Comprehensive Analysis of the Cork Oak (Quercus suber) Transcriptome Involved in the Regulation of Bud Sprouting

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Total RNA Extraction and Sequencing
2.3. Sequence Data and Transcriptome Assembly
2.4. Read Mapping and Differential Expression Analysis
2.5. Validation of Differentially Expressed Genes by Quantitative Real-Time PCR Analysis
2.6. Functional Annotation
3. Results
3.1. Preprocessing of the Sequence Data and De Novo Transcriptome Assembly
3.2. Read Mapping, Differential Expression
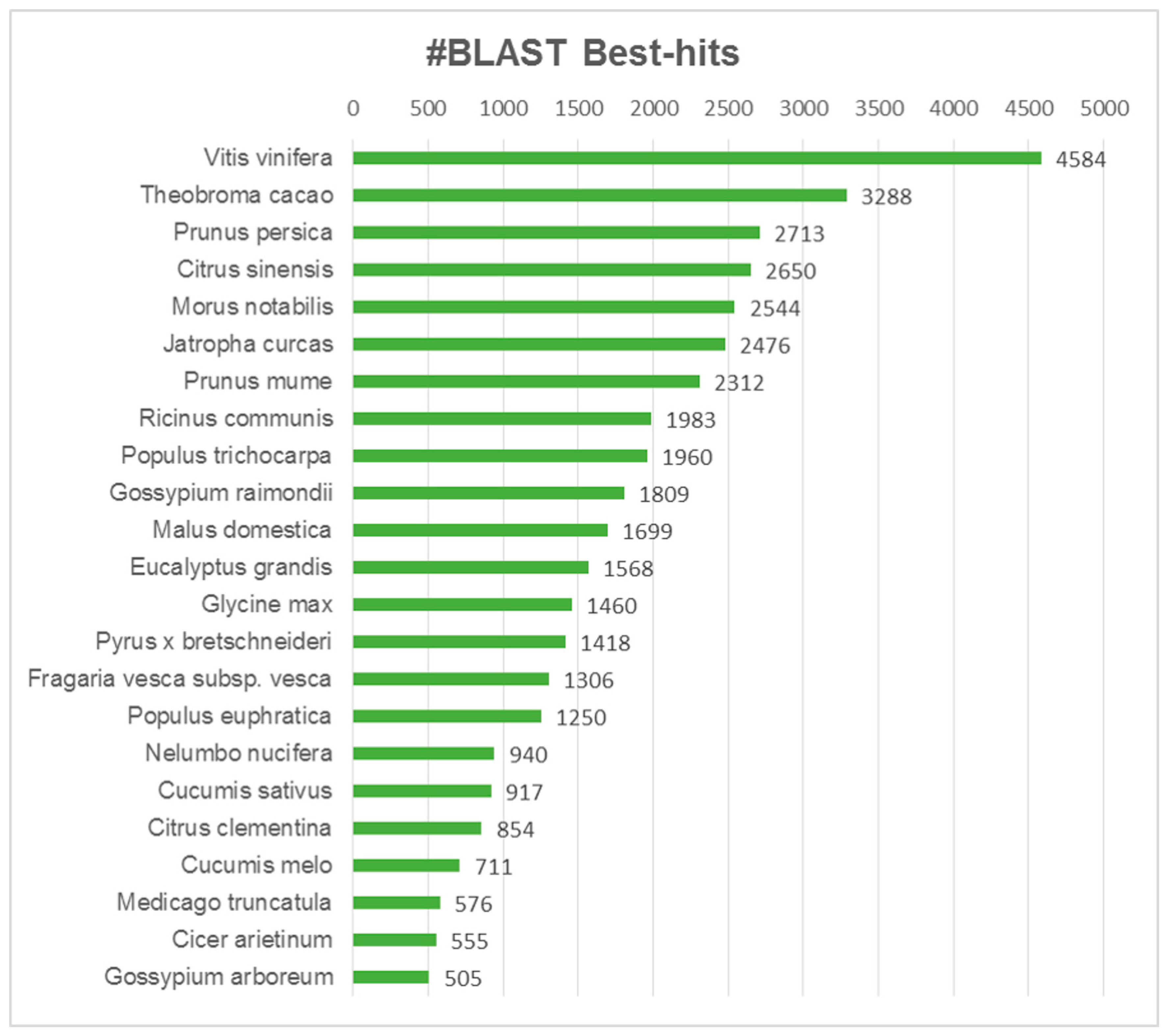
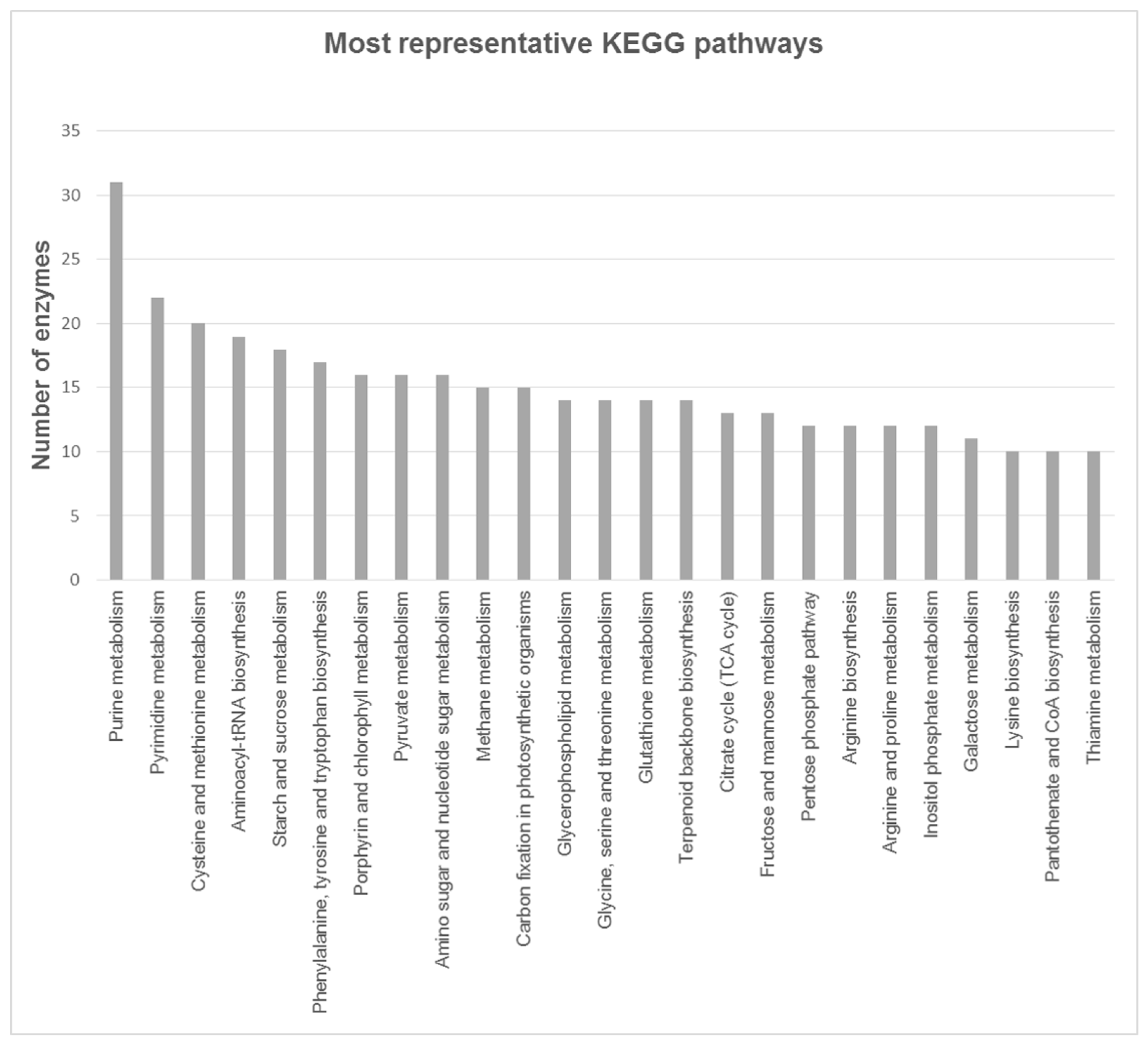
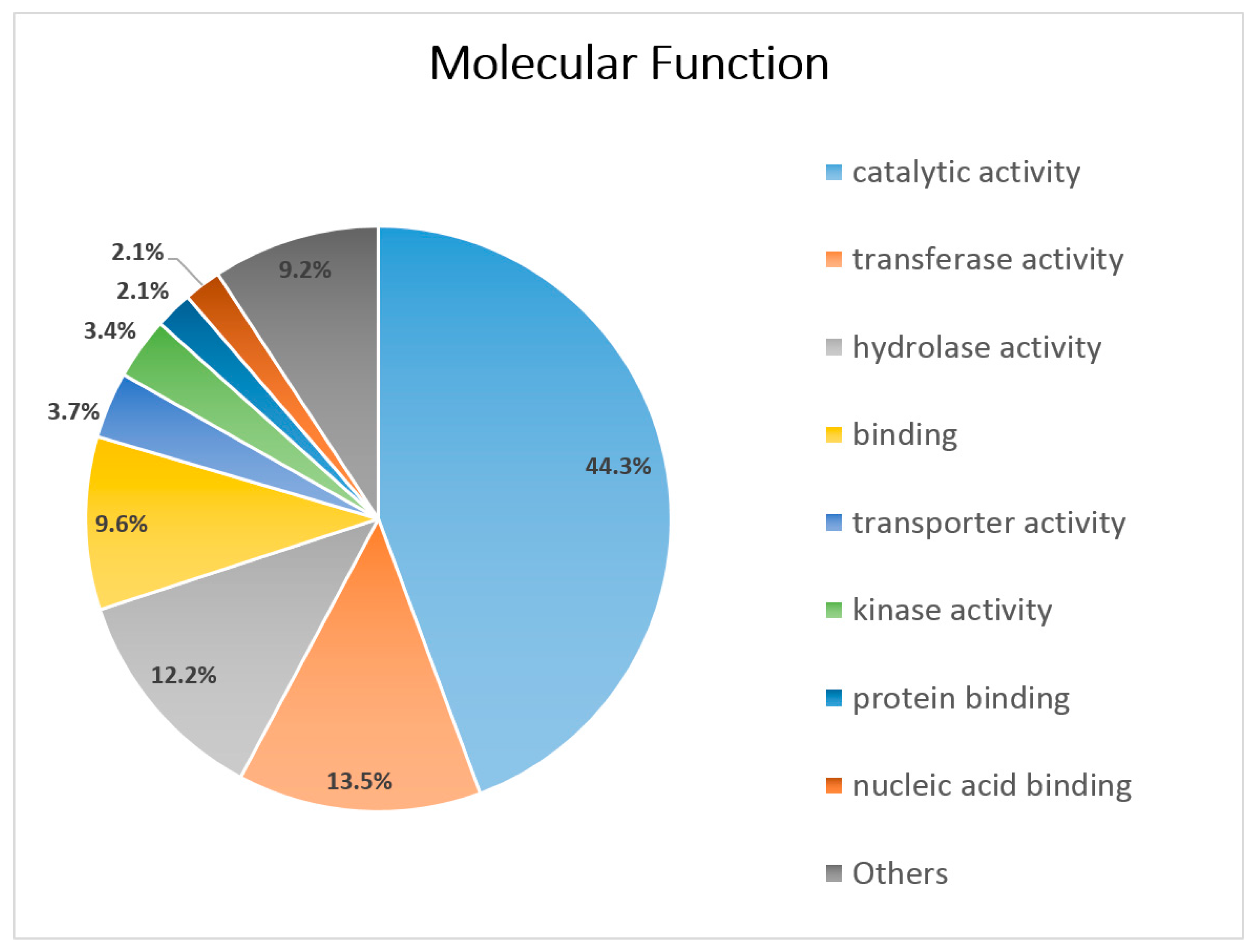
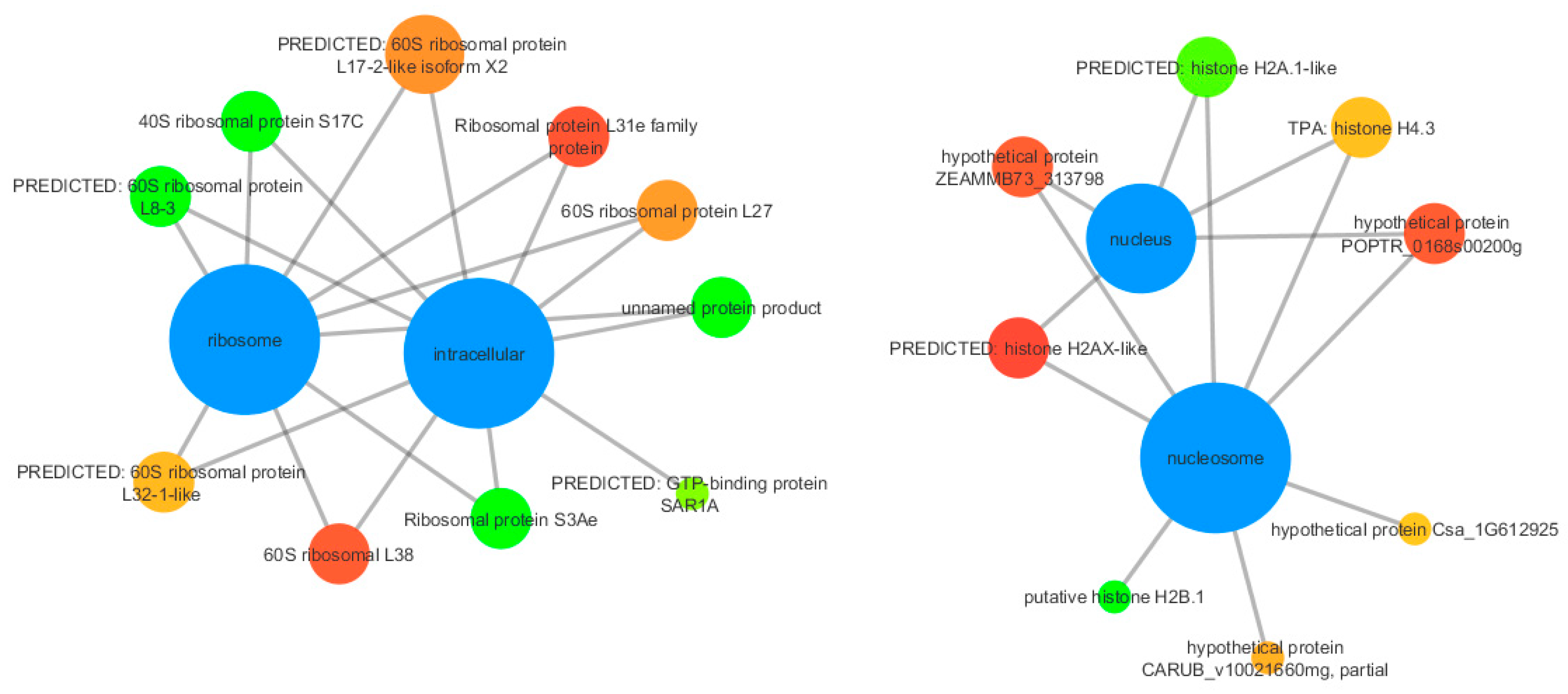
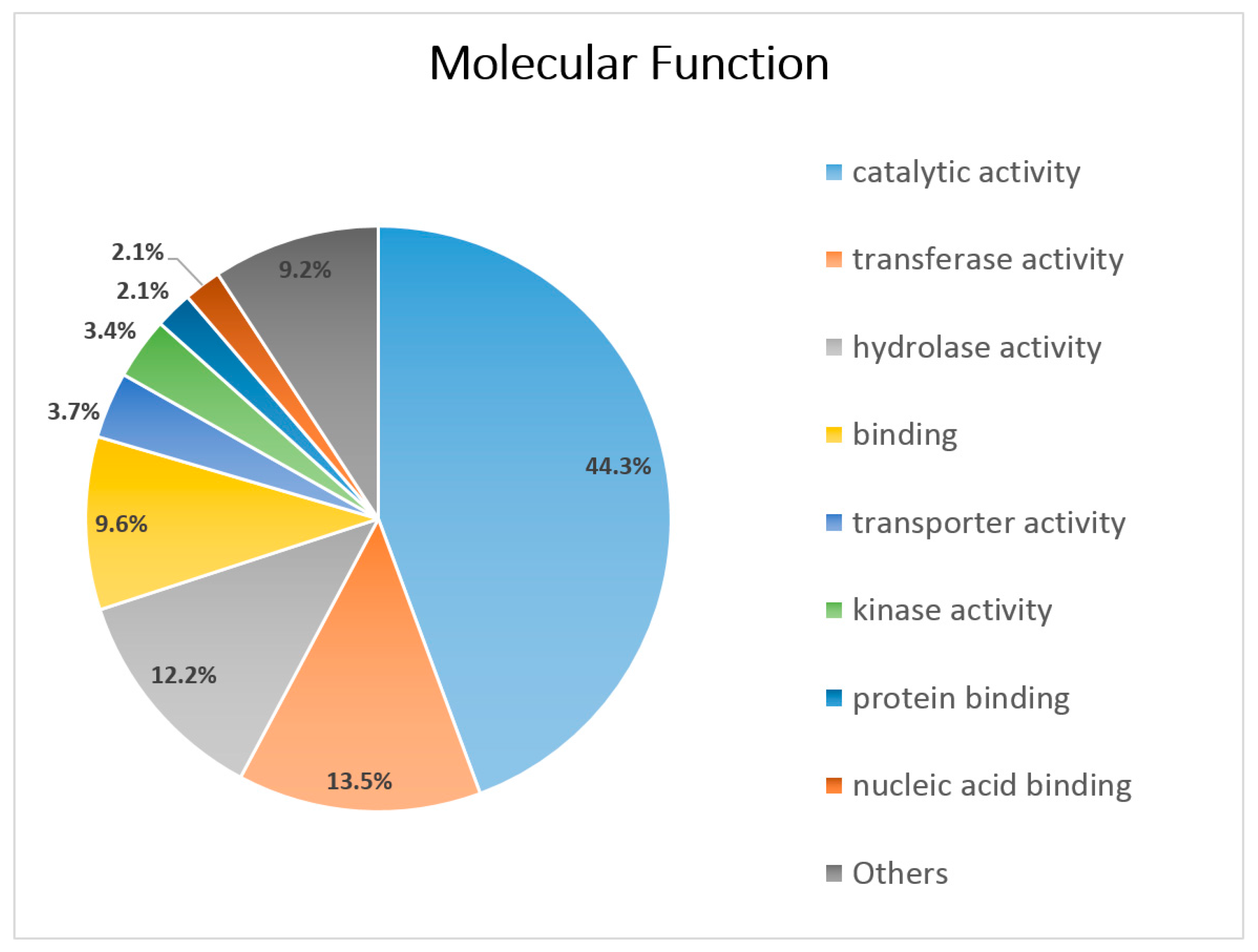
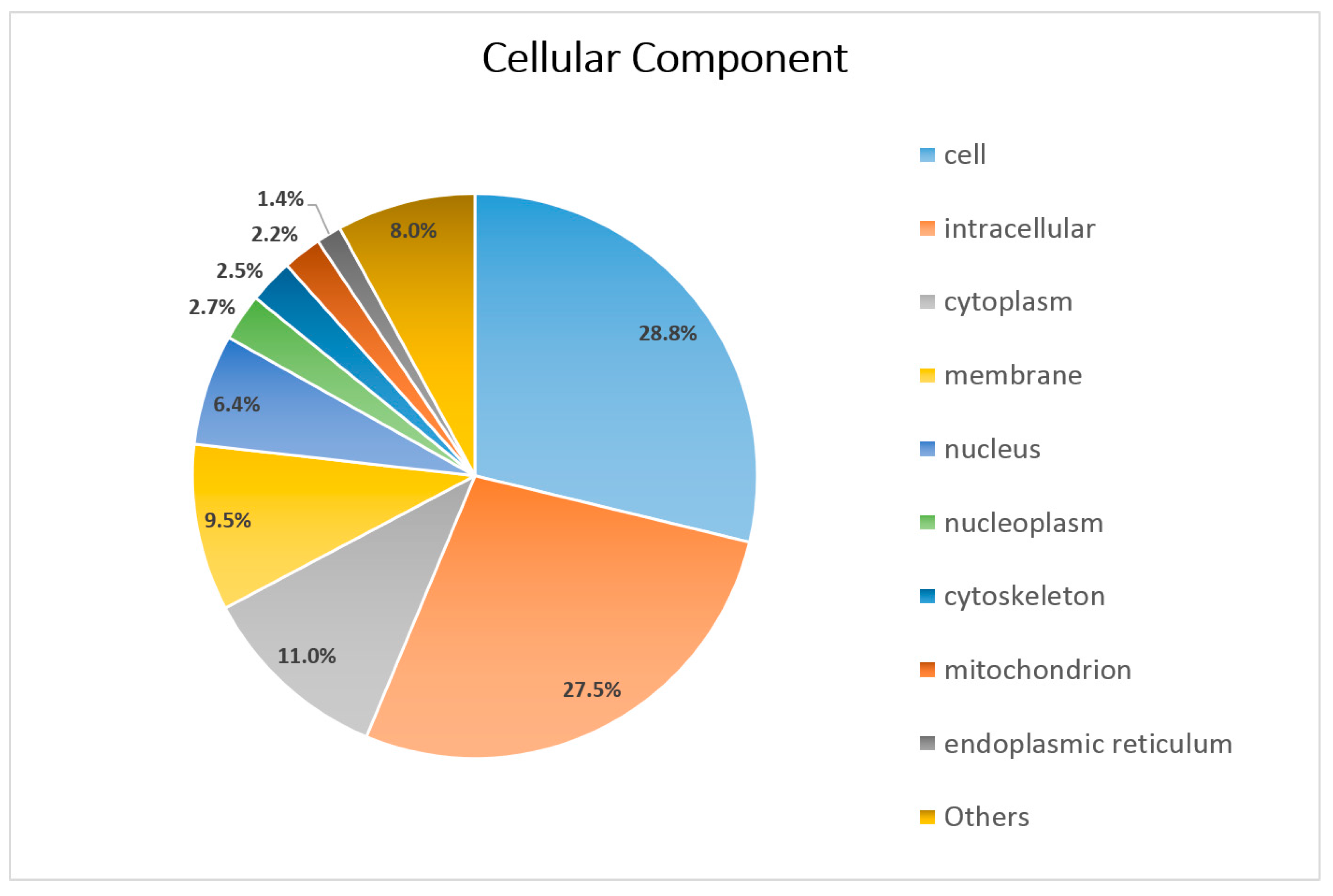
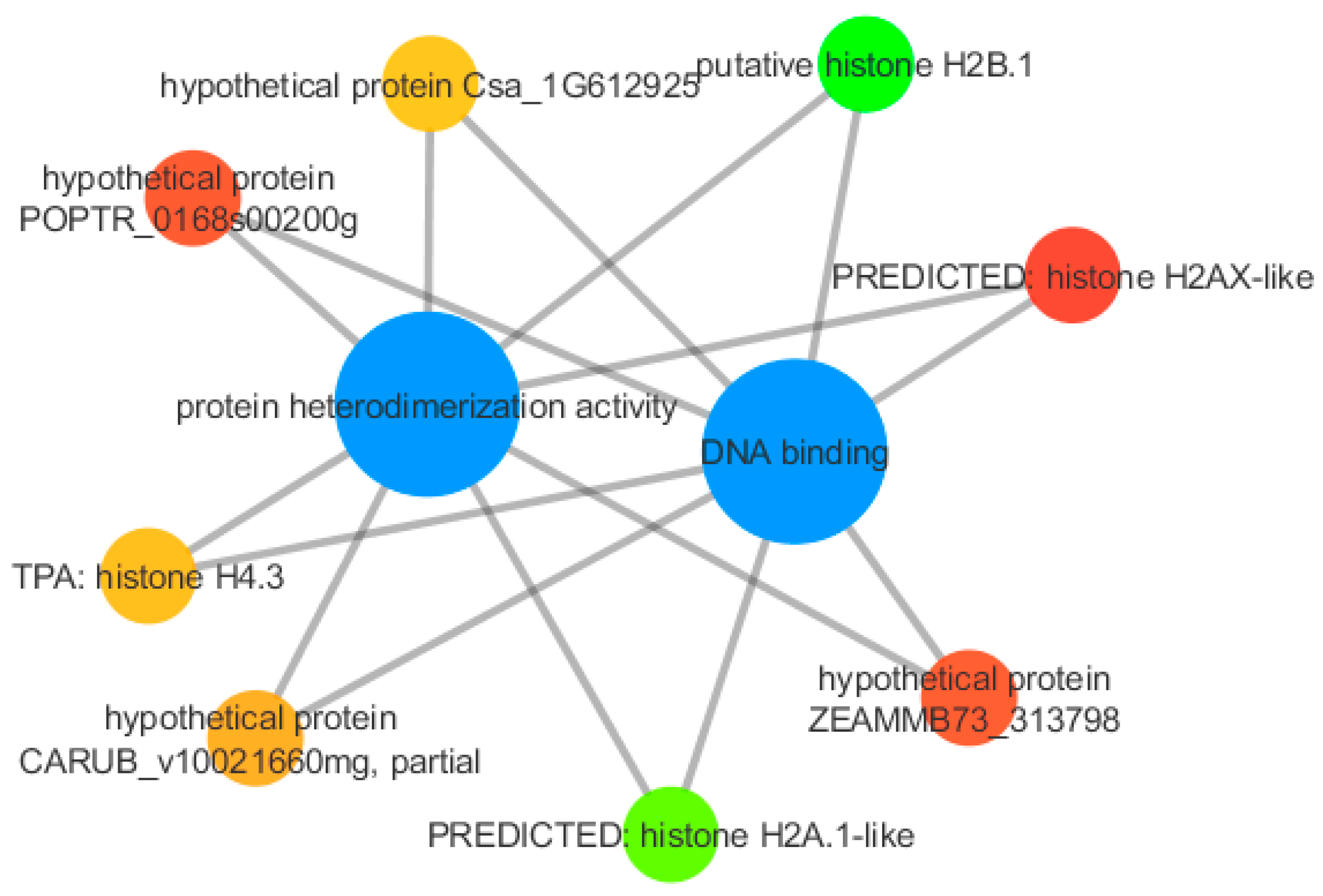
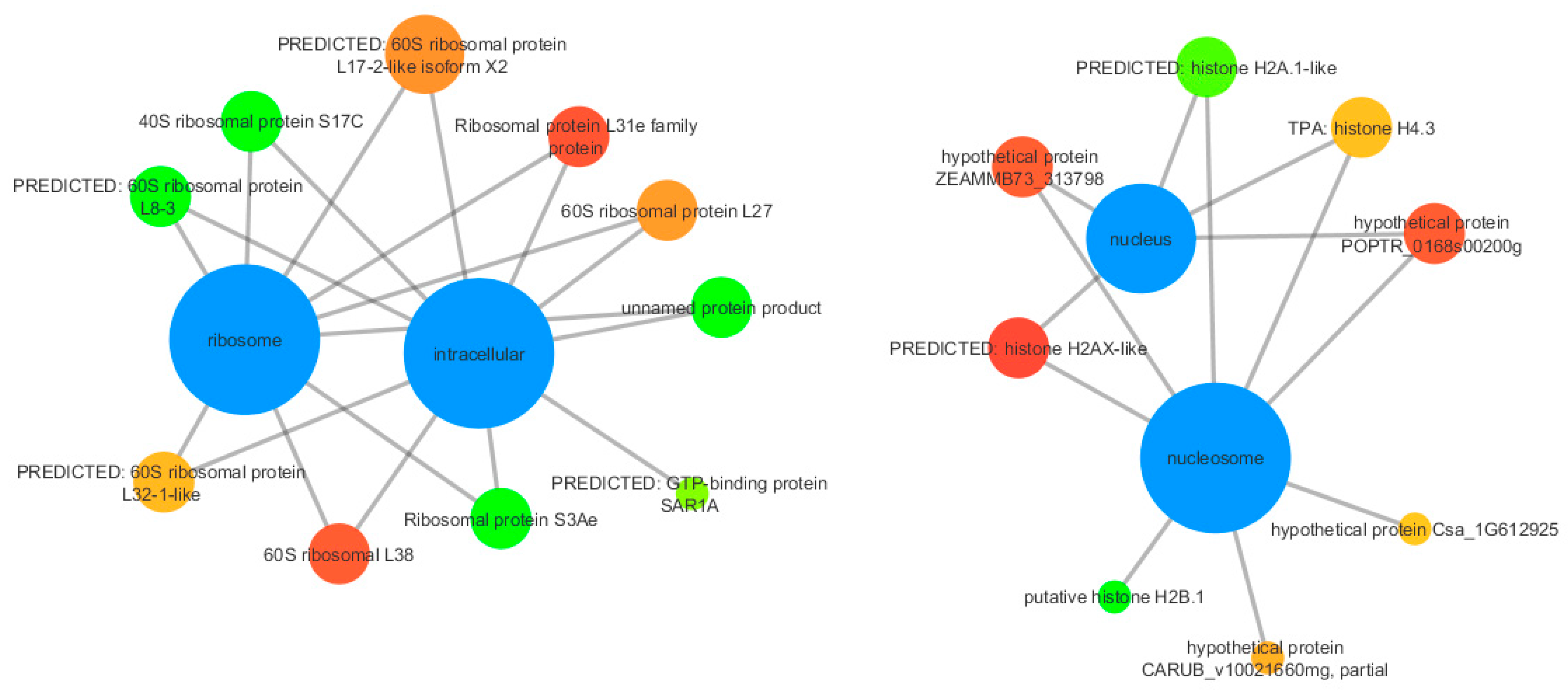
3.3. Functional Annotation
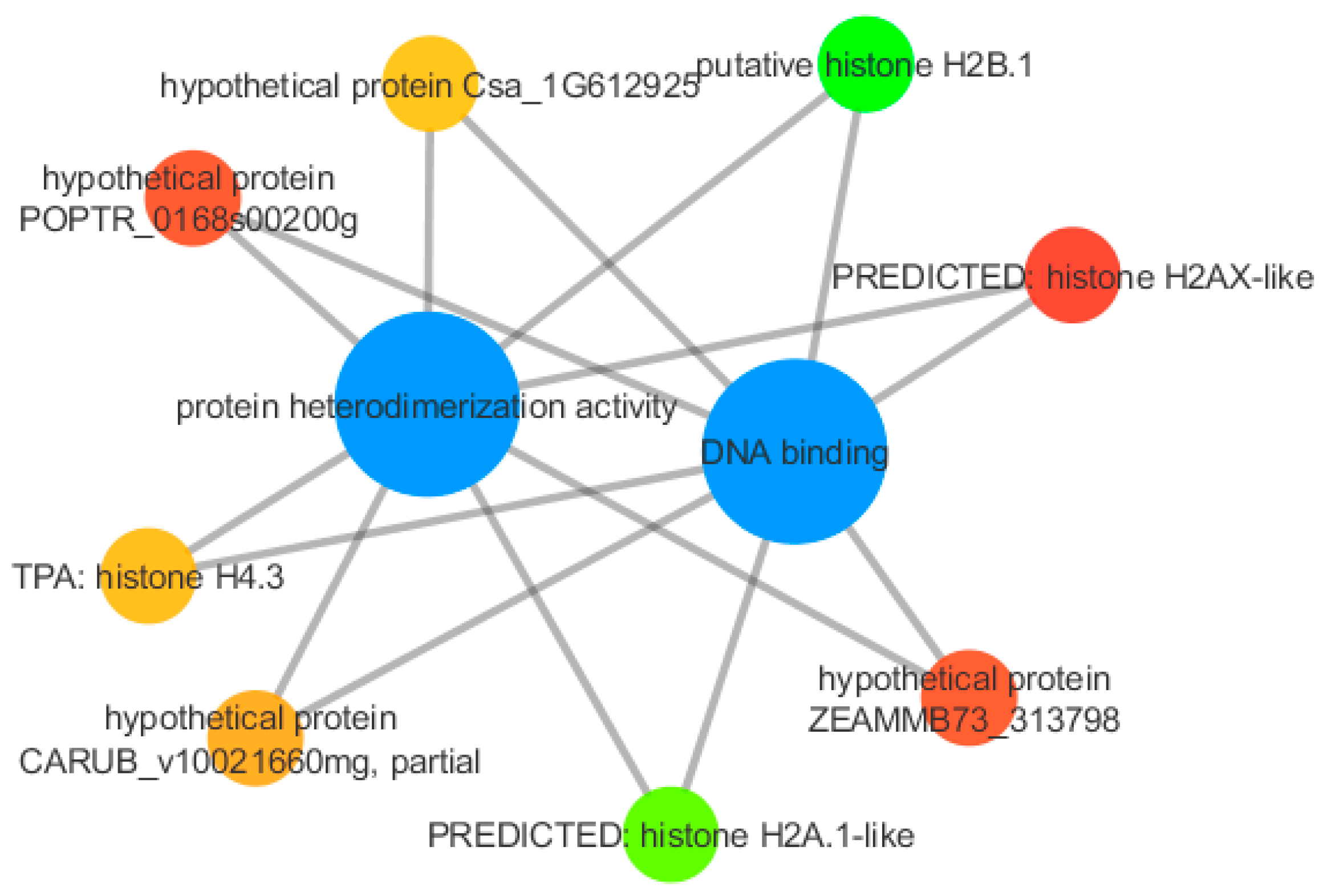
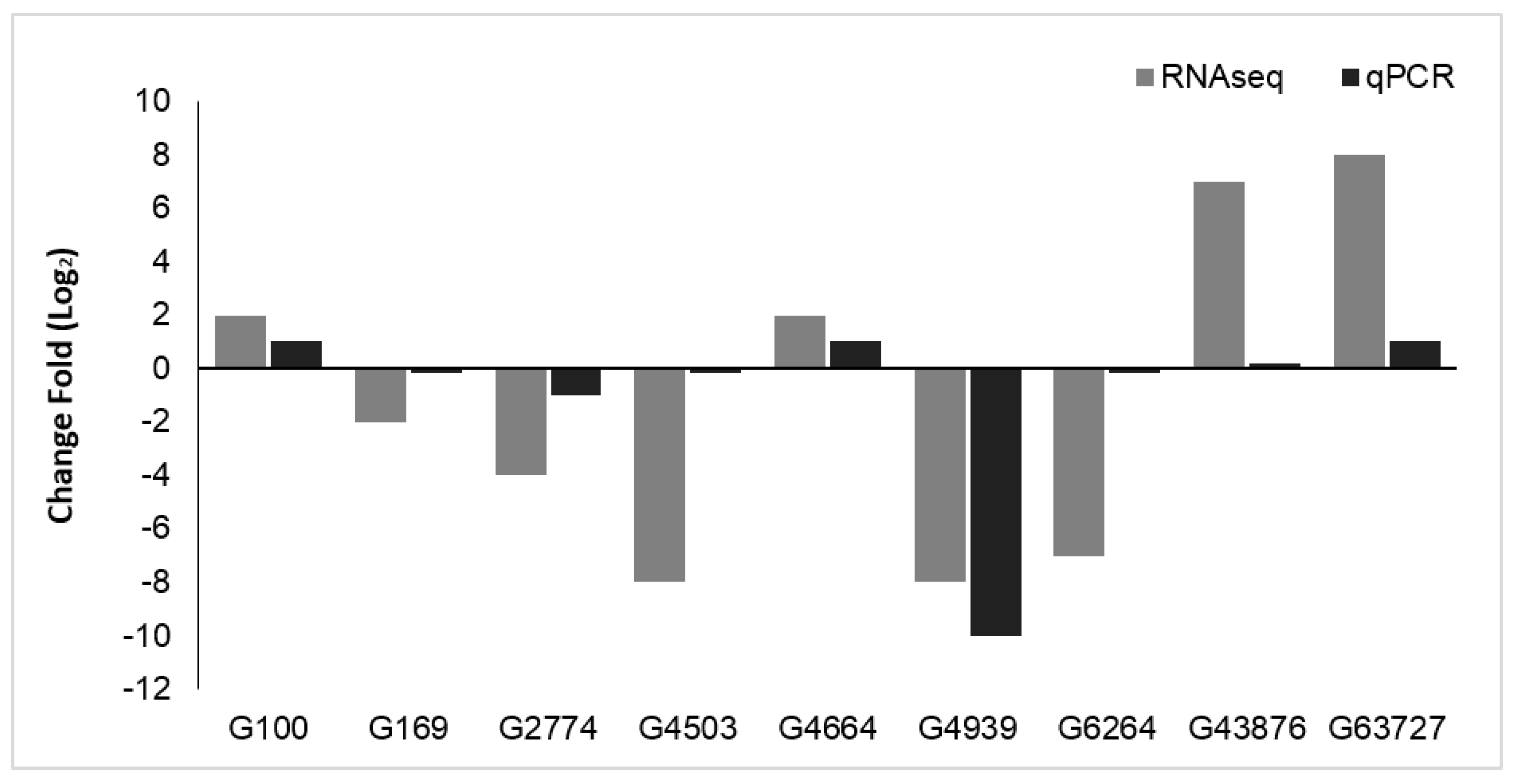
3.4. qPCR Validation
4. Discussion
4.1. A High Input of Energy Is Required during Bud Sprouting in Cork Oak
4.2. Cork Oak Bud Burst and Development Is under Tight Hormonal Regulation
4.3. Genes Related to Water Status Play a Key Role in Cork Oak Bud Sprouting
4.4. An Alternative Pathway for the Synthesis of Polysaccharides
4.5. Stress Response and Development Related Proteins Are Expressed in the Cork Oak Bud Transcriptome
4.6. Common Sets of Genes Used by Different Species in the Regulation of Bud Burst and Development
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berrahmouni, N.; Regato, P.; Stein, C. Beyond Cork—A Wealth of Resources for People and Nature, Lessons from the Mediterranean; WWF Mediterranean: Rome, Italy, 2007; Volume 118. [Google Scholar]
- Chuine, I.; Beaubien, E.G. Phenology is a major determinant of tree species range. Ecol. Lett. 2001, 4, 500–510. [Google Scholar] [CrossRef]
- Pereira, H. Cork: Biology, Production and Uses; Elsevier: Amsterdam, The Netherlands, 2007; ISBN 9780444529671. [Google Scholar]
- Chaves, I.; Passarinho, J.A.P.; Capitão, C.; Chaves, M.M.; Fevereiro, P.; Ricardo, C.P.P. Temperature stress effects in Quercus suber leaf metabolism. J. Plant Physiol. 2011, 168, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Scotti-Saintagne, C.; Bodénès, C.; Barreneche, T.; Bertocchi, E.; Plomion, C.; Kremer, A. Detection of quantitative trait loci controlling bud burst and height growth in Quercus robur L. Theor. Appl. Genet. 2004, 109, 1648–1659. [Google Scholar] [CrossRef] [PubMed]
- Saintagne, C.; Bodénès, C.; Barreneche, T.; Pot, D.; Plomion, C.; Kremer, A. Distribution of genomic regions differentiating oak species assessed by QTL detection. Heredity 2004, 92, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H.H.; Howe, G.T.; Bradshaw, H.D., Jr. Molecular genetic analysis of dormancy-related traits in poplars. Weed Sci. 2002, 50, 232–240. [Google Scholar] [CrossRef]
- Derory, J.; Léger, P.; Garcia, V.; Schaeffer, J.; Hauser, M.-T.; Salin, F.; Luschnig, C.; Plomion, C.; Glössl, J.; Kremer, A. Transcriptome analysis of bud burst in sessile oak (Quercus petraea). New Phytol. 2006, 170, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Lesur, I.; Le Provost, G.; Bento, P.; Da Silva, C.; Leplé, J.-C.; Murat, F.; Ueno, S.; Bartholomé, J.; Lalanne, C.; Ehrenmann, F.; et al. The oak gene expression atlas: Insights into Fagaceae genome evolution and the discovery of genes regulated during bud dormancy release. BMC Genom. 2015, 16, 112. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, R.T.; Fortes, A.M.; Pinheiro, C.; Pereira, H. Comparison of good- and bad-quality cork: Application of high-throughput sequencing of phellogenic tissue. J. Exp. Bot. 2014, 65, 4887–4905. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Leal, J.B.; Abreu, I.A.; Alabaça, C.S.; Almeida, M.H.; Almeida, P.; Almeida, T.; Amorim, M.I.; Araújo, S.; Azevedo, H.; Badia, A.; et al. A comprehensive assessment of the transcriptome of cork oak (Quercus suber) through EST sequencing. BMC Genom. 2014, 15, 371. [Google Scholar] [CrossRef] [PubMed]
- Miguel, A.; de Vega-Bartol, J.; Marum, L.; Chaves, I.; Santo, T.; Leitão, J.; Varela, M.C.; Miguel, C.M. Characterization of the cork oak transcriptome dynamics during acorn development. BMC Plant Biol. 2015, 15, 158. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Müller, W.E.G.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Marum, L.; Miguel, A.; Ricardo, C.P.; Miguel, C. Reference gene selection for quantitative real-time PCR normalization in Quercus suber. PLoS ONE 2012, 7, e35113. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Apweiler, R. InterProScan—An integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-L.; Bao, J.; Reecy, J. CateGOrizer: A web-based program to batch analyze gene ontology classification categories. Online J. Bioinform. 2008, 9, 108–112. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Derory, J.; Scotti-Saintagne, C.; Bertocchi, E.; Le Dantec, L.; Graignic, N.; Jauffres, A.; Casasoli, M.; Chancerel, E.; Bodénès, C.; Alberto, F.; et al. Contrasting relationships between the diversity of candidate genes and variation of bud burst in natural and segregating populations of European oaks. Heredity 2010, 104, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Sweetlove, L.J.; Beard, K.F.M.; Nunes-Nesi, A.; Fernie, A.R.; Ratcliffe, R.G. Not just a circle: Flux modes in the plant TCA cycle. Trends Plant Sci. 2010, 15, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Jardim-Messeder, D.; Caverzan, A.; Rauber, R.; de Souza Ferreira, E.; Margis-Pinheiro, M.; Galina, A. Succinate dehydrogenase (mitochondrial complex II) is a source of reactive oxygen species in plants and regulates development and stress responses. New Phytol. 2015, 208, 776–789. [Google Scholar] [CrossRef] [PubMed]
- Nersissian, A.M.; Immoos, C.; Hill, M.G.; Hart, P.J.; Williams, G.; Herrmann, R.G.; Selverstone Valentine, J. Uclacyanins, stellacyanins, and plantacyanins are distinct subfamilies of phytocyanins: Plant-specific mononuclear blue copper proteins. Protein Sci. 1998, 7, 71915–71929. [Google Scholar] [CrossRef] [PubMed]
- Peer, W.A.; Brown, D.E.; Tague, B.W.; Muday, G.K.; Taiz, L.; Murphy, A.S. Flavonoid accumulation patterns of transparent testa mutants of arabidopsis. Plant Physiol. 2001, 126, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Broun, P. Transcriptional control of flavonoid biosynthesis: A complex network of conserved regulators involved in multiple aspects of differentiation in Arabidopsis. Curr. Opin. Plant Biol. 2005, 8, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.; Rubery, P.H. Naturally occurring auxin transport regulators. Science 1988, 241, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, P. Effect of synthetic and natural protein tyrosine kinase inhibitors on auxin efflux in zucchini (Cucurbita pepo) hypocotyls. Physiol. Plant. 1996, 96, 205–210. [Google Scholar] [CrossRef]
- Murphy, A.; Peer, W.A.; Taiz, L. Regulation of auxin transport by aminopeptidases and endogenous flavonoids. Planta 2000, 211, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.E.; Rashotte, A.M.; Murphy, A.S.; Normanly, J.; Tague, B.W.; Peer, W.A.; Taiz, L.; Muday, G.K. Flavonoids act as negative regulators of auxin transport in vivo in Arabidopsis. Plant Physiol. 2001, 126, 524–535. [Google Scholar] [CrossRef] [PubMed]
- El Zein, R.; Breda, N.; Gerant, D.; Zeller, B.; Maillard, P. Nitrogen sources for current-year shoot growth in 50-year-old sessile oak trees: An in situ 15N labeling approach. Tree Physiol. 2011, 31, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Linz, J.E.; Sypherd, P.S. Expression of three genes for elongation factor 1 alpha during morphogenesis of Mucor racemosus. Mol. Cell. Biol. 1987, 7, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Hovemann, B.; Richter, S.; Walldorf, U.; Cziepluch, C. Two genes encode related cytoplasmic elongation factors 1 alpha (EF-1 alpha) in Drosophila melanogaster with continuous and stage specific expression. Nucleic Acids Res. 1988, 16, 3175–3194. [Google Scholar] [CrossRef] [PubMed]
- Ursin, V.M.; Irvine, J.M.; Hiatt, W.R.; Shewmaker, C.K. Developmental analysis of elongation factor-1 alpha expression in transgenic tobacco. Plant Cell 1991, 3, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cao, D.; Chen, Y.; Wei, D.; Wang, Y.; Stevenson, R.A.; Zhu, Y.; Lin, J. Gene expression and proteomic analysis of shoot apical meristem transition from dormancy to activation in Cunninghamia lanceolata (Lamb.) Hook. Sci. Rep. 2016, 6, 19938. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.J. The plant hormones: Their nature, occurrence, and functions. In Plant Hormones; Springer: Dordrecht, The Netherlands, 2010; pp. 1–15. [Google Scholar]
- O’Neill, D.P.; Davidson, S.E.; Clarke, V.C.; Yamauchi, Y.; Yamaguchi, S.; Kamiya, Y.; Reid, J.B.; Ross, J.J. Regulation of the gibberellin pathway by auxin and DELLA proteins. Planta 2010, 232, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Bajji, M.; M’Hamdi, M.; Gastiny, F.; Rojas-Beltran, J.A.; du Jardin, P. Catalase inhibition accelerates dormancy release and sprouting in potato (Solanum tuberosum L.) tubers. Biotechnol. Agron. Soc. Environ. 2007, 11, 121–131. [Google Scholar]
- Ohmiya, A. Characterization of ABP19/20, sequence homologues of germin-like protein in Prunus persica L. Plant Sci. 2002, 163, 683–689. [Google Scholar] [CrossRef]
- Ongaro, V.; Leyser, O. Hormonal control of shoot branching. J. Exp. Bot. 2008, 59, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Santner, A.; Estelle, M. The ubiquitin-proteasome system regulates plant hormone signaling. Plant J. 2010, 61, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Luong, P.; Huq, E. The F-Box Protein MAX2 Functions as a positive regulator of photomorphogenesis in Arabidopsis. Plant Physiol. 2007, 145, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.; Estelle, M. Ubiquitin-mediated control of plant hormone signaling. Plant Physiol. 2012, 160, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Mayzlish-Gati, E.; LekKala, S.; Resnick, N.; Wininger, S.; Bhattacharya, C.; Lemcoff, J.H.; Kapulnik, Y.; Koltai, H. Strigolactones are positive regulators of light-harvesting genes in tomato. J. Exp. Bot. 2010, 61, 3129–3136. [Google Scholar] [CrossRef] [PubMed]
- Brewera, P.B.; Koltaib, H.; Beveridge, C.A. Diverse roles of strigolactones in plant development. Mol. Plant 2013, 6, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.; Klopp, C.; Leplé, J.C.; Derory, J.; Noirot, C.; Léger, V.; Prince, E.; Kremer, A.; Plomion, C.; Le Provost, G. Transcriptional profiling of bud dormancy induction and release in oak by next-generation sequencing. BMC Genom. 2013, 14, 236. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.R.; Saunders, M.R.; Michler, C.H. Epicormic buds in trees: A review of bud establishment, development and dormancy release. Tree Physiol. 2012, 32, 565–584. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Halaly, T.; Acheampong, A.K.; Takebayashi, Y.; Jikumaru, Y.; Kamiya, Y.; Or, E. Abscisic acid (ABA) regulates grape bud dormancy, and dormancy release stimuli may act through modification of ABA metabolism. J. Exp. Bot. 2015, 66, 1527–1542. [Google Scholar] [CrossRef] [PubMed]
- Dhanaraj, A.L.; Slovin, J.P.; Rowland, L.J. Analysis of gene expression associated with cold acclimation in blueberry floral buds using expressed sequence tags. Plant Sci. 2004, 166, 863–872. [Google Scholar] [CrossRef]
- Kaldenhoff, R.; Fischer, M. Functional aquaporin diversity in plants. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Maurel, C.; Verdoucq, L.; Luu, D.-T.; Santoni, V. Plant Aquaporins: Membrane channels with multiple integrated functions. Annu. Rev. Plant Biol. 2008, 59, 595–624. [Google Scholar] [CrossRef] [PubMed]
- Ludevid, D.; Hofte, H.; Himelblau, E.; Chrispeels, M.J. The expression pattern of the tonoplast intrinsic protein gamma-tip in Arabidopsis-Thaliana is correlated with cell enlargement. Plant Physiol. 1992, 100, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.L.; Huttly, A.K. Cloning of two gibberellin-regulated cDNAs from Arabidopsis thaliana by subtractive hybridization: Expression of the tonoplast water channel, γ-TIP, is increased by GA3. Plant Mol. Biol. 1994, 24, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Chaumont, F.; Moshelion, M.; Daniels, M.J. Regulation of plant aquaporin activity. Biol. Cell 2005, 97, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.P.; Wilson, I.D.; Kim, S.H.; Grierson, D. Inhibiting expression of a tomato ripening-associated membrane protein increases organic acids and reduces sugar levels of fruit. Planta 2001, 212, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Afzal, Z.; Howton, T.; Sun, Y.; Mukhtar, M. The roles of aquaporins in plant stress responses. J. Dev. Biol. 2016, 4, 9. [Google Scholar] [CrossRef]
- Ahamed, A.; Murai-Hatano, M.; Ishikawa-Sakurai, J.; Hayashi, H.; Kawamura, Y.; Uemura, M. Cold stress-induced acclimation in rice is mediated by root-specific aquaporins. Plant Cell Physiol. 2012, 53, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Reiter, W.-D. Biochemical genetics of nucleotide sugar interconversion reactions. Curr. Opin. Plant Biol. 2008, 11, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Kärkönen, A. Biosynthesis of UDP-GlcA: Via UDPGDH or the myo-inositol oxidation pathway? Plant Biosyst. Int. J. Deal. Asp. Plant Biol. 2005, 139, 46–49. [Google Scholar] [CrossRef]
- Loewus, F.A.; Kelly, S.; Neufeld, E.F. Metabolism of myo-inositol in plants: Conversion to pectin, hemicellulose, d-xylose and sugar acids. Proc. Natl. Acad. Sci. USA 1962, 48, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.M.; Loewus, F. The conversion of d-Glucose-6-C to cell wall polysaccharide material in Zea mays in presence of high endogenous levels of myoinositol. Plant Physiol. 1973, 52, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Belknap, W.R.; Garbarino, J.E. The role of ubiquitin in plant senescence and stress responses. Trends Plant Sci. 1996, 10, 331–335. [Google Scholar] [CrossRef]
- Garbarino, J.E.; Rockhold, D.R.; Belknap, W.R. Expression of stress-responsive ubiquitin genes in potato tubers. Plant Mol. Biol. 1992, 20, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Janse, J.D. Chapter: 3 Disease and symptoms caused by plant pathogenic bacteria. In Phytobacteriology: Principles and Practice; Janse, J.D., Ed.; CABI: Wallingford, Oxon, UK; New York, NY, USA, 2005; pp. 91–93. [Google Scholar]
- Kulik, A.; Wawer, I.; Krzywińska, E.; Bucholc, M.; Dobrowolska, G. SnRK2 Protein Kinases—Key regulators of plant response to abiotic stresses. OMICS 2011, 15, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, J.P. Regulation of growth and dormancy in pea axillary buds. In Dormancy in Plants: From Whole Plant Behaviour to Cellular Control; Viémont, J.D., Crabbé, J., Eds.; CABI: Wallingford, Oxon, UK; New York, NY, USA, 2000; pp. 337–339. [Google Scholar]
- Devitt, M.; Stafstrom, J. Cell cycle regulation during growth-dormancy cycles in pea axillary buds. Plant Mol. Biol. 1995, 29, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Horvath, D. Common mechanisms regulate flowering and dormancy. Plant Sci. 2009, 177, 523–531. [Google Scholar] [CrossRef]
- Kryvych, S.; Nikiforova, V.; Herzog, M.; Perazza, D.; Fisahn, J. Gene expression profiling of the different stages of Arabidopsis thaliana trichome development on the single cell level. Plant Physiol. Biochem. 2008, 46, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, C.; Saito, T.; Ubi, B.E.; Shimizu, T.; Itai, A.; Saito, T.; Yamamoto, T.; Moriguchi, T. Transcriptome analysis of Pyrus pyrifolia leaf buds during transition from endodormancy to ecodormancy. Sci. Hortic. 2012, 147, 49–55. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library | Raw Reads | Processed Reads | ||
|---|---|---|---|---|
| Number | <AL> | Number | <AL> | |
| Non-normalized 1 | 566,726 | 574.1 | 452,455 | 519.9 |
| Non-normalized 2 | 513,382 | 577.8 | 404,242 | 522.9 |
| Normalized 1 | 595,388 | 568.4 | 464,611 | 523.8 |
| Normalized 2 | 570,030 | 560.7 | 450,842 | 518.1 |
| Total | 2,245,526 | - | 1,772,150 | - |
| Library | Reads Used | Mapped Reads (%) | UMR (%) |
|---|---|---|---|
| Non-normalized 1 | 452,455 | 430,935 (95.2%) | 197,521 (43.7%) |
| Non-normalized 2 | 404,242 | 384,352 (95.1%) | 161,264 (39.9%) |
| Total | 856,697 | 815,287 (95.2%) | 358,785 (41.9%) |
| Annotation | LogFC | p-Value | Gene Identifier ID |
|---|---|---|---|
| unnamed protein product | 8.48 | 1.22 × 10 −12 | 296082254 |
| Ribosomal protein S3Ae | 7.88 | 4.37 × 10 −5 | 976900419 |
| putative histone H2B.1 | 7.83 | 7.13 × 10 −5 | 703085592 |
| PREDICTED: 60S ribosomal protein L8-3 | 7.45 | 1.67 × 10 −6 | 449434174 |
| basic blue copper family protein | 7.38 | 3.17 × 10 −6 | 224054286 |
| serin/threonine protein kinase | 7.38 | 3.17 × 10 −6 | 38343920 |
| hypothetical protein PRUPE_ppa012332mg | 7.3 | 6.03 × 10 −6 | 595797137 |
| PREDICTED: pentatricopeptide repeat-containing protein At1g74750-like | 7.3 | 6.03 × 10 −6 | 470127288 |
| 40S ribosomal protein S17C | 7.13 | 2.20 × 10 −5 | 313586437 |
| 60S ribosomal L38 | −7.15 | 1.15 × 10 −5 | 728829564 |
| PREDICTED: succinate dehydrogenase | −3.34 | 3.39 × 10 −5 | 449455896 |
| hypothetical protein EUGRSUZ_G02560 (myo-inositol oxynase) | −4.57 | 1.55 × 10 −7 | 629099270 |
| Translation elongation factor 1 alpha | −5.2 | 1.53 × 10 −10 | 110224776 |
| PREDICTED: uncharacterized protein | −7.22 | 6.03 × 10 −6 | 645275588 |
| PREDICTED: polyubiquitin | −7.22 | 6.03 × 10 −6 | 743820616 |
| PREDICTED: protein NUCLEAR FUSION DEFECTIVE 2 | −7.22 | 6.03 × 10 −6 | 470125885 |
| aquaporin TIP2;2 | −7.22 | 6.03 × 10 −6 | 383479044 |
| Ribosomal protein L31e family protein | −7.48 | 4.66 × 10 −7 | 590724775 |
| RecName: Full = Agglutinin; AltName: Full = CCA | −7.7 | 7.00 × 10 −8 | 48428322 |
| PREDICTED: histone H2AX-like | −7.94 | 3.11 × 10 −9 | 658055874 |
| agglutinin isoform | −8.1 | 2.68 × 10 −10 | 85376265 |
| chlorophyll a/b-binding protein | −8.84 | 9.97 × 10 −15 | 2804572 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usié, A.; Simões, F.; Barbosa, P.; Meireles, B.; Chaves, I.; Gonçalves, S.; Folgado, A.; Almeida, M.H.; Matos, J.; Ramos, A.M. Comprehensive Analysis of the Cork Oak (Quercus suber) Transcriptome Involved in the Regulation of Bud Sprouting. Forests 2017, 8, 486. https://doi.org/10.3390/f8120486
Usié A, Simões F, Barbosa P, Meireles B, Chaves I, Gonçalves S, Folgado A, Almeida MH, Matos J, Ramos AM. Comprehensive Analysis of the Cork Oak (Quercus suber) Transcriptome Involved in the Regulation of Bud Sprouting. Forests. 2017; 8(12):486. https://doi.org/10.3390/f8120486
Chicago/Turabian StyleUsié, Ana, Fernanda Simões, Pedro Barbosa, Brígida Meireles, Inês Chaves, Sónia Gonçalves, André Folgado, Maria H. Almeida, José Matos, and António M. Ramos. 2017. "Comprehensive Analysis of the Cork Oak (Quercus suber) Transcriptome Involved in the Regulation of Bud Sprouting" Forests 8, no. 12: 486. https://doi.org/10.3390/f8120486