
Genetic Diversity of Walnut (Juglans Regia L.) in the Eastern Italian Alps

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction
2.3. SSR Genotyping
2.4. Data Analysis
2.4.1. Genetic Variation and Related Metrics
2.4.2. The Comparison among Populations
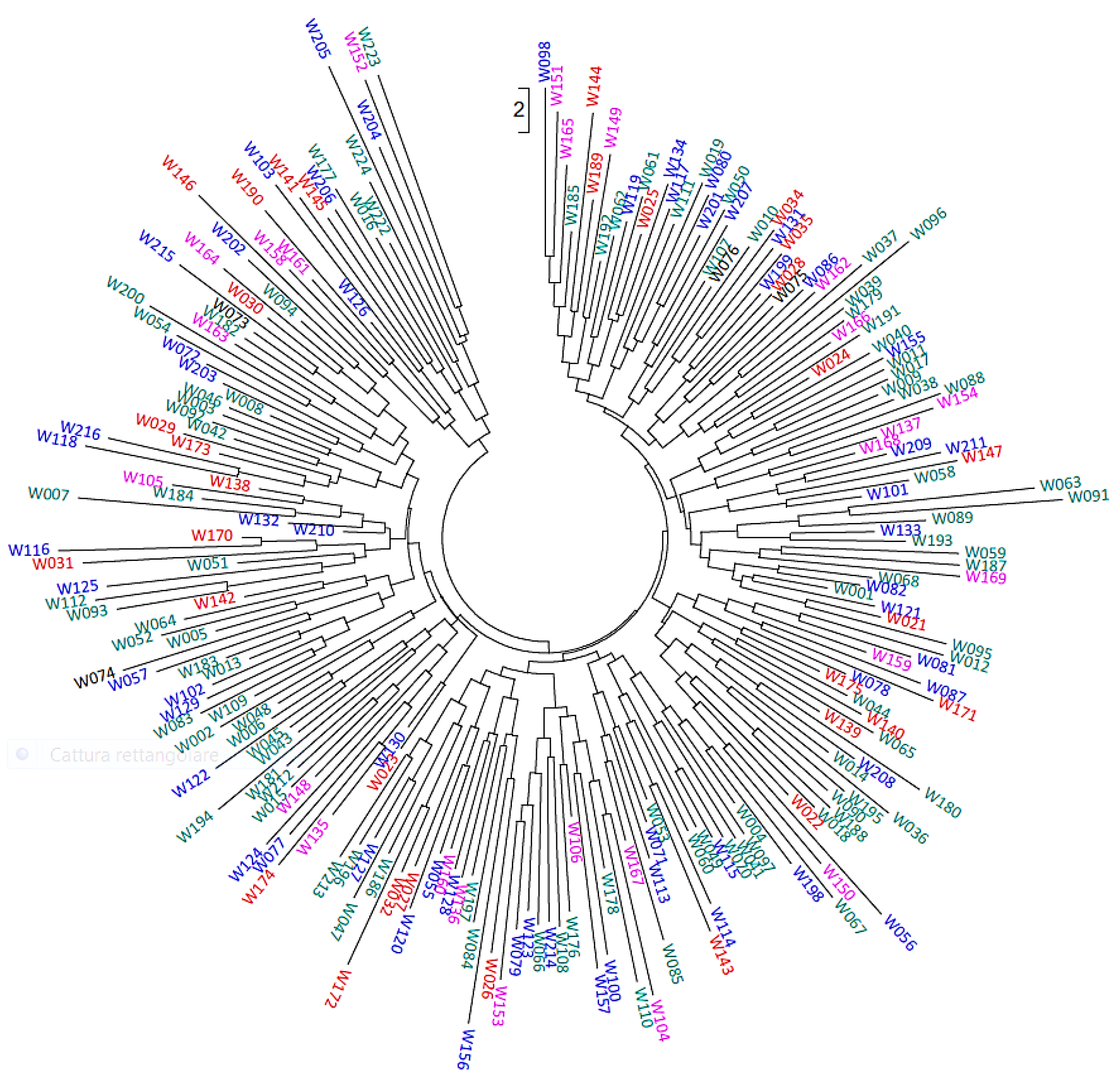
2.4.3. Distance-Based Clustering
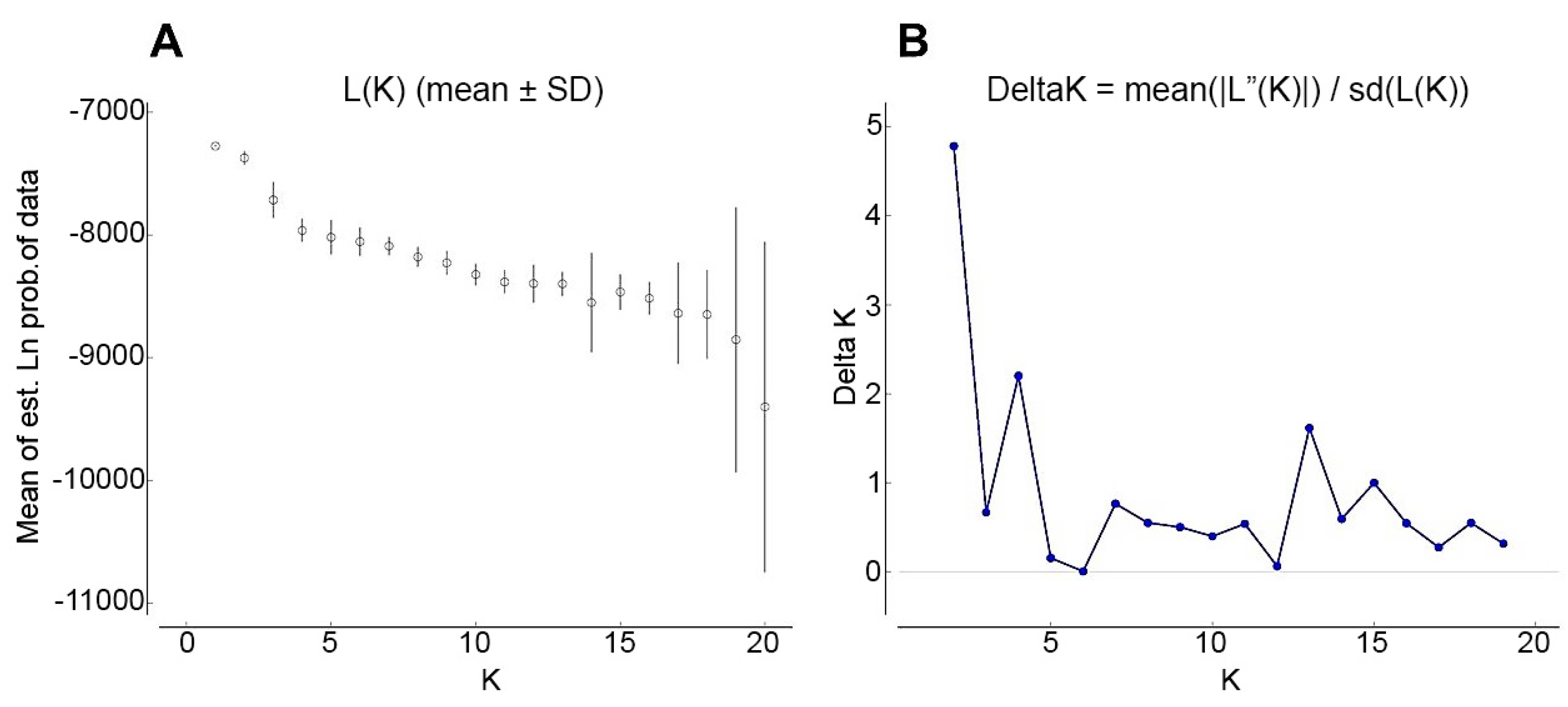
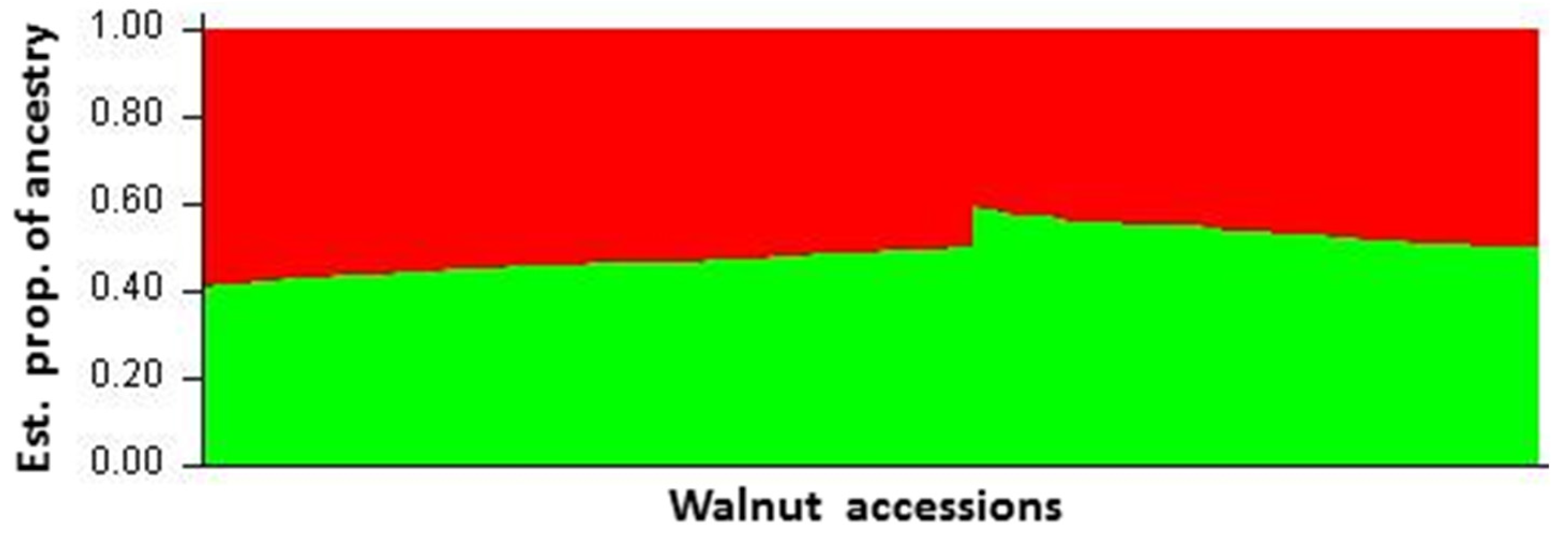
2.4.4. Model-Based Clustering
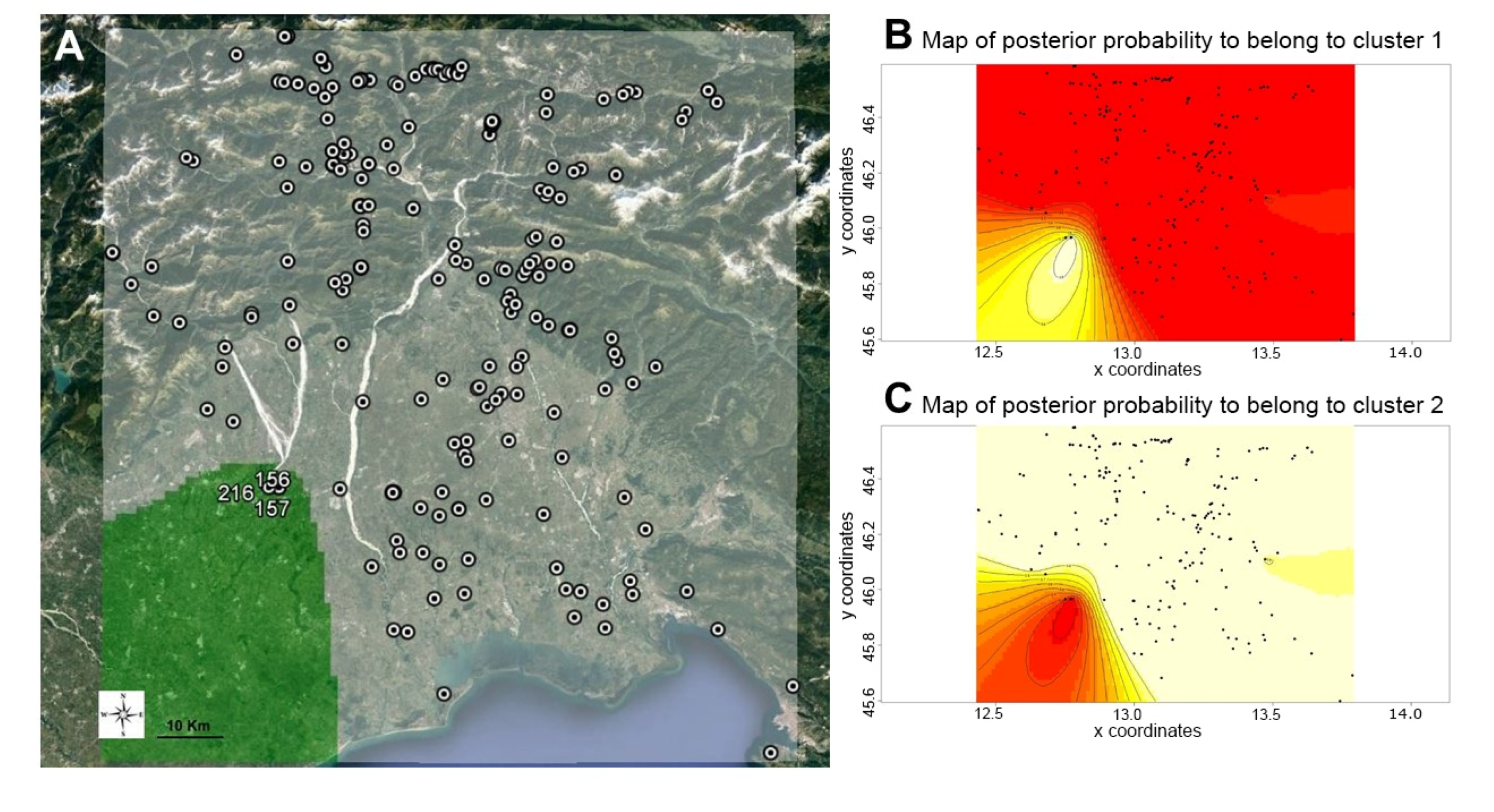
2.4.5. Landscape Genetics
3. Results and Discussion
3.1. SSR Marker Polymorphism and Genetic Metrics
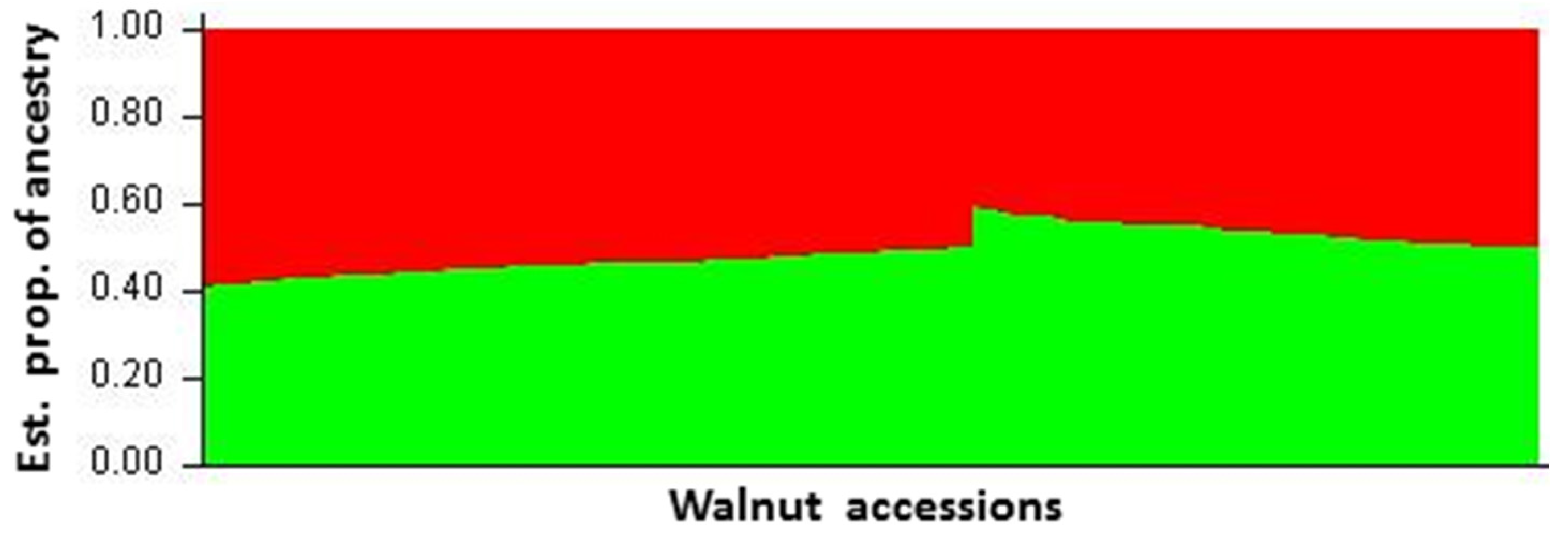
3.2. Hierarchical and Model-Based Clustering of Walnut Accessions
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Manning, W.E. The classification within the Juglandaceae. Ann. MO. Bot. Gard. 1978, 65, 1058–1087. [Google Scholar] [CrossRef]
- Manchester, S.R. The fossil history of the Juglandaceae. In Monographs in Systematic Botany; Missouri Bot Garden: St. Louis, MO, USA, 1987; Volume 21, pp. 1–137. [Google Scholar]
- Willis, K.J.; McElwain, J.C. The Evolution of Plants; Oxford University Press: New York, NY, USA, 2002; p. 378. [Google Scholar]
- McGranahan, G.H.; Leslie, C. Walnuts (Juglans). In Genetic Resources of Temperate Fruit and Nut Crops; Moore, J.N., Ballington, J.R., Eds.; International Society for Horticultural Science: Wageningen, The Netherlands, 1991; pp. 907–951. [Google Scholar]
- Vavilov, N.I. The wild relatives of fruit trees of the Asian part of the USSR and Caucasus and problems of origin of fruit trees. Tr. Po Prikl. Bot. Genet. Sel. 1931, 23, 343–360. (In Russian) [Google Scholar]
- Molnar, T.J.; Zaurov, D.E.; Capik, J.M.; Eisenman, S.W.; Ford, T.; Nikolyi, L.V.; Funk, C.R. Persian walnuts (Juglans regia L.) in Central Asia. Annu. Rep. North Nut. Grow Assoc. 2011, 101, 56–69. [Google Scholar]
- Carrión, J.S.; Sanchez-Gomez, P. Palynological data in support of the survival of walnut (Juglans regia L.) in the western Mediterranean area during last glacial times. J. Biogeogr. 1992, 19, 623–630. [Google Scholar] [CrossRef]
- Figueiral, I.; Terral, J. Late Quaternary refugia of Mediterranean taxa in the Portuguese Estremadura: Charcoal based palaeovegetation and climatic reconstruction. Quat. Sci. Rev. 2002, 21, 549–558. [Google Scholar] [CrossRef]
- Beer, R.; Kaiser, F.; Schmidt, K.; Ammann, B.; Carraro, G.; Grisa, E.; Tinner, W. Vegetation history of the walnut forests in Kyrgyzstan (Central Asia): Natural or anthropogenic origin? Quat. Sci. Rev. 2008, 27, 621–632. [Google Scholar] [CrossRef]
- Gunn, B.F.; Aradhya, M.; Salick, J.M.; Miller, A.J.; Yongping, Y.; Lin, L.; Xian, H. Genetic variation in walnuts (Juglans regia and J sigillata; Juglandaceae): Species distinctions, human impacts, and the conservation of agrobiodiversity in Yunnan, China. Am. J. Bot. 2010. [Google Scholar] [CrossRef] [PubMed]
- Epple, C. A vegetation study in the walnut and fruit-tree forests of Southern Kyrgyzstan. Phytocoenologia 2001, 31, 571–604. [Google Scholar] [CrossRef]
- Pollegioni, P.; Woeste, K.E.; Chiocchini, F.; Olimpieri, I.; Tortolano, V.; Clark, J.; Hemery, G.E.; Mapelli, S.; Maria, E.M. Landscape genetics of Persian walnut (Juglans regia L.) across its Asian range. Tree Genet. Genomes 2014. [Google Scholar] [CrossRef]
- Pollegioni, P.; Woeste, K.E.; Chiocchini, F.; Del Lungo, S.; Olimpieri, I.; Tortolano, V.; Clark, J.; Hemery, G.E.; Mapelli, S.; Malvolti, M.E. Ancient Humans Influenced the Current Spatial Genetic Structure of Common Walnut Populations in Asia. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Bottema, S. The Holocene history of walnut, sweet-chestnut, manna-ash and plane tree in the Eastern Mediterranean. Pallas 2000, 52, 35–59. [Google Scholar]
- Poldini, L. Atlante Corologico Delle Piante Vascolari Nel Friuli-Venezia Giulia: Inventario Floristico Regionale; Arti Grafiche Friulane: Udine, Italy, 1991; p. 899. [Google Scholar]
- Rottoli, M.; Castiglioni, E. Prehistory of plant growing and collecting in northern Italy, based on seed remains from the early Neolithic to the Chalcolithic (c. 5600–2100 cal b.c.). Veg. Hist. Archaeol. 2009, 18, 91–103. [Google Scholar] [CrossRef]
- Woeste, K.; Burns, R.; Rhodes, O.; Michler, C. Thirty polymorphic nuclear microsatellite loci from black walnut. J. Hered. 2002, 93, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Dangl, G.S.; Woeste, K.; Ardhya, M.K.; Koehmstedt, A.; Simon, C. Characterization of 14 microsatellite markers for genetic analysis and cultivar identification of walnut. J. Am. Soc. Hortic. Sci. 2005, 130, 348–354. [Google Scholar]
- Foroni, I.; Rao, R.; Woeste, K.; Gallitelli, M. Characterization of Juglans regia L. through SSR markers and evaluation of genetic relationships among cultivars and the ‘Sorrento’ landrace. J. Hortic. Sci. Biotechnol. 2005, 80, 49–53. [Google Scholar] [CrossRef]
- Wang, H.; Pei, D. Genetic diversity and structure of walnut populations in Central and Southwestern China revealed by microsatellite markers. J. Am. Soc. Hortic. Sci. 2008, 133, 197–203. [Google Scholar]
- Pollegioni, P.; Woeste, K.; Mugnozza, G.S.; Malvolti, M.E. Retrospective identification of hybridogenic walnut plants by SSR fingerprinting and parentage analysis. Mol. Breed. 2009, 24, 321–335. [Google Scholar] [CrossRef]
- Zhang, R.; Zhu, A.D.; Wang, X.L.; Yu, J.; Zhang, H.R.; Gao, J.S.; Cheng, Y.J.; Deng, X.X. Development of Juglans regia SSR Markers by Data Mining of the EST Database. Plant Mol. Biol. Rep. 2010, 28, 646–653. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Fatahi, R.; Zamani, Z. Analysis of genetic diversity among some Persian walnut genotypes (Juglans regia L.) using morphological traits and SSRs markers. Sci. Hortic. 2011, 130, 146–151. [Google Scholar] [CrossRef]
- Pollegioni, P.; Woeste, K.; Olimpieri, I.; Marandola, D.; Cannata, F.; Malvolti, M.E. Long-term human impacts on genetic structure of Italian walnut inferred by SSR markers. Tree Genet. Genomes 2011, 7, 707–723. [Google Scholar] [CrossRef]
- Ruiz-Garcia, L.; Lopez-Ortega, G.; Fuentes Denia, A.; Frutos Tomas, D. Identification of a walnut (Juglans regia L.) germplasm collection and evaluation of their genetic variability by microsatellite markers. Span. J. Agric. Res. 2011, 9, 179–192. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Han, J.W.; Jin, Q.; Wang, Y.; Pang, X.M.; Li, Y.Y. Development and characterization of new microsatellites for walnut (Juglans regia). Genet. Mol. Res. 2013, 12, 4723–4734. [Google Scholar] [CrossRef] [PubMed]
- Manly, B.F.J. The Statistics of Natural Selection on Animal Populations; Chapman and Hall: London, UK, 1985; pp. 272–282. [Google Scholar]
- Yeh, F.C.; Boyle, T.E. Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belg. J. Bot. 1997, 129, 129–157. [Google Scholar]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Boutin-Ganache, I.; Raposo, M.; Raymond, M.; Deschepper, C.F. M13-tailed primers improve the readability and usability of microsatellite analyses performed with two different allele-sizing methods. Biotechniques 2001, 31, 24–28. [Google Scholar] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Waits, L.P.; Luikart, G.; Taberlet, P. Estimating the probability of identity among genotypes in natural populations: Cautions and guidelines. Mol. Ecol. 2001, 10, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Marshall, T.C.; Slate, J.; Kruuk, L.E.B.; Pemberton, J.M. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 1998, 7, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T.; Taper, M.L. Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv. Genet. 2006. [Google Scholar] [CrossRef]
- Loiselle, B.A.; Sork, V.L.; Nason, J.; Graham, C. Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am. J. Bot. 1995, 82, 1420–1425. [Google Scholar] [CrossRef]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef]
- Sokal, R.R.; Rohlf, F.J. Biometry: The Principles and Practice of Statistics in Biological Research, 3rd ed.; W.H. Freeman, and Company: New York, NY, USA, 1995; 887p. [Google Scholar]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Kalinowski, S.T. Counting alleles with rarefaction: Private alleles and hierarchical sampling designs. Conserv. Genet. 2004, 5, 539–543. [Google Scholar] [CrossRef]
- Kalinowski, S.T. HP-Rare: A computer program for performing rarefaction on measures of allelic diversity. Mol. Ecol. Notes 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Mantel, N.A. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Page, R.D. TreeView: An application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 1996, 12, 357–358. [Google Scholar] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure from multi-locus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [PubMed]
- Pritchard, J.K.; Wen, W.; Falush, D. Documentation for Structure Software: Version 2.3; Department of Human Genetics, University of Chicago: Chicago, IL, USA, 2010; p. 38. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Res. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Guillot, G.; Leblois, R.; Coulon, A.; Frantz, A.C. Statistical methods in spatial genetics. Mol. Ecol. 2009, 18, 4734–4756. [Google Scholar] [CrossRef] [PubMed]
- Guillot, G.; Estoup, A.; Mortier, F.; Cosson, J.F. A spatial statistical model for landscape genetics. Genetics 2005, 170, 1261–1280. [Google Scholar] [CrossRef] [PubMed]
- Guillot, G.; Mortier, F.; Estoup, A. Geneland: A computer package for landscape genetics. Mol. Ecol. Notes 2005, 5, 712–715. [Google Scholar] [CrossRef]
- Guillot, G.; Santos, F.; Estoup, A. Analysing georeferenced population genetics data with Geneland: A new algorithm to deal with null alleles and a friendly graphical user interface. Bioinformatics 2008, 24, 1406–1407. [Google Scholar] [CrossRef] [PubMed]
- Guillot, G. Inference of structure in subdivided populations at low levels of genetic differentiation. The correlated allele frequencies model revisited. Bioinformatics 2008, 24, 2222–2228. [Google Scholar] [CrossRef] [PubMed]
- Youssef, J.; Nassimbeni, P.L. La frutticoltura nel Friuli-Venezia Giulia. Origine, situazione e prospettive (Fruit-growing in Friuli-Venezia Giulia. Origin, situation and prospects) (IN ITALIAN). Frutticoltura 1977, 39, 19–45. [Google Scholar]
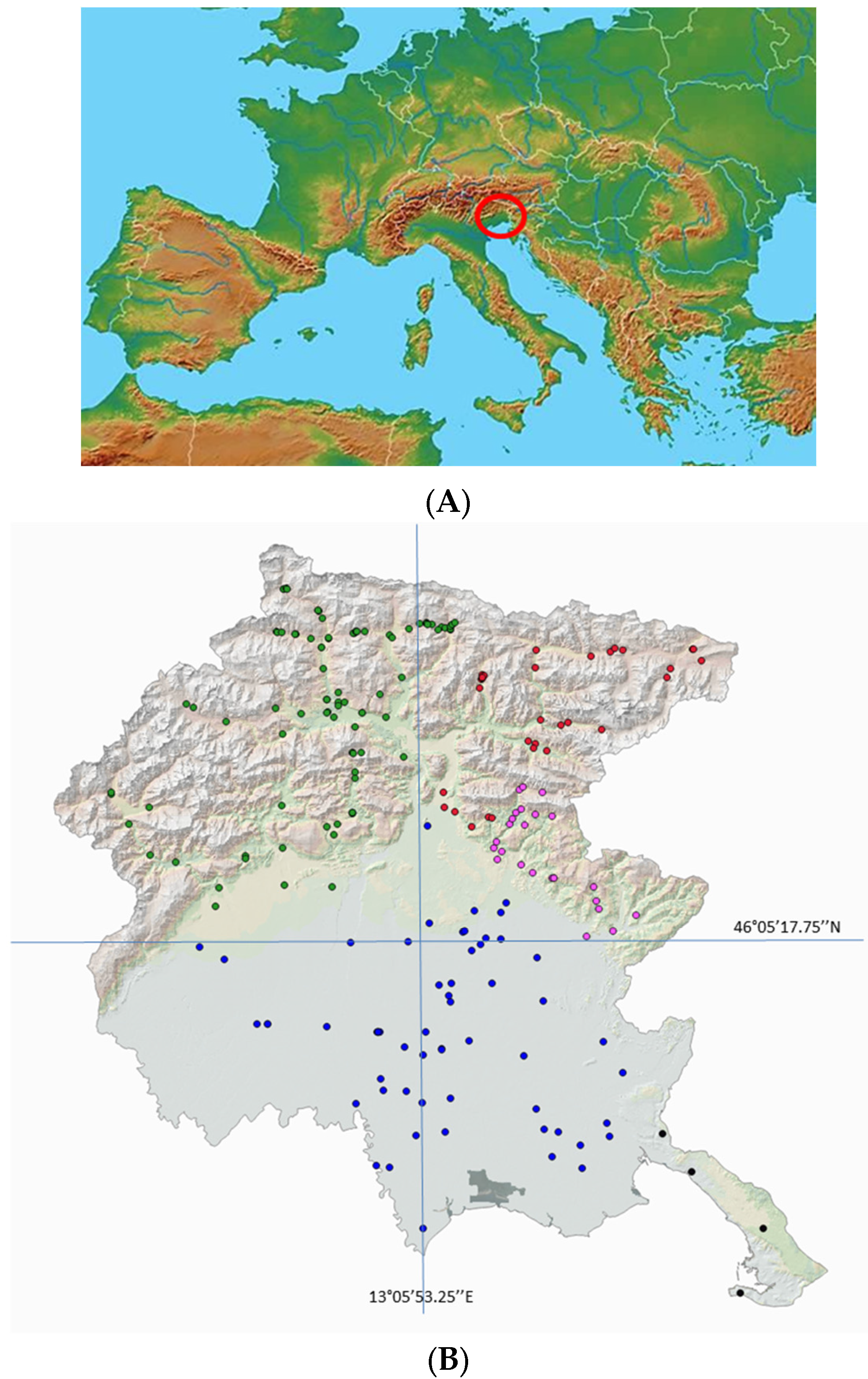
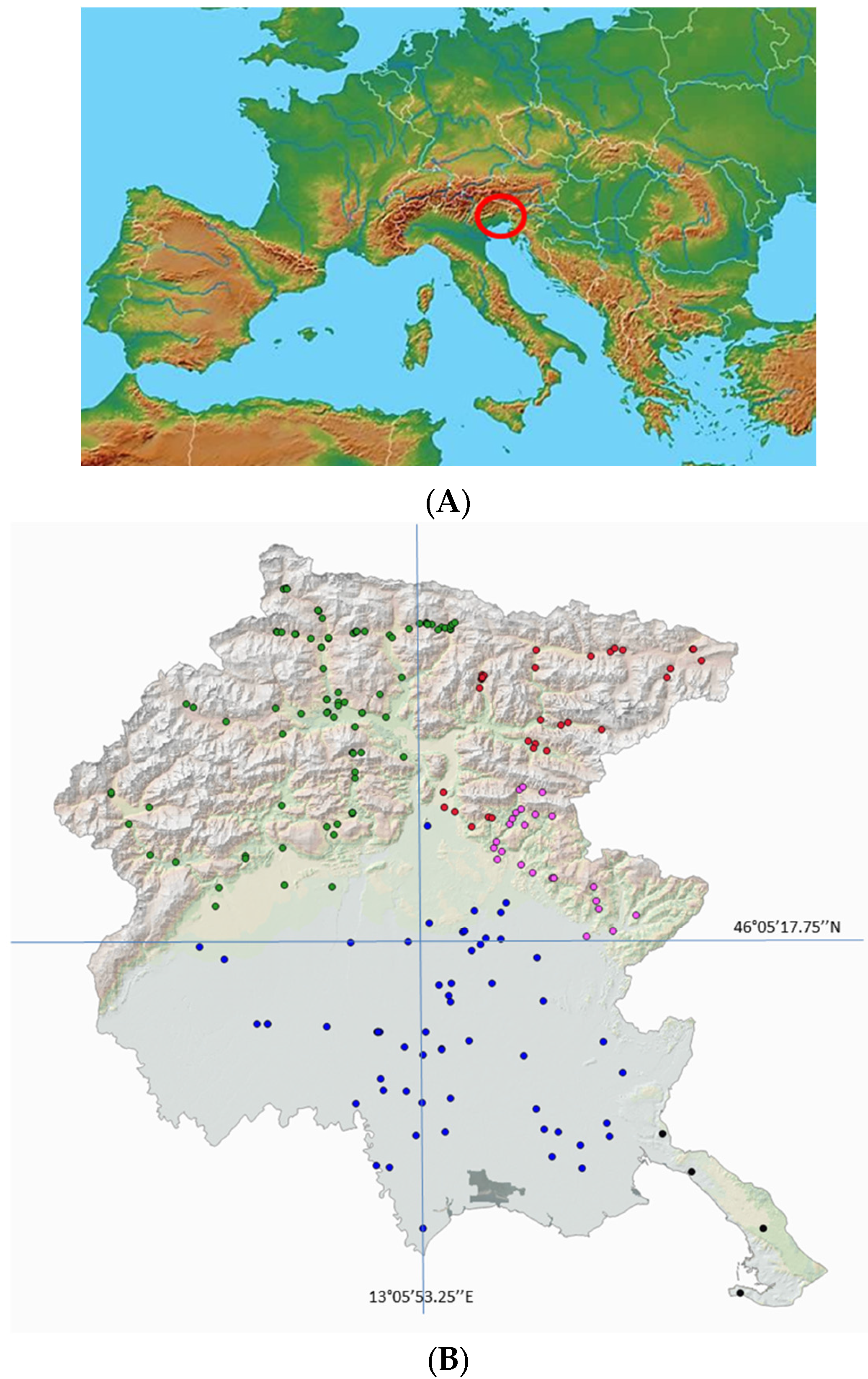
| Color | Geographic Area |
|---|---|
 | Western Alpine Highlands |
 | Eastern Alpine Highlands |
 | Torre Natisone Valleys |
 | Friuli Plains |
 | Trieste Karst |
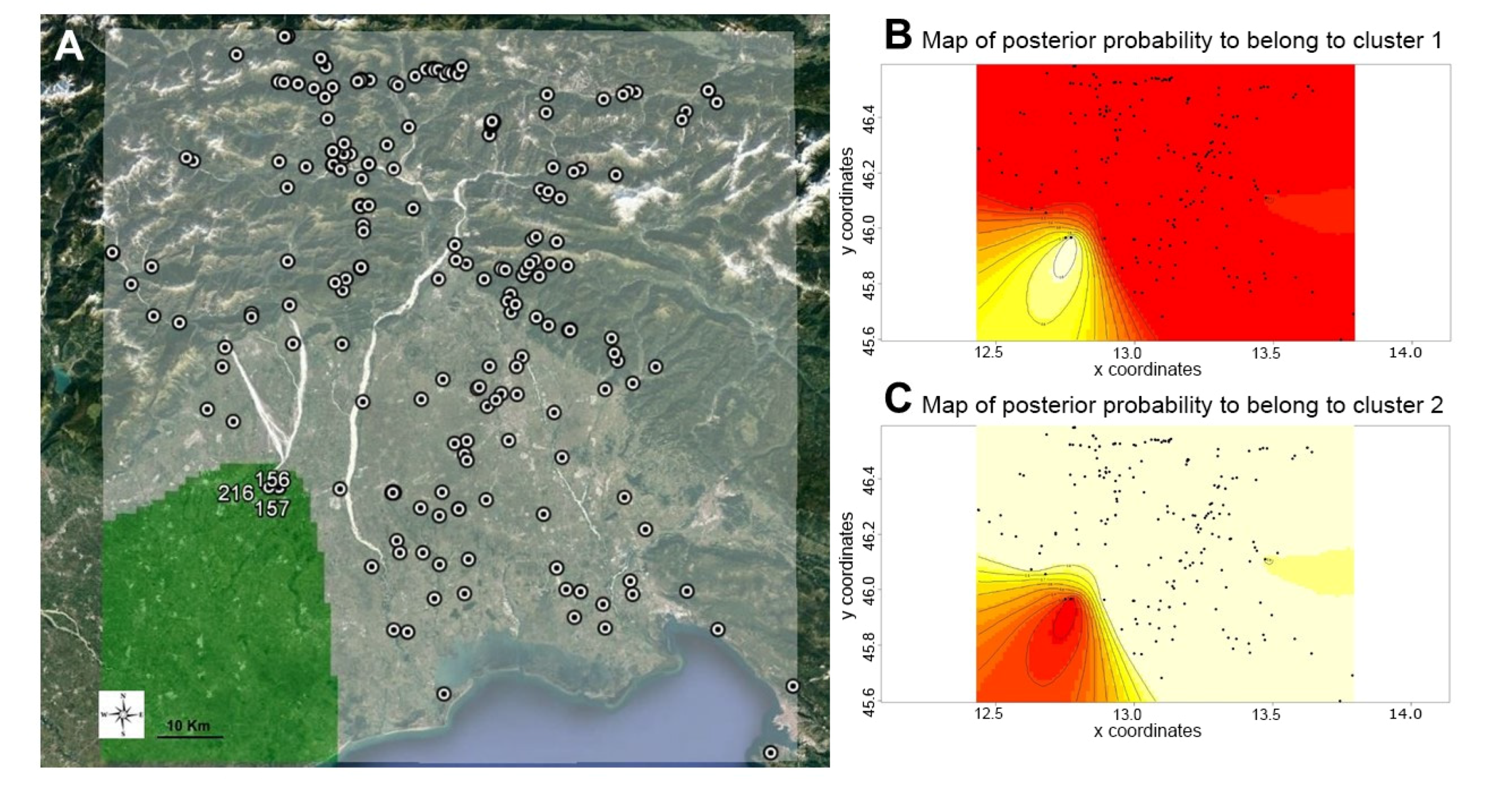

{kind=link}
{kind=link}
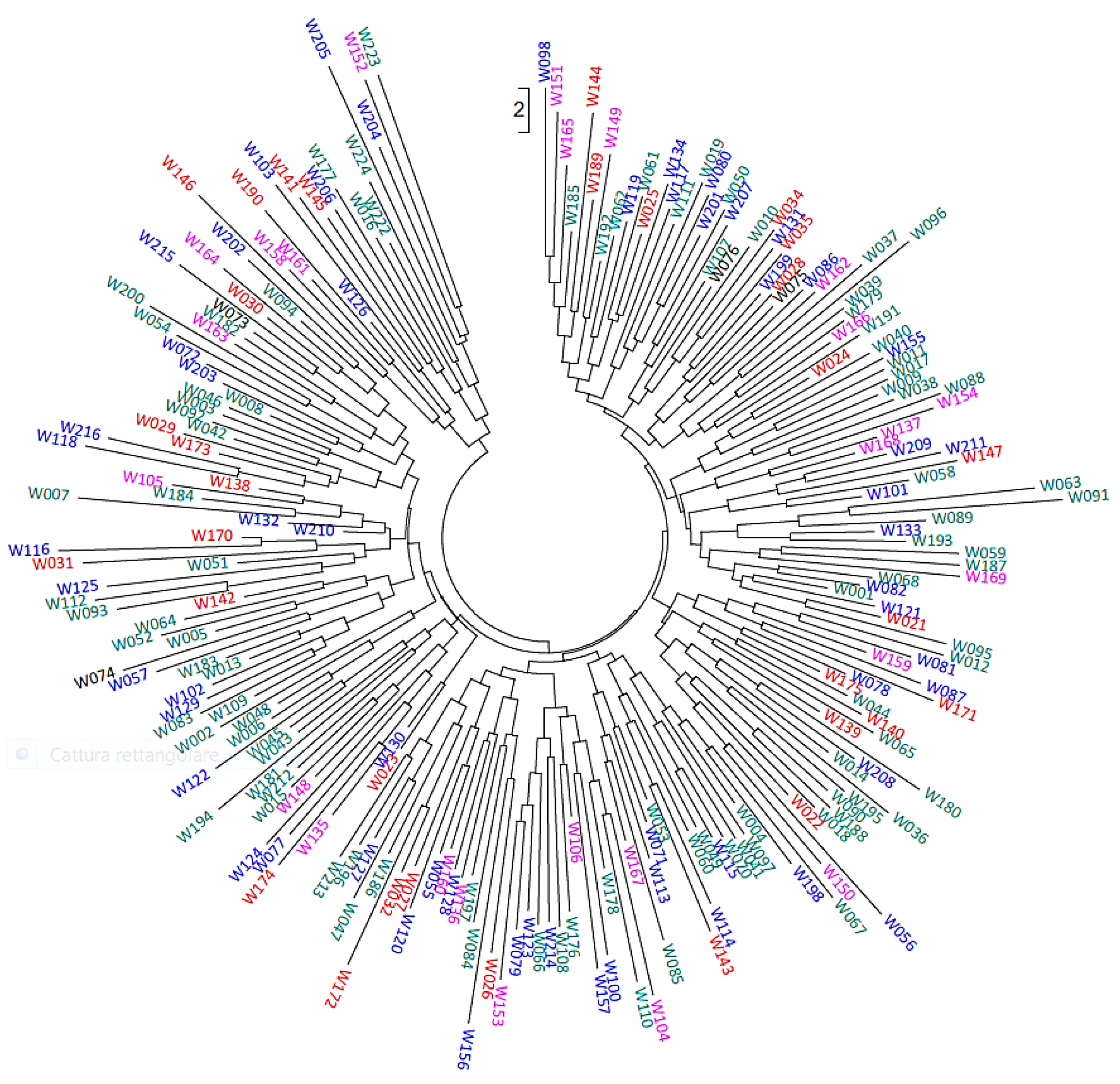
{kind=link}
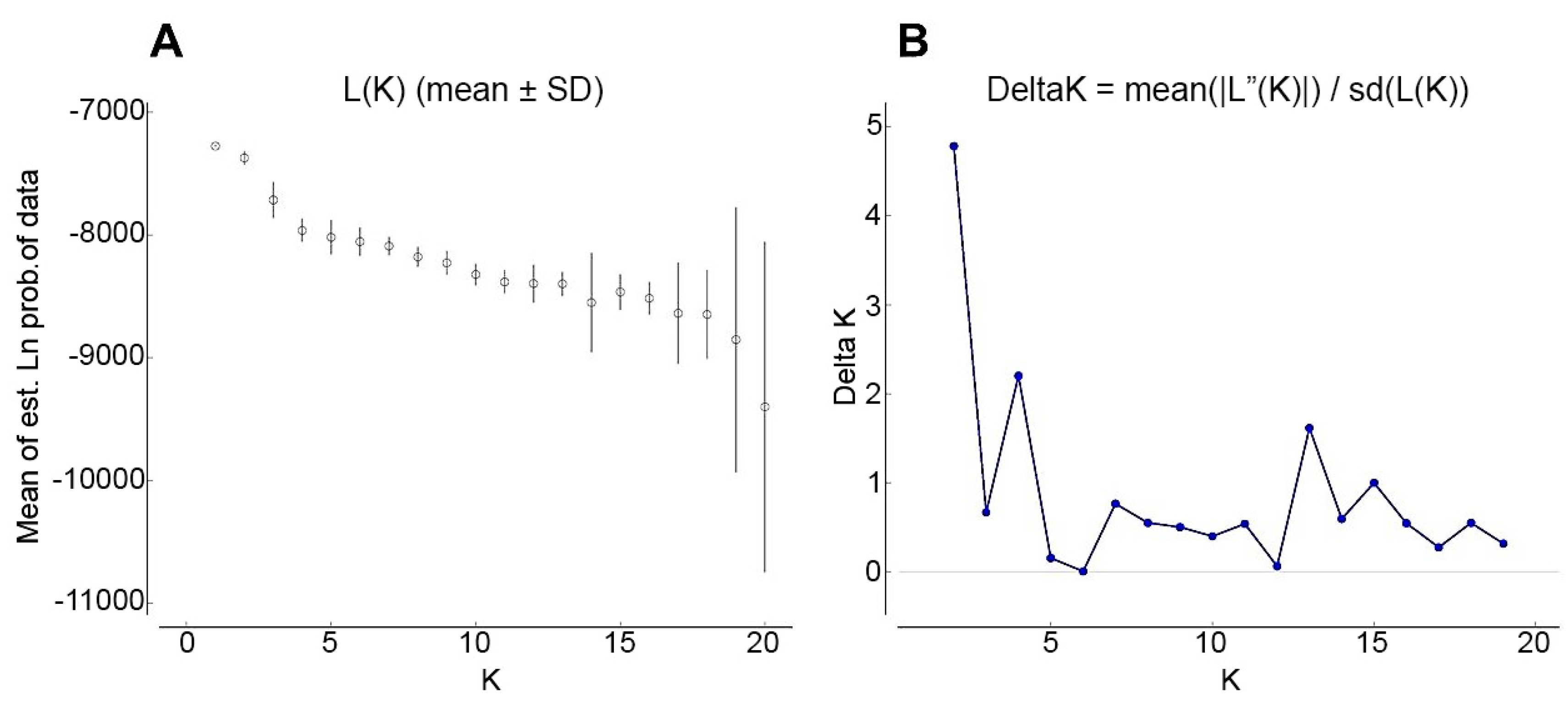
{kind=link}
{kind=link}
| SSR Code | Repeat Array | Forward Primer | Reverse Primer | Tanneal (°C) (a) | Reference |
|---|---|---|---|---|---|
| WGA001 | (GA)5GCA(GA)3GCA(GA)3 | attggaagggaagggaaatg | cgcgcacatacgtaaatcac | 48–58 | Dangl et al., 2005 [18] |
| WGA004 | (GT)5,(GA)15,(GA)11 | tgttgcattgacccacttgt | taagccaacatggtatgcca | 50–58 | Woeste et al., 2002 [17] |
| WGA009 | (GA)16 | catcaaagcaagcaatggg | ccattgctctgtgattggg | 48–58 | Dangl et al., 2005 [18] |
| WGA027 | (GA)30 | aaccctacaacgccttgatg | tgctcaggctccacttcc | 51–55 | Woeste et al., 2002 [17] |
| WGA069 | (GA)4ATATAA(GA)16 | ttagttagcaaacccacccg | agatgcacagaccaaccctc | 45–58 | Woeste et al., 2002 [17] |
| WGA071 | (GA)6,(G)12 | acccgagagatttctgggat | ggacccagctcctcttctct | 45 | Woeste et al., 2002 [17] |
| WGA072 | (CT)14 | aaaccacctaaaaccctgca | acccatccatgatcttccaa | 55–58 | Woeste et al., 2002 [17] |
| WGA089 | (GT)13(GA)21 | acccatctttcacgtgtgtg | tgcctaattagcaatttcca | 53–58 | Dangl et al., 2005 [18] |
| WGA118 | (GA)18(GT)11 | tgtgctctgatctgcctcc | gggtgggtgaaaagtagcaa | 55–62 | Dangl et al., 2005 [18] |
| WGA225 | (AG)14 | caatccctctcctgggcag | tgttccactgaccacttcca | n.r. | Dangl et al., 2005 [18] (b) |
| WGA321 | (GA)14 | tccaatcgaaactccaaagg | tgtccaaagacgatgatgga | 50–58 | Dangl et al., 2005 [18] |
| WGA331 | (GA)13 | tccccctgaaatcttctcct | cggtggtgtaaggcaaatg | 53–58 | Dangl et al., 2005 [18] |
| WGA349 | (CT)14 | gtggcgaaagtttattttttgc | acaaatgcacagcagcaaac | n.r. | Dangl et al., 2005 [18] (b) |
| WGA376 | (AG)2AA(AG)6 | gccctcaaagtgatgaacgt | tcatccatatttacccctttcg | n.r. | Dangl et al., 2005 [18] (b) |
| Contig_40 | (CTGT)5 | tgggctgagctggattgccgt | tccaccgtcatggtttccacg | 59 | Zhang et al., 2010 [22] |
| Contig_156 | (TTTG)6 | tgcaagagtggcgcaggcactg | tggtagcctaatctcatggctcg | 60 | Zhang et al., 2010 [22] |
| Contig_642 | (CAG)7 | tgaaaggttttggcctccaatgg | tgagatcatgggctgcctgtagg | 59 | Zhang et al., 2010 [22] |
| Contig_721 | (CTT)8 | accccttggtttgaactgcgac | agatccaactttcgcgtggaac | 57 | Zhang et al., 2010 [22] |
| Contig_1528 | (CCT)7 | ccgaagagatcctaagctcaacc | gaggtggaaatgatggtggggtg | 59 | Zhang et al., 2010 [22] |
| Contig_1681 | (TTC)8 | agagatttctccaggaaggctcc | tctggtggccaacgatagccga | 62 | Zhang et al., 2010 [22] |
| Contig_1692 | (CCA)6 | caatggtcagtttccgtccgatc | cgagctcgaatacttctcgtcg | 58 | Zhang et al., 2010 [22] |
| SSR Marker | Samples n | Alleles n | Size Range bp (b) | Genotypes n | Ho | GD | PIC | MAF | HW | Fnull | PID Unrelated | PID Full-Sibs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genomic SSR | ||||||||||||
| WGA004 | 215 | 2 | 228–238 | 3 | 0.619 | 0.482 | 0.365 | 0.60 | ** | −0.125 | 0.385 | 0.606 |
| WGA009 | 215 | 3 | 229–245 | 6 | 0.619 | 0.639 | 0.561 | 0.40 | NS | +0.016 | 0.208 | 0.483 |
| WGA027 | 212 | 2 | 206–210 | 3 | 0.533 | 0.488 | 0.368 | 0.58 | NS | −0.046 | 0.382 | 0.602 |
| WGA069 | 209 | 9 | 160–182 | 18 | 0.656 | 0.745 | 0.698 | 0.32 | NS | +0.063 | 0.111 | 0.406 |
| WGA071 | 209 | 3 | 205–209 | 4 | 0.498 | 0.503 | 0.381 | 0.54 | NS | +0.002 | 0.369 | 0.591 |
| WGA072 | 214 | 5 | 128–142 | 6 | 0.411 | 0.467 | 0.368 | 0.64 | NS | +0.058 | 0.383 | 0.613 |
| WGA089 | 215 | 4 | 212–220 | 7 | 0.712 | 0.673 | 0.599 | 0.33 | NS | −0.029 | 0.180 | 0.459 |
| WGA118 | 215 | 7 | 185–207 | 11 | 0.581 | 0.581 | 0.490 | 0.48 | NS | −0.004 | 0.267 | 0.527 |
| WGA225 | 194 | 3 | 190–202 | 6 | 0.567 | 0.644 | 0.566 | 0.40 | NS | +0.063 | 0.204 | 0.480 |
| WGA321 | 215 | 5 | 223–245 | 12 | 0.642 | 0.674 | 0.608 | 0.41 | NS | +0.025 | 0.172 | 0.457 |
| WGA331 | 214 | 4 | 273–279 | 6 | 0.481 | 0.484 | 0.373 | 0.60 | NS | +0.003 | 0.377 | 0.603 |
| WGA349 | 205 | 6 | 262–276 | 11 | 0.405 | 0.646 | 0.591 | 0.51 | *** | +0.226 | 0.180 | 0.473 |
| WGA376 | 204 | 8 | 231–256 | 17 | 0.525 | 0.634 | 0.594 | 0.56 | * | +0.094 | 0.174 | 0.477 |
| Mean | 4.7 | 8.5 | 0.558 | 0.589 | 0.505 | 0.49 | ||||||
| EST-derived SSR | ||||||||||||
| Contig_40 | 215 | 3 | 210–223 | 4 | 0.349 | 0.340 | 0.286 | 0.79 | NS | −0.005 | 0.490 | 0.703 |
| Contig_156 | 215 | 2 | 300–313 | 3 | 0.433 | 0.493 | 0.371 | 0.56 | NS | +0.064 | 0.379 | 0.599 |
| Contig_642 | 215 | 2 | 262–281 | 3 | 0.488 | 0.494 | 0.371 | 0.56 | NS | +0.004 | 0.379 | 0.598 |
| Contig_721 | 215 | 3 | 369–390 | 4 | 0.051 | 0.059 | 0.058 | 0.97 | ND | +0.068 | 0.887 | 0.942 |
| Contig_1528 | 215 | 3 | 154–163 | 6 | 0.553 | 0.562 | 0.463 | 0.50 | NS | +0.008 | 0.290 | 0.542 |
| Contig_1681 | 215 | 4 | 197–210 | 9 | 0.619 | 0.647 | 0.576 | 0.46 | NS | +0.023 | 0.196 | 0.476 |
| Contig_1692 | 215 | 2 | 208–211 | 2 | 0.074 | 0.072 | 0.069 | 0.96 | ND | −0.010 | 0.864 | 0.930 |
| Mean | 2.7 | 4.4 | 0.367 | 0.381 | 0.313 | 0.69 | ||||||
| General Mean (a) | 4.0 | 6.4 | 0.462 | 0.516 | 0.438 | 0.59 | 2.93E-11 | 1.07E-5 | ||||
| Source of Variation | d.f. | Variance Components | % Variation |
|---|---|---|---|
| Among populations | 4 | 28.18 | 2.12 |
| Among individuals | 210 | 1296.91 | 97.87 |
| Total | 214 | 1325.09 |
| Population | WAH | EAH | TNV | FP | TK |
|---|---|---|---|---|---|
| Western Alpine Highlands (WAH) (95) | |||||
| Eastern Alpine Highlands (EAH) (32) | 0.005 | ||||
| Torre-Natisone Valley (TNV)(25) | 0.008 | 0.012 | |||
| Friuli Plains (FP) (59) | 0.005 | 0.005 | 0.010 | ||
| Trieste Karst (TK) (4) | 0.046 | 0.042 | 0.052 | 0.048 |
| Population | A | Ne | Rs | Ho | GD | Pa |
|---|---|---|---|---|---|---|
| Western Alpine Highlands (95) | 3.050 | 2.257 | 2.25 | 0.483 | 0.506 | WGA118 (220), WGA331 (298) |
| Eastern Alpine Highlands (32) | 3.200 | 2.279 | 2.29 | 0.489 | 0.511 | WGA069 (178, 198), WGA118 (212), WGA331 (294), WGA376 (268) |
| Torre-Natisone Valley (25) | 2.950 | 2.301 | 2.29 | 0.519 | 0.514 | WGA072 (156), WGA118 (222) |
| Friuli Plains (59) | 3.400 | 2.256 | 2.27 | 0.488 | 0.515 | WGA069 (186, 188), WGA071 (224), WGA072 (148), WGA349 (284, 291) |
| Trieste Karst (4) | 2.350 | 1.978 | 2.23 | 0.554 | 0.431 | WGA376 (257) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vischi, M.; Chiabà, C.; Raranciuc, S.; Poggetti, L.; Messina, R.; Ermacora, P.; Cipriani, G.; Paffetti, D.; Vettori, C.; Testolin, R. Genetic Diversity of Walnut (Juglans Regia L.) in the Eastern Italian Alps. Forests 2017, 8, 81. https://doi.org/10.3390/f8030081
Vischi M, Chiabà C, Raranciuc S, Poggetti L, Messina R, Ermacora P, Cipriani G, Paffetti D, Vettori C, Testolin R. Genetic Diversity of Walnut (Juglans Regia L.) in the Eastern Italian Alps. Forests. 2017; 8(3):81. https://doi.org/10.3390/f8030081
Chicago/Turabian StyleVischi, Massimo, Cristina Chiabà, Steluta Raranciuc, Luca Poggetti, Rachele Messina, Paolo Ermacora, Guido Cipriani, Donatella Paffetti, Cristina Vettori, and Raffaele Testolin. 2017. "Genetic Diversity of Walnut (Juglans Regia L.) in the Eastern Italian Alps" Forests 8, no. 3: 81. https://doi.org/10.3390/f8030081