
Computational Search for Two-Dimensional MX2 Semiconductors with Possible High Electron Mobility at Room Temperature

Abstract
:1. Introduction
2. Methods
3. Results and Discussion
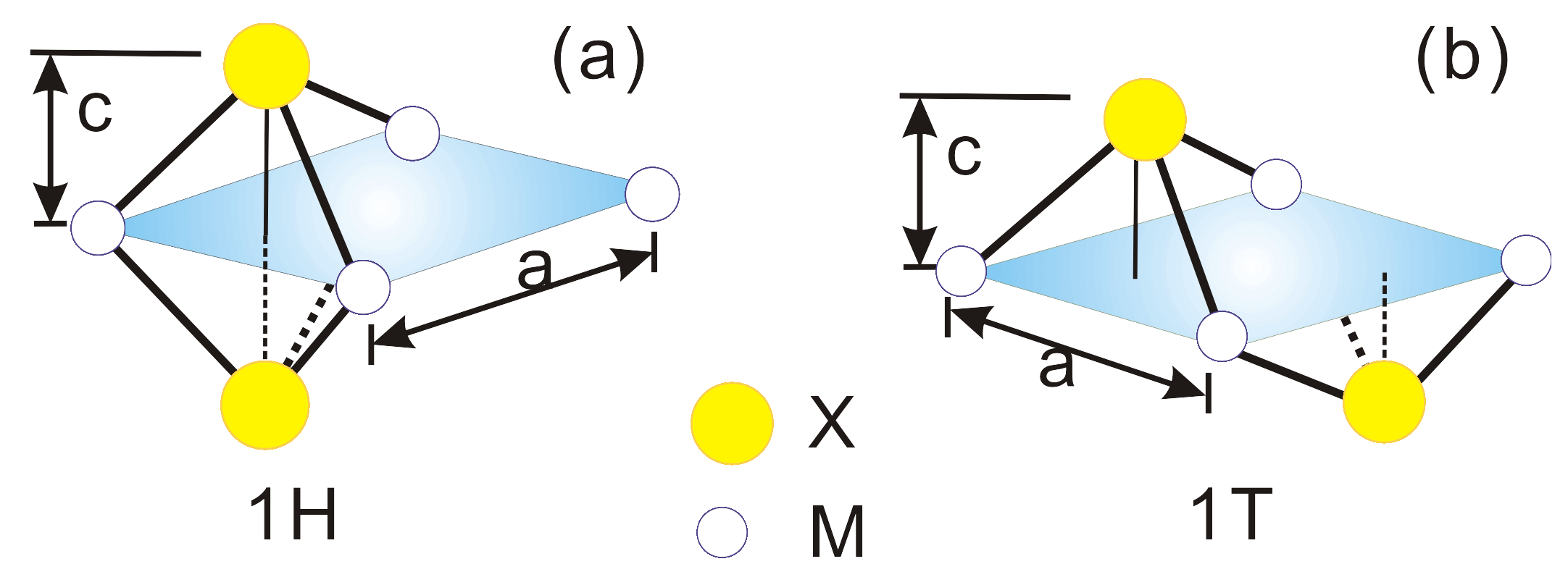
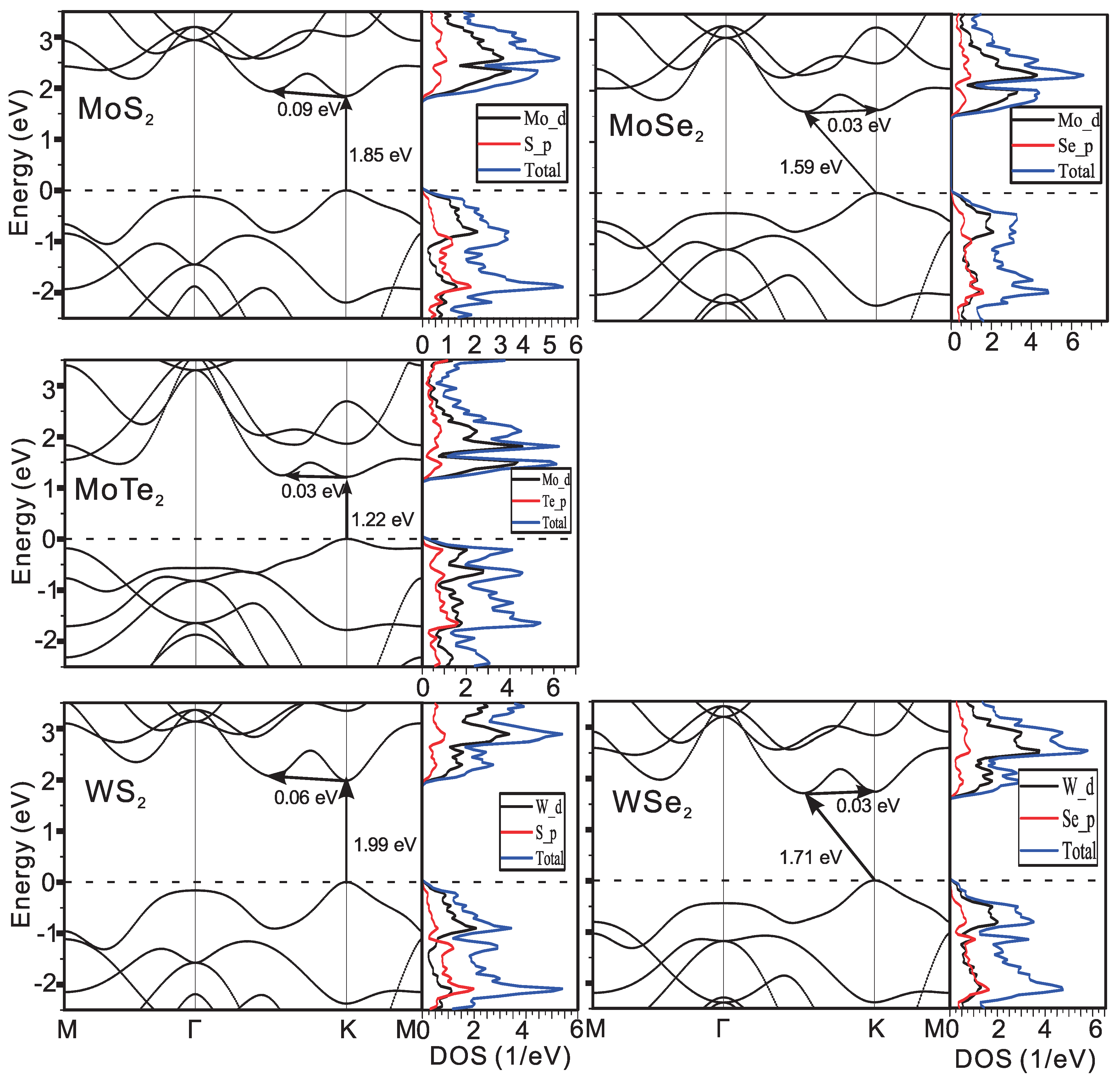
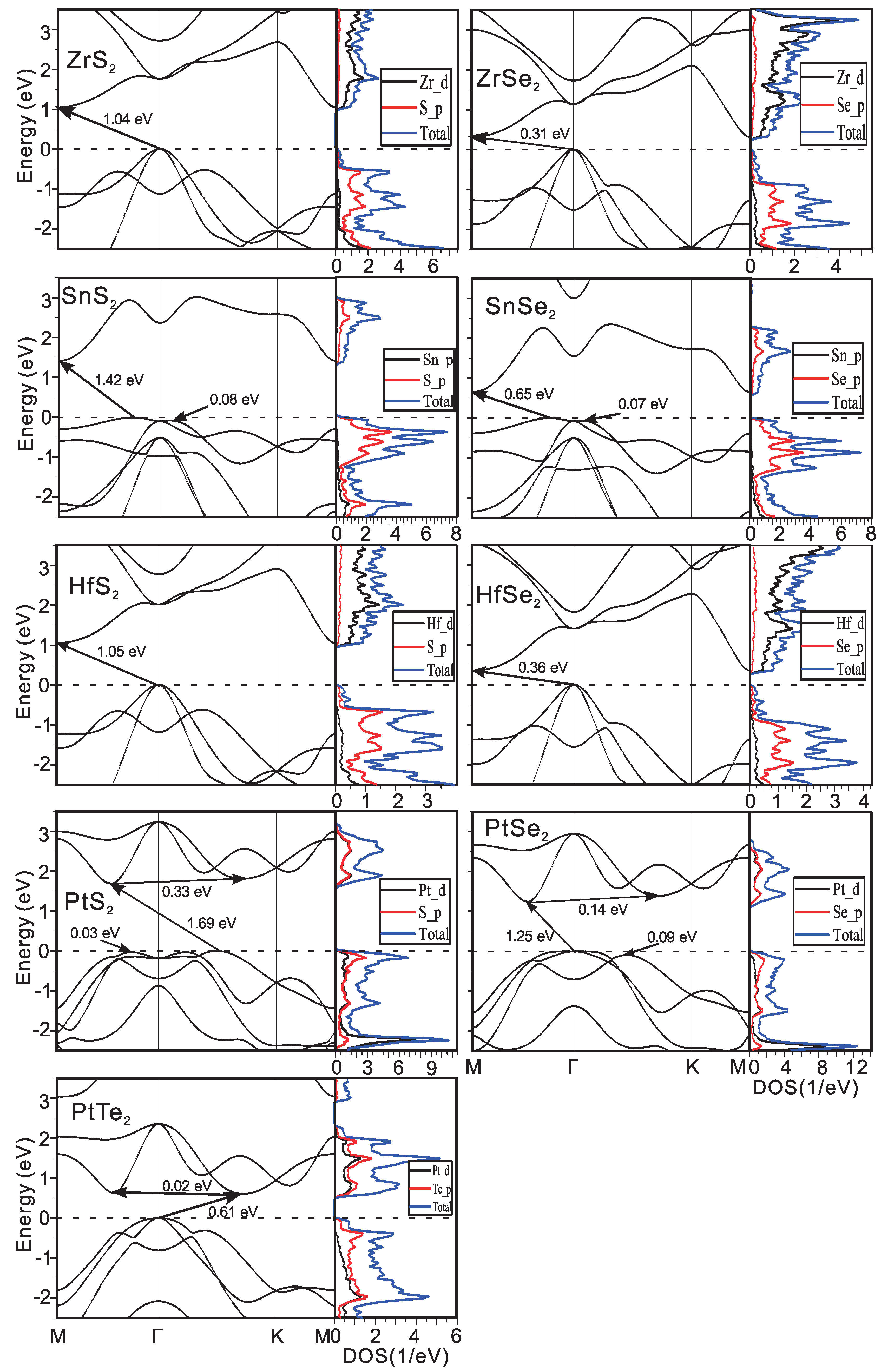
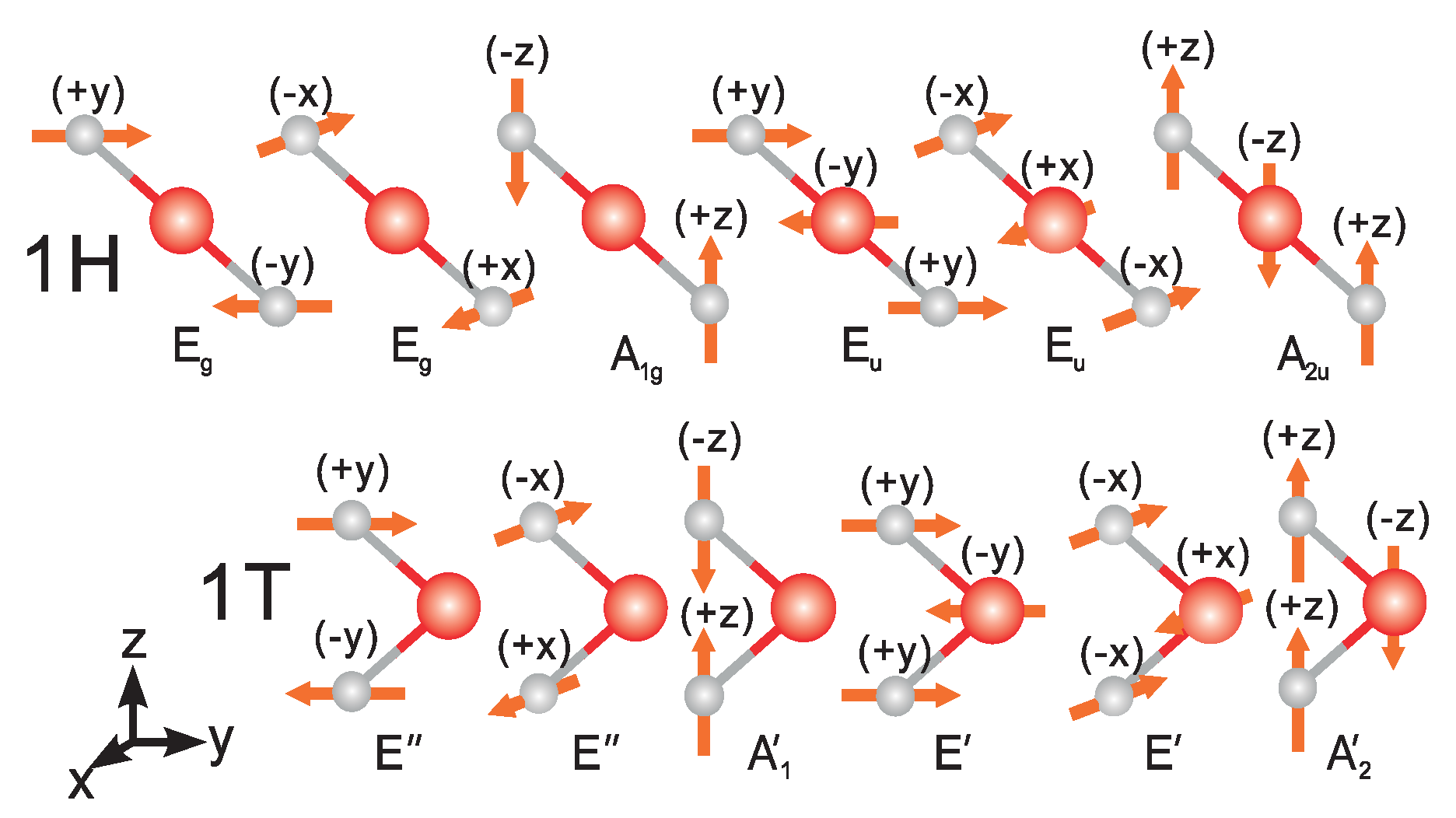
3.1. Electron and Phonon Dispersions
3.2. Acoustic Phonon Scattering
3.3. Optical Phonon Scattering
3.4. Piezoelectric Effects
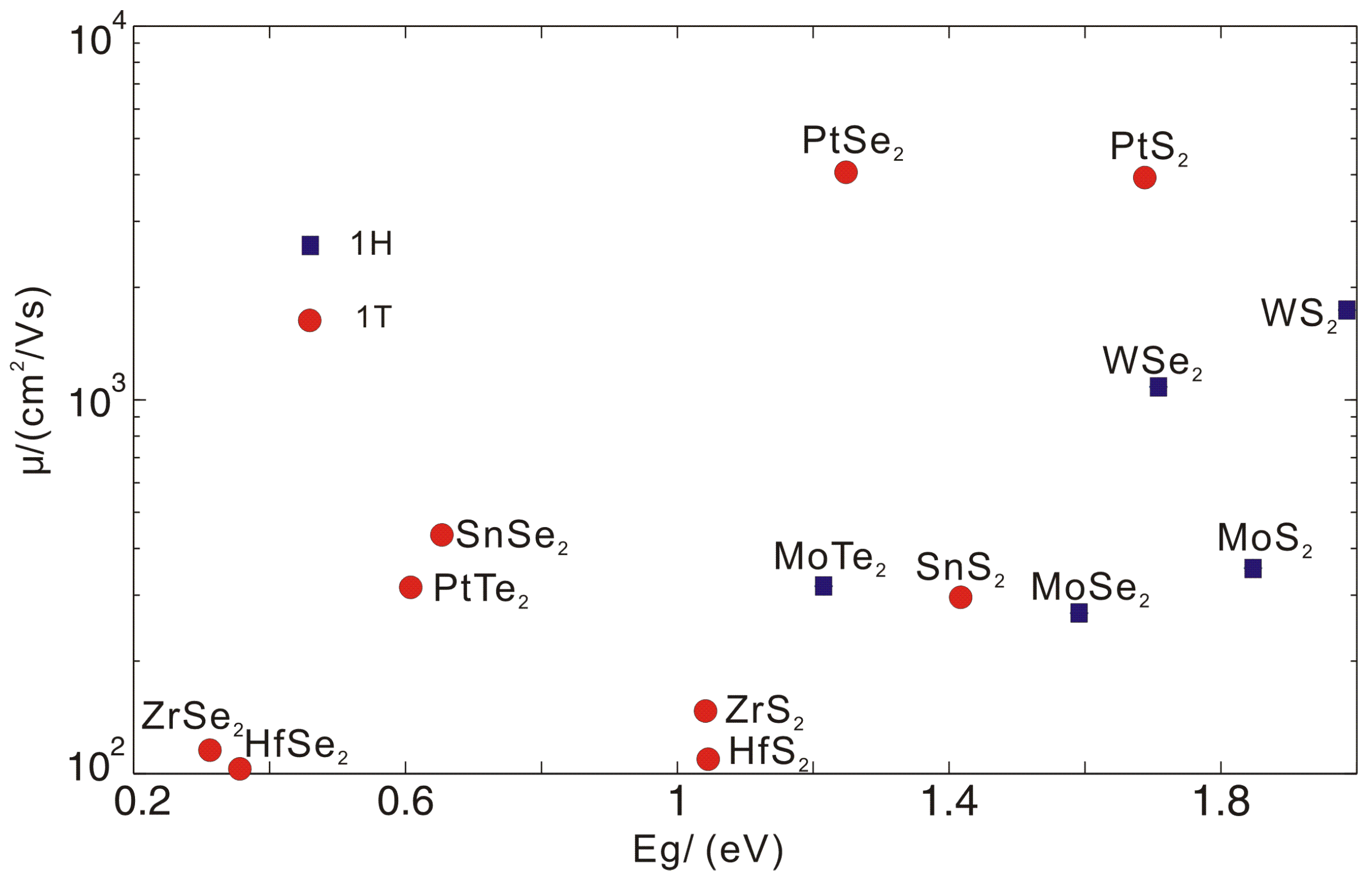
3.5. The Total Mobilities
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Neal, A.T.; Zhu, Z.; Luo, Z.; Xu, X.; Tománek, D.; Ye, P.D. Phosphorene: An Unexplored 2D Semiconductor with a High Hole Mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.; Ganapathi, K.; Salahuddin, S. How Good Can Monolayer MoS2 Transistors Be? Nano Lett. 2011, 11, 3768–3773. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Li, H.; Li, H.; Jiang, L.; Shi, Y.; Sun, Y.; Lu, G.; Zhang, Q.; Chen, X.; Zhang, H. Single-Layer MoS2 Phototransistors. ACS Nano 2012, 6, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Qu, D.; Lee, D.; Liu, X.; Watanabe, K.; Taniguchi, T.; Yoo, W.J. Lateral MoS2 p-n Junction Formed by Chemical Doping for Use in High-Performance Optoelectronics. ACS Nano 2014, 8, 9332–9340. [Google Scholar] [CrossRef] [PubMed]
- Franklin, A.D. Nanomaterials in transistors: From high-performance to thin-film applications. Science 2015, 349, 704. [Google Scholar] [CrossRef] [PubMed]
- Kuc, A.; Heine, Th.; Kis, A. Electronic properties of transitional-metal. MRS Bull. 2015, 40, 577–584. [Google Scholar] [CrossRef]
- Liao, L.; Lin, Y.; Bao, M.; Cheng, R.; Bai, J.; Liu, Y.; Qu, Y.; Wang, K.L.; Huang, Y.; Duan, X. High-speed graphene transistors with a self-aligned nanowire gate. Nature 2010, 467, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Jariwala, D.; Sangwan, V.K.; Late, D.J.; Johns, J.E.; Dravid, V.P.; Marks, T.J.; Lauhon, L.J.; Hersam, M.C. Band-like transport in high mobility unencapsulated single-layer MoS2 transistors. Appl. Phys. Lett. 2013, 102, 173107. [Google Scholar] [CrossRef]
- Bao, W.; Cai, X.; Kim, D.; Sridhara, K.; Fuhrer, M.S. High mobility ambipolar MoS2 field-effect transistors. Appl. Phys. Lett. 2013, 102, 042104. [Google Scholar] [CrossRef]
- Liu, W.; Kang, J.; Sarkar, D.; Khatami, Y.; Jena, D.; Banerjee, K. Role of Metal Contacts in Designing High-Performance Monolayer n-Type WSe2 Field Effect Transistors. Nano Lett. 2013, 13, 1983–2013. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Kong, X.; Hu, Z.; Yang, F.; Ji, W. High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus. Nat. Commum. 2014, 5, 4475. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, Y.; Ye, G.J.; Ge, Q.; Ou, X.; Wu, H.; Feng, D.; Chen, X.H.; Zhang, Y. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Kaasbjerg, K.; Thygesen, K.S.; Jacobsen, K.W. Phonon-limited mobility in n-type single-layer MoS2 from first principles. Phys. Rev. B 2012, 85, 115317. [Google Scholar] [CrossRef]
- Kaasbjerg, K.; Thygesen, K.S.; Jauho, A.P. Acoustic phonon limited mobility in two-dimensional semiconductors: Deformation potential and piezoelectric scattering in monolayer MoS2 from first principles. Phys. Rev. B 2013, 87, 235312. [Google Scholar] [CrossRef]
- Restrepo, O.D.; Krymowski, K.E.; Goldberger, J.; Windl, W. A First Principles Method to Simulate Electron Mobilities in 2D Materials. New J. Phys. 2014, 16, 105009. [Google Scholar] [CrossRef]
- Li, X.; Mullen, J.T.; Jin, Z.; Borysenko, K.M.; Nardelli, M.B.; Kim, K.W. Intrinsic electrical transport properties of monolayer silicene and MoS2 from first principles. Phys. Rev. B 2013, 87, 115418. [Google Scholar] [CrossRef]
- Jin, Z.; Li, X.; Mullen, J.T.; Kim, W.K. Intrinsic transport properties of electrons and holes in monolayer transition-metal dichalcogenides. Phys. Rev. B 2014, 90, 045422. [Google Scholar] [CrossRef]
- Zhang, W.X.; Huang, Z.S.; Zhang, W.L.; Li, Y. Two-dimensional semiconductors with possible high room temperature mobility. Nano Res. 2014, 7, 1731. [Google Scholar] [CrossRef]
- Lebègue, S.; Björkman, T.; Klintenberg, M.; Nieminen, R.M.; Eriksson, O. Two-Dimensional Materials from Data Filtering and Ab Initio Calculations. Phys. Rev. X 2013, 3, 031002. [Google Scholar]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Koepernik, K.; Eschrig, H. Full-potential nonorthogonal local-orbital minimum-basis band-structure scheme. Phys. Rev. B 1999, 59, 1743. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Gu, X.; Yang, R. Phonon transport in single-layer transition metal dichalcogenides: A first-principles study. Appl. Phys. Lett. 2014, 105, 131903. [Google Scholar] [CrossRef]
- Bardeen, J.; Shockey, W. Deformation Potentials and Mobilities in Non-Polar Crystals. Phys. Rev. 1950, 80, 72–80. [Google Scholar] [CrossRef]
- Takagi, S.; Hoyt, J.L.; Welser, J.J.; Gibbons, J.F. Comparative study of phononlimited mobility of two-dimensional electrons in strained and unstrained Si metal-oxide-semiconductor field-effect transistors. J. Appl. Phys. 1996, 80, 1567–1577. [Google Scholar] [CrossRef]
- Bruzzone, S.; Fiori, G. Ab-initio simulations of deformation potentials and electron mobility in chemically modified graphene and two-dimensional hexagonal boron-nitride. Appl. Phys. Lett. 2011, 99, 222108. [Google Scholar] [CrossRef]
- Chakraborty, B.; Bera, A.; Muthu, D.V.S.; Bhowmick, S.; Waghmare, U.V.; Sood, A.K. Symmetry-dependent phonon renormalization in monolayer MoS2 transistor. Phys. Rev. B 2012, 85, 161403. [Google Scholar] [CrossRef]
- Duerloo, K.N.; Ong, M.T.; Reed, E.J. Intrinsic Piezoelectricity in Two-Dimensional Materials. J. Phys. Chem. Lett. 2012, 3, 2871–2876. [Google Scholar] [CrossRef]
- Ashcroft, N.W.; Mermin, N.D. Solid State Physics; Thomson Learning, Inc.: Glendale, CA, USA, 1976. [Google Scholar]
- Ding, Y.; Wang, Y.; Nic, J.; Shi, L.; Shi, S.; Tang, W. First principles study of structural, vibrational and electronic properties of graphene-like MX2 (M=Mo, Nb, W, Ta; X=S, Se, Te) monolayers. Physica B 2011, 406, 2254–2260. [Google Scholar] [CrossRef]
- Zhuang, H.; Hennig, G.R. Computational Search for Single-Layer Transition-Metal Dichalcogenide Photocatalysts. J. Phys. Chem. C 2013, 117, 20440–20445. [Google Scholar] [CrossRef]
- Gong, C.; Zhang, H.; Wang, W.; Colombo, L.; Wallace, R.M.; Cho, K. Band alignment of two-dimensional transition metal dichalcogenides: Application in tunnel field effect transistors. Appl. Phys. Lett. 2013, 103, 053513. [Google Scholar] [CrossRef]
- Fang, H.; Tosun, M.; Seol, G.; Chang, T.C.; Takei, K.; Guo, J.; Javey, A. Degenerate n-Doping of Few-Layer Transition Metal Dichalcogenides by Potassium. Nano Lett. 2013, 13, 1991–1995. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MX | a | c | m | m | E | E (Type) |
|---|---|---|---|---|---|---|
| (a.u.) | (a.u.) | (m) | (m) | (eV) | (eV) | |
| MoS | 5.927 | 2.962 | 0.45 | 0.45 | 3.90 | 1.85 (d) |
| MoSe | 6.168 | 3.156 | 0.52 | 0.52 | 3.65 | 1.59 (i) |
| MoTe | 6.618 | 3.411 | 0.53 | 0.57 | 0.92 | 1.22 (d) |
| WS | 6.047 | 2.992 | 0.24 | 0.26 | 3.92 | 1.99 (d) |
| WSe | 6.166 | 3.164 | 0.33 | 0.31 | 3.78 | 1.71 (i) |
| SnS | 6.879 | 2.797 | 2.11 | 0.21 | 3.55 | 1.42 (i) |
| SnSe | 7.165 | 2.999 | 2.91 | 0.17 | 2.91 | 0.65 (i) |
| HfS | 6.731 | 2.750 | 3.30 | 0.24 | 1.31 | 1.05 (i) |
| HfSe | 6.944 | 2.978 | 3.10 | 0.18 | 1.08 | 0.36 (i) |
| ZrS | 6.817 | 2.771 | 1.62 | 0.31 | 1.52 | 1.04 (i) |
| ZrSe | 7.007 | 3.008 | 2.03 | 0.22 | 1.25 | 0.31 (i) |
| PtS | 6.670 | 2.327 | 0.26 | 0.25 | 3.63 | 1.69 (i) |
| PtSe | 6.978 | 2.464 | 0.21 | 0.19 | 2.86 | 1.25 (i) |
| PtTe | 7.485 | 2.634 | 0.90 | 0.77 | 1.73 | 0.61 (i) |
| MX | V | D | D |
|---|---|---|---|
| (km/s) | (eV) | (10 eV/cm) | |
| MoS | 7.93 | 3.90, 2.4 [18], 4.5 [14] | 1.75, 5.8 [18], 4.1 [14] |
| MoSe | 6.01 | 3.65, 3.4 [18] | 1.10, 5.2 [18] |
| MoTe | 5.04 | 0.92 | 1.34 |
| WS | 6.67 | 3.92, 3.2 [18] | 2.34, 3.1 [18] |
| WSe | 5.55 | 3.78, 3.2 [18] | 1.12, 2.3 [18] |
| SnS | 6.18 | 3.55 | 0.69 |
| SnSe | 4.83 | 2.91 | 0.38 |
| HfS | 5.86 | 1.31 | 0.99 |
| HfSe | 4.72 | 1.08 | 0.62 |
| ZrS | 7.21 | 1.52 | 1.12 |
| ZrSe | 5.42 | 1.25 | 0.75 |
| PtS | 6.61 | 3.63 | 1.06 |
| PtSe | 4.73 | 2.86 | 0.84 |
| PtTe | 4.89 | 1.73 | 0.95 |
| MX | ( C/m) | (eV) | ||
|---|---|---|---|---|
| This Work | Ref. [29] | |||
| MoS | 2.98 | 3.06 | 4.26 | 5.59 |
| MoSe | 2.68 | 2.80 | 4.74 | 4.54 |
| MoTe | 2.57 | 2.98 | 5.76 | 3.56 |
| WS | 1.72 | 2.20 | 4.13 | 3.32 |
| WSe | 1.51 | 1.93 | 4.63 | 2.61 |
| MX | μ | |||
|---|---|---|---|---|
| MoS | 1362 | 1722 | 663 | 354 |
| MoSe | 963 | 929 | 621 | 269 |
| MoTe | 10,104 | 638 | 673 | 317 |
| WS | 4415 | 5374 | 6148 | 1739 |
| WSe | 2822 | 2496 | 5921 | 1083 |
| HfS | 7334 | 110 | - | 109 |
| HfSe | 14,317 | 102 | - | 102 |
| PtS | 4429 | 35,816 | - | 3942 |
| PtSe | 7568 | 8654 | - | 4038 |
| PtTe | 1467 | 401 | - | 315 |
| SnS | 1224 | 389 | - | 295 |
| SnSe | 1788 | 577 | - | 436 |
| ZrS | 4989 | 152 | - | 148 |
| ZrSe | 9264 | 117 | - | 116 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Z.; Zhang, W.; Zhang, W. Computational Search for Two-Dimensional MX2 Semiconductors with Possible High Electron Mobility at Room Temperature. Materials 2016, 9, 716. https://doi.org/10.3390/ma9090716
Huang Z, Zhang W, Zhang W. Computational Search for Two-Dimensional MX2 Semiconductors with Possible High Electron Mobility at Room Temperature. Materials. 2016; 9(9):716. https://doi.org/10.3390/ma9090716
Chicago/Turabian StyleHuang, Zhishuo, Wenxu Zhang, and Wanli Zhang. 2016. "Computational Search for Two-Dimensional MX2 Semiconductors with Possible High Electron Mobility at Room Temperature" Materials 9, no. 9: 716. https://doi.org/10.3390/ma9090716