Hepatitis C Virus Infection: Molecular Pathways to Steatosis, Insulin Resistance and Oxidative Stress

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

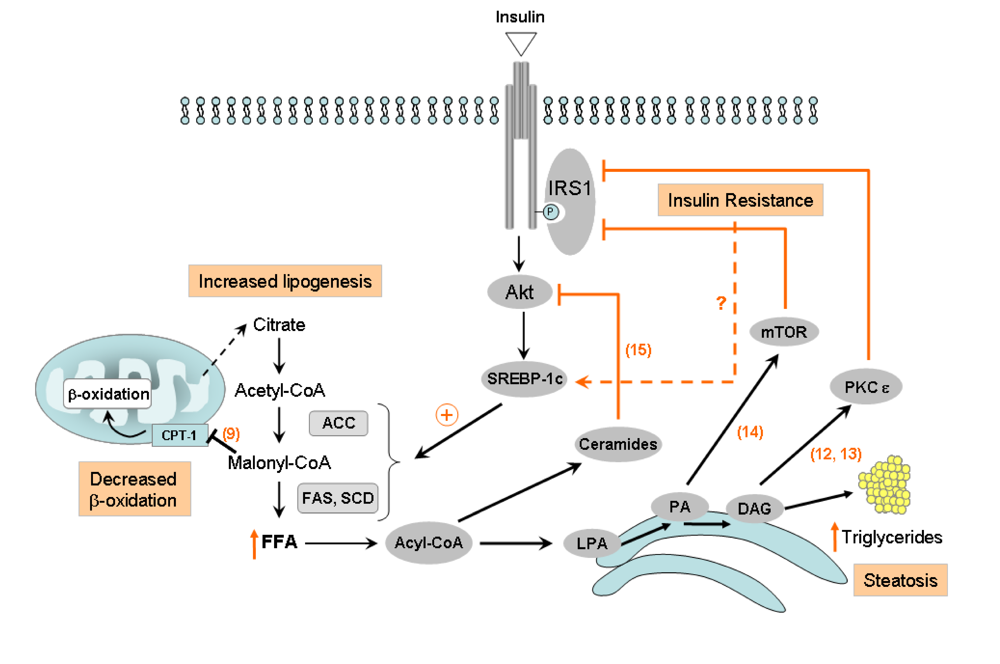
2. HCV interference with insulin signaling

3. HCV and oxidative stress

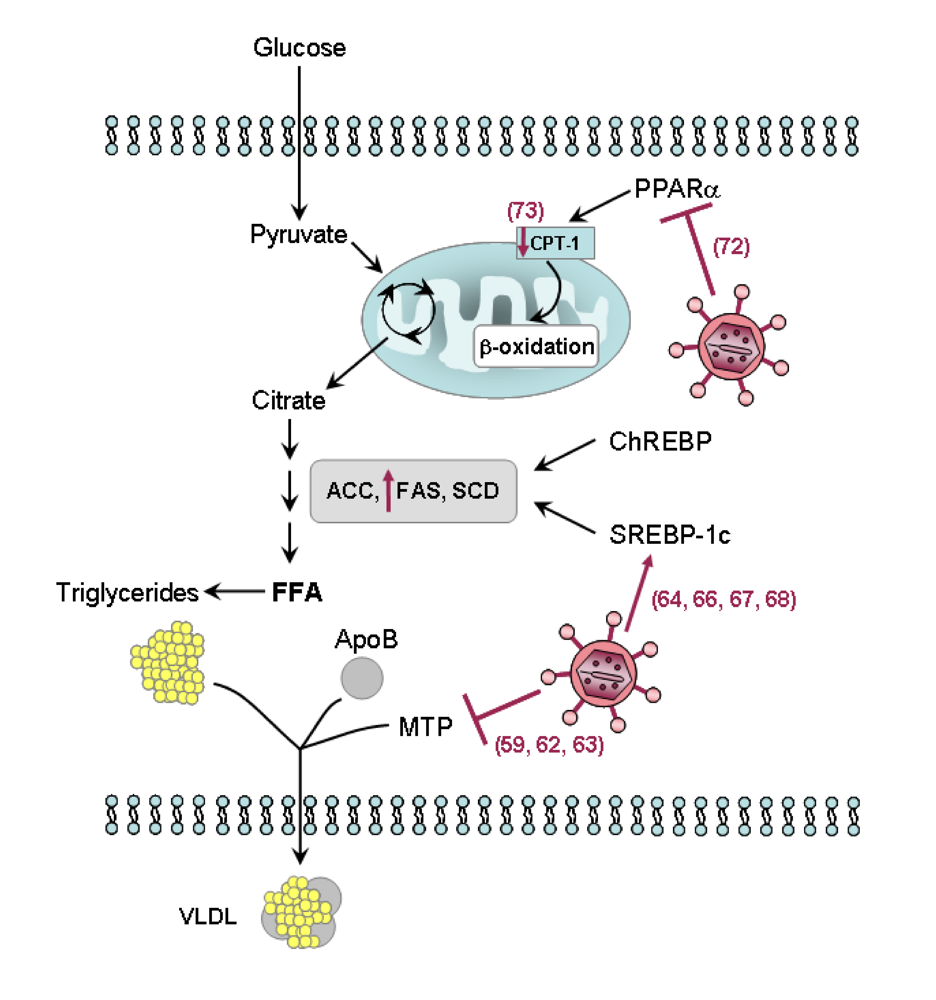
4. Role of the HCV in steatosis development

5. Clinical consequences
6. Conclusions and relevant questions for the future
- What is the exact relationship between virally-induced steatosis and HCV replication? Can we pharmacologically manipulate the lipid metabolism to inhibit HCV replication in vivo?
- What is the long-term clinical impact of purely viral IR? Is this associated with an increased cardiovascular risk, as is the metabolic syndrome-associated IR?
- Are life-style changes – especially increased physical activity – associated with metabolic changes in the liver, and, if this is the case, what would be the consequences for HCV replication, HCV-related steatosis and HCV-related IR?
Acknowledgments
References and Notes
- National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C. Hepatology 2002, s3–s20.
- Alter, M.J. Epidemiology of hepatitis C in the West. Semin. Liver Dis. 1995, 15, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Micallef, J.M.; Kaldor, J.M.; Dore, G.J. Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. J. Viral Hepat. 2006, 13, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene 2006, 25, 3834–3847. [Google Scholar] [CrossRef] [PubMed]
- Fartoux, L.; Poujol-Robert, A.; Guechot, J.; Wendum, D.; Poupon, R.; Serfaty, L. Insulin resistance is a cause of steatosis and fibrosis progression in chronic hepatitis C. Gut 2005, 54, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Pekow, J.R.; Bhan, A.K.; Zheng, H.; Chung, R.T. Hepatic steatosis is associated with increased frequency of hepatocellular carcinoma in patients with hepatitis C-related cirrhosis. Cancer 2007, 109, 2490–2496. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.Y.; Xie, X.; Lai, M.M.; Wu, P.C. Apoptosis and viral hepatitis. Semin. Liver Dis. 1998, 18, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Bieche, I.; Asselah, T.; Laurendeau, I.; Vidaud, D.; Degot, C.; Paradis, V.; Bedossa, P.; Valla, D.-C.; Marcellin, P.; Vidaud, M. Molecular profiling of early stage liver fibrosis in patients with chronic hepatitis C virus infection. Virology 2005, 332, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Hoppel, C.L.; Tomec, R.J. Carnitine palmityltransferase. Location of two enzymatic activities in rat liver mitochondria. J. Biol. Chem. 1972, 247, 832–841. [Google Scholar] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. U S A 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Neschen, S.; Morino, K.; Hammond, L.E.; Zhang, D.; Liu, Z.X.; Romanelli, A.J.; Cline, G.W.; Pongratz, R.L.; Zhang, X.-M.; Choi, C.S.; Coleman, R.A.; Shulman, G.I. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005, 2, 55–65. [Google Scholar] [CrossRef]
- Nagle, C.A.; An, J.; Shiota, M.; Torres, T.P.; Cline, G.W.; Liu, Z.X.; Wang, S.; Catlin, R.L.; Shulman, G.I.; Newgard, C.B.; A. Coleman, R.A. Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase 1 in rats causes insulin resistance. J. Biol. Chem. 2007, 282, 14807–14815. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.A. Regulation of mTOR by phosphatidic acid? Cancer Res. 2007, 67, 1–4. [Google Scholar] [CrossRef]
- Holland, W.L.; Summers, S.A. Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. [Google Scholar] [CrossRef] [PubMed]
- Vidali, M.; Tripodi, M.F.; Ivaldi, A.; Zampino, R.; Occhino, G.; Restivo, L.; Sutti, S.; Marrone, A.; Ruggiero, G.; Albano, E.; Adinolfi, L.E. Interplay between oxidative stress and hepatic steatosis in the progression of chronic hepatitis C. J. Hepatol. 2008, 48, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Aytug, S.; Reich, D.; Sapiro, L.E.; Bernstein, D.; Begum, N. Impaired IRS-1/PI3-kinase signaling in patients with HCV: a mechanism for increased prevalence of type 2 diabetes. Hepatology 2003, 38, 1384–1392. [Google Scholar] [PubMed]
- Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Tsukamoto, K.; Kimura, S.; Moriya, K.; Koike, K. Hepatitis C virus infection and diabetes: direct involvement of the virus in the development of insulin resistance. Gastroenterology 2004, 126, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Yoshida, T.; Harada, M.; Hisamoto, T.; Nagao, Y.; Ide, T.; Taniguchi, E.; Kumemura, H.; Hanada, S.; Maeyama, M.; Baba, S.; Koga, H.; Kumashiro, R.; Ueno, T.; Ogata, H.; Yoshimura, A.; Sata, M. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am. J. Pathol. 2004, 165, 1499–1508. [Google Scholar] [PubMed]
- Pazienza, V.; Clément, S.; Pugnale, P.; Conzelman, S.; Foti, M.; Mangia, A.; Negro, F. The Hepatitis C Virus Core Protein of Genotypes 3a and 1b Down-Regulates Insulin Receptor Substrate 1 via Genotype-Specific Mechanisms. Hepatology 2007, In Press. [Google Scholar]
- Christen, V.; Treves, S.; Duong, F.H.; Heim, M.H. Activation of endoplasmic reticulum stress response by hepatitis viruses up-regulates protein phosphatase 2A. Hepatology 2007, 46, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Bernsmeier, C.; Duong, F.H.; Christen, V.; Pugnale, P.; Negro, F.; Terracciano, L.; Heim, M.H. Virus-induced over-expression of protein phosphatase 2A inhibits insulin signalling in chronic hepatitis C. J. Hepatol. 2008, 49, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Saito, K.; Ait-Goughoulte, M.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream akt/protein kinase B signaling pathway for insulin resistance. J. Virol. 2008, 82, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Sugimoto, R.; Ma, N.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; Kawanishi, S.; Watanabe, S.; Kaito, M.; Takei, Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J. Viral Hepat. 2008, 15, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.J.; Bosch-Morell, F.; Romero, B.; Rodrigo, J.M.; Serra, M.A.; Romero, F.J. Serum malondialdehyde: possible use for the clinical management of chronic hepatitis C patients. Free Radic. Biol. Med. 1998, 25, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Swietek, K.; Juszczyk, J. Reduced glutathione concentration in erythrocytes of patients with acute and chronic viral hepatitis. J. Viral Hepat. 1997, 4, 139–141. [Google Scholar] [PubMed]
- Houglum, K.; Venkataramani, A.; Lyche, K.; Chojkier, M. A pilot study of the effects of d-alpha-tocopherol on hepatic stellate cell activation in chronic hepatitis C. Gastroenterology 1997, 113, 1069–1073. [Google Scholar] [CrossRef] [PubMed]
- Gabbay, E.; Zigmond, E.; Pappo, O.; Hemed, N.; Rowe, M.; Zabrecky, G.; Cohen, R.; Ilan, Y. Antioxidant therapy for chronic hepatitis C after failure of interferon: results of phase II randomized, double-blind placebo controlled clinical trial. World J. Gastroenterol. 2007, 13, 5317–5323. [Google Scholar] [PubMed]
- Mitsuyoshi, H.; Itoh, Y.; Sumida, Y.; Minami, M.; Yasui, K.; Nakashima, T.; Okanoue, T. Evidence of oxidative stress as a cofactor in the development of insulin resistance in patients with chronic hepatitis C. Hepatol. Res. 2008, 38, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Vendemiale, G.; Grattagliano, I.; Portincasa, P.; Serviddio, G.; Palasciamo, G.; Altomare, E. Oxidative stress in symptom-free HCV carriers: relation with ALT flare-up. Eur. J. Clin. Invest. 2001, 31, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Nakagawa, K.; Santa, T.; Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Miyazawa, T.; Ishibashi, K.; Horie, T.; Imai, K.; Todoroki, T.; Kimura, S.; Koike, K. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001, 61, 4365–4370. [Google Scholar] [PubMed]
- Okuda, M.; Li, K.; Beard, M.R.; Showalter, L.A.; Scholle, F.; Lemon, S.M.; Weinman, S.A. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 2002, 122, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.Y.; Ahmad, I.M.; Spitz, D.R.; Schmidt, W.N.; Britigan, B.E. Hepatitis C virus-core and non structural proteins lead to different effects on cellular antioxidant defenses. J. Med. Virol. 2005, 76, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Dionisio, N.; Garcia-Mediavilla, M.V.; Sanchez-Campos, S.; Majano, P.L.; Benedicto, I.; Rosado, J.A.; Salido, G.M.; Gonzalez-Gallego, J. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J. Hepatol. 2009, 50, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; Ren, S.; Pietschmann, T.; Kartenbeck, J.; Kaehlcke, K.; Bartenschlager, R.; Yen, T.S.; Ott, M. Targeting of hepatitis C virus core protein to mitochondria through a novel C-terminal localization motif. J. Virol. 2004, 78, 7958–7968. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Boehning, D.F.; Qian, T.; Popov, V.L.; Weinman, S.A. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. Faseb J. 2007, 21, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. U S A 2001, 98, 9599–9604. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.C.; Marchant, J.S. Enhanced Ca2+ leak from ER Ca2+ stores induced by hepatitis C NS5A protein. Biochem Biophys Res Commun 2008, 368, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.A.; Walters, K.A.; Lamb, S.E.; Yeh, M.M.; Zhu, L.F.; Kneteman, N.; Doyle, J.S.; Katze, M.G.; Tyrrell, D.L. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009, 5, e1000291. [Google Scholar] [CrossRef] [PubMed]
- Bureau, C.; Bernad, J.; Chaouche, N.; Orfila, C.; Beraud, M.; Gonindard, C.; Alric, L.; Vinel, J.P.; Pipy, B. Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J. Biol. Chem. 2001, 276, 23077–23083. [Google Scholar] [CrossRef] [PubMed]
- Thoren, F.; Romero, A.; Lindh, M.; Dahlgren, C.; Hellstrand, K. A hepatitis C virus-encoded, nonstructural protein (NS3) triggers dysfunction and apoptosis in lymphocytes: role of NADPH oxidase-derived oxygen radicals. J. Leukoc. Biol. 2004, 76, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Asselah, T.; Rubbia-Brandt, L.; Marcellin, P.; Negro, F. Steatosis in chronic hepatitis C: why does it really matter? Gut 2006, 55, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Thomopoulos, K.C.; Arvaniti, V.; Tsamantas, A.C.; Dimitropoulou, D.; Gogos, C.A.; Siagris, D.; Theocharis, G.J.; Labropoulou-Karatza, C. Prevalence of liver steatosis in patients with chronic hepatitis B: a study of associated factors and of relationship with fibrosis. Eur. J. Gastroenterol. Hepatol. 2006, 18, 233–237. [Google Scholar] [CrossRef]
- Rubbia-Brandt, L.; Fabris, P.; Paganin, S.; Leandro, G.; Male, P.J.; Giostra, E.; Carlotto, A.; Bozzola, L.; Smedile, A.; Negro, F. Steatosis affects chronic hepatitis C progression in a genotype specific way. Gut 2004, 53, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Gambardella, M.; Andreana, A.; Tripodi, M.F.; Utili, R.; Ruggiero, G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology 2001, 33, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.M.; Kench, J.; Farrell, G.C.; Lin, R.; Samarasinghe, D.; Liddle, C.; Byth, K.; George, J. Genotype-specific mechanisms for hepatic steatosis in chronic hepatitis C infection. J. Gastroenterol. Hepatol. 2002, 17, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Farrell, G.C.; Fung, C.; George, J. Hepatitis C virus genotype 3 is cytopathic to hepatocytes: Reversal of hepatic steatosis after sustained therapeutic response. Hepatology 2002, 36, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Poynard, T.; Ratziu, V.; McHutchison, J.; Manns, M.; Goodman, Z.; Zeuzem, S.; Younossi, Z.; Albrecht, J. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology 2003, 38, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Rubbia-Brandt, L.; Giostra, E.; Mentha, G.; Quadri, R.; Negro, F. Expression of liver steatosis in hepatitis C virus infection and pattern of response to alpha-interferon. J. Hepatol. 2001, 35, 307. [Google Scholar] [CrossRef] [PubMed]
- Barba, G.; Harper, F.; Harada, T.; Kohara, M.; Goulinet, S.; Matsuura, Y.; Eder, G.; Schaff, Z.; Chapman, M.J.; Miyamura, T.; Bréchot, C. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. U S A 1997, 94, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Yotsuyanagi, H.; Shintani, Y.; Fujie, H.; Ishibashi, K.; Matsuura, Y.; Miyamura, T.; Koike, K. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 1997, 78, 1527–1531. [Google Scholar] [PubMed]
- Abid, K.; Pazienza, V.; de Gottardi, A.; Rubbia-Brandt, L.; Conne, B.; Pugnale, P.; Rossi, C.; Mangiab, A.; Negro, F. An in vitro model of hepatitis C virus genotype 3a-associated triglycerides accumulation. J. Hepatol. 2005, 42, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Hourioux, C.; Patient, R.; Morin, A.; Blanchard, E.; Moreau, A.; Trassard, S.; Giraudeau, B.; Roingeard, P. The genotype 3-specific hepatitis C virus core protein residue phenylalanine 164 increases steatosis in an in vitro cellular model. Gut 2007, 56, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Serfaty, L.; Andreani, T.; Giral, P.; Carbonell, N.; Chazouilleres, O.; Poupon, R. Hepatitis C virus induced hypobetalipoproteinemia: a possible mechanism for steatosis in chronic hepatitis C. J. Hepatol. 2001, 34, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Hofer, H.; Bankl, H.C.; Wrba, F.; Steindl-Munda, P.; Peck-Radosavljevic, M.; Osterreicher, C.; Mueller, C.; Gangl, A.; Ferenci, P. Hepatocellular fat accumulation and low serum cholesterol in patients infected with HCV-3a. Am. J. Gastroenterol. 2002, 97, 2880–2885. [Google Scholar] [CrossRef] [PubMed]
- Perlemuter, G.; Sabile, A.; Letteron, P.; Vona, G.; Topilco, A.; Chretien, Y.; Koike, K.; Pessayre, D.; Chapman, J.; Barba, G.; Bréchot, C. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. Faseb J. 2002, 16, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Wetterau, J.R.; Lin, M.C.; Jamil, H. Microsomal triglyceride transfer protein. Biochim. Biophys. Acta 1997, 1345, 136–150. [Google Scholar] [PubMed]
- Hussain, M.M.; Shi, J.; Dreizen, P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res 2003, 44, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, S.; Realdon, S.; Iqbal, J.; Gerotto, M.; Dal Pero, F.; Bortoletto, G.; Marcolongo, M.; Vario, A.; Datz, C.; Hussain, M.M.; Alberti, A. Liver microsomal triglyceride transfer protein is involved in hepatitis C liver steatosis. Gastroenterology 2006, 130, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, S.; Osterreicher, C.H.; Marcolongo, M.; Datz, C.; Aigner, E.; Schlabrakowski, A.; Realdon, S.; Gerotto, M.; Alberti, A.; Stickel, F. Microsomal triglyceride transfer protein polymorphism (-493G/T) is associated with hepatic steatosis in patients with chronic hepatitis C. Liver Int. 2009, 29, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; Chisari, F.V. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. U S A 2002, 99, 15669–15674. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Felmlee, D.J.; Negro, F.; Siddiqui, A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 2007, 81, 8122–8130. [Google Scholar] [CrossRef] [PubMed]
- Oem, J.K.; Jackel-Cram, C.; Li, Y.P.; Zhou, Y.; Zhong, J.; Shimano, H.; Babiuk, L.A.; Liu, Q. Activation of sterol regulatory element-binding protein 1c and fatty acid synthase transcription by hepatitis C virus non-structural protein 2. J. Gen. Virol. 2008, 89, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Jun, H.J.; Wakita, T.; Cheong, J.H.; Hwang, S.B. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J. Biol. Chem. 2009, 284, 9237–9246. [Google Scholar] [CrossRef] [PubMed]
- Jackel-Cram, C.; Babiuk, L.A.; Liu, Q. Up-regulation of fatty acid synthase promoter by hepatitis C virus core protein: genotype-3a core has a stronger effect than genotype-1b core. J. Hepatol. 2007, 46, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hood, B.L.; Chadwick, S.L.; Liu, S.; Watkins, S.C.; Luo, G.; Conrads, T.P.; Wang, T. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology 2008, 48, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, T.; Suzuki, T.; Shimoike, T.; Suzuki, R.; Moriya, K.; Shintani, Y.; Fujie, H.; Koike, K.; Miyamura, T. Interaction of hepatitis C virus core protein with retinoid X receptor alpha modulates its transcriptional activity . Hepatology 2002, 35 , 937–946. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Dharancy, S.; Malapel, M.; Desreumaux, P. Hepatitis C virus infection down-regulates the expression of peroxisome proliferator-activated receptor alpha and carnitine palmitoyl acyl-CoA transferase 1A. World J. Gastroenterol. 2005, 11, 7591–7596. [Google Scholar] [PubMed]
- Yamaguchi, A.; Tazuma, S.; Nishioka, T.; Ohishi, W.; Hyogo, H.; Nomura, S.; Chayama, K. Hepatitis C virus core protein modulates fatty acid metabolism and thereby causes lipid accumulation in the liver. Dig. Dis. Sci. 2005, 50, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Dharancy, S.; Malapel, M.; Perlemuter, G.; Roskams, T.; Cheng, Y.; Dubuquoy, L.; Podevin, P.; Conti, F.; Canva, V.; Philippe, D.; Gambiez, L.; Mathurin, P.; Paris, J.C.; Schoonjans, K.; Calmus, Y.; Pol, S.; Auwerx, J.; Desreumaux, P. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology 2005, 128, 334–342. [Google Scholar] [CrossRef] [PubMed]
- de Gottardi, A.; Pazienza, V.; Pugnale, P.; Bruttin, F.; Rubbia-Brandt, L.; Juge-Aubry, C.E.; Meier, C.A.; Hadengue, A.; Negro, F. Peroxisome proliferator-activated receptor-alpha and -gamma mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Aliment. Pharmacol. Ther. 2006, 23, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Groenbaek, K.; Friis, H.; Hansen, M.; Ring-Larsen, H.; Krarup, H.B. The effect of antioxidant supplementation on hepatitis C viral load, transaminases and oxidative status: a randomized trial among chronic hepatitis C virus-infected patients. Eur. J. Gastroenterol. Hepatol. 2006, 18, 985–989. [Google Scholar] [CrossRef]
- Negro, F.; Sanyal, A.J. Hepatitis C virus, steatosis and lipid abnormalities: clinical and pathogenic data. Liver Int. 2009, 29, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Leandro, G.; Mangia, A.; Hui, J.; Fabris, P.; Rubbia-Brandt, L.; Colloredo, G.; Adinolfi, L.E.; Jonsson, J.R.; Smedile, A.; Terrault, N.; Pazienza, V.; Giordani, M.T.; Giostra, E.; Sonzogni, A.; Ruggiero, G.; Marcellin, P.; Powell, E.E.; George, J.; Negro, F. Relationship between steatosis, inflammation, and fibrosis in chronic hepatitis C: a meta-analysis of individual patient data. Gastroenterology 2006, 130, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.M.; Sud, A.; Farrell, G.C.; Bandara, P.; Byth, K.; Kench, J.G.; McCaughan, G.W.; George, J. Insulin resistance is associated with chronic hepatitis C virus infection and fibrosis progression [corrected]. Gastroenterology 2003, 125, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Conjeevaram, H.S.; Kleiner, D.E.; Everhart, J.E.; Hoofnagle, J.H.; Zacks, S.; Afdhal, N.H.; Wahed, A.S. Race, insulin resistance and hepatic steatosis in chronic hepatitis C. Hepatology 2007, 45, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.F.; Yu, M.L.; Da, i.C.Y.; Hsieh, M.Y.; Lee, L.P.; Lin, Z.Y.; Chen, S.C.; Chang, W.Y.; Chuang, W.L. Pretreatment insulin sensitivity contributes to the treatment response to peginterferon plus ribavirin combination therapy for patients with chronic hepatitis C. Hepatology 2007, 46, 349A. [Google Scholar]
- Bortoletto, G.; Realdon, S.; Dal Pero, F.; Gerotto, M.; Scribano, L.; Boninsegna, S.; Martines, D.; Alberti, A. Insulin resistance (IR) defined by the homeostasis model of assessment insulin resistance (HOMA-IR) index has a direct effect on early viral kinetics during pegylated-interferon therapy for chronic hepatitis C. Hepatology 2007, 46, 361A. [Google Scholar]
- Nasta, P.; Gatti, F.; Puoti, M.; Cologni, G.; Bergamaschi, V.; Borghi, F.; Matti, A.; Carosi, G. Insulin resistance impairs rapid virologic response in HIV/hepatitis C virus coinfected patients on peginterferon-alfa-2a. Aids 2008, 22, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Romero-Gomez, M.; Del Mar Viloria, M.; Andrade, R.J.; Salmeron, J.; Diago, M.; Fernandez-Rodriguez, C.M.; Corpas, R.; Cruz, M.; Grande, L. Insulin resistance impairs sustained response rate to peginterferon plus ribavirin in chronic hepatitis C patients. Gastroenterology 2005, 128, 636–641. [Google Scholar] [CrossRef] [PubMed]
- D'Souza, R.; Sabin, C.A.; Foster, G.R. Insulin resistance plays a significant role in liver fibrosis in chronic hepatitis C and in the response to antiviral therapy. Am. J. Gastroenterol. 2005, 100, 1509–1515. [Google Scholar] [CrossRef] [PubMed]
- Persico, M.; Capasso, M.; Persico, E.; Svelto, M.; Russo, R.; Spano, D.; Croce, L.; Moschella, F.; Masutti, F.; Torella, R.; Tiribelli, C.; Iolascon, A. Suppressor of cytokine signaling 3 (SOCS3) expression and hepatitis C virus-related chronic hepatitis: Insulin resistance and response to antiviral therapy. Hepatology 2007, 46, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Poustchi, H.; Negro, F.; Hui, J.; Cua, I.H.; Brandt, L.R.; Kench, J.G.; George, J. Insulin resistance and response to therapy in patients infected with chronic hepatitis C virus genotypes 2 and 3. J. Hepatol. 2008, 48, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.J.; Lee, S.D.; Hung, T.H.; Lin, H.C.; Hwang, S.J.; Lee, F.Y.; Lu, R.H.; Chu, C.J.; Lee, S.D.; Hung, T.H.; Lin, H.C.; Hwang, S.J.; Lee, F.Y.; Lu, R.H.; Yu, M. I.; Chang, C. Y.; Yang, P. L.; Lee, C.Y.; Chang, F.Y. Insulin resistance is a major determinant of sustained virological response in genotype 1 chronic hepatitis C patients receiving peginterferon alpha-2b plus ribavirin. Aliment. Pharmacol.Ther. 2009, 29, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.J.; Jonsson, J.R.; Richardson, M.M.; Lipka, G.M.; Purdie, D.M.; Clouston, A.D.; Powell, E.E. Non-response to antiviral therapy is associated with obesity and increased hepatic expression of suppressor of cytokine signalling 3 (SOCS-3) in patients with chronic hepatitis C, viral genotype 1. Gut 2006, 55, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Hickman, I.J.; Clouston, A.D.; Macdonald, G.A.; Purdie, D.M.; Prins, J.B.; Ash, S.; Jonsson, J.R.; Powell, E.E. Effect of weight reduction on liver histology and biochemistry in patients with chronic hepatitis C. Gut 2002, 51, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Overbeck, K.; Genne, D.; Golay, A.; Negro, F. Pioglitazone in chronic hepatitis C not responding to pegylated interferon-alpha and ribavirin. J. Hepatol. 2008, 49, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Conjeevaram, H.; Burant, C.F.; McKenna Harsh, D.; Kang, H.; Das, A.K.; Everett, L.; White, D.; Lok, A.S.F. A randomized, double-blind, placebo-controlled study of PPARgamma agonist pioglitazone given in combination with peginterferon and ribavirin in patients with genotype-1 chronic hepatitis C. Hepatology 2008, 48, 384A. [Google Scholar]
- Elgouhari, H.M.; Cesario, K.B.; Lopez, R.; Zein, N.N. Pioglitazone improves early virologic kinetic response to PEG IFN/RBV combination therapy in hepatitis C genotype 1 naïve pts. Hepatology 2008, 48, 383A. [Google Scholar] [CrossRef]
- Romero-Gomez, M.; Diago, M.; Andrade, R.J.; Calleja, J.L.; Salmeron, J.F.-R.; Solà, R.; Herrerias, J.M.; Moreno-Otero, R; Olveira, A.; Núñez, O.; de la Mata, M.; Jorquera, F.; Morillas, R.M; Dalmau, B.; Martin-Vivaldi, R.; Arenas-Ruiz, J.I.; Rodriguez, E.; Duran, S.; Giner, P. Metformin with peginterferon alfa-2a and ribavirin in the treatment of naïve genotype 1 chronic hepatitis C patients with insulin resistance (TRIC-1): final results of a randomized and double-blinded trial. Hepatology 2008, 48, 380A. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article isy an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Clément, S.; Pascarella, S.; Negro, F. Hepatitis C Virus Infection: Molecular Pathways to Steatosis, Insulin Resistance and Oxidative Stress. Viruses 2009, 1, 126-143. https://doi.org/10.3390/v1020126
Clément S, Pascarella S, Negro F. Hepatitis C Virus Infection: Molecular Pathways to Steatosis, Insulin Resistance and Oxidative Stress. Viruses. 2009; 1(2):126-143. https://doi.org/10.3390/v1020126
Chicago/Turabian StyleClément, Sophie, Stéphanie Pascarella, and Francesco Negro. 2009. "Hepatitis C Virus Infection: Molecular Pathways to Steatosis, Insulin Resistance and Oxidative Stress" Viruses 1, no. 2: 126-143. https://doi.org/10.3390/v1020126