Structural Fingerprints of Transcription Factor Binding Site Regions


Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Description |
|---|---|
| twist3, roll3, slide3, shift3 | the values of the four 3-step parameters, 3-step twist, roll, slide, shift at the octamer central step |
| groove | the minor groove width, measured as the minimum phosphate-phosphate distance |
| RMSD | RMSD from a notional straight path through the centres of the base-pair triads |
| Bistability | possessing 2 distinct energy minima |
| flexibility force constants, k-Roll, k +Roll, k -Twist, k +Twist | for twist, roll, the force constant required to move the parameter from its minimum energy value. Low values are flexible. |
| 3-step flexibility force constants, 3k-Roll, 3k +Roll, 3k -Twist, 3k +Twist | for 3-step twist, 3-step roll, the force constant required to move the parameter from its minimum energy value. Low values are flexible. |
| flexibility partition coefficients, Q-Roll, Q+Roll, Q-Twist, Q+Twist | flexibility force constants, converted to partition coefficients using Boltzmann’s equation. . Low values are inflexible. |
| 3-step flexibility partition coefficients, 3Q-Roll, 3Q+Roll, 3Q-Twist, 3Q+Twist | decreasing 3-step force constants, converted to partition coefficients using Boltzmann’s equation. Low values are inflexible. |
2. Methods
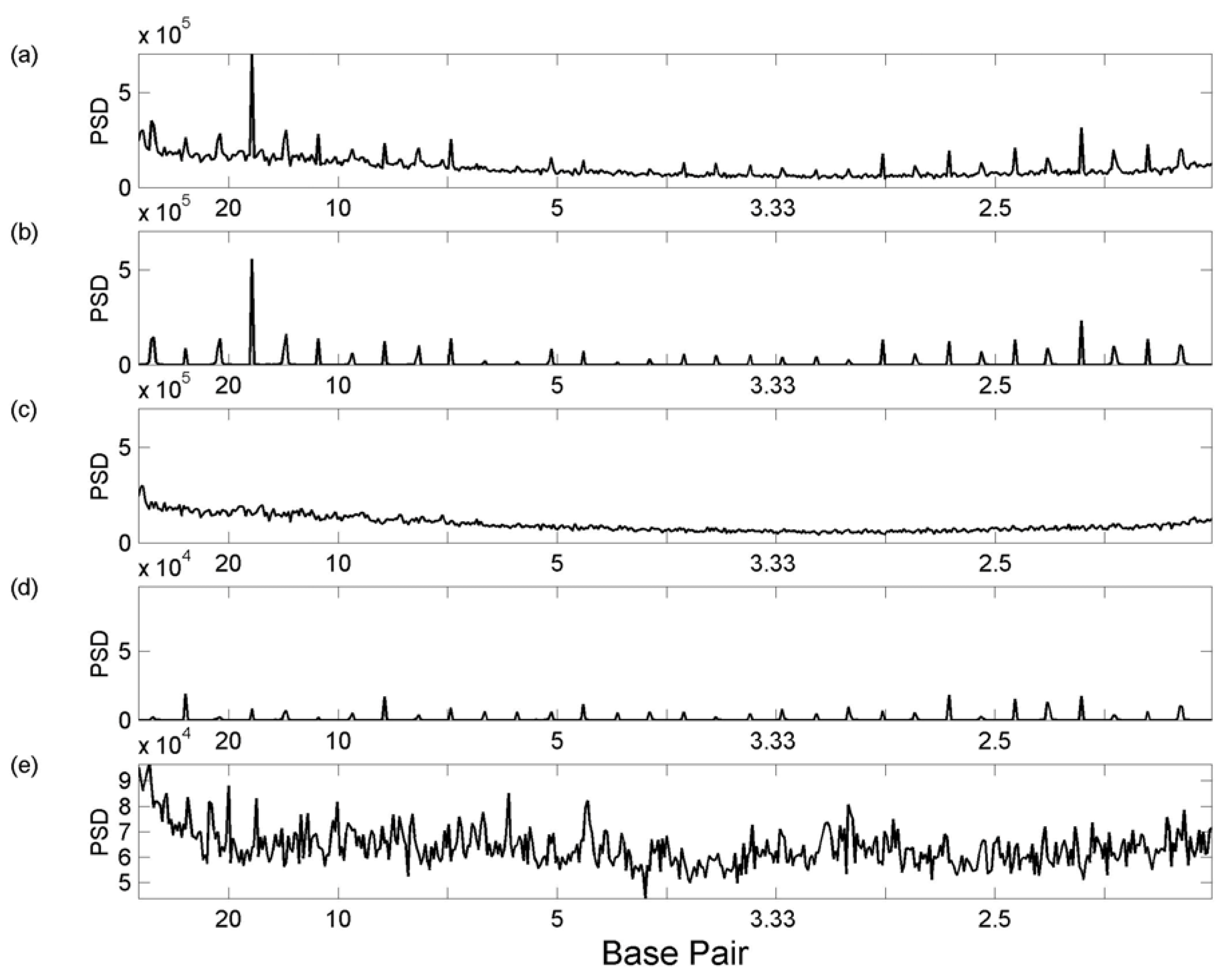
2.1. Fourier Transform Methods
- 1)
- Take a long DNA sequence S, such as an entire chromosome, of length N bases and also a structural parameter, p, (for example roll 3).
- 2)
- First pre-process the DNA sequence. For simplicity, any bases represented by N’s are deleted. (There are relatively very few of these with 140 N’s in the euchromatic portion of chromosome 21 and 333 in the euchromatic portion of chromosome 22. ) All lower case entries are replaced by their upper case equivalent.
- 3)
- Consider S as a set of N-7 overlapping octamers. Divide the sequence of octamers into blocks of size M. (M is 1024 in all the work described here. N.B. 1024 octamer comprise 1031 nucleotides. In preliminary work values of M = 512 and M = 2048 gave very similar results.)
- 4)
- For each block,
- Replace the sequence of letters by a numeric vector, consisting of the value of p determined by the minimum energy structure of each octamer.
- Take the M-step Fourier transform of the structural property vector.
- Obtain the power spectrum. (NB Although the power spectra are of length 1024, they are symmetric about the centre and so only the first 512 elements need be considered).
- 5)
- Sum the power spectra.
- 6)
- Optionally, normalise by dividing each element of the total spectrum by the number of blocks, to obtain a mean structural power spectrum representing the entire DNA length.
2.2. Transcription Factor Binding Site Regions
2.3. Tfbs Retrieval Experiments
3. Results and Discussion
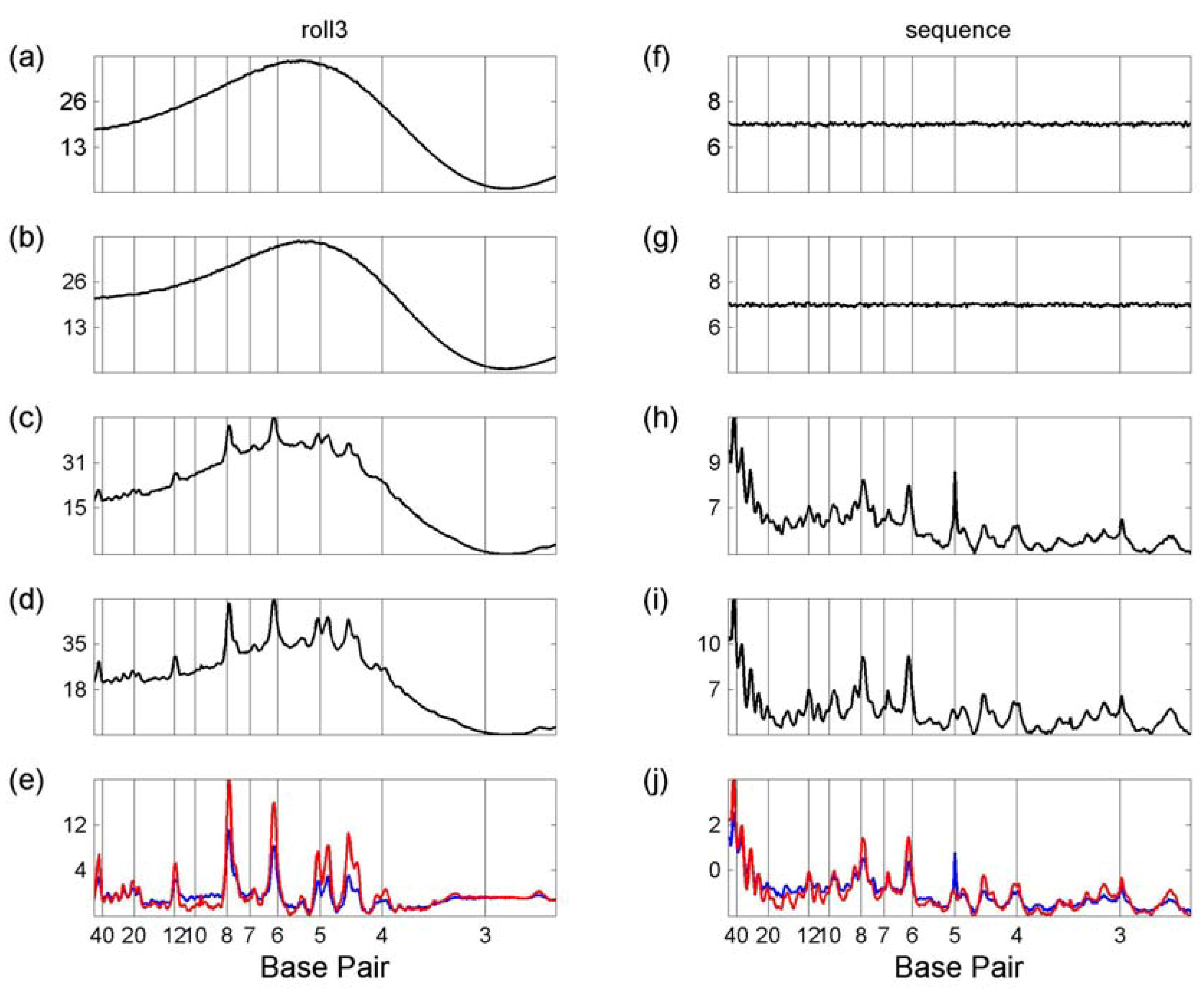
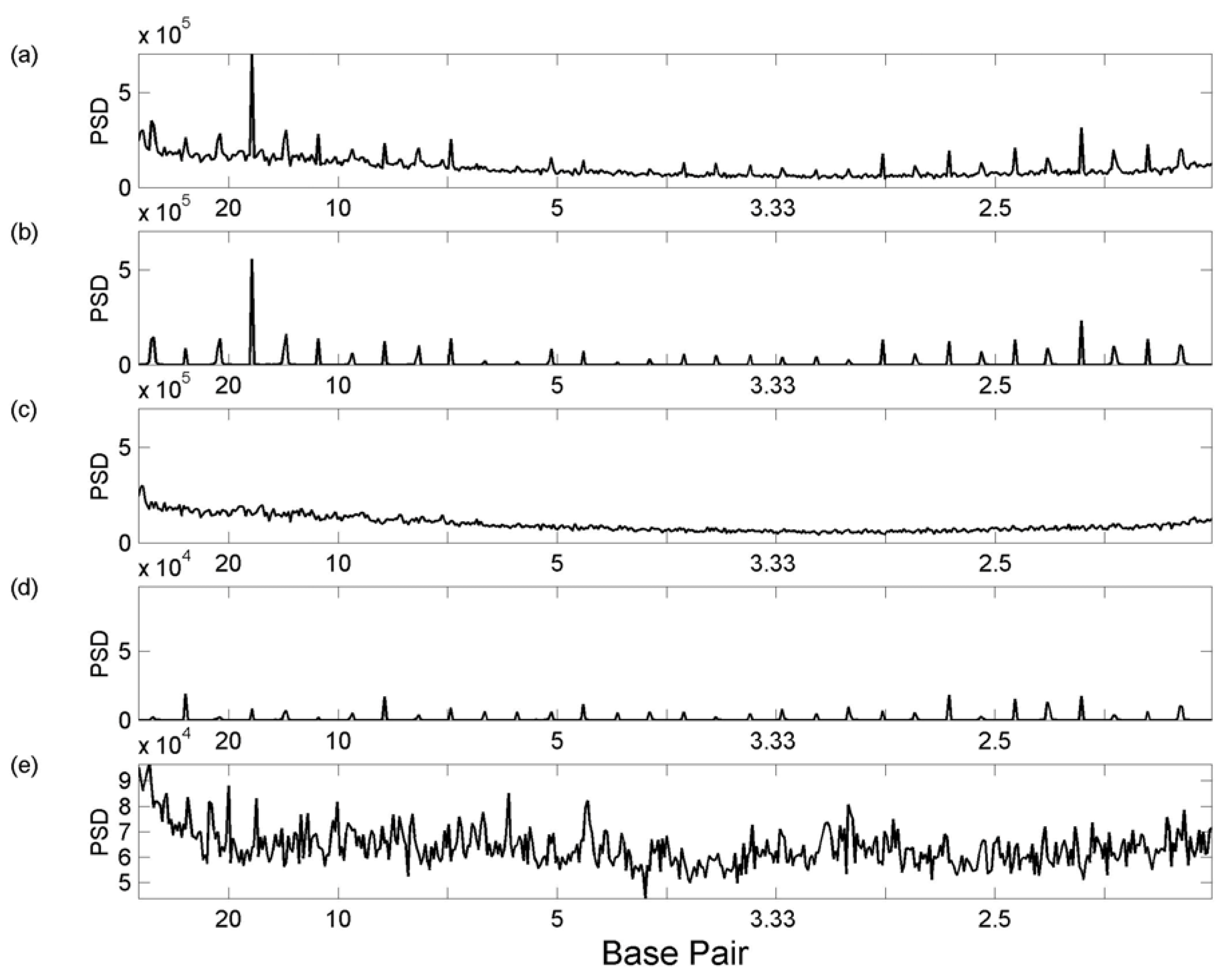
3.1 Whole Genome Transforms
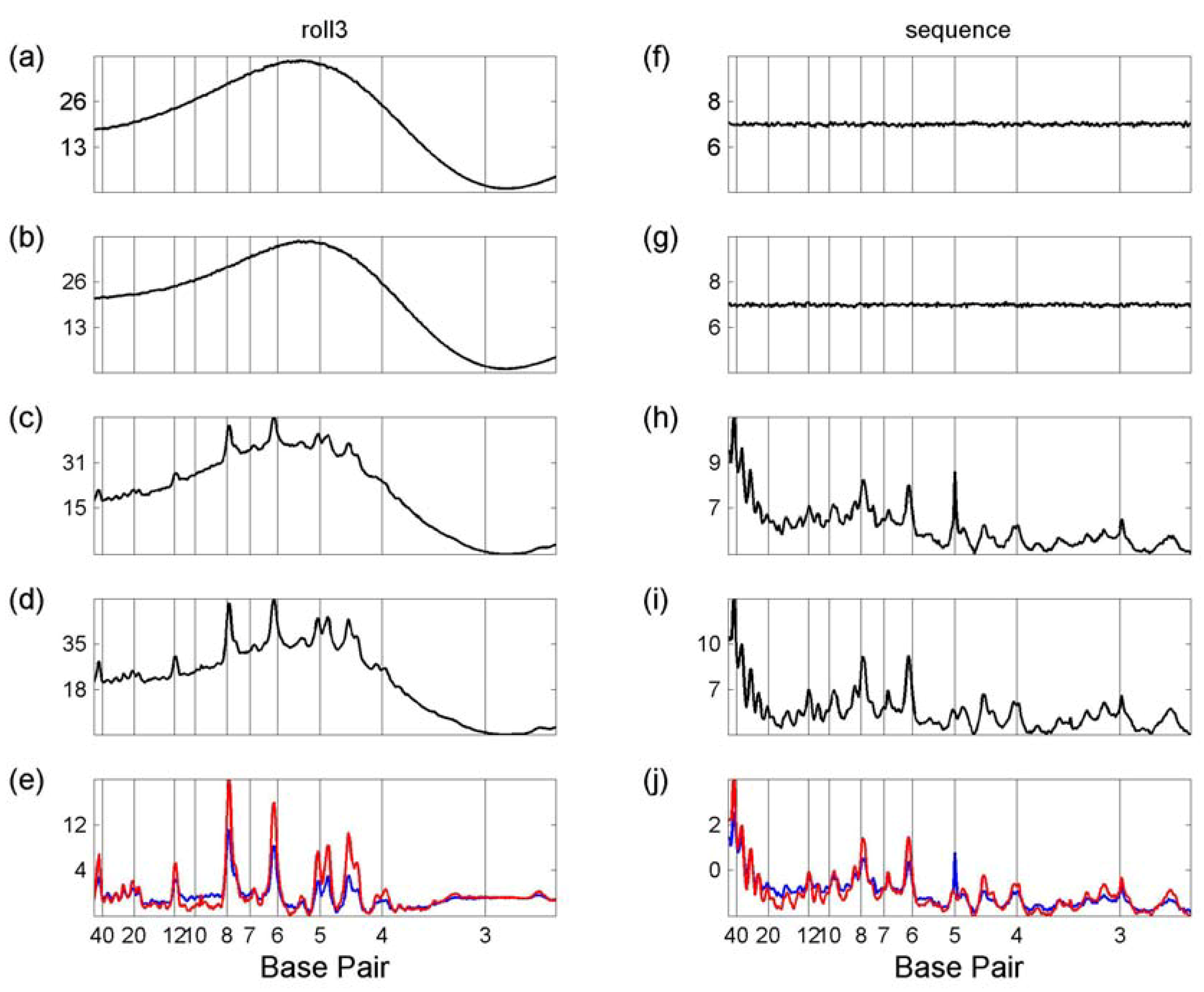
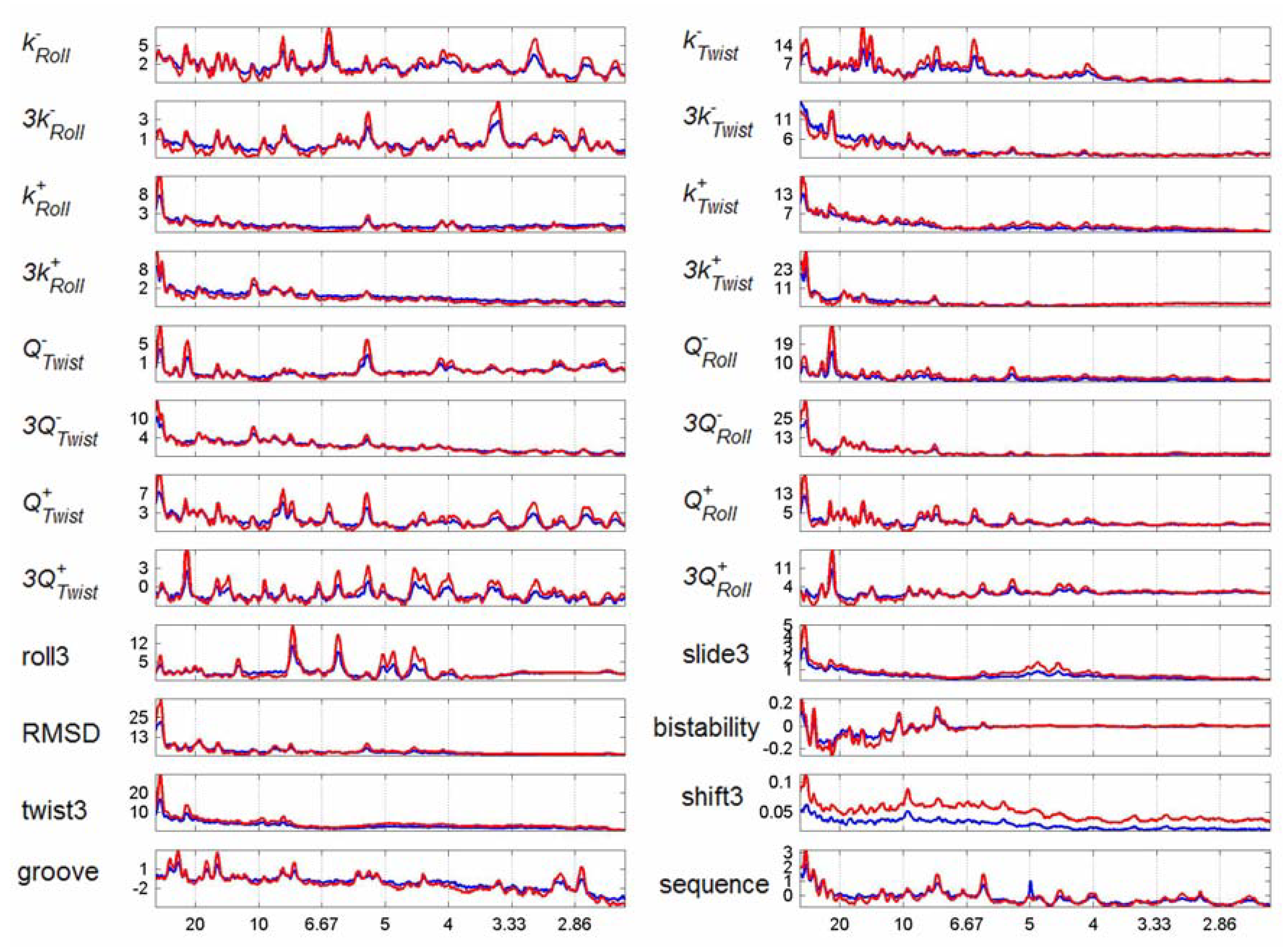
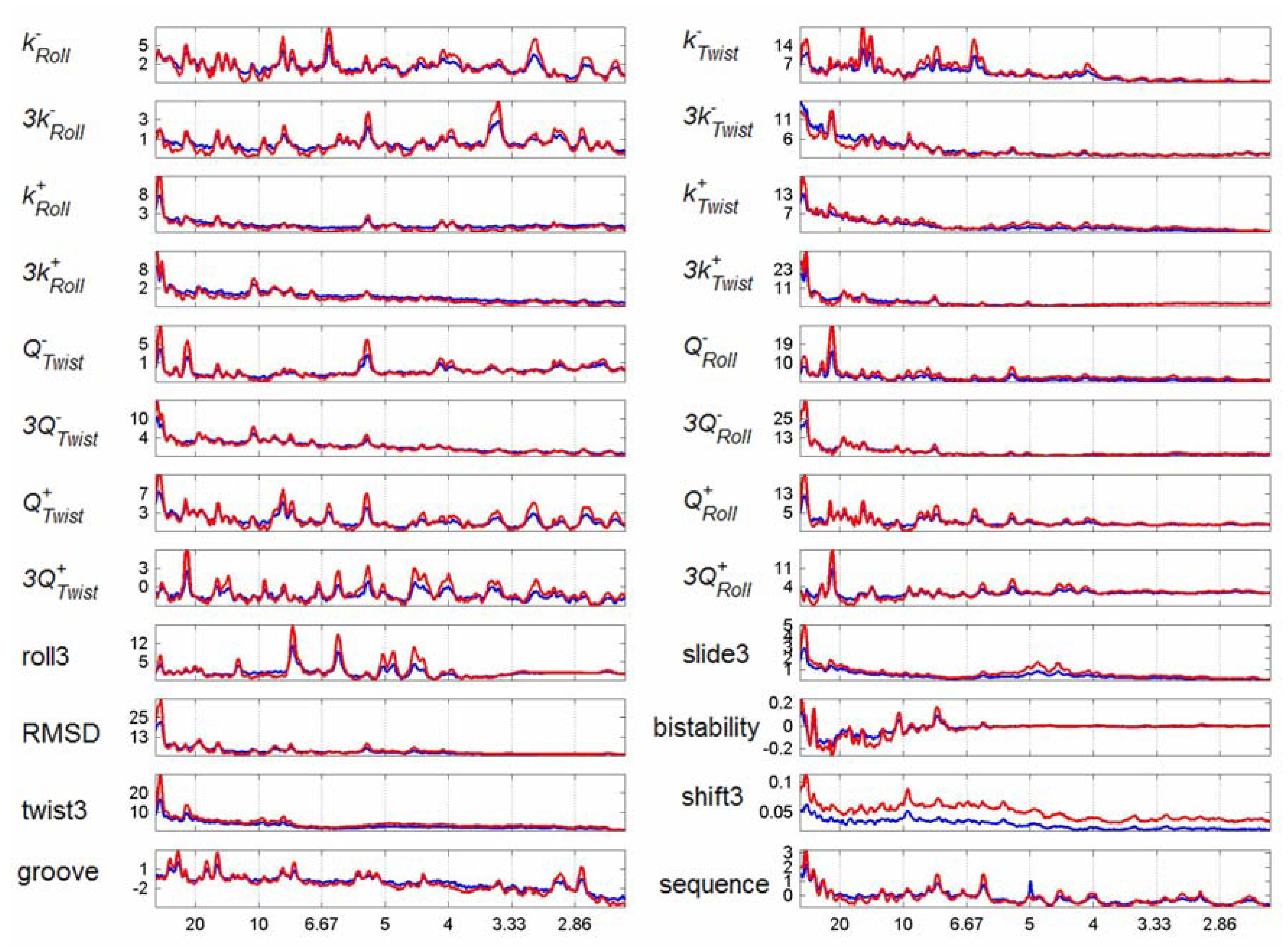
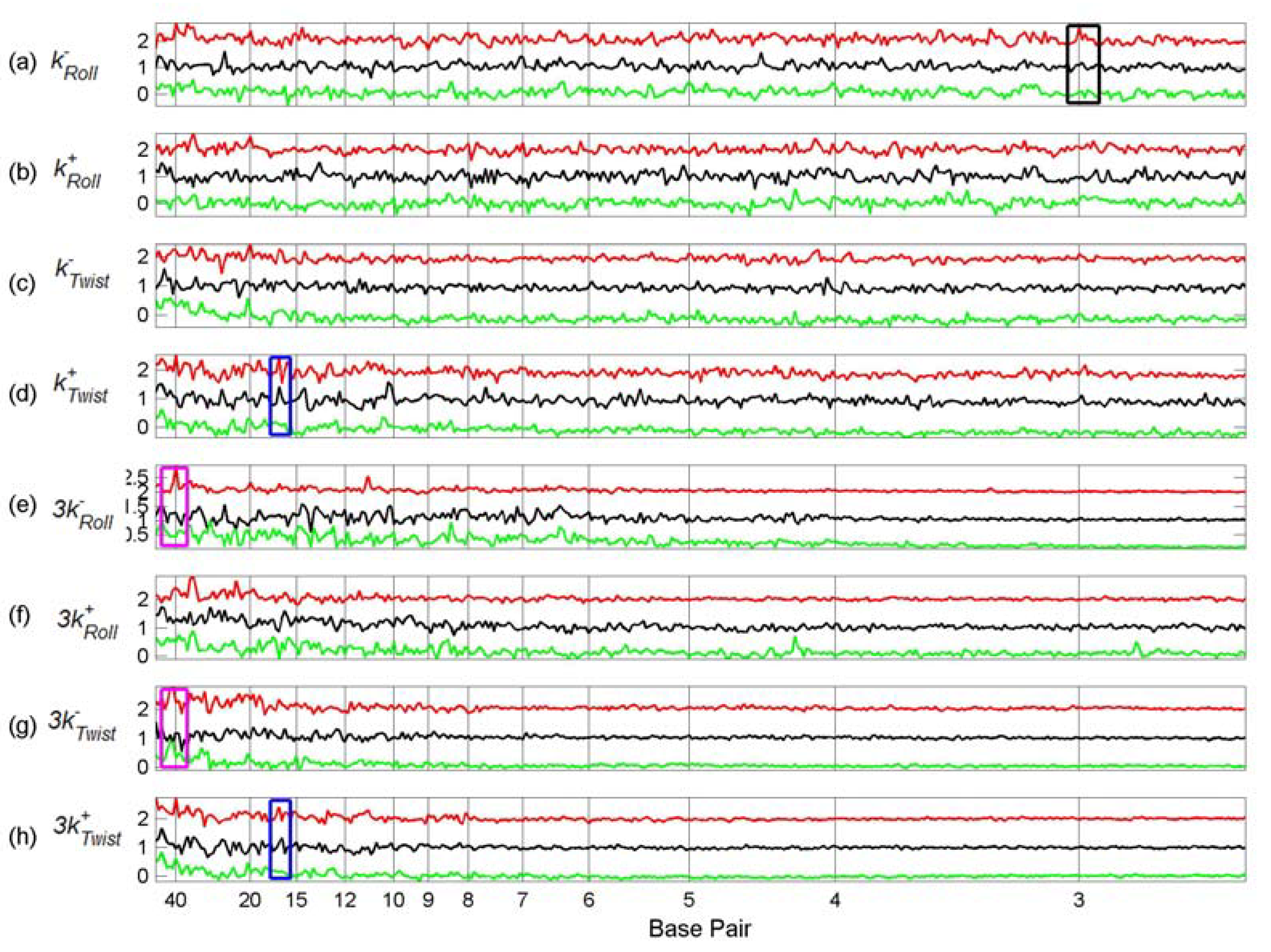
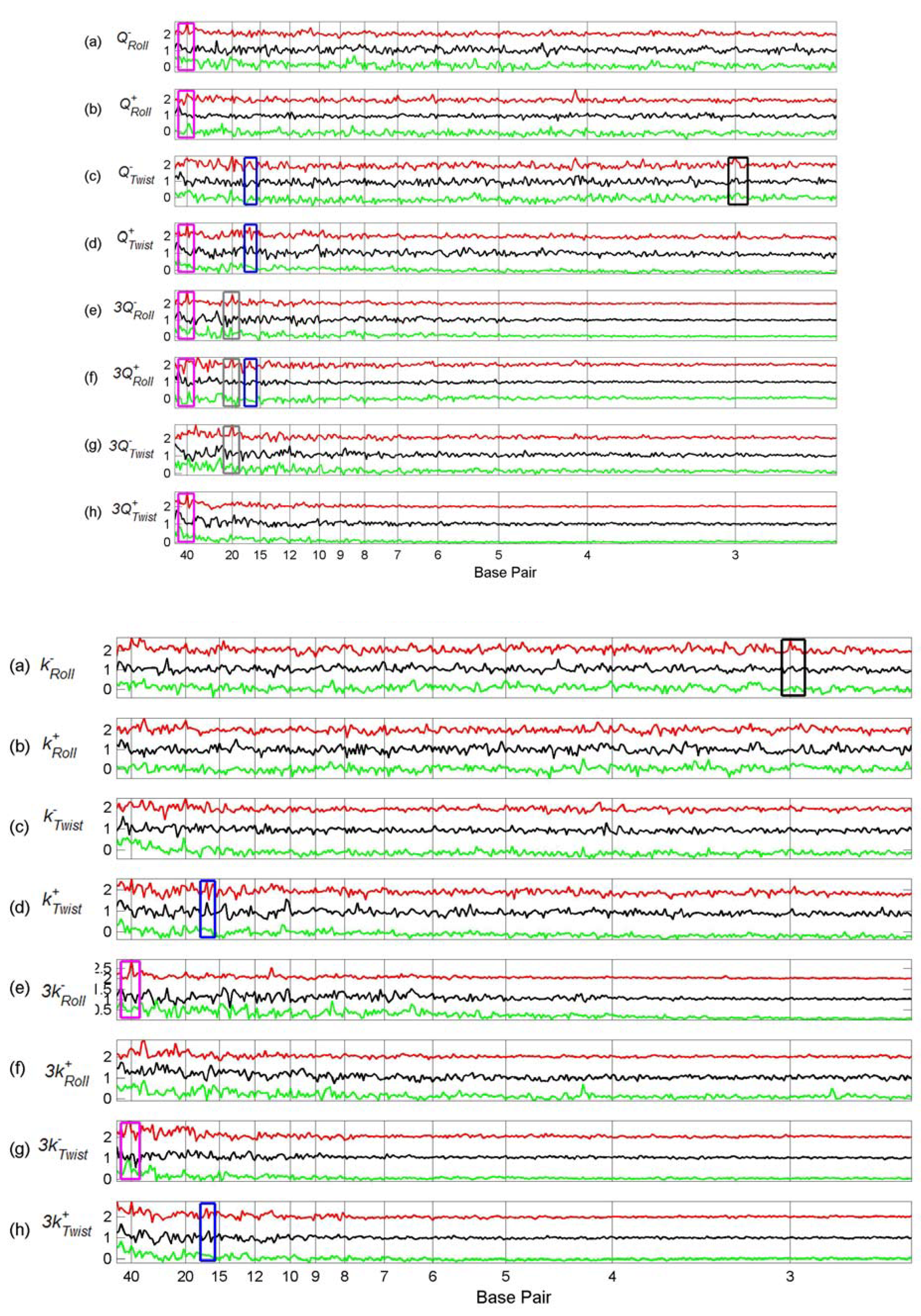
3.2. Transcription Factor Binding Site Transforms
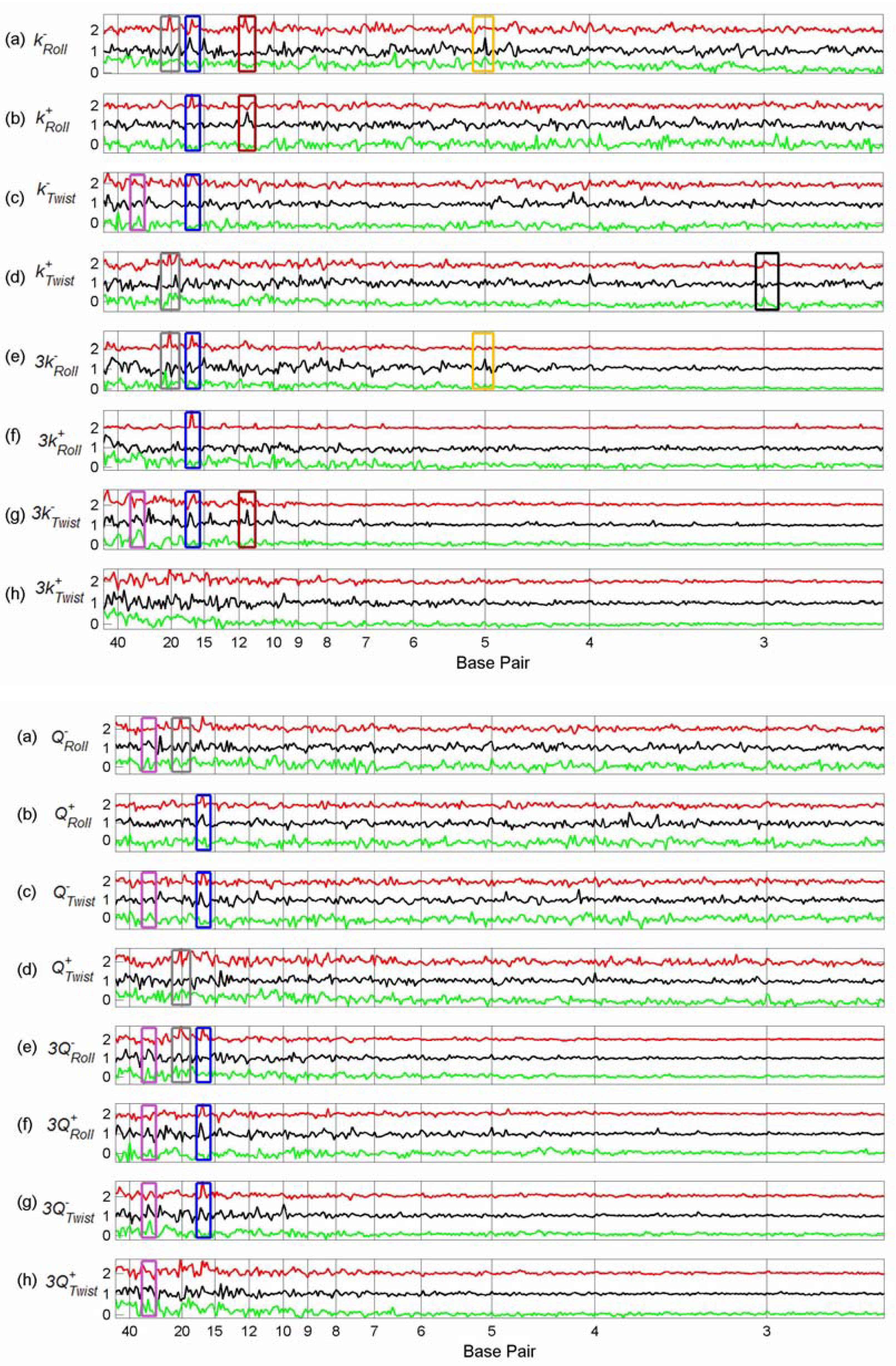
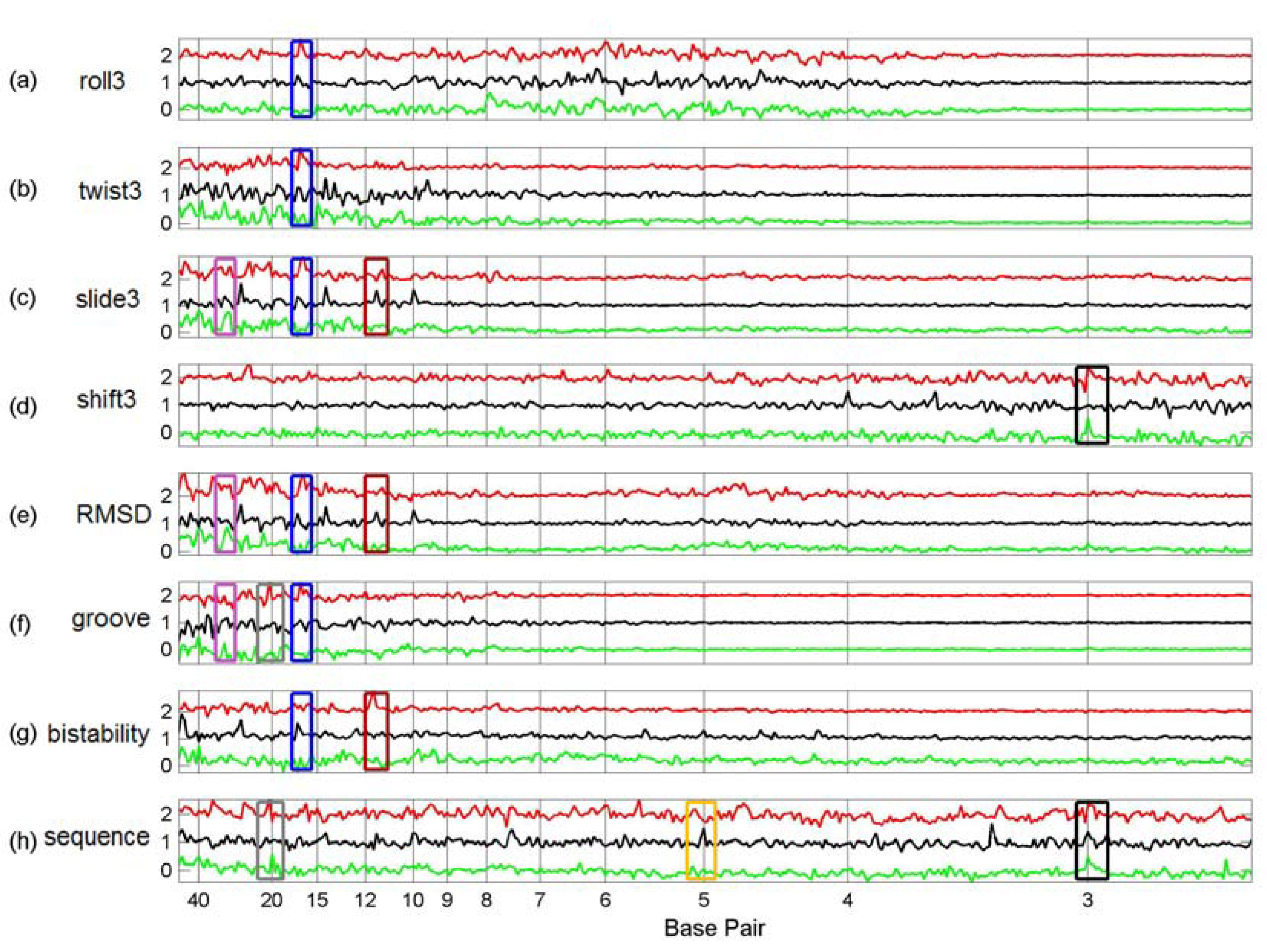

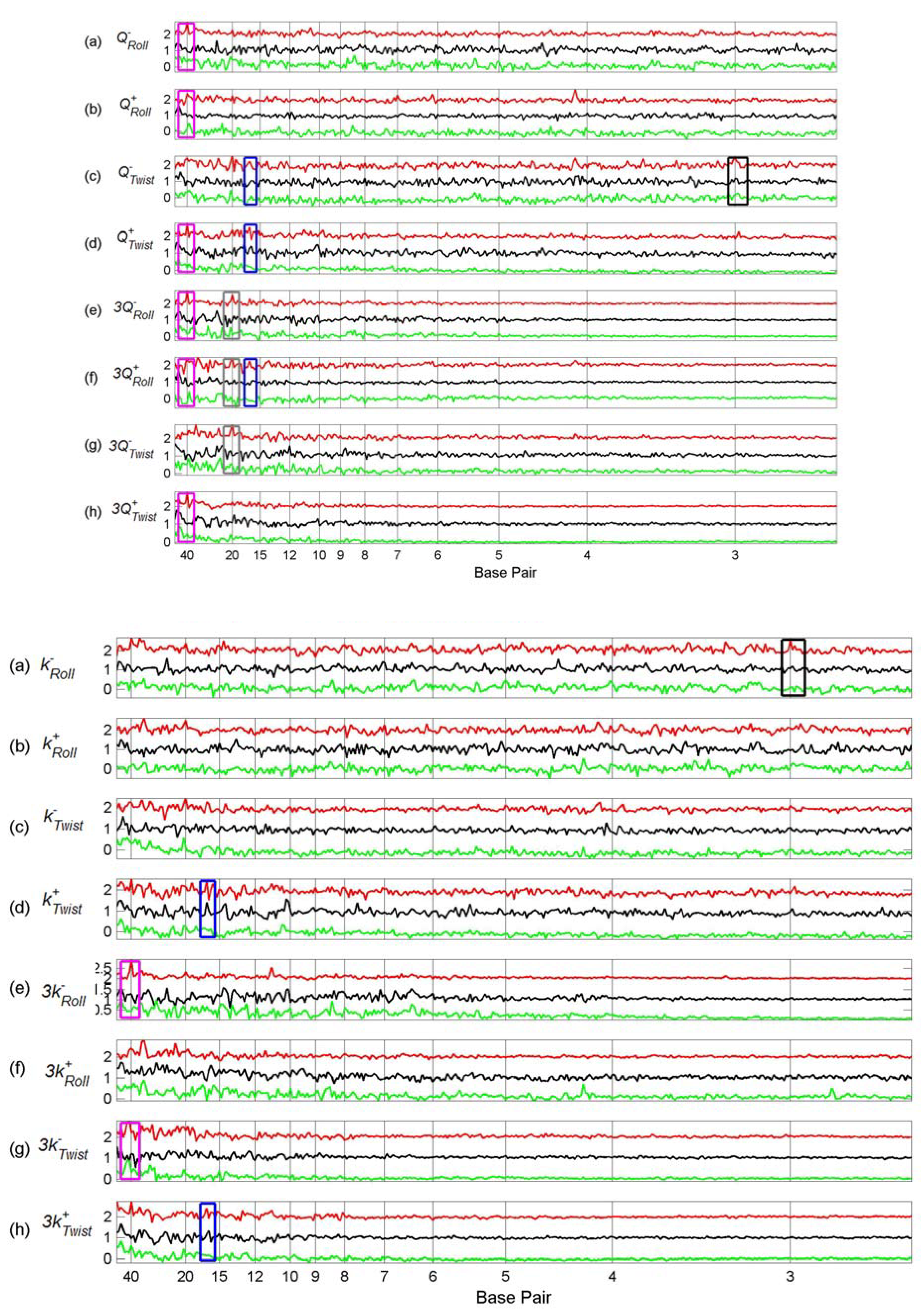
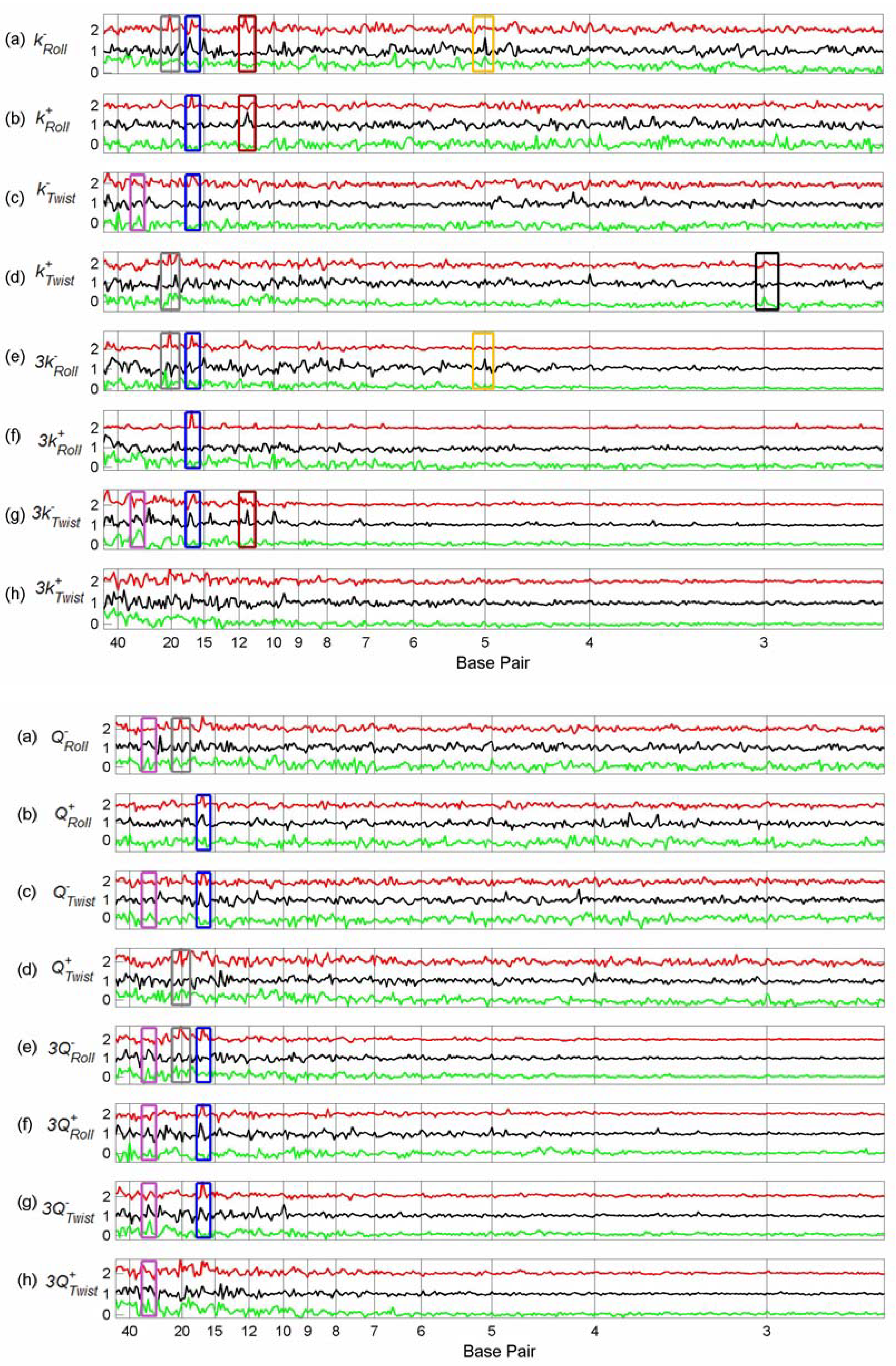
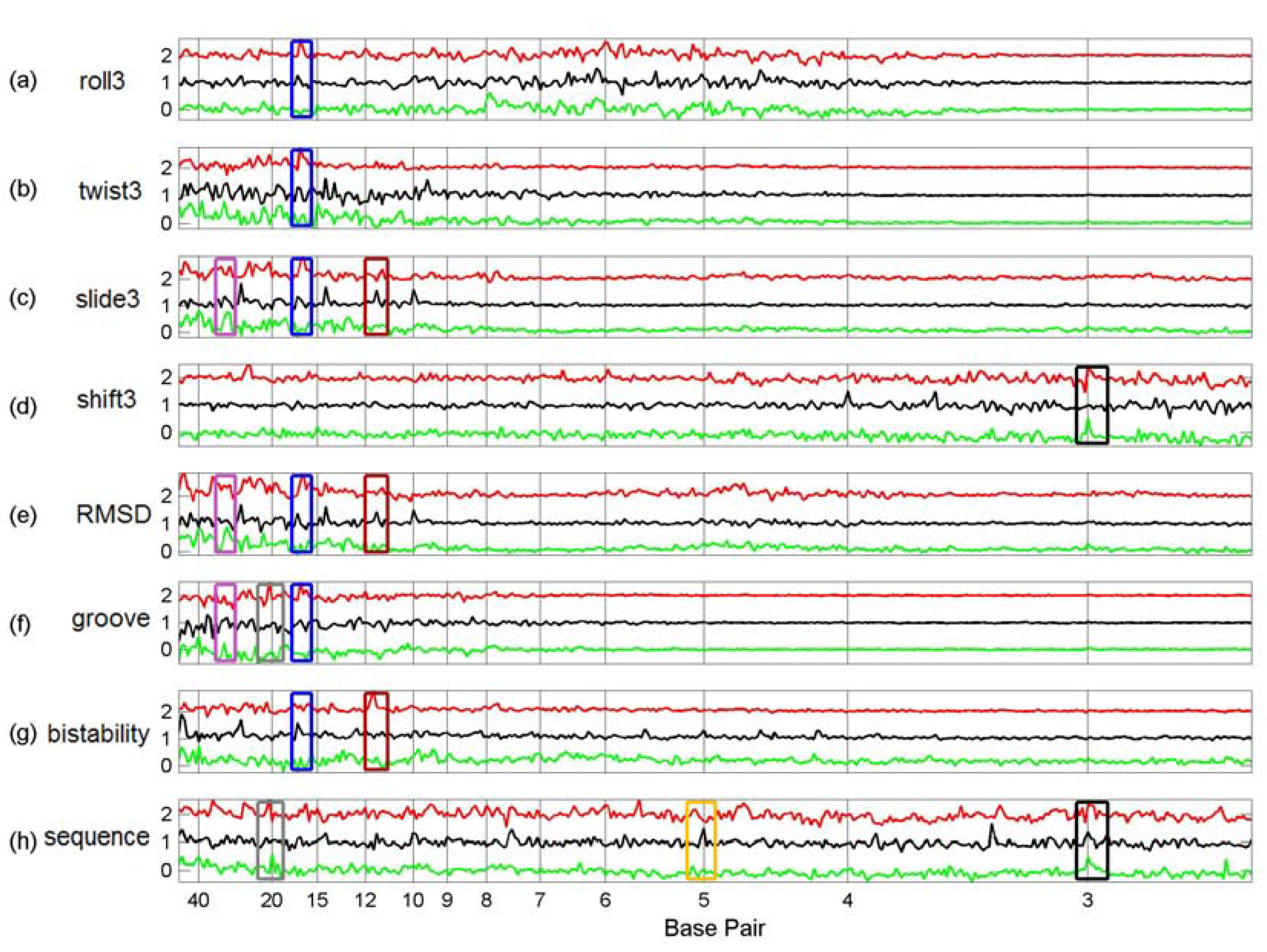
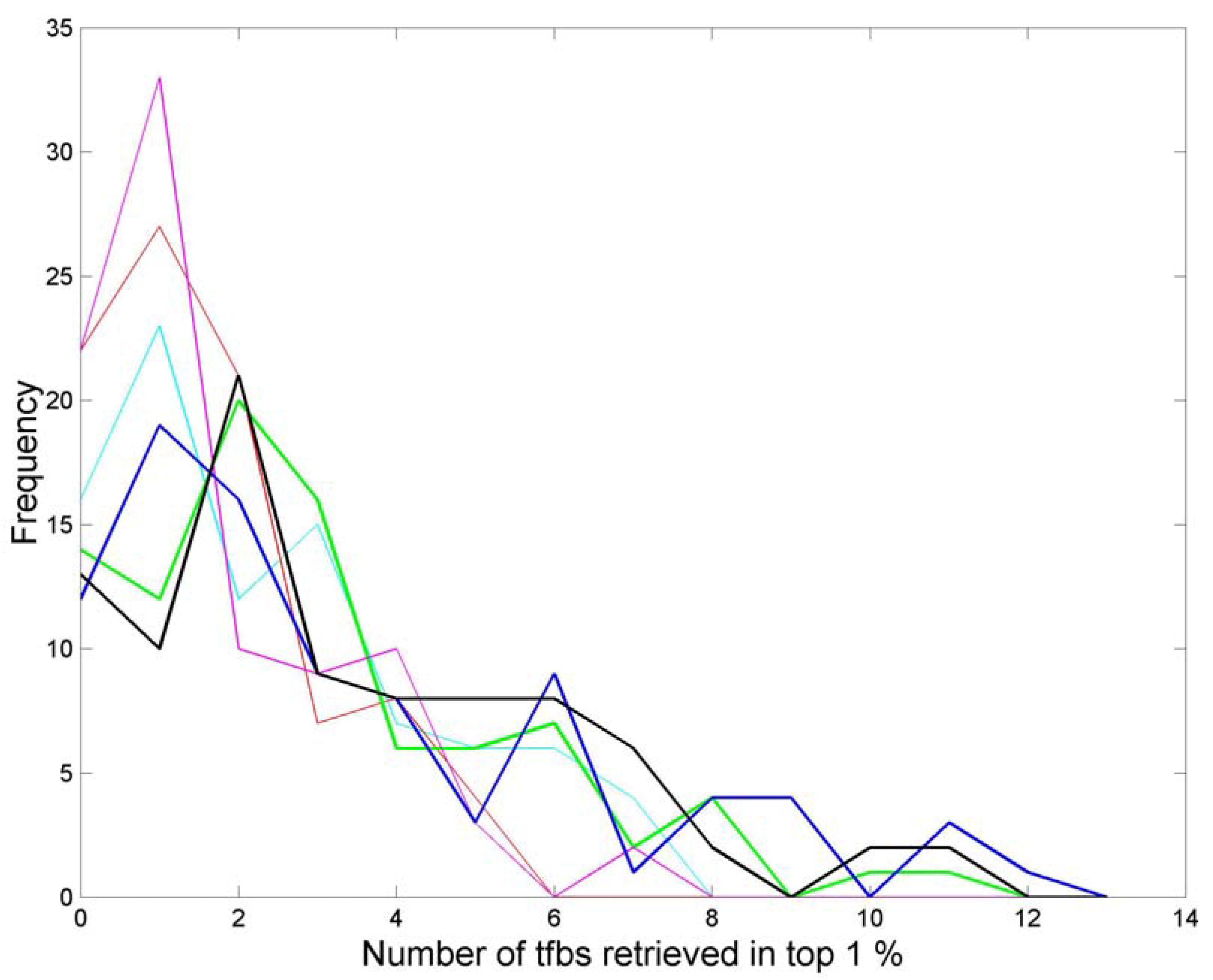
3.3. Finding tfbs Using Power Spectra as Fingerprints
| parameter | Sp1 | Random | Shuffled | Zscore |
|---|---|---|---|---|
| k -Twist | 1.60 | 1.16 | 1.01 | 2.8 |
| k +Twist | 0.85 | 0.61 | 1.02 | -1.2 |
| k-Roll | 0.90 | 0.71 | 0.78 | 0.64 |
| k+Roll | 0.67 | 0.81 | 0.83 | -1.1 |
| 3k -Twist | 1.15 | 0.94 | 0.97 | 1.1 |
| 3k +Twist | 1.10 | 0.86 | 0.67 | 1.1 |
| 3k-Roll | 1.13 | 0.94 | 0.94 | 1.5 |
| 3k+Roll | 1.14 | 0.86 | 1.09 | 0.48 |
| Q -Twist | 1.18 | 0.84 | 0.66 | 3.0 |
| Q +Twist | 0.90 | 0.62 | 0.87 | 0.40 |
| Q-Roll | 0.88 | 0.69 | 0.74 | 1.1 |
| Q+Roll | 0.92 | 0.74 | 0.71 | 1.0 |
| 3Q -Twist | 0.92 | 0.78 | 0.99 | -0.85 |
| 3Q +Twist | 1.45 | 0.93 | 0.82 | 3.6 |
| 3Q-Roll | 1.41 | 0.95 | 0.65 | 4.8 |
| 3Q+Roll | 1.07 | 0.82 | 0.61 | 2.5 |
| twist3 | 1.25 | 0.86 | 0.67 | 2.4 |
| roll3 | 1.35 | 0.83 | 0.82 | 2.6 |
| slide3 | 1.12 | 0.82 | 0.80 | 2.1 |
| shift3 | 0.79 | 0.68 | 0.67 | 1.2 |
| RMSD | 0.97 | 0.78 | 0.79 | 1.7 |
| groove | 2.40 | 1.16 | 0.88 | 5.2 |
| bistability | 0.38 | 0.11 | 0.00 | 2.1 |
| Sequence | 1.67 | 1.05 | 0.67 | 4.4 |
| Parameter combination | 1% retrieval rate |
|---|---|
| groove + k –Twist | 3.0 |
| groove + k –Twist + roll3 | 3.2 |
| groove + k –Twist + roll3 + 3Q +Twist | 3.2 |
| groove + k –Twist + roll3 + 3Q +Twist +3Q-Roll | 2.9 |
| groove + k –Twist +3Q-Roll | 3.2 |
| groove + sequence | 2.5 |
| k –Twist + sequence | 2.4 |
| groove + k –Twist + sequence | 3.3 |
| groove + k –Twist + roll3+sequence | 3.4 |
| groove + k –Twist + roll3+sequence + 3Q +Twist | 3.4 |
| All parameters better then random | 1.3 |
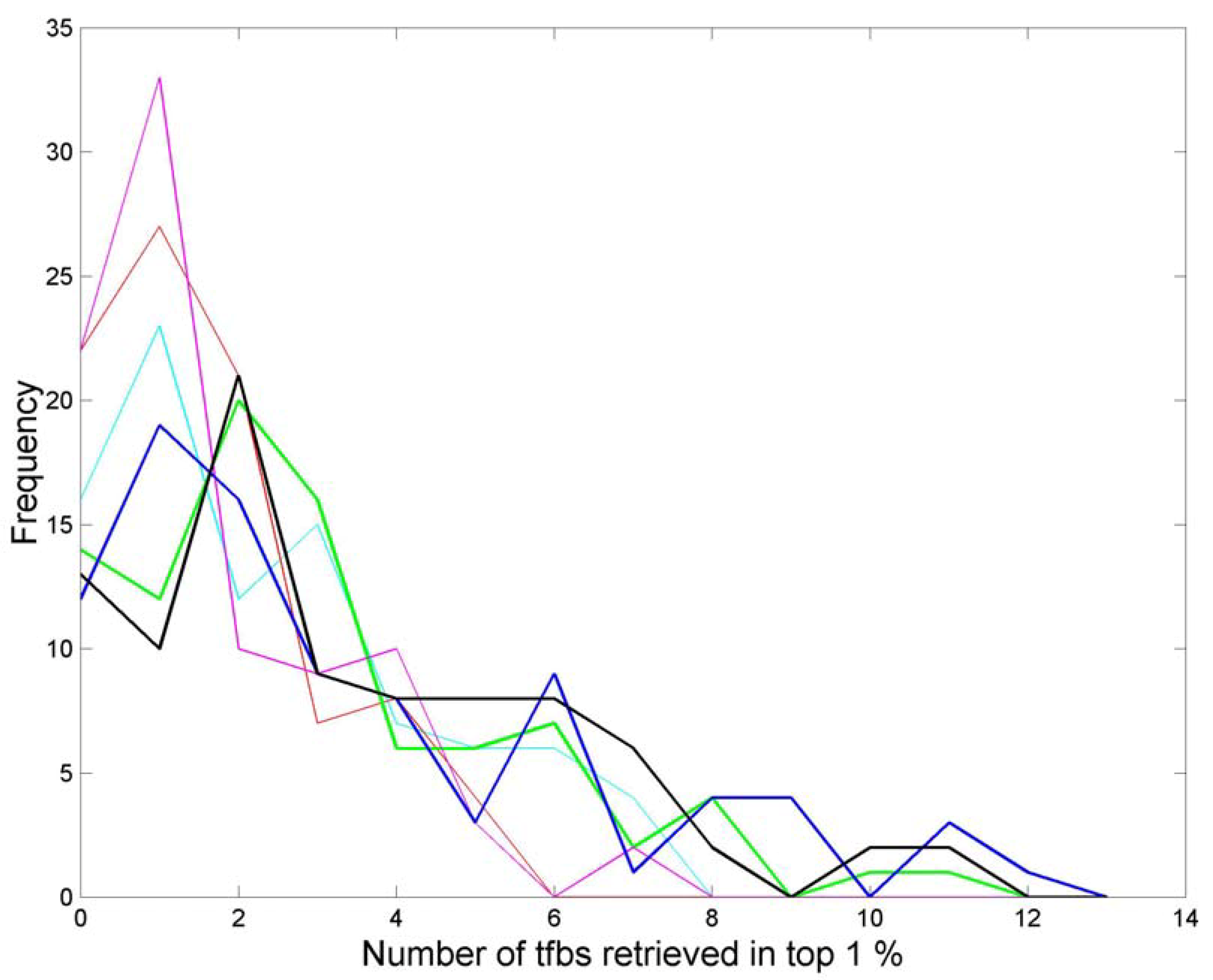
4. Conclusions
Acknowledgements
References and Notes
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; Funke, R.; Gage, D.; Harris, K.; Heaford, A.; Howland, J.; Kann, L.; Lehoczky, J.; LeVine, R.; McEwan, P.; McKernan, K.; Meldrim, J.; Mesirov, J.P.; Miranda, C.; Morris, W.; Naylor, J.; Raymond, C.; Rosetti, M.; Santos, R.; Sheridan, A.; Sougnez, C.; Stange-Thomann, N.; Stojanovic, N.; Subramanian, A.; Wyman, D.; Rogers, J.; Sulston, J.; Ainscough, R.; Beck, S.; Bentley, D.; Burton, J.; Clee, C.; Carter, N.; Coulson, A.; Deadman, R.; Deloukas, P.; Dunham, A.; Dunham, I.; Durbin, R.; French, L.; Grafham, D.; Gregory, S.; Hubbard, T.; Humphray, S.; Hunt, A.; Jones, M.; Lloyd, C.; McMurray, A.; Matthews, L.; Mercer, S.; Milne, S.; Mullikin, J.C.; Mungall, A.; Plumb, R.; Ross, M.; Shownkeen, R.; Sims, S.; Waterston, R.H.; Wilson, R.K.; Hillier, L.W.; McPherson, J.D.; Marra, M.A.; Mardis, E.R.; Fulton, L.A.; Chinwalla, A.T.; Pepin, K.H.; Gish, W.R.; Chissoe, S.L.; Wendl, M.C.; Delehaunty, K.D.; Miner, T.L.; Delehaunty, A.; Kramer, J.B.; Cook, L.L.; Fulton, R.S.; Johnson, D.L.; Minx, P.J.; Clifton, S.W.; Hawkins, T.; Branscomb, E.; Predki, P.; Richardson, P.; Wenning, S.; Slezak, T.; Doggett, N.; Cheng, J.F.; Olsen, A.; Lucas, S.; Elkin, C.; Uberbacher, E.; Frazier, M.; Gibbs, R.A.; Muzny, D.M.; Scherer, S.E.; Bouck, J.B.; Sodergren, E.J.; Worley, K.C.; Rives, C.M.; Gorrell, J.H.; Metzker, M.L.; Naylor, S.L.; Kucherlapati, R.S.; Nelson, D.L.; Weinstock, G.M.; Sakaki, Y.; Fujiyama, A.; Hattori, M.; Yada, T.; Toyoda, A.; Itoh, T.; Kawagoe, C.; Watanabe, H.; Totoki, Y.; Taylor, T.; Weissenbach, J.; Heilig, R.; Saurin, W.; Artiguenave, F.; Brottier, P.; Bruls, T.; Pelletier, E.; Robert, C.; Wincker, P.; Rosenthal, A.; Platzer, M.; Nyakatura, G.; Taudien, S.; Rump, A.; Yang, H.M.; Yu, J.; Wang, J.; Huang, G.Y.; Gu, J.; Hood, L.; Rowen, L.; Madan, A.; Qin, S.Z.; Davis, R.W.; Federspiel, N.A.; Abola, A.P.; Proctor, M.J.; Myers, R.M.; Schmutz, J.; Dickson, M.; Grimwood, J.; Cox, D.R.; Olson, M.V.; Kaul, R.; Shimizu, N.; Kawasaki, K.; Minoshima, S.; Evans, G.A.; Athanasiou, M.; Schultz, R.; Roe, B.A.; Chen, F.; Pan, H.Q.; Ramser, J.; Lehrach, H.; Reinhardt, R.; McCombie, W.R.; de la Bastide, M.; Dedhia, N.; Blocker, H.; Hornischer, K.; Nordsiek, G.; Agarwala, R.; Aravind, L.; Bailey, J.A.; Bateman, A.; Batzoglou, S.; Birney, E.; Bork, P.; Brown, D.G.; Burge, C.B.; Cerutti, L.; Chen, H.C.; Church, D.; Clamp, M.; Copley, R.R.; Doerks, T.; Eddy, S.R.; Eichler, E.E.; Furey, T.S.; Galagan, J.; Gilbert, J.G.R.; Harmon, C.; Hayashizaki, Y.; Haussler, D.; Hermjakob, H.; Hokamp, K.; Jang, W.H.; Johnson, L.S.; Jones, T.A.; Kasif, S.; Kaspryzk, A.; Kennedy, S.; Kent, W.J.; Kitts, P.; Koonin, E.V.; Korf, I.; Kulp, D.; Lancet, D.; Lowe, T.M.; McLysaght, A.; Mikkelsen, T.; Moran, J.V.; Mulder, N.; Pollara, V.J.; Ponting, C.P.; Schuler, G.; Schultz, J.R.; Slater, G.; Smit, A.F.A.; Stupka, E.; Szustakowki, J.; Thierry-Mieg, D.; Thierry-Mieg, J.; Wagner, L.; Wallis, J.; Wheeler, R.; Williams, A.; Wolf, Y.I.; Wolfe, K.H.; Yang, S.P.; Yeh, R.F.; Collins, F.; Guyer, M.S.; Peterson, J.; Felsenfeld, A.; Wetterstrand, K.A.; Patrinos, A.; Morgan, M.J. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Lander, E.S.; Rogers, J.; Waterston, R.H. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar]
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; Antonarakis, S.E.; Attwood, J.; Baertsch, R.; Bailey, J.; Barlow, K.; Beck, S.; Berry, E.; Birren, B.; Bloom, T.; Bork, P.; Botcherby, M.; Bray, N.; Brent, M.R.; Brown, D.G.; Brown, S.D.; Bult, C.; Burton, J.; Butler, J.; Campbell, R.D.; Carninci, P.; Cawley, S.; Chiaromonte, F.; Chinwalla, A.T.; Church, D.M.; Clamp, M.; Clee, C.; Collins, F.S.; Cook, L.L.; Copley, R.R.; Coulson, A.; Couronne, O.; Cuff, J.; Curwen, V.; Cutts, T.; Daly, M.; David, R.; Davies, J.; Delehaunty, K.D.; Deri, J.; Dermitzakis, E.T.; Dewey, C.; Dickens, N.J.; Diekhans, M.; Dodge, S.; Dubchak, I.; Dunn, D.M.; Eddy, S.R.; Elnitski, L.; Emes, R.D.; Eswara, P.; Eyras, E.; Felsenfeld, A.; Fewell, G.A.; Flicek, P.; Foley, K.; Frankel, W.N.; Fulton, L.A.; Fulton, R.S.; Furey, T.S.; Gage, D.; Gibbs, R.A.; Glusman, G.; Gnerre, S.; Goldman, N.; Goodstadt, L.; Grafham, D.; Graves, T.A.; Green, E.D.; Gregory, S.; Guigo, R.; Guyer, M.; Hardison, R.C.; Haussler, D.; Hayashizaki, Y.; Hillier, L.W.; Hinrichs, A.; Hlavina, W.; Holzer, T.; Hsu, F.; Hua, A.; Hubbard, T.; Hunt, A.; Jackson, I.; Jaffe, D.B.; Johnson, L.S.; Jones, M.; Jones, T.A.; Joy, A.; Kamal, M.; Karlsson, E.K.; Karolchik, D.; Kasprzyk, A.; Kawai, J.; Keibler, E.; Kells, C.; Kent, W.J.; Kirby, A.; Kolbe, D.L.; Korf, I.; Kucherlapati, R.S.; Kulbokas, E.J.; Kulp, D.; Landers, T.; Leger, J.P.; Leonard, S.; Letunic, I.; Levine, R.; Li, J.; Li, M.; Lloyd, C.; Lucas, S.; Ma, B.; Maglott, D.R.; Mardis, E.R.; Matthews, L.; Mauceli, E.; Mayer, J.H.; McCarthy, M.; McCombie, W.R.; McLaren, S.; McLay, K.; McPherson, J.D.; Meldrim, J.; Meredith, B.; Mesirov, J.P.; Miller, W.; Miner, T.L.; Mongin, E.; Montgomery, K.T.; Morgan, M.; Mott, R.; Mullikin, J.C.; Muzny, D.M.; Nash, W.E.; Nelson, J.O.; Nhan, M.N.; Nicol, R.; Ning, Z.; Nusbaum, C.; O'Connor, M.J.; Okazaki, Y.; Oliver, K.; Larty, E.O.; Pachter, L.; Parra, G.; Pepin, K.H.; Peterson, J.; Pevzner, P.; Plumb, R.; Pohl, C.S.; Poliakov, A.; Ponce, T.C.; Ponting, C.P.; Potter, S.; Quail, M.; Reymond, A.; Roe, B.A.; Roskin, K.M.; Rubin, E.M.; Rust, A.G.; Santos, R.; Sapojnikov, V.; Schultz, B.; Schultz, J.; Schwartz, M.S.; Schwartz, S.; Scott, C.; Seaman, S.; Searle, S.; Sharpe, T.; Sheridan, A.; Shownkeen, R.; Sims, S.; Singer, J.B.; Slater, G.; Smit, A.; Smith, D.R.; Spencer, B.; Stabenau, A.; Strange-Thomann, N.S.; Sugnet, C.; Suyama, M.; Tesler, G.; Thompson, J.; Torrents, D.; Trevaskis, E.; Tromp, J.; Ucla, C.; Vidal, A.U.; Vinson, J.P.; von Niederhausern, A.C.; Wade, C.M.; Wall, M.; Weber, R.J.; Weiss, R.B.; Wendl, M.C.; West, A.P.; Wetterstrand, K.; Wheeler, R.; Whelan, S.; Wierzbowski, J.; Willey, D.; Williams, S.; Wilson, R.K.; Winter, E.; Worley, K.C.; Wyman, D.; Yang, S.; Yang, S.P.; Zdobnov, E.M.; Zody, M.C.; Lander, E.S. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [PubMed]
- Johnston, M.; Stormo, G.D. Heirlooms in the attic. Science 2003, 302, 997–998. [Google Scholar] [CrossRef] [PubMed]
- Fickett, J.W.; Hatzigeorgiou, A.C. Eukaryotic promoter recognition. Genome Res. 1997, 7, 861–878. [Google Scholar] [PubMed]
- Silverman, B.D.; Linsker, R. A measure of DNA periodicity. J. Theor. Biol. 1986, 118, 295–300. [Google Scholar] [CrossRef]
- Tiwari, S.; Ramachandran, S.; Bhattacharya, A.; Bhattacharya, S.; Ramaswamy, R. Prediction of probable genes by Fourier analysis of genomic sequences. Comp. App. Biosci. 1997, 13, 263–270. [Google Scholar]
- Fukushima, A.; Ikemura, T.; Kinouchi, M.; Oshima, T.; Kudo, Y.; Mori, H.; Kanaya, S. Periodicity in prokaryotic and eukaryotic genomes identified by power spectrum analysis. Gene 2002, 300, 203–211. [Google Scholar] [CrossRef]
- Trifonov, E.N. 3-, 10.5-, 200- and 400-base periodicities in genome sequences. Physica a-Stat. Mech. App. 1998, 249, 511–516. [Google Scholar] [CrossRef]
- Widom, J. Short-range order in two eukaryotic genomes: Relation to chromosome structure. J. Mol. Biol. 1996, 259, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.Z.; Widom, J. Improved alignment of nucleosome DNA sequences using a mixture model. Nucleic Acids Res. 2005, 33, 6743–6755. [Google Scholar] [CrossRef] [PubMed]
- Dalal, Y.; Fleury, T.J.; Cioffi, A.; Stein, A. Long-range oscillation in a periodic DNA sequence motif may influence nucleosome array formation. Nucleic Acids Res. 2005, 33, 934–945. [Google Scholar] [CrossRef] [PubMed]
- D’Avenio, G.; Grigioni, M.; Orefici, G.; Creti, R. SWIFT (sequence-wide investigation with Fourier transform): a software tool for identifying proteins of a given class from the unannotated genome sequence. Bioinf. 2005, 21, 2943–2949. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Issac, B.; Raghava, G.P.S.; Ramaswamy, R. Spectral Repeat Finder (SRF): identification of repetitive sequences using Fourier transformation. Bioinf. 2004, 20, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Bejerano, G.; Pheasant, M.; Makunin, I.; Stephen, S.; Kent, W.J.; Mattick, J.S.; Haussler, D. Ultraconserved elements in the human genome. Science 2004, 304, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Dermitzakis, E.T.; Kirkness, E.; Schwarz, S.; Birney, E.; Reymond, A.; Antonarakis, S.E. Comparison of human chromosome 21 conserved nongenic sequences (CNGs) with the mouse and dog genomes shows that their selective constraint is independent of their genic environment. Genome Res. 2004, 14, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Dermitzakis, E.T.; Reymond, A.; Lyle, R.; Scamuffa, N.; Ucla, C.; Deutsch, S.; Stevenson, B.J.; Flegel, V.; Bucher, P.; Jongeneel, C.V.; Antonarakis, S.E. Numerous potentially functional but non-genic conserved sequences on human chromosome 21. Nature 2002, 420, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, E.J.; Hirons, L.; Hunter, C.A.; Willett, P. Genomic data analysis using DNA structure: an analysis of Conserved Non-Genic sequences and Ultra-Conserved Elements. J. Chem. Inf. Model. 2006, 46, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A. Sequence-dependent DNA-structure - the role of base stacking interactions. J. Mol. Biol. 1993, 230, 1025–1054. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Lu, X.J. Construction of double-helical DNA structures based on dinucleotide building blocks. J. Biomol. Struct. Dyn. 1997, 14, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Lu, X.J. DNA base-stacking interactions: A comparison of theoretical calculations with oligonucleotide X-ray crystal structures. J. Mol. Biol. 1997, 265, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.J.; Dauncey, M.P.; Hunter, C.A. Sequence-dependent DNA structure: Dinucleotide conformational maps. J. Mol. Biol. 2000, 295, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.J.; Dauncey, M.P.; Hunter, C.A. Sequence-dependent DNA structure: Tetranucleotide conformational maps. J. Mol. Biol. 2000, 295, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.J.; Hunter, C.A. Sequence-structure relationships in DNA oligomers: A computational approach. J. Am. Chem. Soc. 2001, 123, 7399–7406. [Google Scholar]
- Gardiner, E.J.; Hunter, C.A.; Lu, X.J.; Willett, P. A structural similarity analysis of double-helical DNA. J. Mol. Biol. 2004, 343, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, E.J.; Hunter, C.A.; Packer, M.J.; Palmer, D.S.; Willett, P. Sequence-dependent DNA structure: A database of octamer structural parameters. J. Mol. Biol. 2003, 332, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Paulsen, M.; Tierling, S.; Mikesa, T.; Lengauer, T.; Walter, J. CpG island methylation in human lymphocytes is highly correlated with DNA sequence patters, repeat frequencies and predicted DNA structure. PLoS Genetics 2006, 2, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Fickett, J.W.; Tung, C.S. Assessment of protein coding measures. Nucleic Acids Res. 1992, 20, 6441–6450. [Google Scholar] [CrossRef] [PubMed]
- Cawley, S.; Bekiranov, S.; Ng, H.H.; Kapranov, P.; Sekinger, E.A.; Kampa, D.; Piccolboni, A.; Sementchenko, V.; Cheng, J.; Williams, A.J.; Wheeler, R.; Wong, B.; Drenkow, J.; Yamanaka, M.; Patel, S.; Brubaker, S.; Tammana, H.; Helt, G.; Struhl, K.; Gingeras, T.R. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 2004, 116, 499–509. [Google Scholar] [CrossRef]
- Kampa, D.; Cheng, J.; Kapranov, P.; Yamanaka, M.; Brubaker, S.; Cawley, S.; Drenkow, J.; Piccolboni, A.; Bekiranov, S.; Helt, G.; Tammana, H.; Gingeras, T.R. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 2004, 14, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Kampa, D.; Kapranov, P.; Cawley, S.; Bekiranov, S.; Ng, H.H.; Sekinger, E.A.; Piccolboni, A.; Sementchenko, V.; Cheng, J.; Drenkow, J.; Yamanaka, M.; Patel, S.; Brubaker, S.; Tammana, H.; Narayanan, B.; Helt, G.; Struhl, K.; Gingeras, T.R. Global mapping of functionally-important and regulatory regions on human chromosomes 21 and 22 reveal novel regulatory networks in the human genome. Am. J. Hum. Genet. 2003, 73, 118. [Google Scholar]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.T.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J.; Weber, R.J.; Haussler, D.; Kent, W.J. The UCSC Genome Browser Database. Nucleic Acids Res. 2003, 31, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT - The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Ginn, C.M.R.; Willett, P.; Bradshaw, J. Combination of molecular similarity measures using data fusion. Perspect. Drug Discov. Des. 2000, 20, 1–16. [Google Scholar] [CrossRef]
- Salim, N.; Holliday, J.; Willett, P. Combination of fingerprint-based similarity coefficients using data fusion. J. Chem. Inf. Comput. Sci. 2003, 43, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gardiner, E.J.J.; Hunter, C.A.; Willett, P. Structural Fingerprints of Transcription Factor Binding Site Regions. Algorithms 2009, 2, 448-469. https://doi.org/10.3390/a2010448
Gardiner EJJ, Hunter CA, Willett P. Structural Fingerprints of Transcription Factor Binding Site Regions. Algorithms. 2009; 2(1):448-469. https://doi.org/10.3390/a2010448
Chicago/Turabian StyleGardiner, Eleanor J. J., Christopher A. Hunter, and Peter Willett. 2009. "Structural Fingerprints of Transcription Factor Binding Site Regions" Algorithms 2, no. 1: 448-469. https://doi.org/10.3390/a2010448