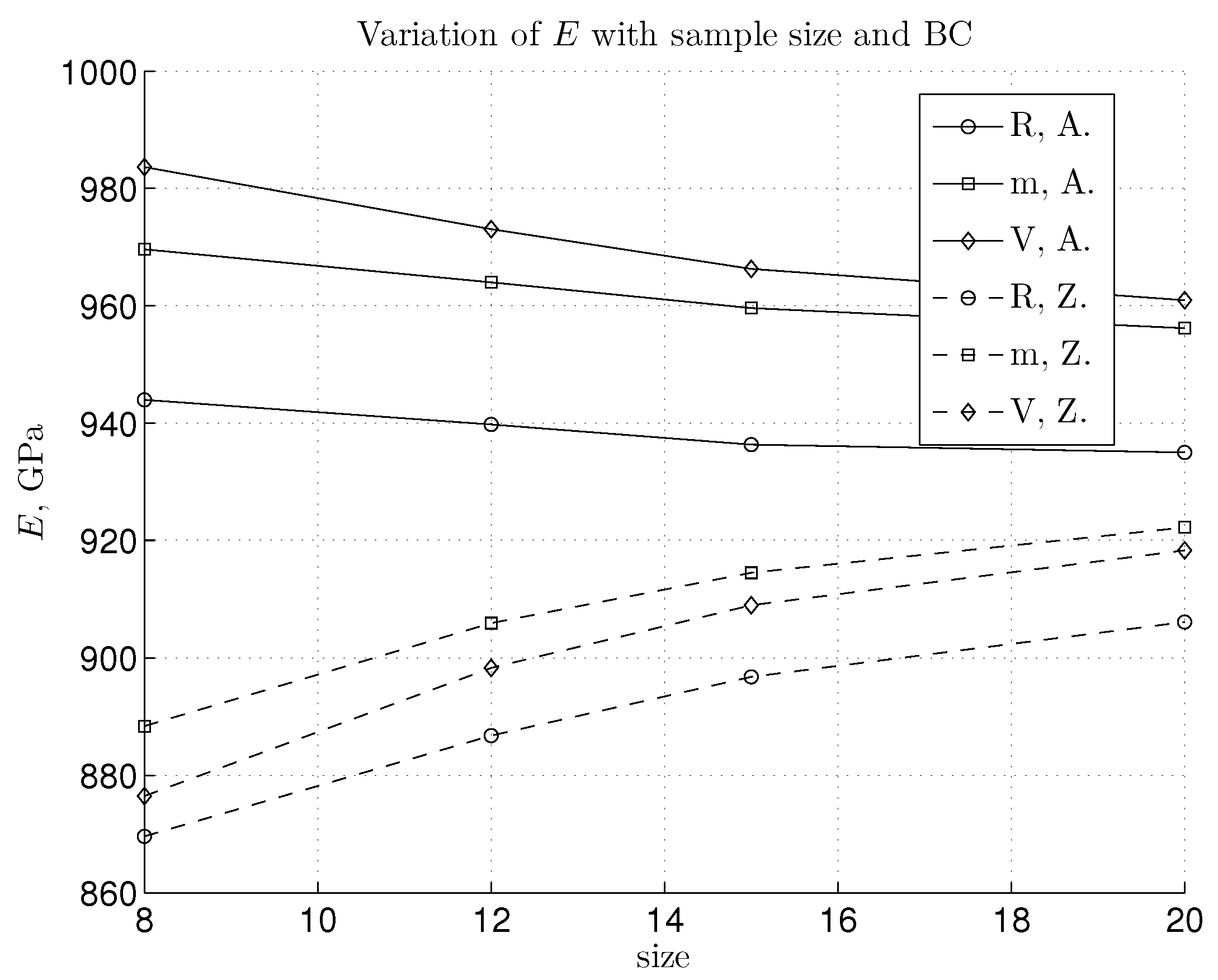
3.1. Linear Regime and Young’s Modulus Value
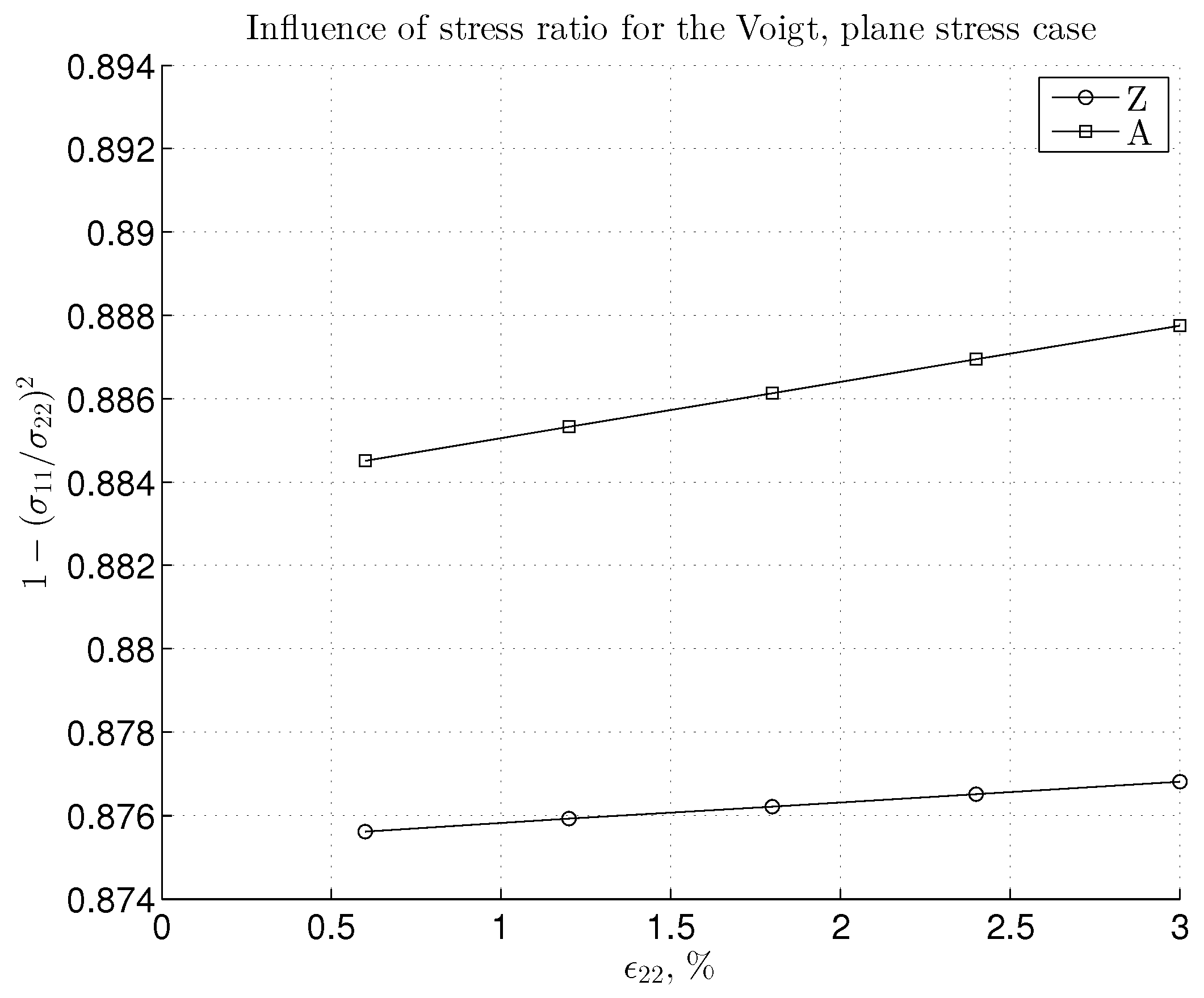
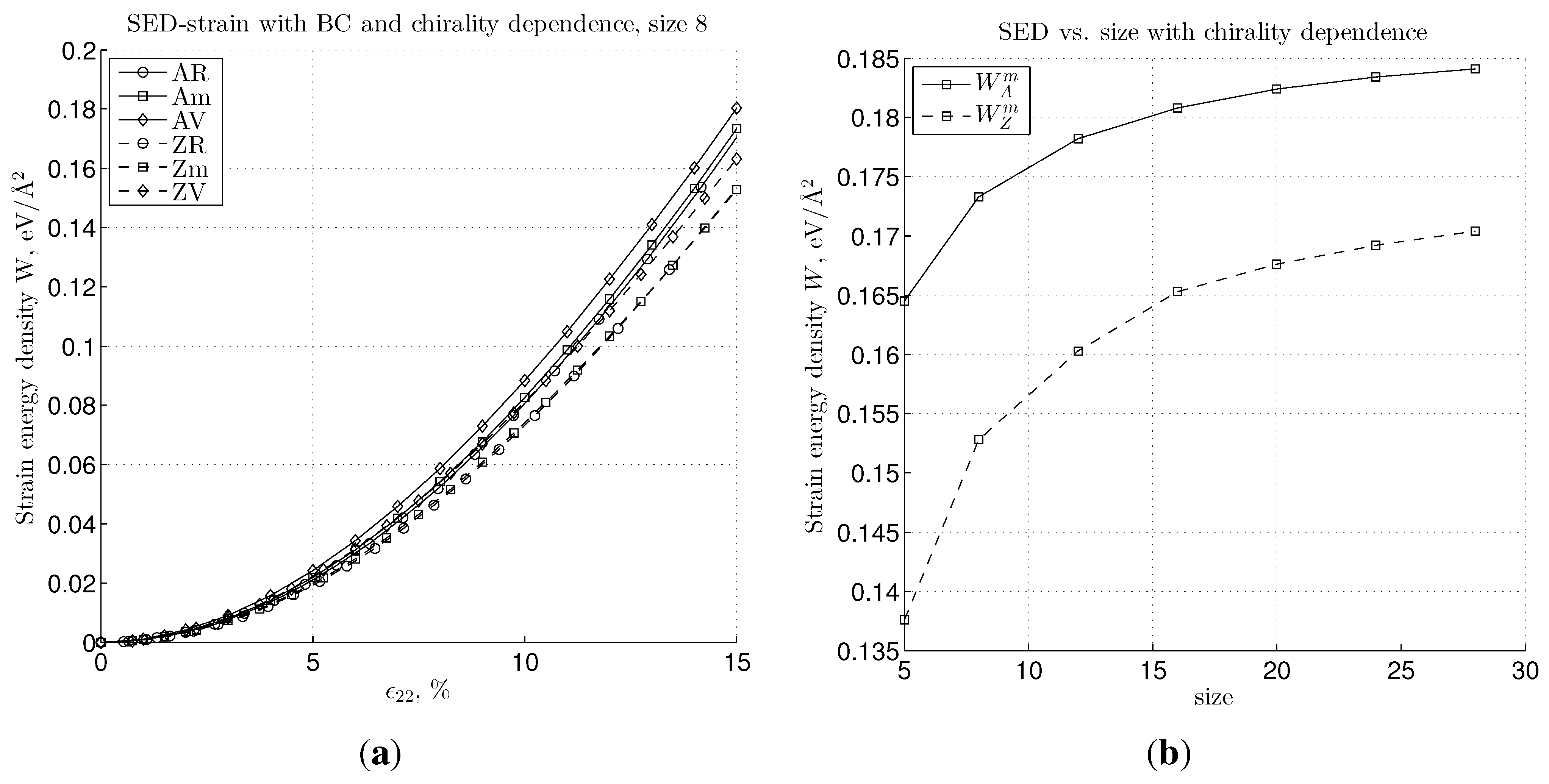
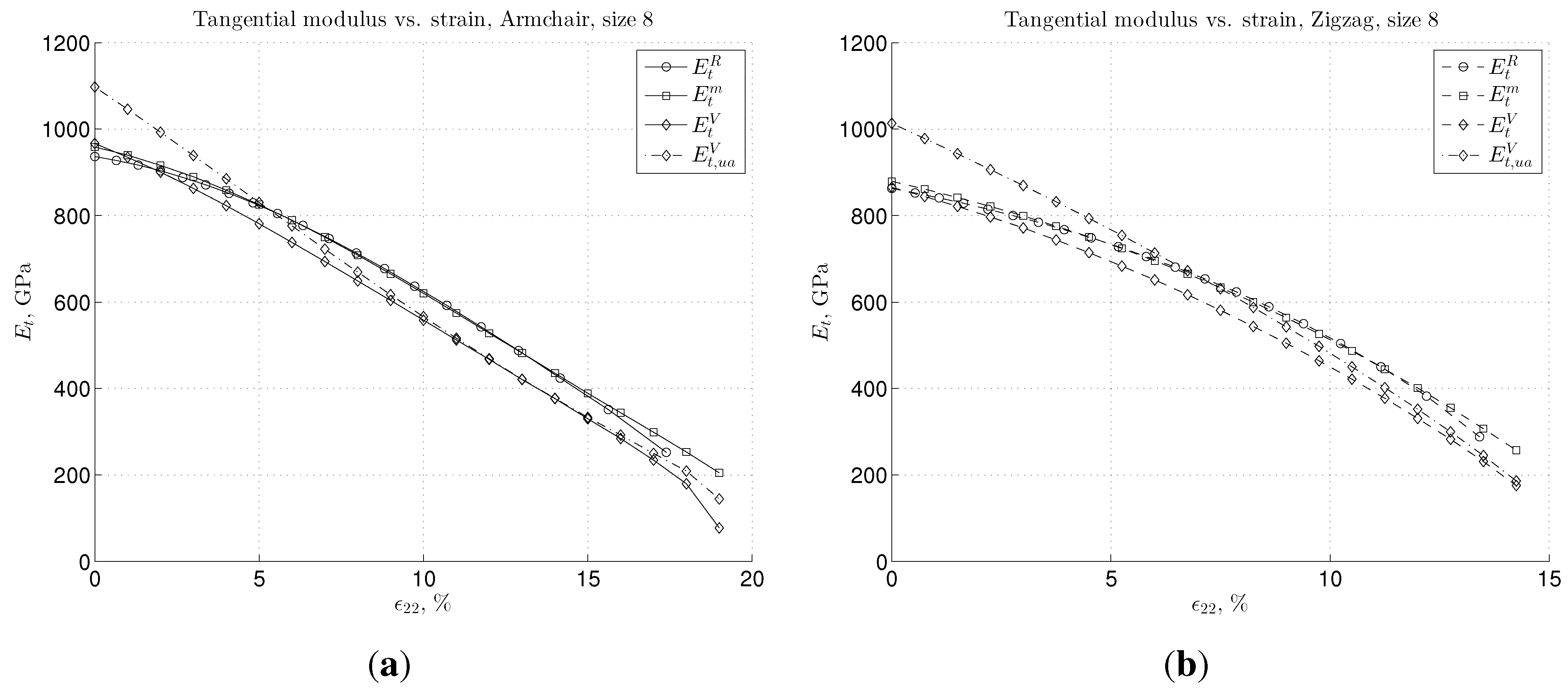
3.2. Nonlinear Regime and Tangential Modulus
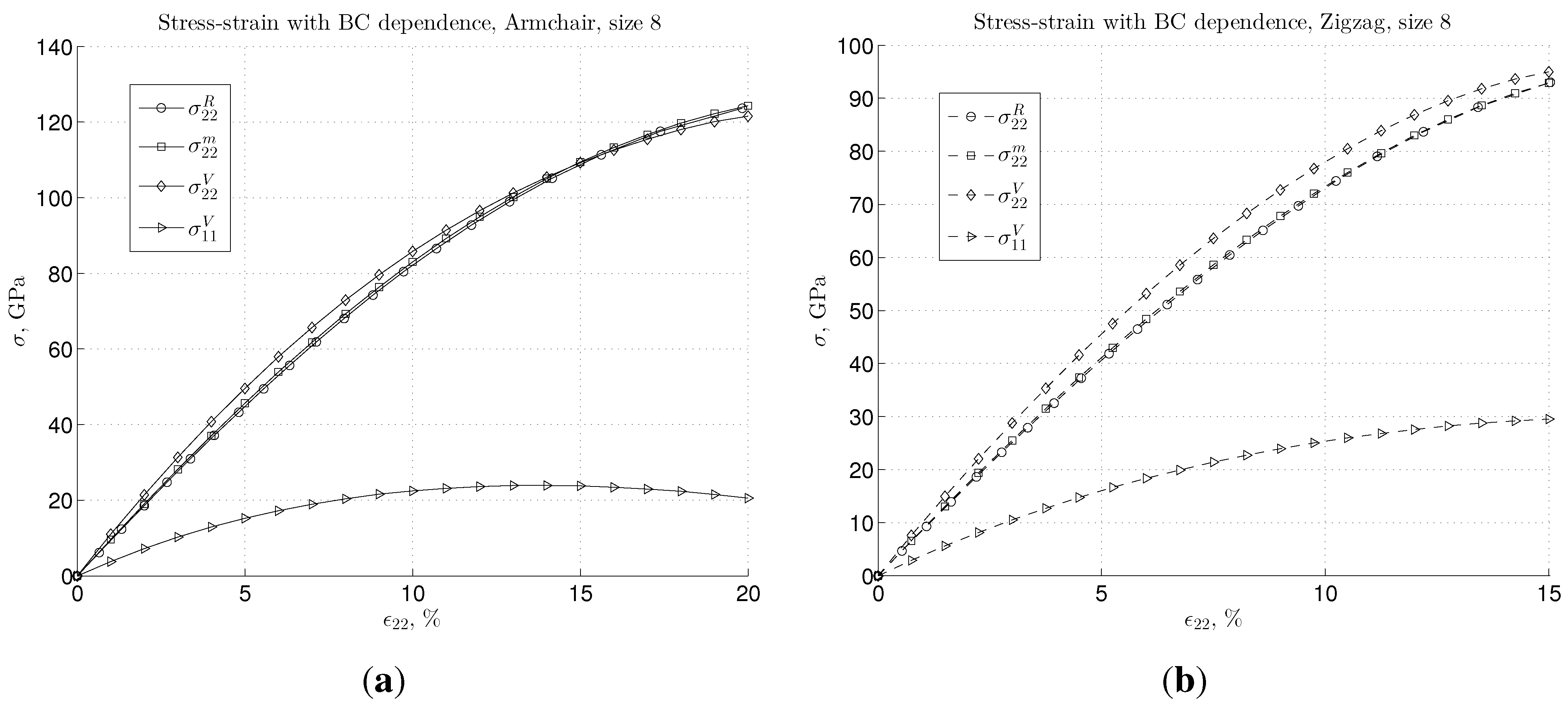
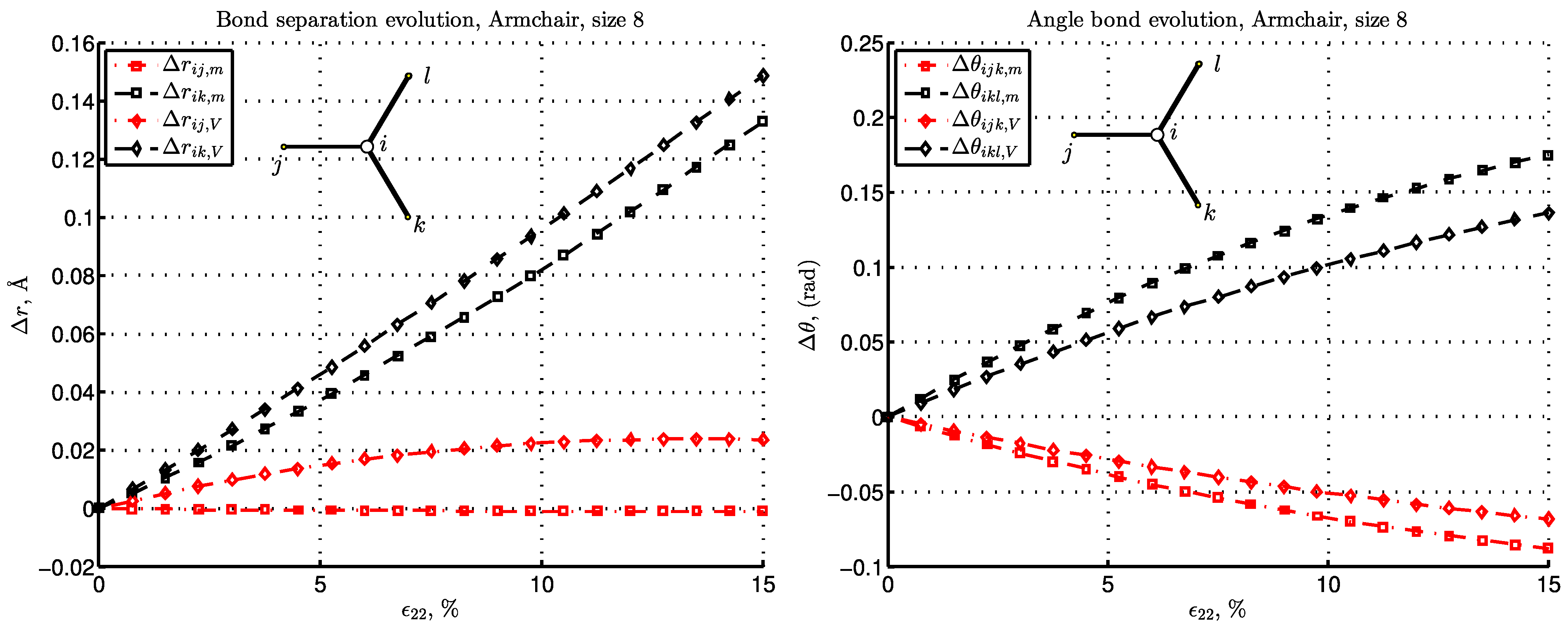
3.3. Energy and Deformation of Bonds

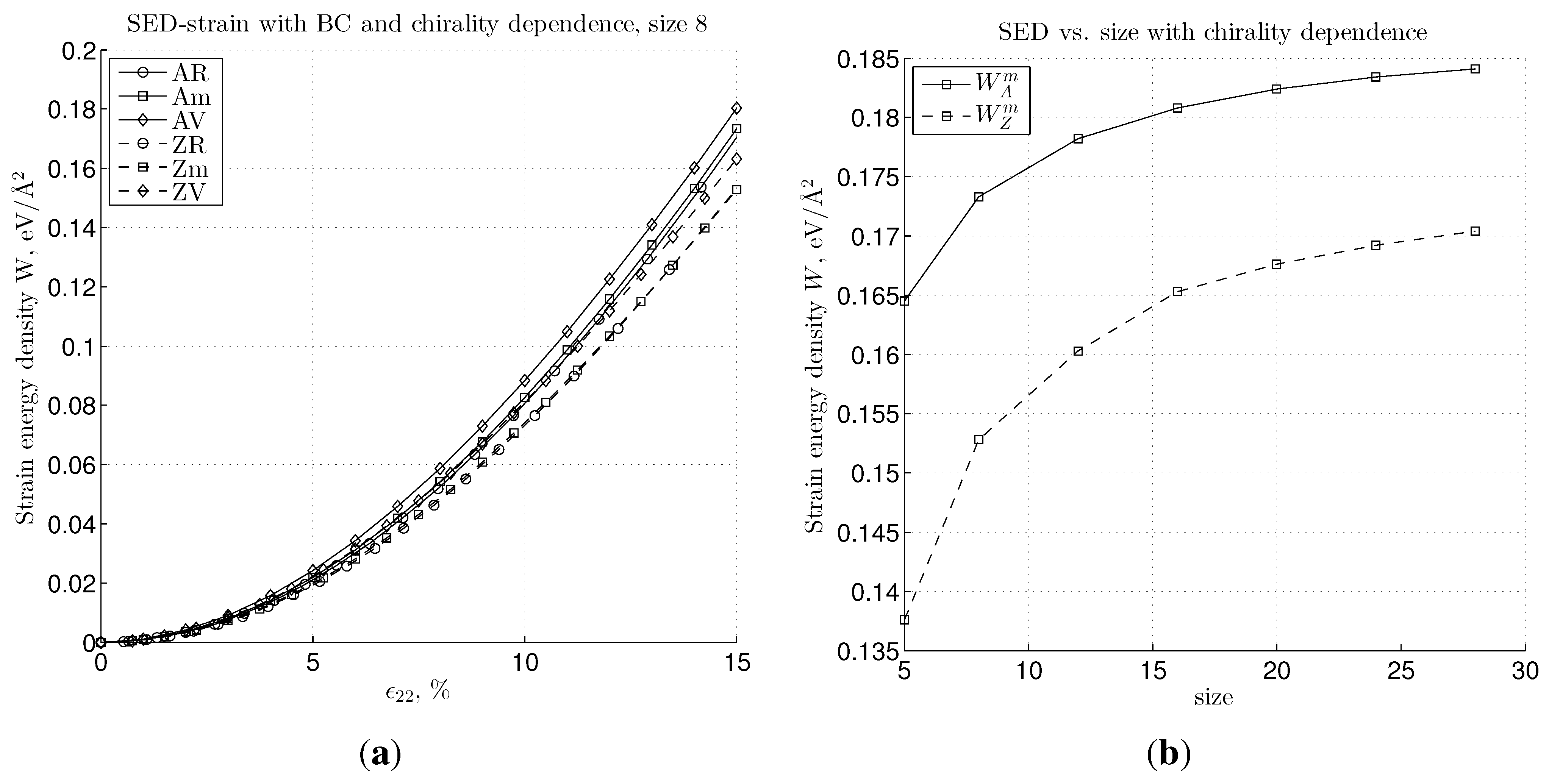
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Jiang, D.; Schedin, F.; Booth, T.J.; Khotkevich, V.V.; Morozov, S.V.; Geim, A.K. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451–10453. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Frank, I.W.; Tanenbaum, D.M.; van der Zande, A.M.; McEuen, P.L. Mechanical properties of suspended graphene sheets. J. Vac. Sci. Technol. B Microelectron. Nanometer Struct. 2007, 25, 2558–2562. [Google Scholar] [CrossRef]
- Lee, C.; Wei, X.; Kysar, J.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Ruoff, R.S.; Qian, D.; Liu, W.K. Mechanical properties of carbon nanotubes: Theoretical predictions and experimental measurements. Comptes Rendus Phys. 2003, 4, 993–1008. [Google Scholar] [CrossRef]
- Atkins, P.; de Paula, J. Physical Chemistry, 8th ed.; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Zhou, Q.; Zettl, A. Electrostatic Graphene Loudspeaker. Appl. Phys. Lett. 2003, 4, 223109. [Google Scholar] [CrossRef]
- Singh, V.; Joung, D.; Zhai, L.; Das, S.; Khondaker, S.I.; Seal, S. Graphene based materials: Past, present and future. Prog. Mater. Sci. 2011, 56, 1178–1271. [Google Scholar] [CrossRef]
- Reddy, C.D.; Rajendran, S.; Liew, K.M. Equilibrium configuration and continuum elastic properties of finite sized graphene. Nanotechnology 2006, 17, 864. [Google Scholar] [CrossRef]
- Brenner, D.W. Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films. Phys. Rev. B 1990, 42, 9458–9471. [Google Scholar] [CrossRef]
- Tersoff, J. New empirical model for the structural properties of silicon. Phys. Rev. Lett. 1986, 56, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, M.; Belytschko, T. Finite crystal elasticity of carbon nanotubes based on the exponential Cauchy-Born rule. Phys. Rev. B 2004, 69, 115415. [Google Scholar] [CrossRef]
- Zhao, H.; Min, K.; Aluru, N.R. Size and chirality dependent elastic properties of graphene nanoribbons under uniaxial tension. Nanoletters 2009, 9, 3012–3015. [Google Scholar] [CrossRef] [PubMed]
- Lier, G.V.; Alsenoy, C.V.; Doren, V.V.; Geerlings, P. Ab initio study of the elastic properties of single-walled carbon nanotubes and graphene. Chem. Phys. Lett. 2000, 326, 181–185. [Google Scholar] [CrossRef]
- Kudin, K.N.; Scuseria, G.E.; Yakobson, B.I. C2F, BN, and C nanoshell elasticity from ab initio computations. Phys. Rev. B 2001, 64, 235406. [Google Scholar] [CrossRef]
- Xu, Z. Graphene nano-ribbons under tension. J. Comput. Theor. Nanosci. 2009, 6, 625–628. [Google Scholar] [CrossRef]
- Lu, Q.; Huang, R. Excess energy and deformation along free edges of graphene nanoribbons. Phys. Rev. B 2010, 81, 155410. [Google Scholar] [CrossRef]
- Lu, Q.; Gao, W.; Huang, R. Atomistic simulation and continuum modeling of graphene nanoribbons under uniaxial tension. Modell. Simul. Mater. Sci. Eng. 2011, 19, 054006. [Google Scholar] [CrossRef]
- Odegard, G.M.; Gates, T.S.; Nicholson, L.M.; Wise, K.E. Equivalent-continuum modeling of nano-structured materials. Compos. Sci. Technol. 2002, 62, 1869–1880. [Google Scholar] [CrossRef]
- Scarpa, F.; Adhikari, S.; Phani, A.S. Effective elastic mechanical properties of single layer graphene sheets. Nanotechnology 2009, 20, 065709. [Google Scholar] [CrossRef] [PubMed]
- Georgantzinos, S.; Giannopoulos, G.; Katsareas, D.; Kakavas, P.; Anifantis, N. Size-dependent non-linear mechanical properties of graphene nanoribbons. Comput. Mater. Sci. 2011, 50, 2057–2062. [Google Scholar] [CrossRef]
- Georgantzinos, S.; Katsareas, D.; Anifantis, N. Graphene characterization: A fully non-linear spring-based finite element prediction. Phys. E Low-dimens. Syst. Nanostruct. 2011, 43, 1833–1839. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, J.; Hwang, K.C. Thickness of graphene and single-wall carbon nanotubes. Phys. Rev. B 2006, 74, 245413. [Google Scholar] [CrossRef]
- Georgantzinos, S.; Katsareas, D.; Anifantis, N. Limit load analysis of graphene with pinhole defects: A nonlinear structural mechanics approach. Int. J. Mech. Sci. 2012, 55, 85–94. [Google Scholar] [CrossRef]
- Lu, Q.; Huang, R. Nonlinear mechanics of Single-atomic-layer graphene sheets. Int. J. Appl. Mech. 2009, 1, 443–467. [Google Scholar] [CrossRef]
- Arroyo, M.; Belytschko, T. Finite element methods for the non-linear mechanics of crystalline sheets and nanotubes. Int. J. Numer. Methods Eng. 2004, 59, 419–456. [Google Scholar] [CrossRef]
- Caillerie, D.; Mourat, A.; Raoult, A. Discrete homogenization in graphene sheet modeling. J. Elast. 2006, 84, 33–68. [Google Scholar] [CrossRef]
- Markovic, D.; Ibrahimbegovic, A. On micro-macro interface conditions for micro scale based FEM for inelastic behavior of heterogeneous materials. Comput. Methods Appl. Mech. Eng. 2004, 193, 5503–5523. [Google Scholar] [CrossRef]
- Huet, C. Application of variational concepts to size effects in elastic heterogeneous bodies. J. Mech. Phys. Solids 1990, 38, 813–841. [Google Scholar] [CrossRef]
- Ibrahimbegovic, A.; Herve, G.; Villon, P. Nonlinear impact dynamics and field transfer suitable for parametric design studies. Eng. Comput. 2009, 26, 185–204. [Google Scholar] [CrossRef]
- Liu, B.; Huang, Y.; Jiang, H.; Qu, S.; Hwang, K.C. The atomic-scale finite element method. Comput. Methods Appl. Mech. Eng. 2004, 193, 1849–1864. [Google Scholar] [CrossRef]
- Belytschko, T.; Xiao, S.P.; Schatz, G.C.; Ruoff, R.S. Atomistic simulations of nanotube fracture. Phys. Rev. B 2002, 65, 235430. [Google Scholar] [CrossRef]
- Liu, B.; Jiang, H.; Huang, Y.; Qu, S.; Yu, M.F.; Hwang, K.C. Atomic-scale finite element method in multiscale computation with applications to carbon nanotubes. Phys. Rev. B 2005, 72, 035435. [Google Scholar] [CrossRef]
- Wackerfuss, J. Molecular mechanics in the context of the finite element method. Int. J. Numer. Methods Eng. 2009, 77, 969–997. [Google Scholar] [CrossRef]
- Xiao, J.; Staniszewski, J.; Gillespie, J.G., Jr. Fracture and progressive failure of defective graphene sheets and carbon nanotubes. Compos. Struct. 2009, 88, 602–609. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Marenić, E.; Ibrahimbegovic, A.; Sorić, J.; Guidault, P.-A. Homogenized Elastic Properties of Graphene for Small Deformations. Materials 2013, 6, 3764-3782. https://doi.org/10.3390/ma6093764
Marenić E, Ibrahimbegovic A, Sorić J, Guidault P-A. Homogenized Elastic Properties of Graphene for Small Deformations. Materials. 2013; 6(9):3764-3782. https://doi.org/10.3390/ma6093764
Chicago/Turabian StyleMarenić, Eduard, Adnan Ibrahimbegovic, Jurica Sorić, and Pierre-Alain Guidault. 2013. "Homogenized Elastic Properties of Graphene for Small Deformations" Materials 6, no. 9: 3764-3782. https://doi.org/10.3390/ma6093764