
Transcriptome Sequencing and Comparative Analysis of Piptoporus betulinus in Response to Birch Sawdust Induction

1
Inner Mongolia Key Laboratory of Meadow Steppe Ecosystem and Global Change, College of Life and Environmental Science, Hulunbuir University, Inner Mongolia 021000, China
2
College of Life Science, Northeast Forestry University, Harbin 150040, China
3
Key Laboratory of Saline-alkali Vegetation Ecology Restoration in Oil Field (SAVER), Alkali Soil Natural Environmental Science Center (ASNESC), Northeast Forestry University, Harbin 150040, China
4
Key Laboratory of Vegetation Ecology, Ministry of Education, Institute of Grassland Science, Northeast Normal University, Changchun 130024, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Forests 2017, 8(10), 374; https://doi.org/10.3390/f8100374
Submission received: 18 August 2017
/
Revised: 25 September 2017
/
Accepted: 27 September 2017
/
Published: 7 October 2017
(This article belongs to the Special Issue Genetics and Genomics of Forest Trees)
Abstract
:Piptoporus betulinus, a brown-rot parasitic fungus of birch trees (Betula species), has been used as a common anti-parasitic and antibacterial agent. The lack of genetic resource data for P. betulinus has limited the exploration of this species. In this present study, we used Illumina Hiseq 2500 technology to examine the transcriptome assembly of P. betulinus in response to birch sawdust induction. By de novo assembly, 21,882 non-redundant unigenes were yielded, and 21,255 (97.1%) were annotated with known gene sequences. A total of 340 responsive unigenes were highly homologous with putative lignocellulose-degrading enzyme candidates. Additionally, 86 unigenes might be involved in the chemical reaction in xenobiotics biodegradation and metabolism, which suggests that this fungus could convert xenobiotic materials and has the potential ability to clean up environmental pollutants. To our knowledge, this was the first study on transcriptome sequencing and comparative analysis of P. betulinus, which provided a better understanding of molecular mechanisms underlying birch sawdust induction and identified lignocelluloses degrading enzymes.
1. Introduction
Piptoporus betulinus, a brown-rot parasitic fungus of birch trees (Betula species), has been used as a common anti-parasitic and antibacterial agent for the treatment of wounds and various diseases, such as cancer, inflammation and so on [1,2]. Its extract has been demonstrated to be effective in preventing fatigue, strengthen immunity and relieving pain. Previous findings have suggested that this fungus has the ability to degrade of wood components, including cellulose and hemicellulose [3,4,5].
Lignocellulose represents an abundant carbon-neutral renewable resource that can be used for the production of bioenergy and biomaterials. It could be efficiently converted into ethanol using fungi as biological pre-treatment [6]. Furthermore, published findings have shown that brown-rot fungi have the potential as a new biocatalyst with unprecedented fermentability and a microbial starter [7]. Moreover, brown-rot fungi primarily break down cellulose and hemicellulose with carbohydrate active enzymes (CAZy), which catalyze cellulose into 6-carbon sugars [8,9]. In addition, it could greatly balance the conversion system of bioethanol production without genetic engineering support or external hydrolase. Therefore, it was considered as a promoter in lignocellulose biodegradation.
Although P. betulinus has various potential properties, the gene sequences in the publicly available databases for this fungus are rare. Only about 78 nucleotide sequences of P. betulinus to date have been deposited in National Center for Biotechnology Information’s (NCBI) GenBank database, and most of them were isolated and cloned from ribosomal RNA genes [10,11]. In recent years, the lignocellulose-decaying transcriptomes of white-rot fungi have been thoroughly studied [12,13], while only few studies describing the transcriptomes of brown-rot fungi on wood have been reported [14,15]. This present study used Illumina Hiseq 2500 technology to examine the transcriptome of P. betulinus in response to birch sawdust induction. Transcriptome profiling of P. betulinus provides a better understanding of molecular mechanisms underlying birch sawdust induction and could identify lignocellulose-degrading enzymes in this species.
2. Materials and Methods
2.1. Fungal Strain
In August 2014, the fruiting body of the fungal strain was collected from Betula platyphylla at Liangshui Nature Reserve, Lesser Xing’an Mountains in Yichun city, Heilongjiang Province, China. The fruiting body was sterilized in sodium hypochlorite for 1 min, before being rinsed with sterile deionized water 3–5 times. This was cultured on potato dextrose agar plate and kept in the dark at 28 °C for 10 days. Following this, this strain was identified as P. betulinus according to Internal Transcribed Spacer (ITS) sequencing and alignment in NCBI (GenBank accession number MF967582).
2.2. RNA Extraction
The strain was inoculated into different liquid media (Potato dextrose broth (PDB) and PDB + birch sawdust (Betula platyphlla) (5 g/L)), which were cultured with continuous shaking (150 rpm) at 28 °C for 10 days. For each treatment (PDB and PDB + birch sawdust, respectively), the mycelia of P. betulinus were harvested by filtration, before being frozen in liquid nitrogen and prepared for RNA extraction. Total RNA was extracted using TRNzol reagent according to the manufacturer’s protocol (TIANGEN, Beijing, China). Total RNA was extracted from each sample in triplicate, before synthesized cDNA were pooled together for sequencing. A quantitative real-time PCR (qRT-PCR) using RNA samples as templates was performed to detect genomic DNA. Samples with no amplified qRT-PCR products were used as a template for cDNA synthesis. The extracted RNA yield and purity were checked by NanoDrop 2000 (Thermo Scientific, Hudson, NH, USA). The qualified RNA samples were used for cDNA synthesis using PrimeScript™ RT reagent Kit (TakaRa). Subsequently, cDNA fragments were selected for PCR amplification and cDNA library were used for sequence analysis via Illumina HiSeq™ 2500(Illumina, San Diego, CA, USA).
2.3. Transcriptome Analysis
Raw reads were cleaned by removing adaptor sequences, short sequences and low-quality reads (reads containing Ns >5). The remaining clean reads were assembled into unigenes using short reads assembling program, which is also known as SOAPdenovo [16]. TIGR Gene Indices clustering tool (TGICL) was used to acquire a set of non-redundant unigenes [17]. After this, all the non-redundant unigenes were used for blast search (E-value < 10−5 or 10−3) and annotation in various databases, including NCBI Nr database, SwissProt database, Kyoto Encyclopedia of Genes and Genomes (KEGG) database and Cluster of Orthologous Groups (COG) database. For the functional annotation, gene ontology (GO) terms were analyzed using the Blast2GO program [18]. Finally, Web Gene Ontology Annotation Plot (WEGO) was used to classify GO function for all unigenes [19].
A differential expression analysis between two treatments was performed using the DESeq2 R package (http://www.bioconductor.org/package/DESeq2/). The false discovery rate (FDR) control method was applied in the Benjamini and Hochberg method to correct the results for p-values. An FDR <0.01 and FC (fold change) ≥2 was set as the threshold to determine the significance of gene expression differences. The fragments per kilo bases per million reads (FPKM) were used to evaluate expressed values and quantify transcript levels, which normalizes and eliminates the influence of gene length and sequencing depth in calculating gene expression, allowing for direct comparison of gene expression between different treatments [20]. Genes with an adjusted p-value < 0.05 was assigned as differentially expressed according to the reports of Deng et al. [21]. For pathway enrichment analysis, all differentially expressed unigenes were submitted in the online KEGG Automatic Annotation Server (http://www.genome.jp/kegg/) and we searched for significantly enriched KEGG terms to obtain the pathway annotation (adjusted p-value < 0.05).
2.4. Quantitative Real-Time PCR Validation
A total of 12 representative birch sawdust induced-relevant unigenes (carbohydrate-active enzyme genes and lignocellulose-degrading enzyme genes) with significantly differential expression (adjusted p-value < 0.05) identified by RNA-seq were chosen for experimental validation using qRT-PCR with gene specific primers (Table S1). The qRT-PCR was performed using SYBR® Premix Ex Taq™ II Kit (Tli RNaseH Plus) (TakaRa, Tokyo, Japan) in a volume of 20 μL, which contained 10 μL of SYBR Premix Ex Taq (2×), 0.4 μL of ROX Reference Dye II (10×), 2 μL of cDNA template and 0.5 μM of each primer. The amplification was performed as follows: 95 °C for 30 s, 45 cycles of 95 °C for 5 s and 60 °C for 40 s. The qRT-PCR amplifications were conducted in an ABI 7500 detection system (Applied Biosystems, Carlsbad, CA, USA). All samples and reactions were performed in triplicate and the results were expressed relative to the expression levels of actin in each gene by using the 2−△△Ct method [22].
2.5. Availability of Data
All transcriptome data files were submitted to the Sequence Read Archive database with the accession number SRP117136.
3. Results
3.1. Raw Reads Processing and de novo Assembly
For a better understanding of molecular mechanisms underlying birch sawdust induction and comparative transcriptome analysis in P. betulinus, two cDNA treatments prepared from PDB media and PDB + sawdust were sequenced with Illumina HiSeq™ 2500. An overview of the sequencing and assembly is given in Table 1 and Table S2. After filtering adapters and short sequences or low-quality bases, a total of 94,942,136 clean reads with 9,510,783,105 nucleotides were obtained in the two treatments (Table S2). Finally, de novo assembly yielded 21,882 non-redundant unigenes with an average length of 1949 bp and an N50 of 3007 bp (Table 1). Of these unigenes, 17,081 (88.1%) were >500 bp and 10,670 (48.8%) were >1500 bp. These obtained sequences provided abundant information on transcriptomes for further analysis of the birch sawdust-induced genes in P. betulinus.
3.2. Functional Annotation
To identify the putative functions of unigenes in P. betulinus, we annotated all the assembled unigenes against the Nr, Swiss-port, KEGG and COG databases. With comparison against those four databases, a total of 21,255 (97.1%) unigenes were successfully annotated with known gene sequences. The number of unigenes with significant similarity to sequences in Nr, Swissport, KEGG, and COG databases were 21,255 (97.1%), 3351 (15.3%), 6256 (28.6%) and 7955 (36.4%), respectively (Table 2).
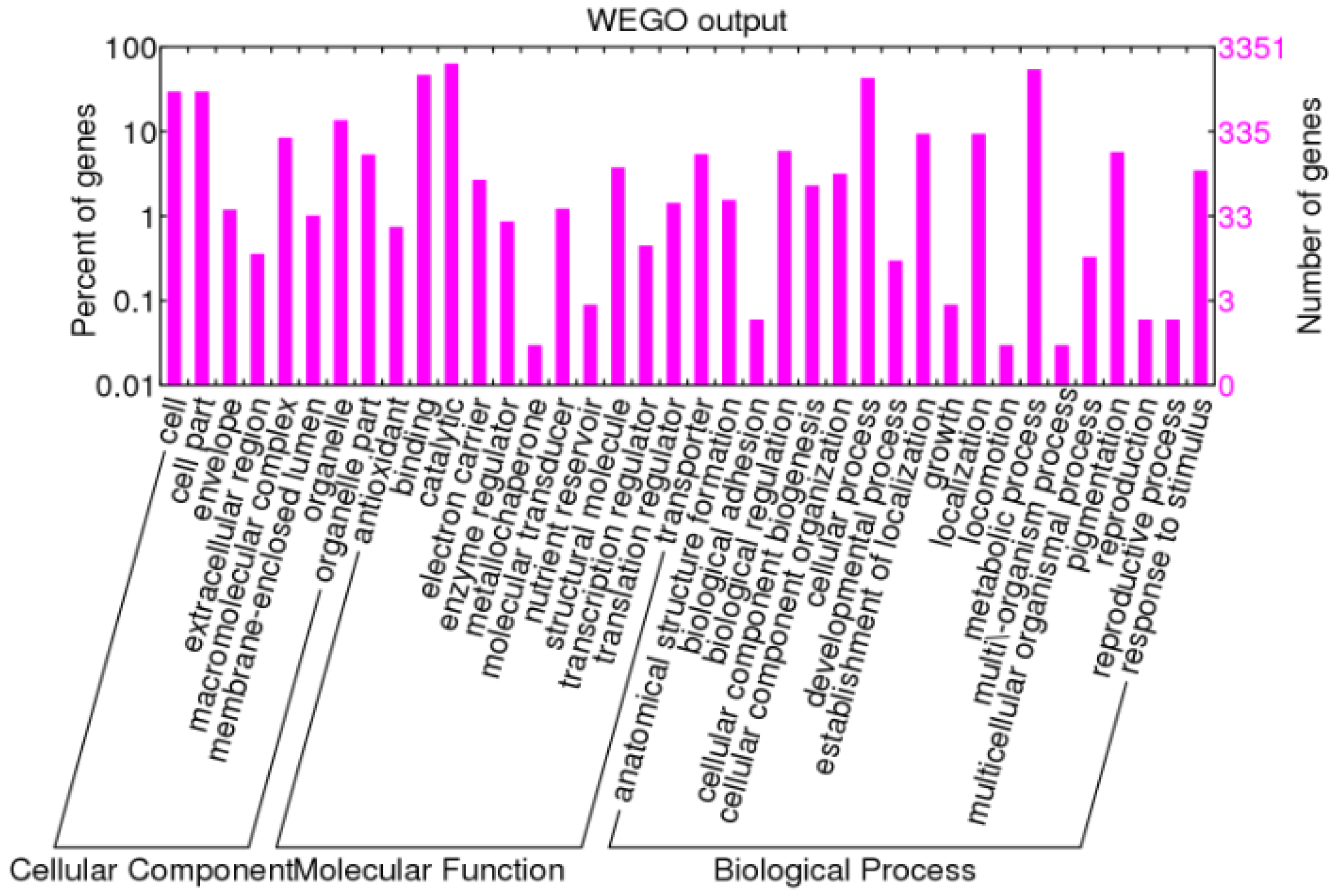
The GO analysis classified the functions of predicted unigenes into three main categories: biological processes, molecular function and cellular components (Figure 1). A total of 3351 sequences were assigned with 9111 GO terms, among which 2000 unigenes (22.0%) were assigned at least one GO term in the biological processes, 4253 (46.7%) in the cellular components and 4658 (51.1%) in molecular functions. Additionally, these unigenes were further classified into functional subcategories. In cellular components, the largest subcategory was cells (29.8%) and the second largest was cell parts (13.6%). Regarding molecular function, the largest number were found in catalytic activity (63.5%) and binding (46.8%). According to biological processes, genes involved in metabolic process and cellular process were highly represented, which accounted for 54.3% and 42.9% of the matched unigenes in the subcategory, respectively.
The genes belonging to the same biological pathway synergistically participated to accomplish the biological functions. The KEGG database was used to identify and predict the metabolism pathways in P. betulinus. Details of sequences involved in KEGG annotation are listed in Table S3. In KEGG metabolic pathways, a total of 6256 unigenes were matched in 309 different KEGG pathways (Table S3). Among these pathways, the most common was amino acid metabolic pathways with 240 members, followed by ribosome biogenesis (202) and energy metabolism (197). These might be involved in maintaining the basic metabolic process of P. betulinus.
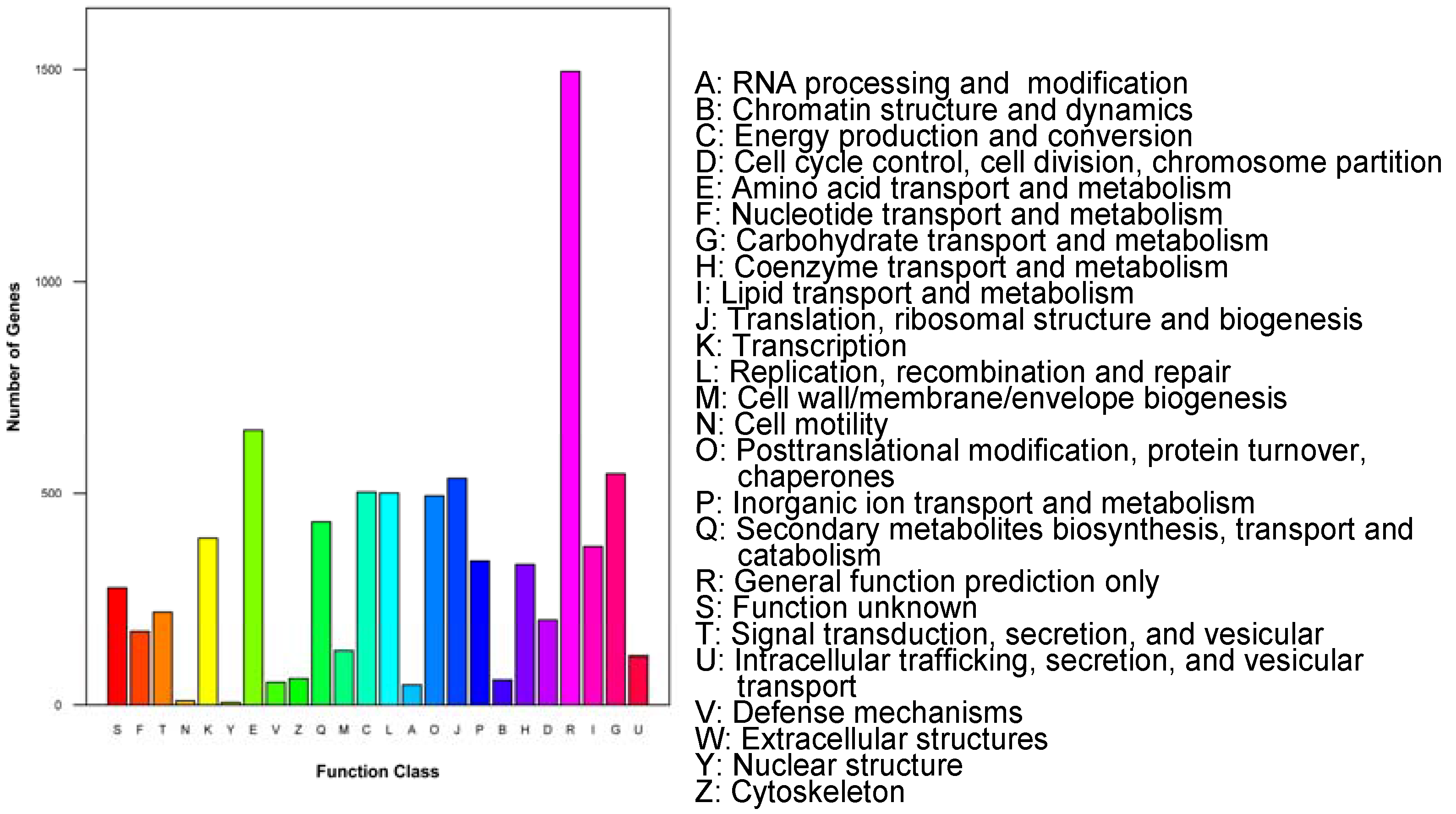
In addition, in COG classification analysis, out of 21,882 Nr hits, 7955 unigenes were grouped into 25 COG categories. Among them, the prediction of the general function was the most populated group (18.8%), followed by amino acid transport and metabolism (8.16%), and carbohydrate transport and metabolism (6.9%). Two hundred and seventy-six unigenes were mapped into unknown functions, which might be involved in a specific gene in P. betulinus. The smallest groups were found in cell wall/membrane/envelope biogenesis and nuclear structures (Figure 2).
Based on GO classification, KEGG pathway and COG annotation, the catalytic function accounted for the highest percentage in the molecular functions, while amino acid transport and metabolism, carbohydrate transport and metabolism were the predominant categories. Therefore, further analysis was needed with specific focus on those pathways.
3.3. Changes in Gene Expression under Sawdust Induction
To characterize the differences of molecular response to the PDB (control) and PDB + birch sawdust medias, unigene expression levels were calculated by the FPKM method. Based on FPKM values, 776 unigenes were identified as differentially expressed genes (Table S4). Among them, 444 genes were up-regulated and 332 were down-regulated, which were identified by DESeq2 (http://www.bioconductor.org/package/DESeq2/) with adjusted p-value ≤ 0.05 and fold change value >2. Following this, all differentially expressed genes were mapped into 26 different metabolic pathway categories (Table S5). Of these annotated pathways, phenylpropanoid biosynthesis, starch and sucrose metabolism and lysosome were the main pathways that accounted for 7.8%, 7.3% and 6.4%, respectively. Notably, the pathway enrichment analysis revealed that phenylpropanoid biosynthesis as well as starch and sucrose metabolism were the important pathways in the degradation of wood components by P. betulinus.
3.4. Detection of Lignocellulose-Degrading Enzyme-Relevant Gene Sequences and qRT-PCR Analysis
For further insight into birch sawdust induction in P. betulinus, we detected and analyzed induction-related genes in this study. These matched enzymes were mainly involved in the degradation of cellulose, hemicellulose and starch. There were 340 highly homologous unigenes in putative wood-degrading enzymes (Table 3 and Table S6). Among them, 109 unigenes were identified as glycoside hydrolases (GHs), 37 belonged to glycosyl transferases (GTs), while 12 and 13 genes were assigned to carbohydrate esterases (CEs) and polysaccharide lyases (PLs), respectively. Fifty-three unigenes shared a similar identity to glucosidase, among which 52 homologs belonged to beta-glucosidase. Twenty-three unigenes were similar to 3-(3-hydroxy-phenyl) propionate hydroxylase, 20 unigenes to chitinase, 16 unigenes to mannosidase, 11 unigenes to polygalacturonase, 9 unigenes to mannosidase, 9 unigenes to pectinesterase.
Several putative unigenes were involved in xenobiotic biodegradation and metabolism (Table S6). Enzymes involved in chloroalkane and chloroalkene degradation were S-(hydroxymethyl) glutathione dehydrogenase/alcohol dehydrogenase and aldehyde dehydrogenase with 7 and 41 unigenes, respectively [23]. One unigene was identified as carboxymethylenebutenolidase, which catalyzed the chemical reaction in toluene and chlorobenzene degradation [24]. Two unigenes, 1 and 23 unigenes, were found to exhibit a striking homology to amidase, nitrilase and 3-(3-hydroxy-phenyl) propionate hydroxylase, respectively. These above enzymes were related to styrene degradation. Alcohol dehydrogenase, which mainly degrade naphthalene compounds, were detected in P. betulinus transcriptome with 7 unigenes. Four unigenes belonged to aflatoxin B1 aldehyde reductase, which regulated the metabolism of xenobiotics by cytochrome P450.
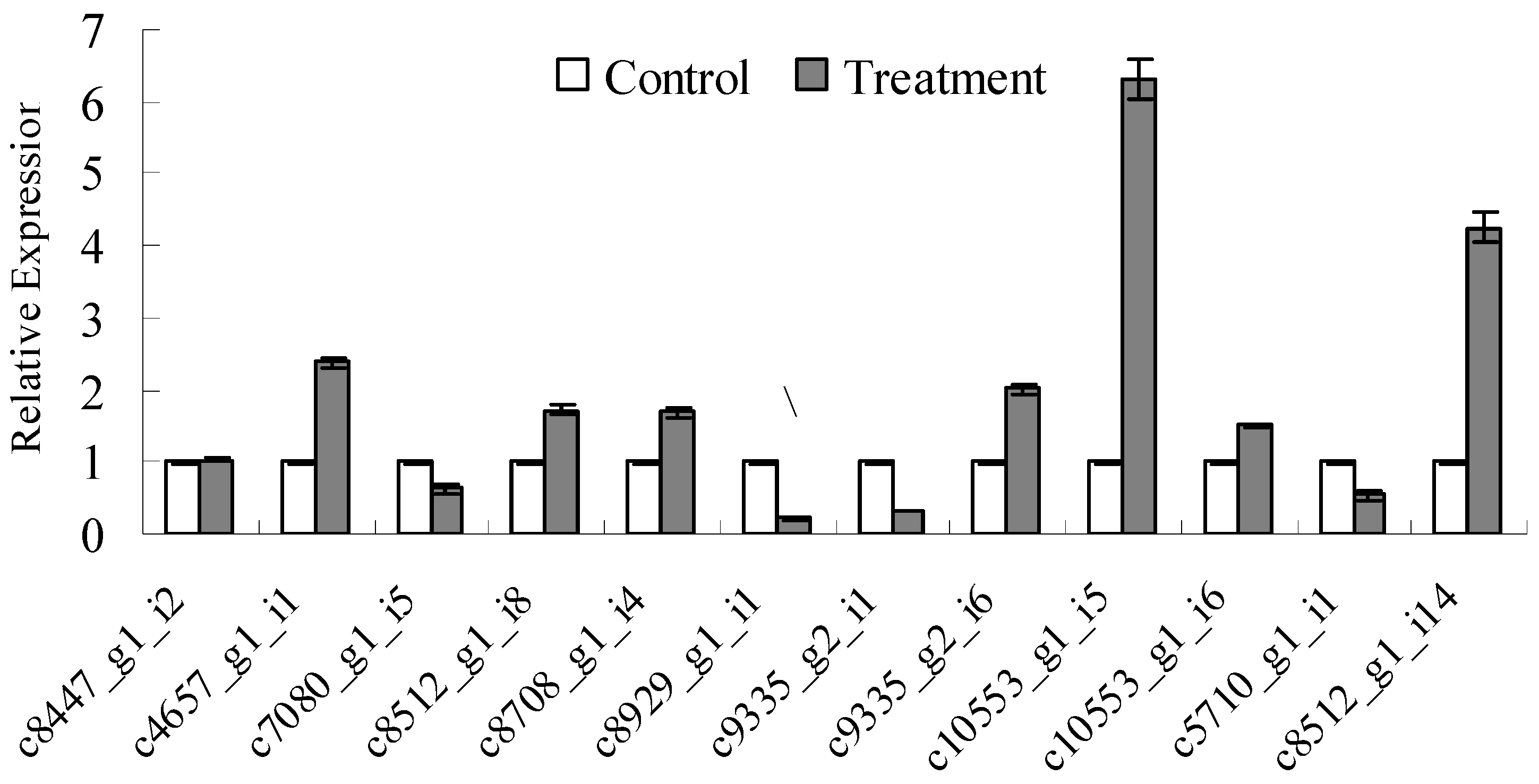
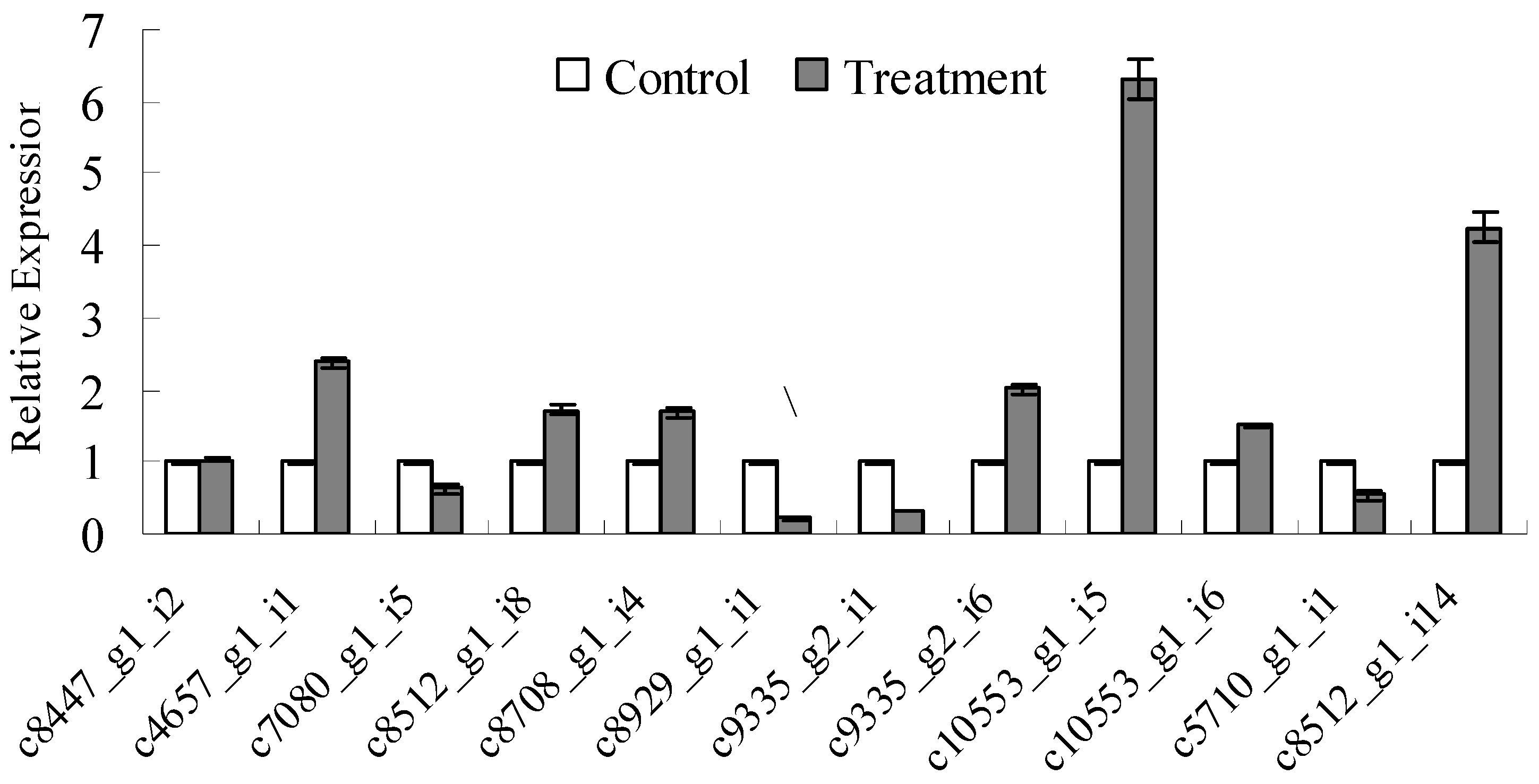
To verify that the unigenes from sequencing were indeed differentially expressed genes and to analyze the difference of gene expression profile between control samples and induced samples, twelve unigenes related to lignocellulose-degrading enzyme-encoding genes were selected for qRT-PCR analysis (Figure 3). The results showed that 8 unigenes were up-regulated and 4 unigenes were down-regulated. Interestingly, the expression of c10553_g1_i5 and c10553_g1_i6 (mannosidase, EC: 3.2.1.21) under sawdust-induced samples increased by 6.3-fold and 1.5-fold compared to controls, suggesting that most of mannosidases were induced by sawdust in the lignocellulose-degrading process. Similarly, the expression of pectinesterase (c8512_g1_i14 and c8512_g1_i8) was also increased after being cultivated in birch sawdust. Several lignocellulose-degrading enzymes were detected to be down-regulated, including c7080_g1_i5 and c8929_g1_i1 (polygalacturonase); as well as c9335_g2_i1 and c5710_g1_i1 [3-(3-hydroxy-phenyl) propionate hydroxylase]. Therefore, those enzymes were jointly involved in the degradation of lignocellulose.
4. Discussion
Transcriptomics, an important part of functional genomics, clarify the genetic transcription of cells at the overall level [25]. Data obtained by Illumina high-throughput sequencing has many advantages due to its large storage size, high efficiency and low cost, which is suitable for a species that has not had its whole genome sequenced. In this study, we firstly sequenced the P. betulinus transcriptome using Illumina Hiseq 2500 technology. After de novo assembly, 21,882 non-redundant unigenes were obtained, among which 21,255 were annotated into known functions involved in the lignocellulose-degrading process. These findings showed that a considerable number of genes were successfully identified through high-throughput sequencing and would be useful for the further functional analysis.
Based on KEGG pathway database, 6256 unigenes were mapped into 309 types of enzyme-encoding genes. Among them, many unigenes participated in degradation of wood components, which would provide a better platform for cloning of cellulose-degrading genes and related functional validation. The KEGG pathway results showed that amino acid transport and metabolism, carbohydrate transport and metabolism as the predominant pathways were closely associated with cellulose and hemicellulose degradation.
Annotation analysis of unigenes in the P. betulinus transcriptome showed that 340 transcripts were identified as putative wood-degrading genes, which is less than other brown-rot fungi [26,27]. Additionally, 53 unigenes were found to be similar to glucosidase, demonstrating that P. betulinus could degrade wood components. Interestingly, 2 unigenes were identified as pectinesterase in our transcriptome, which is comparable with other brown-rot fungi [28] and suggests that this enzyme was relatively conservative. Although we found that 20 unigenes had homology with chitinase, further confirmation of its ability to degrade chitin is required.
Lignin biodegradation was mainly detected in white-rot fungi, which produces multiple isoenzymes of lignin peroxidase, manganese peroxidase and laccase [29]. Although P. betulinus is unable to degrade lignin, it could secrete enzymes which may modify lignin or break lignin seal by the methylation pathway and sufficiently expose cellulose and hemicellulose for enzymatic action. Recently, most of the studies were focused on Postia placenta, such as genome, transcriptome, secretome analysis and nuclear magnetic resonance analysis [27,30]. Martinez et al. first reported transcriptional profile of brown-rot fungus P. placenta, and their results revealed that this fungus possessed unique extracellular enzyme systems, including an unusual repertoire of glycoside hydrolases, while exocellobiohydrolases and cellulose-binding domains were absent in this fungus [27]. When P. placenta was grown in medium containing cellulose as the sole carbon source, transcripts corresponding to many hemicellulases and endoglucanases were expressed at high levels compared to the glucose-grown culture [31]. It is well known that although brown-rot and white-rot fungi are both basidiomycetes, white-rot fungi can produce complex ligninolytic systems to degrade lignin [32]. The different degradation pathway mechanisms in these two fungi have attracted the attention of scientists. However, lignin peroxidase, manganese peroxidase, versatile peroxidase and laccase were not detected in the P. placenta genome [31], suggesting that this fungus could not induce lignin depolymerization, which was consistent with our finding.
In addition, several metabolic pathways might be involved in polycyclic aromatic hydrocarbon degradation, including phenylpropanoid biosynthesis, polycyclic aromatic hydrocarbon degradation, limonene and pinene degradation. However, in the past published papers, P. betulinus was thought to modify the structure of lignin instead of degrading lignin, which involve complex phenolic polymers composed of cinnamyl alcohol subunits, phenylpropanol and its derivatives [33]. Therefore, our paper speculated that those metabolic pathways in P. betulinus transcriptome were likely to participate in lignin modification. Importantly, we also found 8 potential wood-degrading related genes consisting of 3 laccases, 1 cellobiose dehydrogenase and 4 glucose oxidases. This indicated that P. betulinus could efficiently degrade wood components (Table S6), which is consistent with a previous report [34]. Although we detected wood-degrading enzymes encoding genes in transcriptome, enzymatic activity should be measured in a future experiment to confirm its enzyme activity level.
Eighty-six unigenes were matched with xenobiotics biodegradation and metabolism, including chloroalkane and chloroalkene degradation, toluene and chlorobenzene degradation, styrene degradation, naphthalene degradation and metabolism of xenobiotics by cytochrome P450. Prince and Roger [35] reported bioremediation using microbiological processes to clean-up organic and inorganic environmental pollutants by chloroalkane and chloroalkene degradation. Published findings have evaluated the effect of white-rot fungi on removal of gaseous chlorobenzene and naphthalene, while no reports have been published regarding this degradation using brown-rot fungi. Therefore, our next experiment would determine the ability of gaseous chlorobenzene or naphthalene in P. betulinus [36,37,38]. The findings of Krueger et al. [38] indicated that brown-rot fungus Gloeophyllum trabeum might be suitable for the biodegradation of styrene. Although white-rot fungi degrading xenobiotic compounds was originally attributed to its lignin-degrading enzyme system, Ide et al. [39] suggested that cytochrome P450 showed a novel catalytic property in xenobiotic metabolism. All annotated enzymes or degradation processes could be found in P. betulinus transcriptome, demonstrating that P. betulinus could not only harbor metabolic xenobiotic materials but also potentially degradation of environmental pollutants.
5. Conclusions
To our knowledge, this was the first study to describe the changes in transcriptome of brown-rot fungus P. betulinus when growing on birch saw dust. After an in-depth RNA-seq analysis, we obtained and annotated 3351 assembled unigenes to at least one database. A large number of potential lignicellulose-degrading enzyme-relevant genes were identified, suggesting that this species is suitable for wood degradation. In summary, our analysis results obtained from the transcriptome data provide an insight into the genetic background of brown-rot fungus P. betulinus.
Supplementary Materials
The following are available online at www.mdpi.com/1999-4907/8/10/374/s1, Table S1: The primers designed for qRT-PCR analysis, Table S2: Overview of output statistics on P. betulinus transcriptome sequencing, Table S3: The top 20 representative metabolic pathways of P. betulunus, Table S4: The list of significantly differently expressed genes, Table S5: The metabolic pathway of differently expressed genes in samples, Table S6: All the assembled unigenes functionally annotated in Nr database.
Acknowledgments
This study was supported by the National Natural Science Fund Project (31700410), Inner Mongolia Natural Science Fund Project (2016BS0303), Inner Mongolia Science and Technology Research Project of Higher Education (NJZY16298), the Fundamental Research Funds for the Central Universities (2572017AA23), the National Natural Science Foundation of China (31170568).
Author Contributions
Qiuyu Wang conceived and designed the experiments; Mu Peng and Syed Sadaqat Shah performed the experiments and analyzed the data; Lixia Yang wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lemieszek, M.; Langner, E.; Kaczor, J.; Kandefer-Szerszen, M.; Sanecka, B.; Mazurkiewicz, W.; Rzeski, W. Anticancer effect of fraction isolated from medicinal birch polypore mushroom, Piptoporus betulinus (bull.: Fr.) p. Karst. (aphyllophoromycetideae): In Vitro studies. Int. J. Med. Mushrooms 2009, 11, 351–364. [Google Scholar] [CrossRef]
- Wangun, H.V.K.; Berg, A.; Hertel, W.; Nkengfack, A.E.; Hertweck, C. Anti-inflammatory and anti-hyaluronate lyase activities of lanostanoids from Piptoporus betulinus. J. Antibiot. 2004, 57, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Liu, X.; Wang, Q. Identification of wood decay related genes from Piptoporus betulinus (bull. Fr.) karsten using differential display reverse transcription pcr (ddrt-pcr). Biotechnol. Biotechnol. Equip. 2012, 26, 2961–2965. [Google Scholar] [CrossRef]
- Shang, J.; Yan, S.; Wang, Q. Degradation mechanism and chemical component changes in Betula platyphylla wood by wood-rot fungi. BioResources 2013, 8, 6066–6077. [Google Scholar] [CrossRef]
- Wymelenberg, A.V.; Gaskell, J.; Mozuch, M.; Bondurant, S.S.; Sabat, G.; Ralph, J.; Skyba, O.; Mansfield, S.D.; Blanchette, R.A.; Grigoriev, I.V.; Kersten, P.J.; Cullen, D. Significant alteration of gene expression in wood decay fungi Postia placenta and Phanerochaete chrysosporium by plant species. Appl. Environ. Microbiol. 2011, 77, 4499–4507. [Google Scholar] [CrossRef] [PubMed]
- Klinke, H.B.; Thomsen, A.B.; Ahring, B.K. Inhibition of ethanol-producing yeast and bacteria by degradation products produced during pre-treatment of biomass. Appl. Microbiol. Biotechnol. 2004, 66, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kanawaku, R.; Masumoto, M.; Yanase, H. Efficient xylose fermentation by the brown rot fungus Neolentinus lepideus. Enzym. Microb. Technol. 2012, 50, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Dashtban, M.; Schraft, H.; Qin, W. Fungal bioconversion of lignocellulosic residues; opportunities & perspectives. Int. J. Biol. Sci. 2009, 5, 578–595. [Google Scholar] [PubMed]
- Canam, T.; Town, J.; Iroba, K.; Tabil, L.; Dumonceaux, T. Pretreatment of lignocellulosic biomass using microorganisms: Approaches, advantages, and limitations. Sustain. Degrad. Lignocellul. Biomass-Techn. Appl. Commer. 2013. [Google Scholar] [CrossRef]
- Zeng, X.X.; Tang, J.X.; Yin, H.Q.; Liu, X.D.; Pei, J.; Liu, H.W. Isolation, identification and cadmium adsorption of a high cadmium-resistant Paecilomyces lilacinus. Afr. J. Biotechnol. 2010, 9, 6525–6533. [Google Scholar]
- Kim, S.Y.; Park, S.Y.; Ko, K.S.; Jung, H.S. Phylogenetic analysis of antrodia and related taxa based on partial mitochondrial ssu rDNA sequences. Antonie Leeuwenhoek 2003, 83, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Korripally, P.; Hunt, C.G.; Houtman, C.J.; Jones, D.C.; Kitin, P.J.; Cullen, D.; Hammel, K.E. Regulation of gene expression during the onset of ligninolytic oxidation by Phanerochaete chrysosporium on spruce wood. Appl. Environ. Microbiol. 2015, 81, 7802–7812. [Google Scholar] [CrossRef] [PubMed]
- Kuuskeri, J.; Häkkinen, M.; Laine, P.; Smolander, O.P.; Tamene, F.; Miettinen, S.; Nousiainen, P.; Kemell, M.; Auvinen, P.; Lundell, T. Time-scale dynamics of proteome and transcriptome of the white-rot fungus Phlebia radiata: Growth on spruce wood and decay effect on lignocellulose. Biotechnol. Biofuels 2016, 9, 192. [Google Scholar] [CrossRef] [PubMed]
- Gaskell, J.; Blanchette, R.A.; Stewart, P.E.; Bondurant, S.; Adams, M.; Sabat, G.; Kersten, P.; Cullen, D. Transcriptome and secretome analyses of the wood decay fungus Wolfiporia cocos support alternative mechanisms of lignocellulose conversion. Appl. Environ. Microbiol. 2016, 82, 3979–3987. [Google Scholar] [CrossRef] [PubMed]
- Skyba, O.; Cullen, D.; Douglas, C.J.; Mansfield, D. Gene expression patterns of wood decay fungi Postia placenta and Phanerochaete chrysosporium are influenced by wood substrate composition during degradation. Appl. Environ. Microbiol. 2016, 82, 4387–4400. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B. Tigr gene indices clustering tools (TGICL): A software system for fast clustering of large est datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2Go: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L. Wego: A web tool for plotting go annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with tophat and cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Yao, J.; Wang, X.; Guo, H.; Duan, D. Transcriptome sequencing and comparative analysis of Saccharina japonica (laminariales, phaeophyceae) under blue light induction. PLoS ONE 2012, 7, e39704. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔδCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Cong, B.; Wang, N.; Liu, S.; Liu, F.; Yin, X.; Shen, J. Isolation, characterization and transcriptome analysis of a novel Antarctic Aspergillus sydowii strain MS-19 as a potential lignocellulosic enzyme source. BMC Microbiol. 2017, 17, 129. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.; Knackmuss, H.J. Chemical structure and biodegradability of halogenated aromatic compounds. Conversion of chlorinated muconic acids into maleoylacetic acid. Biochem. J. 1980, 192, 339. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, S.C.; Coemans, B.; Podevin, N.; Laukens, K.; Witters, E.; Matsumura, H. Functional genomics in a non-model crop: Transcriptomics or proteomics? Physiol. Plant. 2008, 133, 117. [Google Scholar] [CrossRef] [PubMed]
- Hori, C.; Gaskell, J.; Igarashi, K.; Samejima, M.; Hibbett, D.; Henrissat, B.; Cullen, D. Genomewide analysis of polysaccharides degrading enzymes in 11 white-and brown-rot Polyporales provides insight into mechanisms of wood decay. Mycologia 2013, 105, 1412–1427. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.; Challacombe, J.; Morgenstern, I.; Hibbett, D.; Schmoll, M.; Kubicek, C.P.; Ferreira, P.; Ruiz-Duenas, F.J.; Martinez, A.T.; Kersten, P. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc. Natl. Acad. Sci. USA 2009, 106, 1954–1959. [Google Scholar] [CrossRef] [PubMed]
- Rytioja, J.; Hildén, K.; Yuzon, J.; Hatakka, A.; de Vries, R.P.; Mäkelä, M.R. Plant-polysaccharide-degrading enzymes from basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef] [PubMed]
- Yelle, D.J.; Wei, D.; John, R.; Hammel, K.E. Multidimensional nmr analysis reveals truncated lignin structures in wood decayed by the brown rot basidiomycete Postia placenta. Environ. Microbiol. 2011, 13, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Presley, G.N.; Hammel, K.E.; Ryu, J.S.; Menke, J.R.; Figueroa, M.; Hu, D.; Orr, G.; Schilling, J.S. Localizing gene regulation reveals a staggered wood decay mechanism for the brown rot fungus Postia placenta. Proc. Natl. Acad. Sci. USA 2016, 113, 10968. [Google Scholar] [CrossRef] [PubMed]
- Tuor, U.; Winterhalter, K.; Fiechter, A. Enzymes of white-rot fungi involved in lignin degradation and ecological determinants for wood decay. J. Biotechnol. 1995, 41, 1–17. [Google Scholar] [CrossRef]
- Lee, D.; Meyer, K.; Chapple, C.; Douglas, C.J. Antisense suppression of 4-coumarate:Coenzyme a ligase activity in Arabidopsis leads to altered lignin subunit composition. Plant Cell 1997, 9, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- An, H.D.; Wei, D.S.; Xiao, T.T. Transcriptional profiles of laccase genes in the brown rot fungus Postia placenta mad-r-698. J. Microbiol. 2015, 53, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Prince, R.C. Bioremediation: An overview of how microbiological processes can be applied to the cleanup of organic and inorganic environmental pollutants. In Encyclopedia of Environmental Microbiology; John Wiley & Sons, lnc.: Hoboken, NJ, USA, 2003. [Google Scholar]
- Wang, C.; Jin-Ying, X.I.; Hong-Ying, H.U.; Wen, X.H. Biodegradation of gaseous chlorobenzene by white-rot fungus Phanerochaete chrysosporium. Biomed. Environ. Sci. 2008, 21, 474–478. [Google Scholar] [CrossRef]
- Hadibarata, T.; Teh, Z.C.; Rubiyatno; Zubir, M.M.; Khudhair, A.B.; Yusoff, A.R.; Salim, M.R.; Hidayat, T. Identification of naphthalene metabolism by white rot fungus Pleurotus eryngii. Bioprocess Biosyst. Eng. 2013, 36, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Hadibarata, T.; Yusoff, A.R.M.; Aris, A.; Kristanti, R.A. Identification of naphthalene metabolism by white rot fungus Armillaria sp.F022. J. Environ. Sci. 2012, 24, 728–732. [Google Scholar] [CrossRef]
- Krueger, M.C.; Hofmann, U.; Moeder, M.; Schlosser, D. Potential of wood-rotting fungi to attack polystyrene sulfonate and its depolymerisation by Gloeophyllum trabeum via hydroquinone-driven fenton chemistry. PLoS ONE 2015, 10, e0131773. [Google Scholar] [CrossRef] [PubMed]
- Ide, M.; Ichinose, H.; Wariishi, H. Molecular identification and functional characterization of cytochrome P450 monooxygenases from the brown-rot basidiomycete Postia placenta. Arch. Microbiol. 2012, 194, 243–253. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Functional annotation of non-redundant unigenes based on Gene Ontology (GO) classification. The results are summarized in three functional categories: cellular components, molecular function and biological process.
Figure 1.
Functional annotation of non-redundant unigenes based on Gene Ontology (GO) classification. The results are summarized in three functional categories: cellular components, molecular function and biological process.
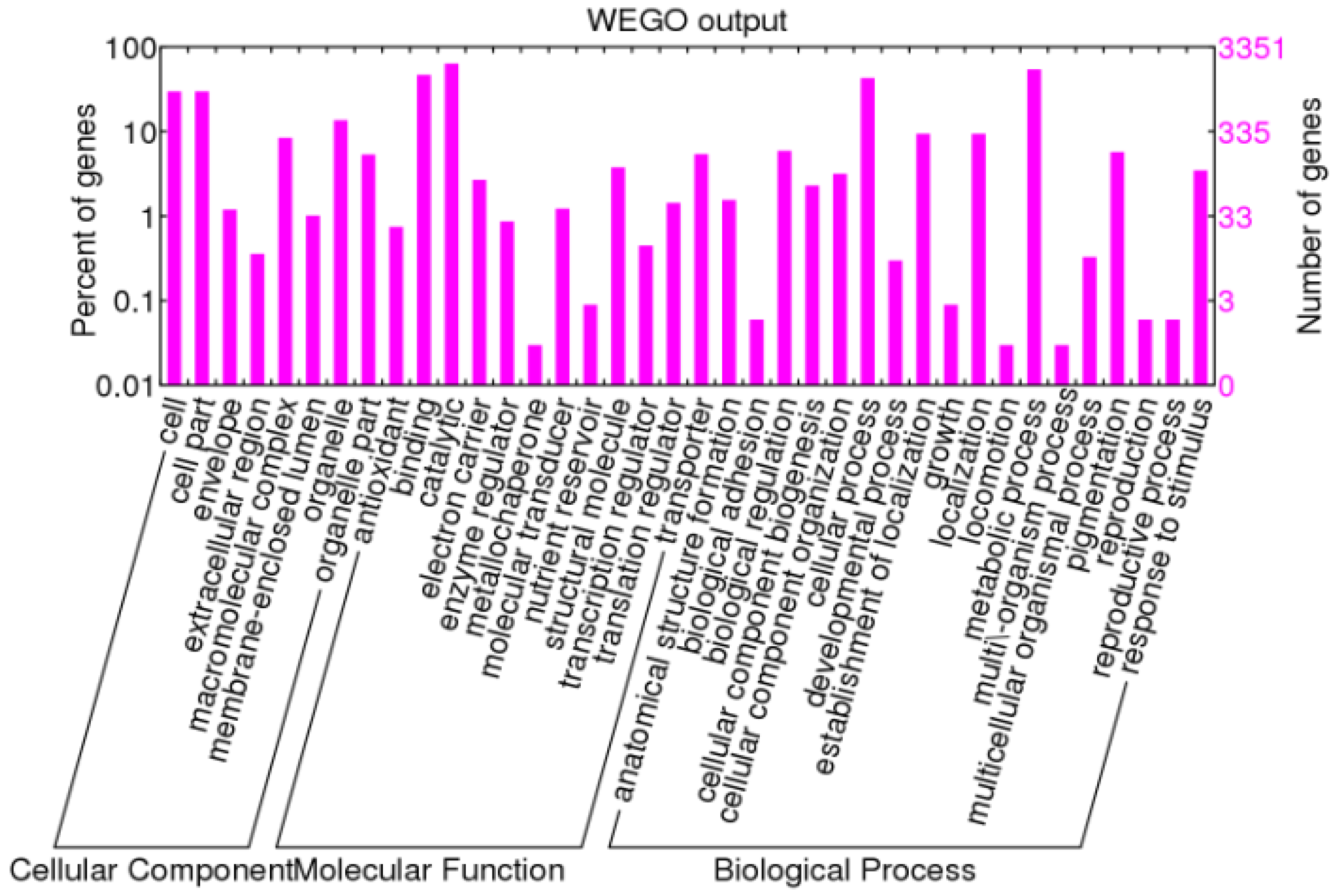
Figure 2.
Histogram presentation of clusters of orthologous groups (COG) classification (7955 sequences have a COG classification among the 25 categories).
Figure 2.
Histogram presentation of clusters of orthologous groups (COG) classification (7955 sequences have a COG classification among the 25 categories).
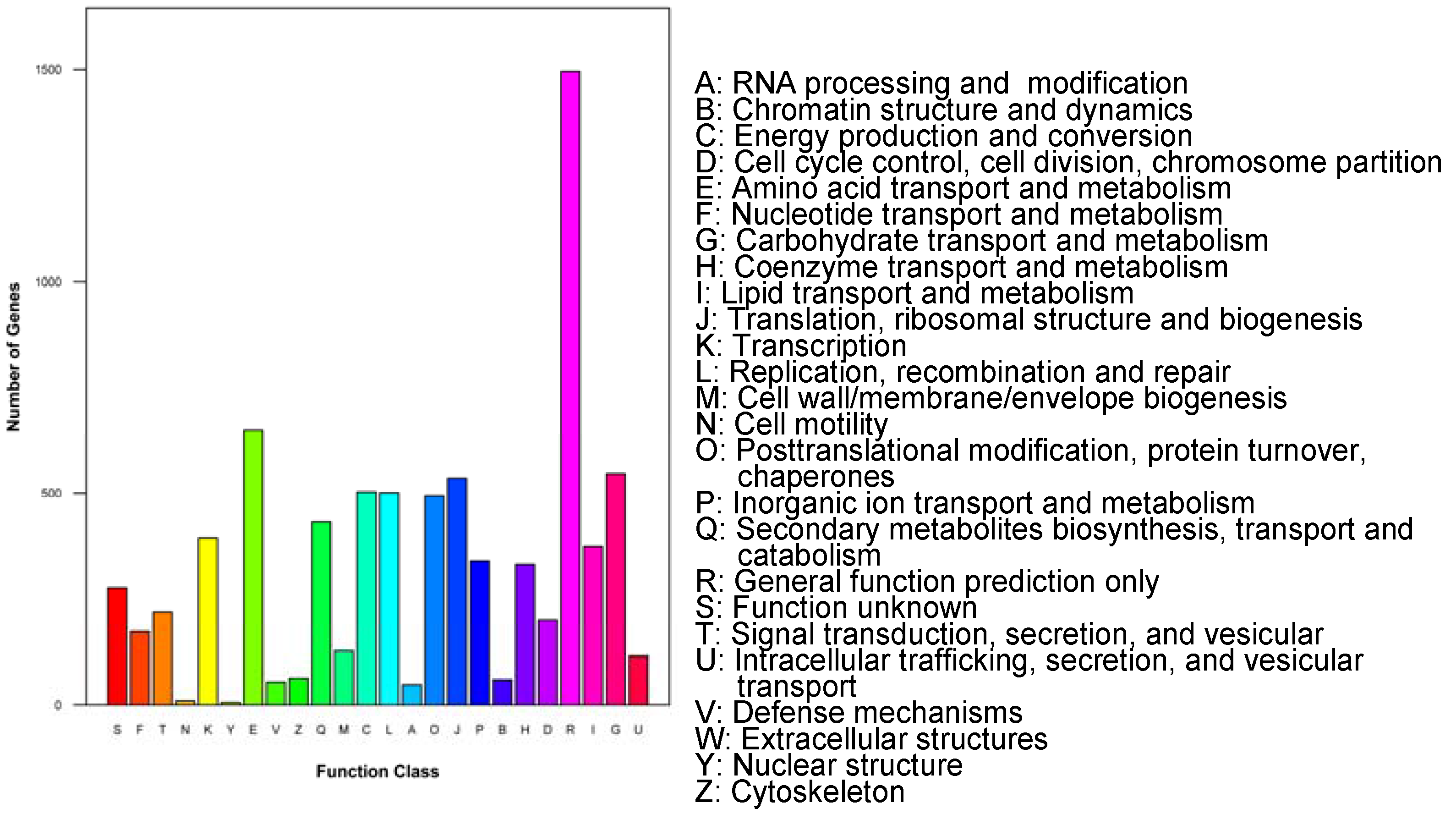
Figure 3.
Quantitative real-time PCR validation of differentially expressed unigenes in the P. betulinus transcriptome. All reactions were performed in biological triplicate, and the error bars represent the standard deviations. This figure included c8447_g1_i2, c9355_g2_i6 β-d-Glucosidase; c10553_g1_i6, c10553_g1_i5,β-mannosidase; c8512_g1_i14, c8512_g1_i8, pectinesterase c4657_g1_i1, c8708_g1_i4, α-glycosidase; c7080_g1_i5, c8929_g1_i1,polygalacturonase; c9335_g2_i1 and c5710_g1_i1 [3-(3-hydroxy-phenyl) propionate hydroxylase].
Figure 3.
Quantitative real-time PCR validation of differentially expressed unigenes in the P. betulinus transcriptome. All reactions were performed in biological triplicate, and the error bars represent the standard deviations. This figure included c8447_g1_i2, c9355_g2_i6 β-d-Glucosidase; c10553_g1_i6, c10553_g1_i5,β-mannosidase; c8512_g1_i14, c8512_g1_i8, pectinesterase c4657_g1_i1, c8708_g1_i4, α-glycosidase; c7080_g1_i5, c8929_g1_i1,polygalacturonase; c9335_g2_i1 and c5710_g1_i1 [3-(3-hydroxy-phenyl) propionate hydroxylase].

{kind=link}
{kind=link}
{kind=link}
Table 1.
Overview of transcriptome sequencing and assembly for P. betulinus.
| Length (bp) | Total Number | Percentage (%) |
|---|---|---|
| <200 | 0 | 0.0 |
| 200–500 | 4800 | 21.9 |
| 500–1000 | 3362 | 15.4 |
| 1000–1500 | 3049 | 23.9 |
| 1500–2000 | 2626 | 12.0 |
| ≥2000 | 8044 | 36.8 |
| Total | 21,882 | |
| Total length of all unigenes | 42,652,276 | |
| Median length of all unigenes (N50) | 3007 | |
| Average length of all unigenes | 1949.17 | |
Table 2.
Annotation of non-redundant unigenes.
| Database | Number of Annotated Unigenes | Percentage of Annotated Unigenes |
|---|---|---|
| Nr | 21,255 | 97.1% |
| Swissport | 3351 | 15.3% |
| KEGG | 6256 | 28.6% |
| COG | 7955 | 36.4% |
Table 3.
Putative wood-degrading genes in the P. betulinus transcriptome.
| Enzymes | Number of Unigenes |
|---|---|
| glycoside hydrolases | 109 |
| glycosyl transferases | 37 |
| carbohydrate esterases | 12 |
| polysaccharide lyases | 13 |
| beta-glucosidase | 52 |
| alpha-glucosidase | 1 |
| mannosidase | 16 |
| glycosyltransferase | 4 |
| polygalacturonase | 11 |
| alpha-galactosidase | 7 |
| D-xylose 1-dehydrogenase (NADP) | 4 |
| N-glycosylase | 2 |
| Pectinesterase | 9 |
| 3-(3-hydroxy-phenyl)propionate hydroxylase | 22 |
| alpha-amylase | 5 |
| xylose isomerase | 7 |
| beta-xylosidase | 2 |
| xyloglucan:xyloglucosyl transferase | 2 |
| glucosylceramidase | 5 |
| chitinase | 20 |
| Total | 340 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yang, L.; Peng, M.; Shah, S.S.; Wang, Q. Transcriptome Sequencing and Comparative Analysis of Piptoporus betulinus in Response to Birch Sawdust Induction. Forests 2017, 8, 374. https://doi.org/10.3390/f8100374
AMA Style
Yang L, Peng M, Shah SS, Wang Q. Transcriptome Sequencing and Comparative Analysis of Piptoporus betulinus in Response to Birch Sawdust Induction. Forests. 2017; 8(10):374. https://doi.org/10.3390/f8100374
Chicago/Turabian StyleYang, Lixia, Mu Peng, Syed Sadaqat Shah, and Qiuyu Wang. 2017. "Transcriptome Sequencing and Comparative Analysis of Piptoporus betulinus in Response to Birch Sawdust Induction" Forests 8, no. 10: 374. https://doi.org/10.3390/f8100374
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.