
Photoresponsive Polymeric Reversible Nanoparticles via Self-Assembly of Reactive ABA Triblock Copolymers and Their Transformation to Permanent Nanostructures

Abstract
:1. Introduction
2. Results and Discussion
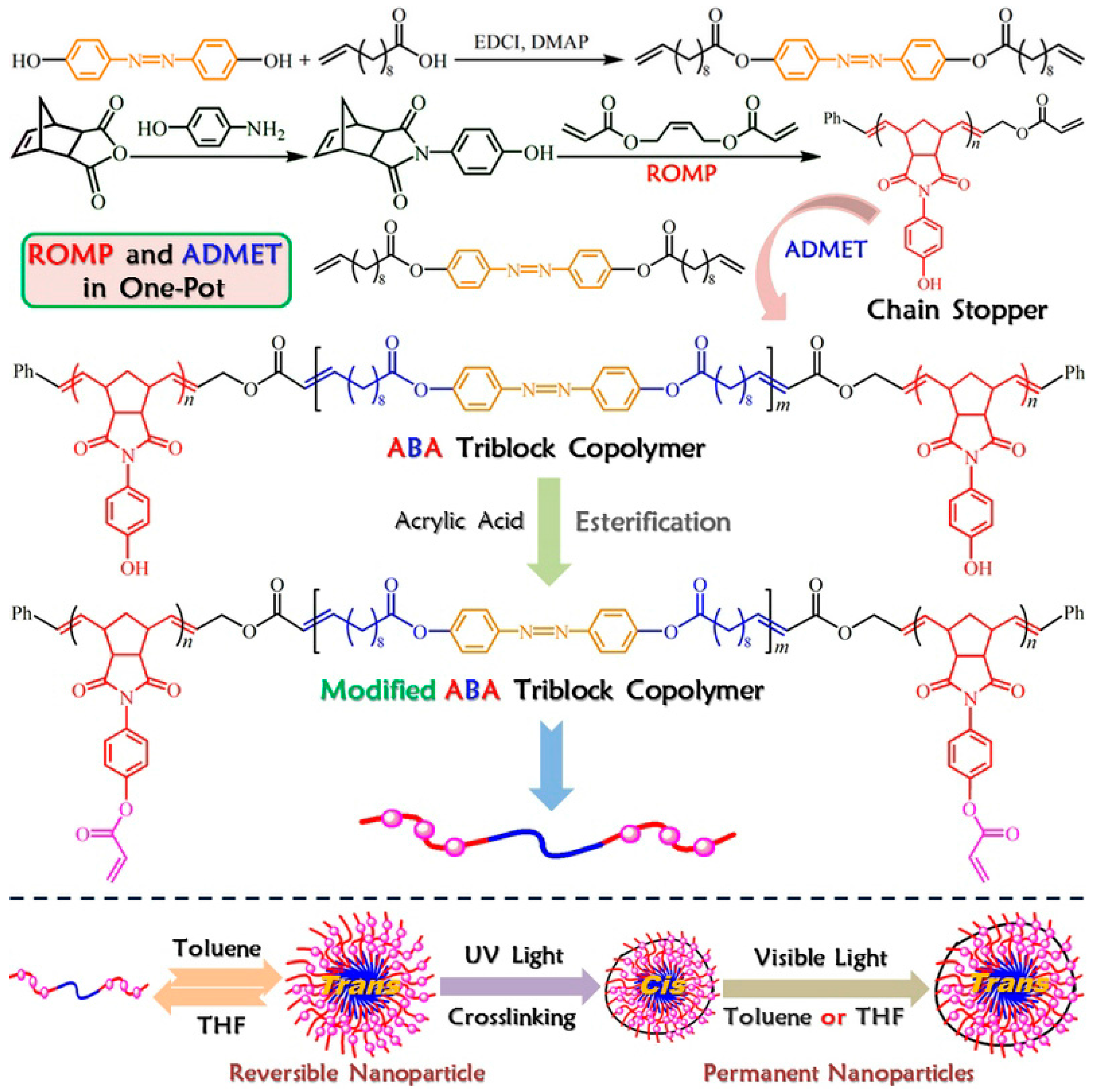
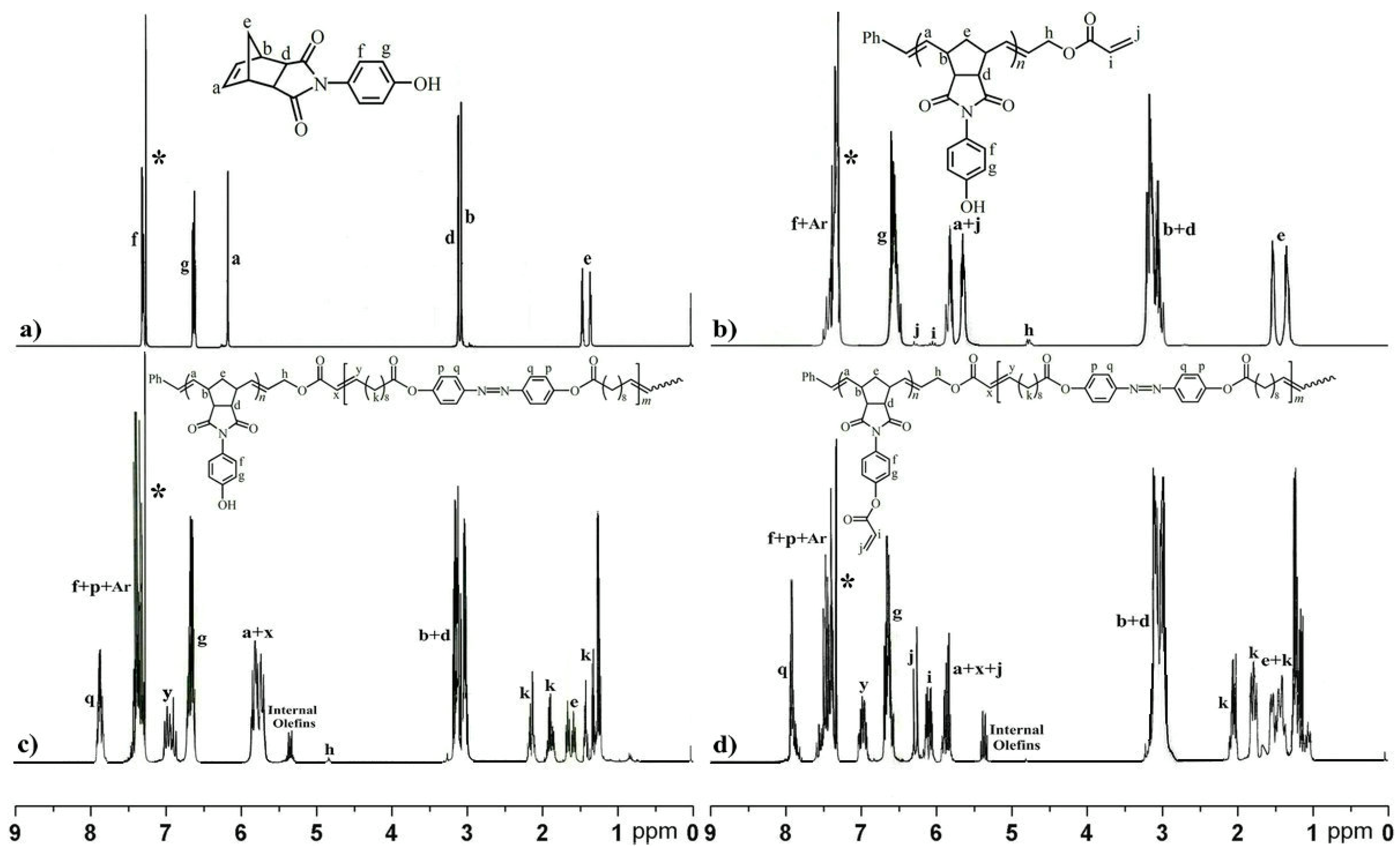
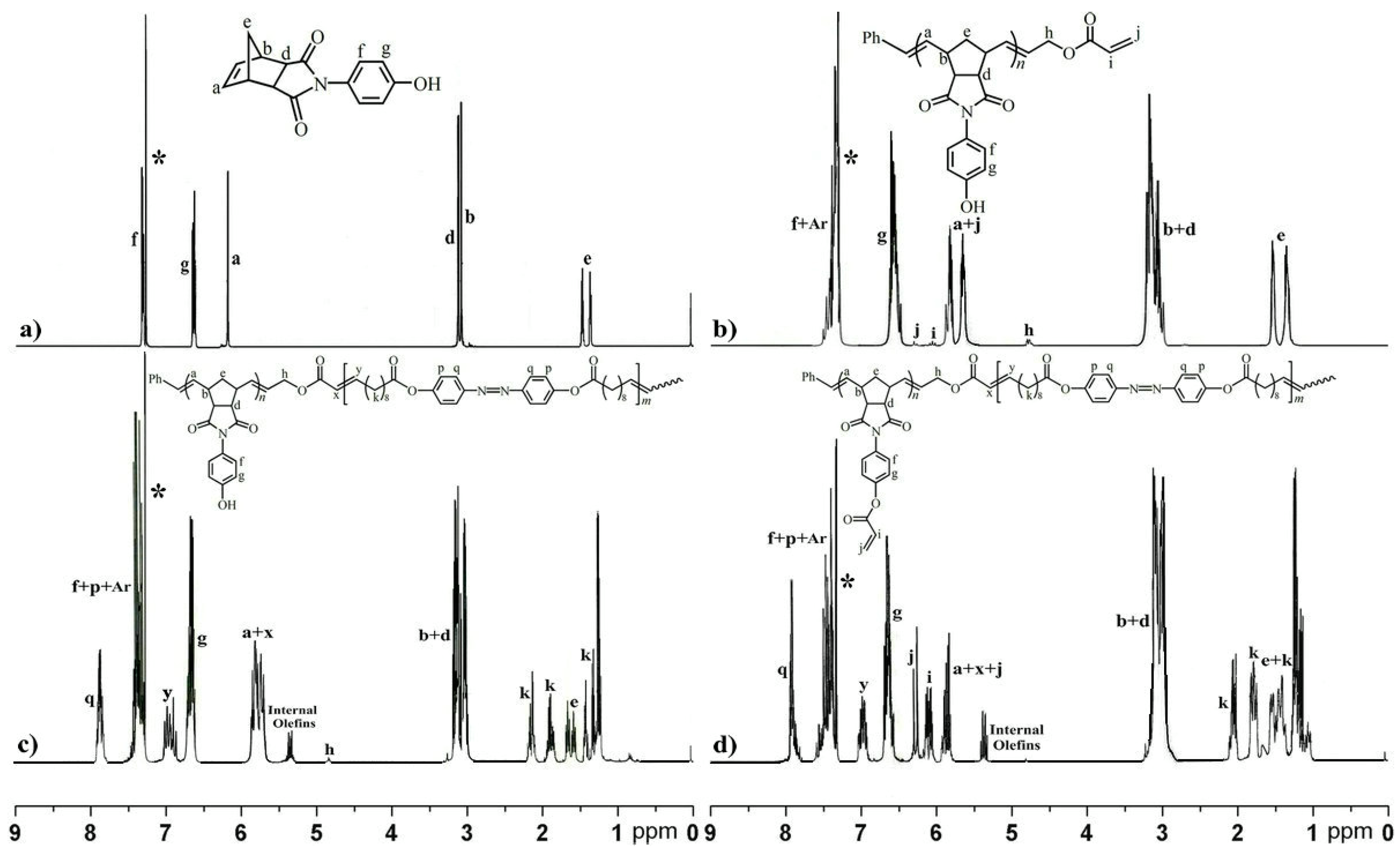
2.1. Preparation of the Reactive ABA Triblock Copolymer Precursor via One-Pot Sequential ROMP and ADMET Polymerization
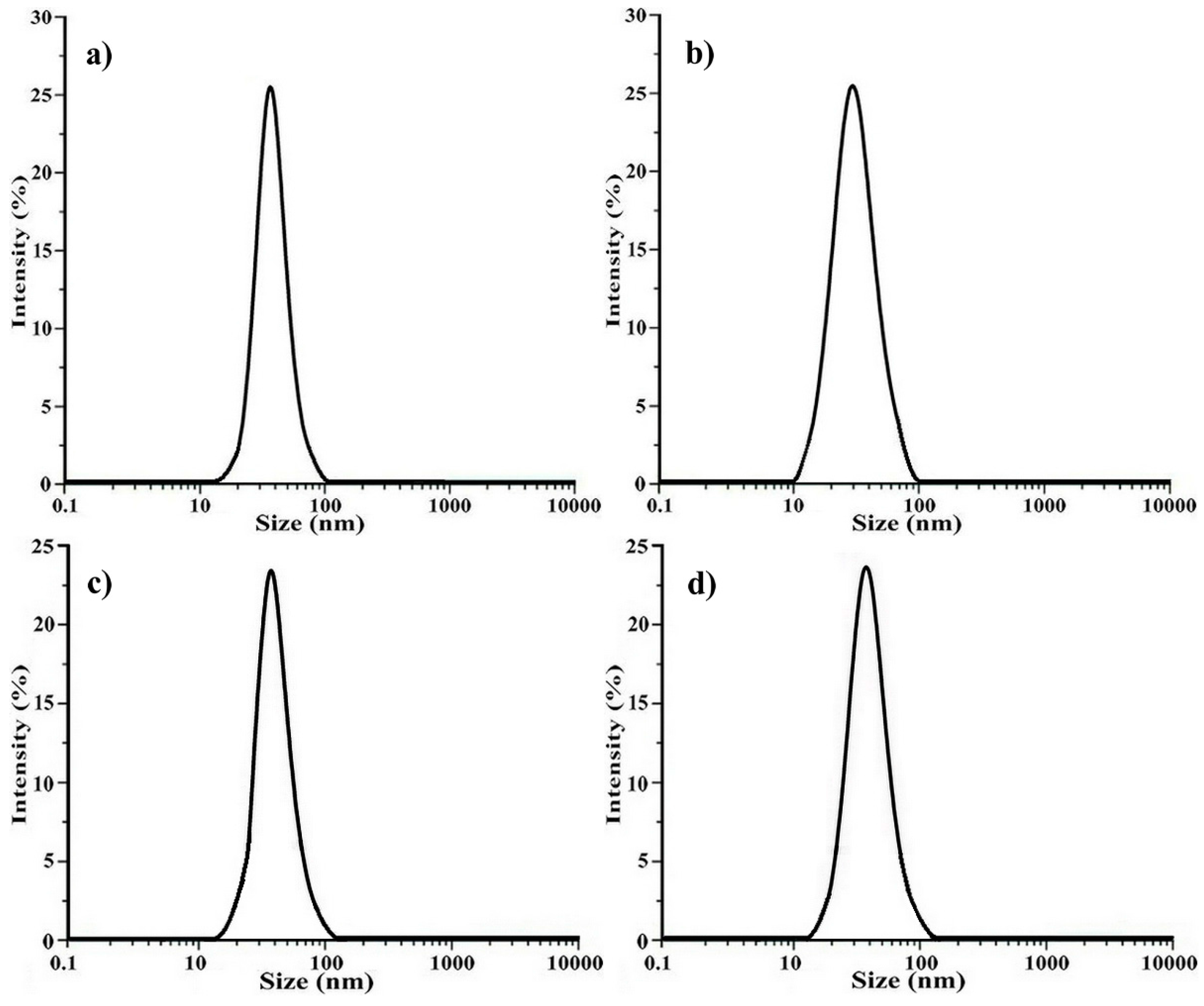
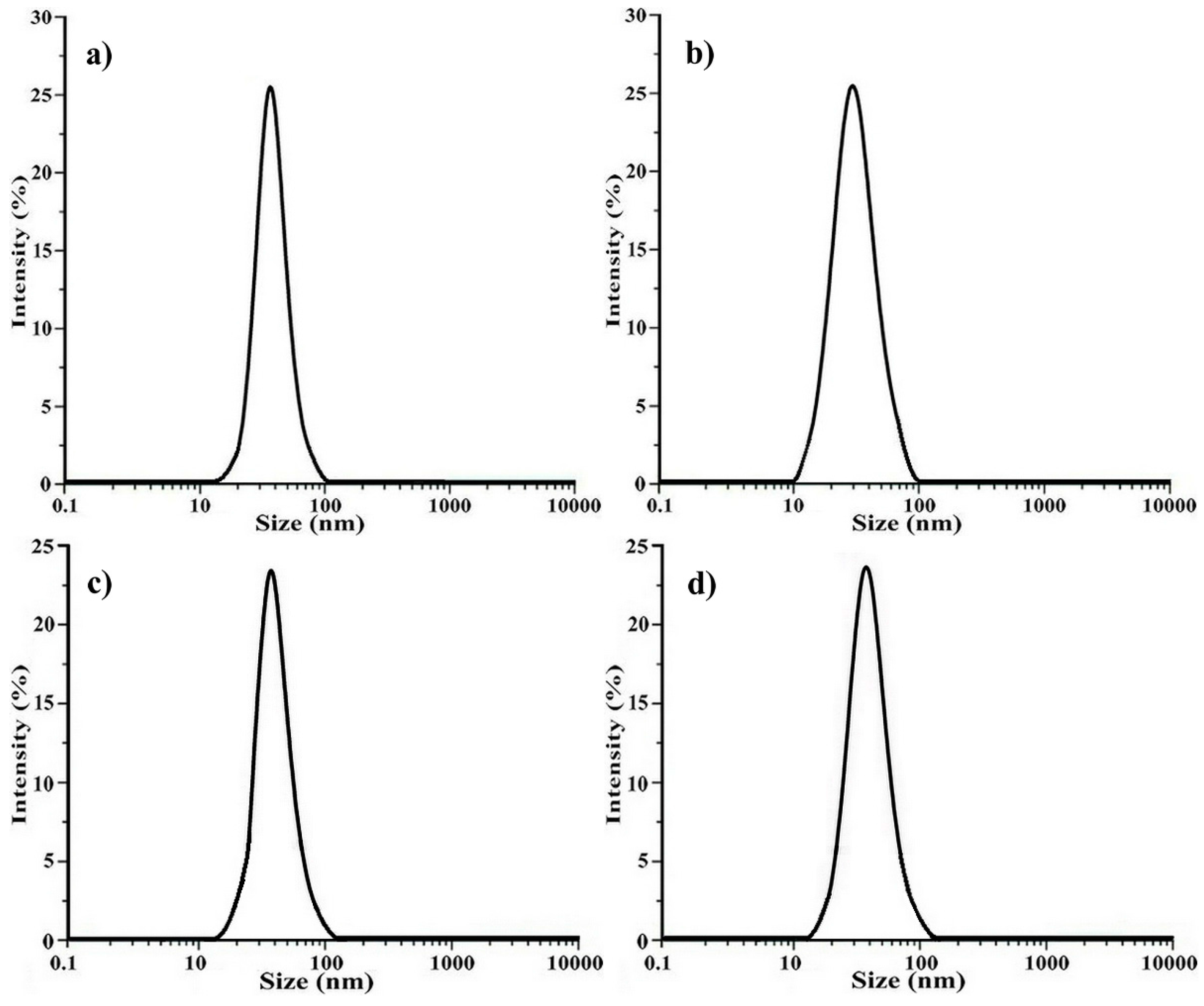
2.2. Determination of the Polymeric Nanoparticles Formed in a Selective Solvent
2.3. Crosslinking of the Shell of Polymeric Core-Shell Nanoparticles by UV Light Irradiation
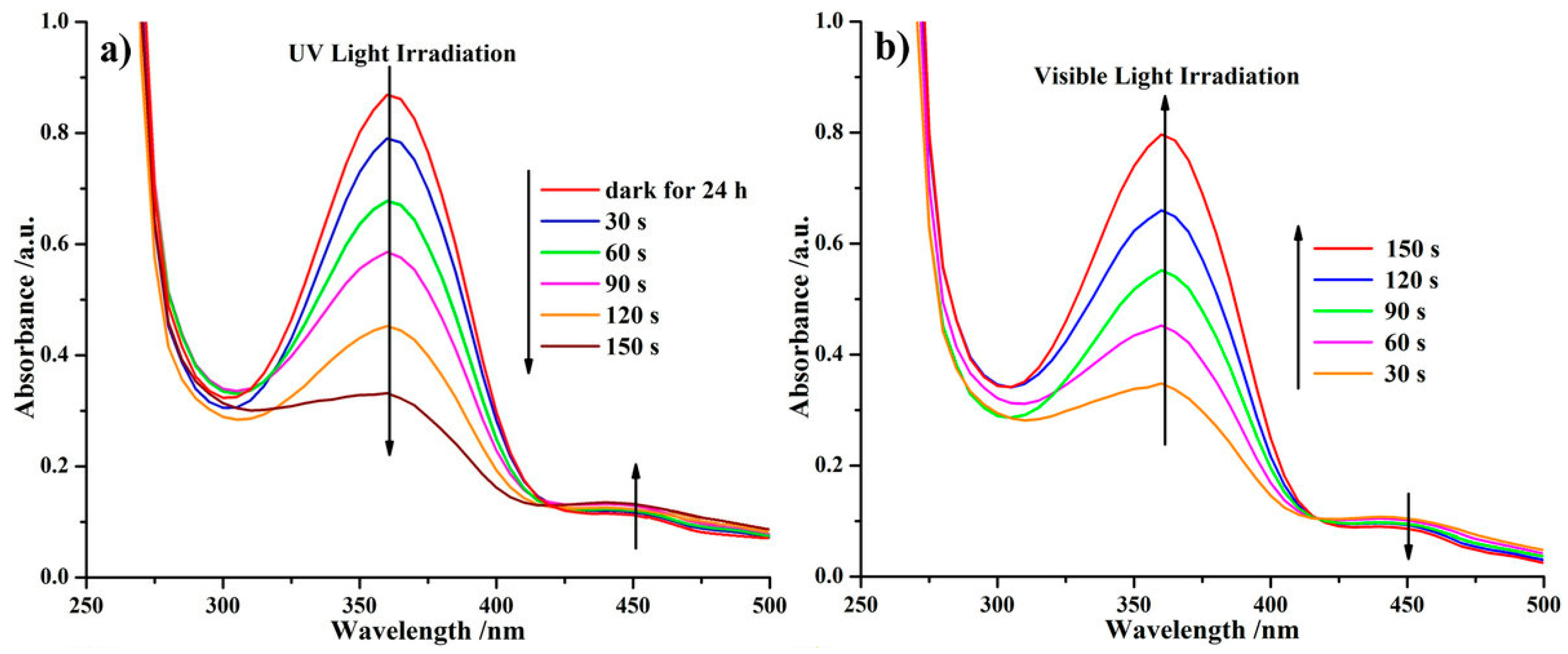
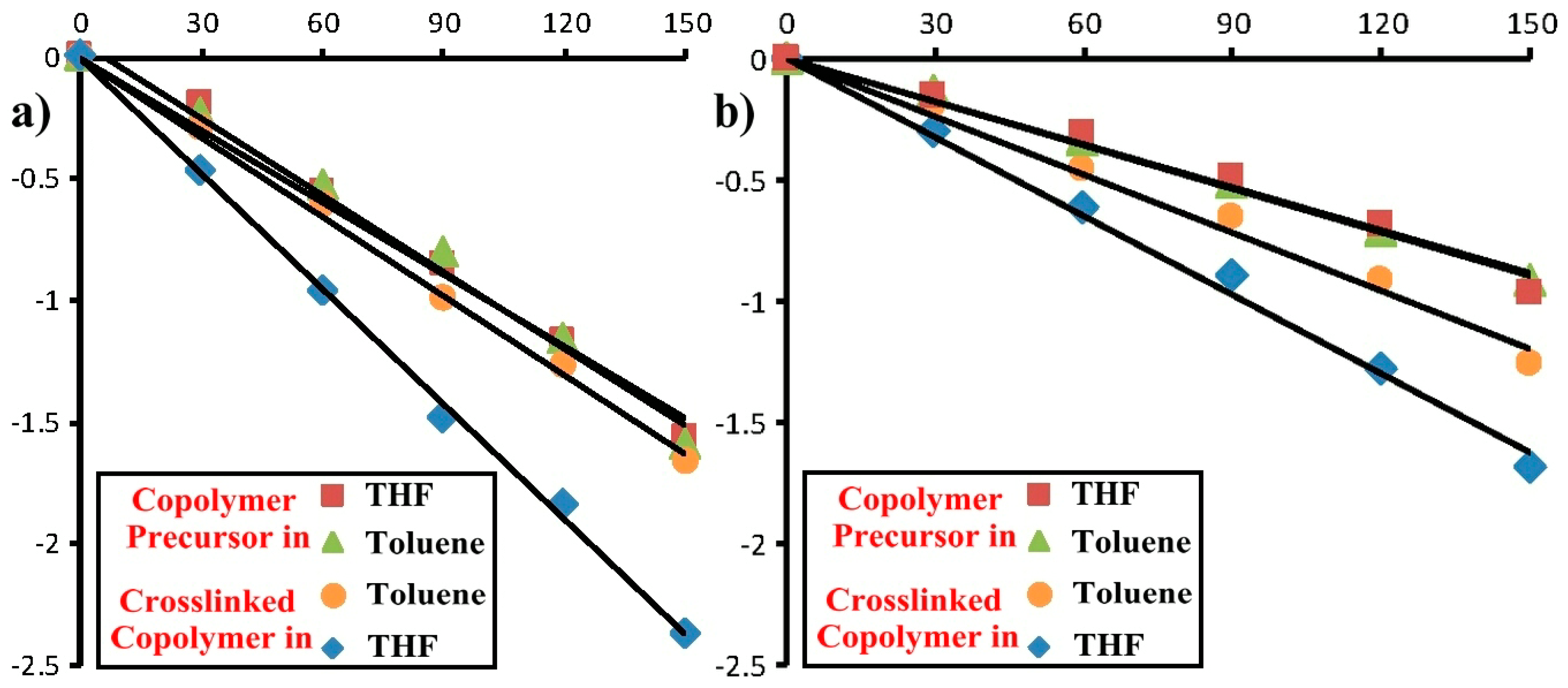
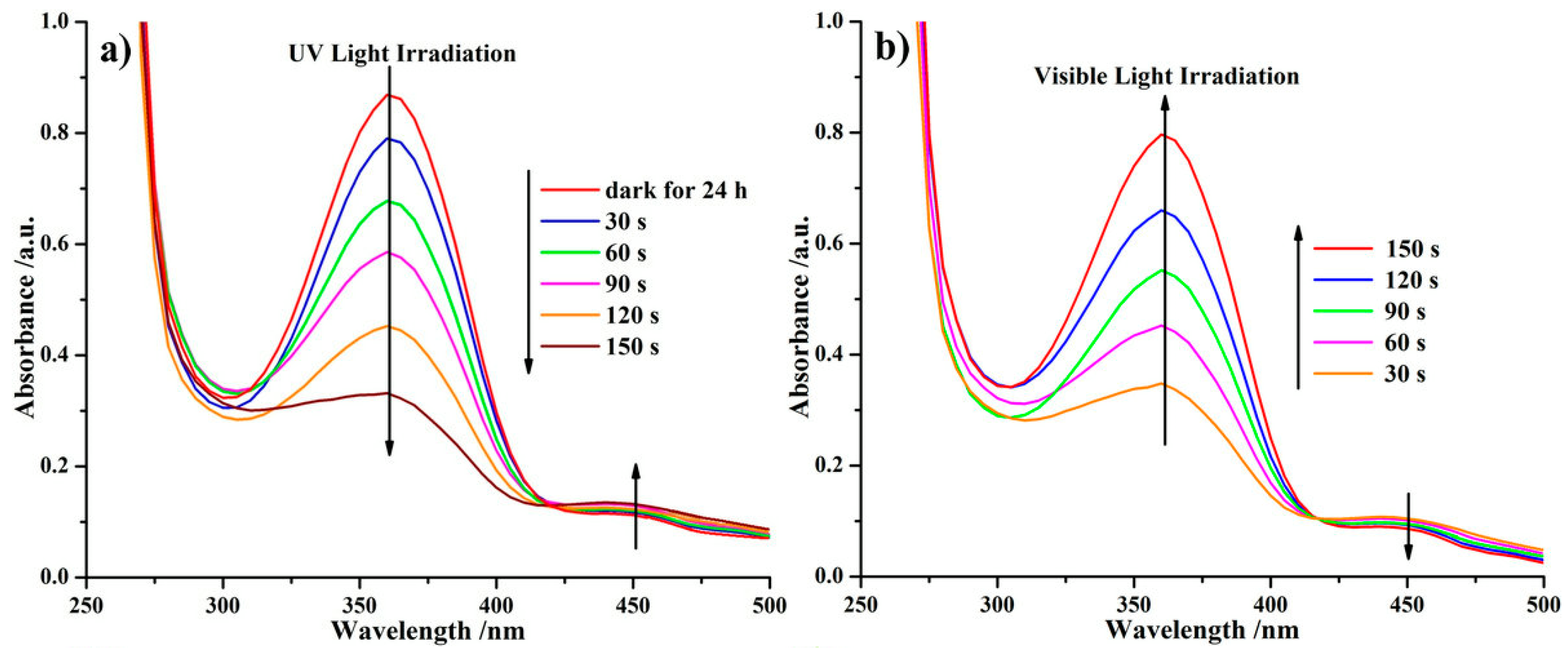
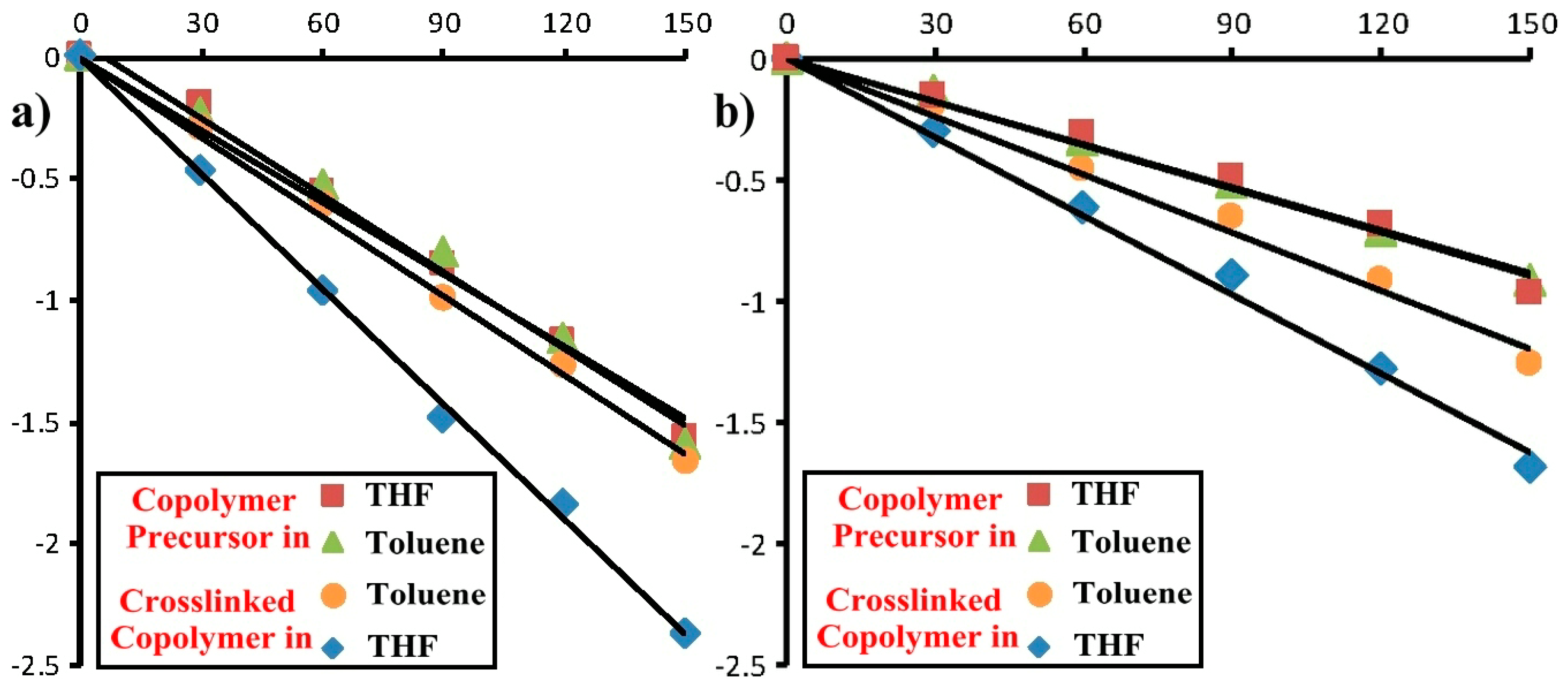
2.4. Photoresponsive Property of the Reactive ABA Triblock Copolymer Precursor and the Polymeric Shell-Crosslinked Nanoparticles in Different Selective Solvents
3. Materials and Methods
3.1. Materials
3.2. Characterization
3.3. Sample Preparation for AFM and TEM
3.4. Synthesis of 4,4′-bis(Undecylenyloxycarbonyl) Azobenzene (Monomer for ADMET Polymerization)
3.5. Synthesis of N-4-Phenol-Norbornene-Dicarboximide (Monomer for ROMP)
3.6. One-Pot Synthesis of Acrylate-Terminated Monotelechelic Polymer (Macromolecular Chain Stopper) and ABA Triblock Copolymer
3.7. Transformation of Hydroxyl Group of ABA Triblock Copolymer via Esterification to Reactive Triblock Copolymer Precursor
3.8. UV-Crosslinking of Polymeric Core-Shell Nanoparticle Solution from Reactive Triblock Copolymer Precursor
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chaudhuri, R.G.; Paria, S. Core/shell nanoparticles: Classes, properties, synthesis mechanisms, characterization, and applications. Chem. Rev. 2012, 112, 2373–2433. [Google Scholar] [CrossRef] [PubMed]
- Elsabahy, M.; Heo, G.S.; Lim, S.-M.; Sun, G.R.; Wooley, K.L. Polymeric nanostructures for imaging and therapy. Chem. Rev. 2015, 115, 10967–11011. [Google Scholar] [CrossRef] [PubMed]
- Kamps, A.C.; Sanchez-Gaytan, B.L.; Hickey, R.J.; Clarke, N.; Fryd, M.; Park, S.-J. Nanoparticle-directed self-assembly of amphiphilic block copolymers. Langmuir 2010, 26, 14345–14350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.S.; Lin, J.P.; Lin, S.L. Self-assembly behavior of amphiphilic block copolymer/nanoparticle mixture in dilute solution studied by self-consistent-field theory/density functional theory. Macromolecules 2007, 40, 5582–5592. [Google Scholar] [CrossRef]
- Klaikherd, A.; Nagamani, C.; Thayumanavan, S. Multi-stimuli sensitive amphiphilic block copolymer assemblies. J. Am. Chem. Soc. 2009, 131, 4830–4838. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sakai, F.; Su, L.; Liu, Y.J.; Wei, K.C.; Chen, G.S.; Jiang, M. Progressive macromolecular self-assembly: From biomimetic chemistry to bio-inspired materials. Adv. Mater. 2013, 25, 5215–5256. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Y.J.; Yin, J.-J.; Nie, Z.H. Self-assembly of amphiphilic block copolymer-tethered nanoparticles: A new approach to nanoscale design of functional materials. Macromol. Rapid Commun. 2015, 36, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Charleux, B.; Delaittre, G.; Rieger, J.; D’Agosto, F. Polymerization-induced self-assembly: From soluble macromolecules to block copolymer nano-objects in one step. Macromolecules 2012, 45, 6753–6765. [Google Scholar] [CrossRef]
- Zheng, G.H.; Pan, C.Y. Reversible addition–fragmentation transfer polymerization in nanosized micelles formed in situ. Macromolecules 2006, 39, 95–102. [Google Scholar] [CrossRef]
- Gao, C.Q.; Zhou, H.; Qu, Y.Q.; Wang, W.; Khan, H.; Zhang, W.Q. In situ synthesis of block copolymer nanoassemblies via polymerization-induced self-assembly in poly(ethylene glycol). Macromolecules 2016, 49, 3789–3798. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Song, C.M.; Yu, J.H.; Yang, D.; Xie, M.R. One-pot synthesis of polymeric nanoparticle by ring-opening metathesis polymerization. J. Polym. Sci. A Polym. Chem. 2010, 48, 5231–5238. [Google Scholar] [CrossRef]
- Zhao, Y. Light-responsive block copolymer micelles. Macromolecules 2012, 45, 3647–3657. [Google Scholar] [CrossRef]
- Canning, S.L.; Smith, G.N.; Armes, S.P. A critical appraisal of RAFT-mediated polymerization-induced self-assembly. Macromolecules 2016, 49, 1985–2001. [Google Scholar] [CrossRef] [PubMed]
- Ladmiral, V.; Semsarilar, M.; Canton, I.; Armes, S.P. Polymerization-induced self-assembly of galactose-functionalized biocompatible diblock copolymers for intracellular delivery. J. Am. Chem. Soc. 2013, 135, 13574–13581. [Google Scholar] [CrossRef] [PubMed]
- Warren, N.J.; Armes, S.P. Polymerization-induced self-assembly of block copolymer nano-objects via RAFT aqueous dispersion polymerization. J. Am. Chem. Soc. 2014, 136, 10174–10185. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wang, C.S.; Lin, L.; Zhu, Z.S. One-pot sequential ring-opening metathesis polymerization and acyclic diene metathesis polymerization synthesis of unsaturated block polyphosphoesters. Macromol. Chem. Phys. 2015, 216, 761–769. [Google Scholar] [CrossRef]
- Ding, L.; Qiu, J.; Li, J.; Wang, C.S.; Wang, L.F. Novel photoresponsive linear, graft, and comb-like copolymers with azobenzene chromophores in the main-chain and/or side-chain: Facile one-pot synthesis and photoresponse properties. Macromol. Rapid Commun. 2015, 36, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.B.; Grubbs, R.H. Monotelechelic poly(oxa)norbornenes by ring-opening metathesis polymerization using direct end-capping and cross-metathesis. Macromolecules 2010, 43, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Montero de Espinosa, L.; Meier, M.A.R. Synthesis of star- and block-copolymers using ADMET: Head-to-tail selectivity during step-growth polymerization. Chem. Commun. 2011, 47, 1908–1910. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.; Mueller, J.O.; Oehlenschlaeger, K.K.; Montero de Espinosa, L.; Meier, M.A.R.; Barner-Kowollik, C. Highly orthogonal functionalization of ADMET polymers via photo-induced diels–alder reactions. Macromolecules 2012, 45, 5012–5019. [Google Scholar] [CrossRef]
- Montero de Espinosa, L.; Winkler, M.; Meier, M.A.R. Acyclic diene metathesis polymerization and heck polymer–polymer conjugation for the synthesis of star-shaped block copolymers. Macromol. Rapid Commun. 2013, 34, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.R.; Han, H.J.; Wang, W.Z.; He, X.H.; Zhang, Y.Q. Synthesis and self-assembly of functionalized cyclooctene block copolymers via ROMP. Macromol. Chem. Phys. 2008, 209, 544–550. [Google Scholar] [CrossRef]
- Ding, L.; Qiu, J.; Lu, R.; Zheng, X.Q.; An, J. Hyperbranched polyphosphoesters with reactive end groups synthesized via acyclic diene metathesis polymerization and their transformation to crosslinked nanoparticles. J. Polym. Sci. A Polym. Chem. 2013, 51, 4331–4340. [Google Scholar] [CrossRef]
- Melinte, V.; Buruiana, T.; Balan, L.; Buruiana, E.C. Photocrosslinkable acid urethane dimethacrylates from renewable natural oil and their use in the design of silver/gold polymeric nanocomposites. React. Funct. Polym. 2012, 72, 252–259. [Google Scholar] [CrossRef]
- Willenbacher, J.; Wuest, K.N.R.; Mueller, J.O.; Kaupp, M.; Wagenknecht, H.A.; Barner-Kowollik, C. Photochemical design of functional fluorescent single-chain nanoparticles. ACS Macro Lett. 2014, 3, 574–579. [Google Scholar] [CrossRef]
- Natansohn, A. Photoinduced motions in azo-containing polymers. Chem. Rev. 2002, 102, 4139–4175. [Google Scholar] [CrossRef] [PubMed]
- Egawa, Y.; Miki, R.; Seki, T. Colorimetric sugar sensing using boronic acid-substituted azobenzenes. Materials 2014, 7, 1201–1220. [Google Scholar] [CrossRef]
- Cai, Y.; Lu, J.J.; Zhou, F.; Zhou, X.J.; Zhou, N.C.; Zhang, Z.B.; Zhu, X.L. Cyclic amphiphilic random copolymers bearing azobenzene side chains: Facile synthesis and topological effects on self-assembly and photoisomerization. Macromol. Rapid Commun. 2014, 35, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, S.; Martella, D.; Parmeggiani, C.; Wiersma, D.S. Photoresist design for elastomeric light tunable photonic devices. Materials 2016, 9, 525. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Yield 1 (%) | [M]/[C] ([M]/[CS]) 2 | Mn,GPC 3 | Mw/Mn 3 | Mn,NMR | DPn,ROMP 6 | DPn,ADMET 7 |
|---|---|---|---|---|---|---|---|
| Homopolymer | 92 | 25:1 | 8500 | 1.68 | 7100 4 | 27.2 | – |
| 93 | 50:1 | 13,700 | 1.59 | 12,800 4 | 49.3 | ||
| 89 | 100:1 | 26,300 | 1.65 | 24,200 4 | 94.2 | ||
| Copolymer | 86 | 10:1 | 34,900 | 1.76 | 32,300 5 | 49.3 | 11.8 |
| Precursor | 96 | – | 36,200 | 1.75 | – | 49.3 | 11.8 |
| Polymer | Solvent | ki 6 | kr 7 | DLS 8 | AFM (nm) | TEM (nm) | |
|---|---|---|---|---|---|---|---|
| (S-1/10-2) | Dh (nm) | PDI | |||||
| Copolymer Precursor | THF 1 | 1.59 | 1.06 | – | – | ||
| Toluene 2 | 1.08 | 0.74 | 92 | 0.165 | 85 | 82 | |
| Crosslinked Copolymer | Toluene 3 | – | 64 | 0.182 | 56 | 50 | |
| Toluene 4 | 0.99 | 0.58 | 108 | 0.189 | – | ||
| THF 5 | 1.02 | 0.60 | 110 | 0.191 | 93 | 98 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, L.; Li, J.; Jiang, R.; Song, W. Photoresponsive Polymeric Reversible Nanoparticles via Self-Assembly of Reactive ABA Triblock Copolymers and Their Transformation to Permanent Nanostructures. Materials 2016, 9, 980. https://doi.org/10.3390/ma9120980
Ding L, Li J, Jiang R, Song W. Photoresponsive Polymeric Reversible Nanoparticles via Self-Assembly of Reactive ABA Triblock Copolymers and Their Transformation to Permanent Nanostructures. Materials. 2016; 9(12):980. https://doi.org/10.3390/ma9120980
Chicago/Turabian StyleDing, Liang, Juan Li, Ruiyu Jiang, and Wei Song. 2016. "Photoresponsive Polymeric Reversible Nanoparticles via Self-Assembly of Reactive ABA Triblock Copolymers and Their Transformation to Permanent Nanostructures" Materials 9, no. 12: 980. https://doi.org/10.3390/ma9120980