Adaptive Immunity to Hepatitis C Virus

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The humoral responses to HCV infection
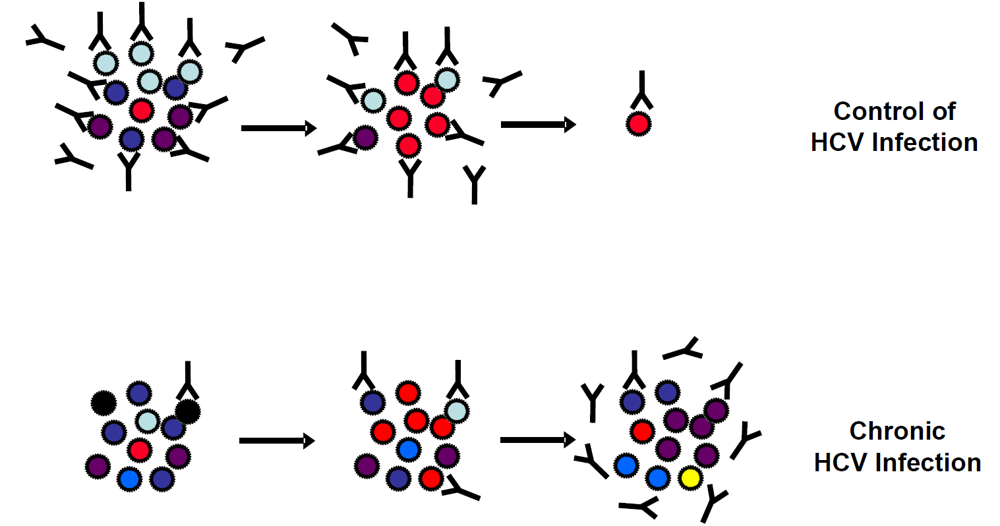
2.1. Neutralizing antibodies and control of viral infection

2.2. Viral escape from neutralizing antibodies
3. T cell responses to HCV infection
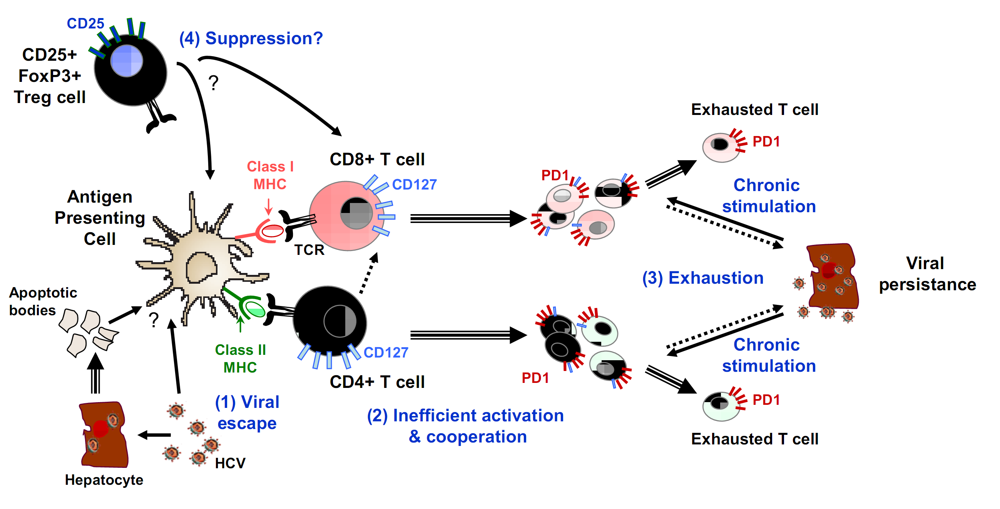
3.2. Mechanisms of T cell failure

4. Conclusions and Perspectives
Acknowledgments
References and Notes
- Shepard, C.W.; Finelli, L.; Alter, M.J. Global epidemiology of hepatitis C virus infection. Lancet. Infect. Dis. 2005, 5, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, J.T.; Diepolder, H.M.; Zachoval, R.; Gruener, N.H.; Jung, M.C.; Ulsenheimer, A.; Schraut, W.W.; Schirren, C.A.; Waechtler, M.; Backmund, M.; Pape, G.R. Acute hepatitis C: high rate of both spontaneous and treatment-induced viral clearance. Gastroenterology 2003, 125, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Micallef, J.M.; Kaldor, J.M.; Dore, G.J. Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. J. Viral. Hepat. 2006, 13, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Colin, C.; Lanoir, D.; Touzet, S.; Meyaud-Kraemer, L.; Bailly, F.; Trepo, C. Sensitivity and specificity of third-generation hepatitis C virus antibody detection assays: an analysis of the literature. J. Viral. Hepat. 2001, 8, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J. A critical role for the chimpanzee model in the study of hepatitis C. Hepatology 2004, 39, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Bukh, J.; Meunier, J.C.; Granier, C.; Engle, R.E.; Blackwelder, W.C.; Emerson, S.U.; Cosset, F.L.; Purcell, R.H. In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc. Natl. Acad. Sci. U S A 2003, 100, 14199–14204. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U S A 2003, 100, 7271–7276. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumert, T.F.; Ito, S.; Wong, D.T.; Liang, T.J. Hepatitis C virus structural proteins assemble into viruslike particles in insect cells. J. Virol. 1998, 72, 3827–3836. [Google Scholar] [PubMed]
- Baumert, T.F.; Wellnitz, S.; Aono, S.; Satoi, J.; Herion, D.; Tilman Gerlach, J.; Pape, G.R.; Lau, J.Y.; Hoofnagle, J.H.; Blum, H.E.; Liang, T.J. Antibodies against hepatitis C virus-like particles and viral clearance in acute and chronic hepatitis C. Hepatology 2000, 32, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, D.; Barth, H.; Gissler, B.; Schürmann, P.; Adah, M.I.; Gerlach, J.T.; Pape, G.R.; Depla, E.; Jacobs, D.; Maertens, G.; Patel, A.H.; Inchauspé, G.; Liang, T.J.; Blum, H.E.; Baumert, T.F. Inhibition of hepatitis C virus-like particle binding to target cells by antiviral antibodies in acute and chronic hepatitis C. J. Virol. 2004, 78, 9030–9040. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; Bartenschlager, R.; Liang, T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U S A 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; Rice, C.M. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Haberstroh, A.; Schnober, E.K.; Zeisel, M.B.; Carolla, P.; Barth, H.; Blum, H.E.; Cosset, F.L.; Koutsoudakis, G.; Bartenschlager, R.; Union, A.; Depla, E.; Owsianka, A.; Patel, A.H.; Schuster, C.; Stoll-Keller, F.; Doffoel, M.; Dreux, M.; Baumert, T.F. Neutralizing host responses in hepatitis C virus infection target viral entry at postbinding steps and membrane fusion. Gastroenterology 2008, 135, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Nielsen, S.; Zhong, J.; Bassendine, M.F.; Drummer, H.E.; Balfe, P.; McKeating, J.A. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J. Virol. 2008, 82, 12020–12029. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.C.; Russell, R.S.; Goossens, V.; Priem, S.; Walter, H.; Depla, E.; Union, A.; Faulk, K.N.; Bukh, J.; Emerson, S.U.; Purcell, R.H. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J. Virol. 2008, 82, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.Y.; Li, T.K.; Xia, J.; Gal-Tanamy, M.; Olson, O.; Li, S.H.; Patel, A.H.; Ball, J.K.; Lemon, S.M.; Foung, S.K. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J. Virol. 2008, 82, 6061–6066. [Google Scholar] [CrossRef] [PubMed]
- Owsianka, A.M.; Tarr, A.W.; Keck, Z.Y.; Li, T.K.; Witteveldt, J.; Adair, R.; Foung, S.K.; Ball, J.K.; Patel, A.H. Broadly neutralizing human monoclonal antibodies to the hepatitis C virus E2 glycoprotein. J. Gen. Virol. 2008, 89, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Libbrecht, L.; De Vos, R.; de Hemptinne, B.; Gevaert, K.; Vandekerckhove, J.; Roskams, T.; Leroux-Roels, G. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology 2005, 41, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Maruyama, T.; Lewis, J.; Giang, E.; Tarr, A.W.; Stamataki, Z.; Gastaminza, P.; Chisari, F.V.; Jones, I.M.; Fox, R.I.; Ball, J.K.; McKeating, J.A.; Kneteman, N.M.; Burton, D.R. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Vanwolleghem, T.; Bukh, J.; Meuleman, P.; Desombere, I.; Meunier, J.C.; Alter, H.; Purcell, R.H.; Leroux-Roels, G. Polyclonal immunoglobulins from a chronic hepatitis C virus patient protect human liver-chimeric mice from infection with a homologous hepatitis C virus strain. Hepatology 2008, 47, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Bukh, J.; Spangenberg, H.C.; Wieland, S.; Pemberton, J.; Steiger, C.; Govindarajan, S.; Purcell, R.H.; Chisari, F.V. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc. Natl. Acad. Sci. U S A 2002, 99, 15661–15668. [Google Scholar] [CrossRef] [PubMed]
- Lechner, F.; Wong, D.K.; Dunbar, P.R.; Chapman, R.; Chung, R.T.; Dohrenwend, P.; Robbins, G.; Phillips, R.; Klenerman, P.; Walker, B.D. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 2000, 191, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Lechner, F.; Gruener, N.H.; Urbani, S.; Uggeri, J.; Santantonio, T.; Kammer, A.R.; Cerny, A.; Phillips, R.; Ferrari, C.; Pape, G.R.; Klenerman, P. CD8+ T lymphocyte responses are induced during acute hepatitis C virus infection but are not sustained. Eur. J. Immunol. 2000, 30, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Logvinoff, C.; Major, M.E.; Oldach, D.; Heyward, S.; Talal, A.; Balfe, P.; Feinstone, S.M.; Alter, H.; Rice, C.M.; McKeating, J.A. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl. Acad. Sci. U S A 2004, 101, 10149–10154. [Google Scholar] [CrossRef] [PubMed]
- Dustin, L.B.; Rice, C.M. Flying under the radar: the immunobiology of hepatitis C. Annu. Rev. Immunol. 2007, 25, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.H.; Cox, A.; Hoover, D.R.; Wang, X.H.; Mao, Q.; Ray, S.; Strathdee, S.A.; Vlahov, D.; Thomas, D.L. Protection against persistence of hepatitis C. Lancet 2002, 359, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Bassett, S.E.; Guerra, B.; Brasky, K.; Miskovsky, E.; Houghton, M.; Klimpel, G.R.; Lanford, R.E. Protective immune response to hepatitis C virus in chimpanzees rechallenged following clearance of primary infection. Hepatology 2001, 33, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Major, M.E.; Mihalik, K.; Puig, M.; Rehermann, B.; Nascimbeni, M.; Rice, C.M.; Feinstone, S.M. Previously infected and recovered chimpanzees exhibit rapid responses that control hepatitis C virus replication upon rechallenge. J. Virol. 2002, 76, 6586–6595. [Google Scholar] [CrossRef] [PubMed]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; Abrignani, S. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, D.; Date, T.; Morikawa, K.; Murayama, A.; Miyamoto, M.; Kaga, M.; Barth, H.; Baumert, T.F.; Dubuisson, J.; Wakita, T. Cd81 Expression Is Important For Heterogeneous Hcv Permissiveness Of Huh7 Cell Clones. J. Virol. 2007, 81, 5036–5045. [Google Scholar] [CrossRef] [PubMed]
- Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Koutsoudakis, G.; Schnober, E.K.; Haberstroh, A.; Blum, H.E.; Cosset, F.-L.; Wakita, T.; Jaeck, D.; Doffoel, M.; Royer, C.; Soulier, E.; Schvoerer, E.; Schuster, C.; Stoll-Keller, F.; Bartenschlager, R.; Pietschmann, T.; Barth, H.; Baumert, T.F. Scavenger receptor BI is a key host factor for Hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 2007, 46, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Bertaux, C.; Cukierman, L.; Cormier, E.; Lavillette, D.; Cosset, F.L.; Dragic, T. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J. Virol. 2008, 82, 3555–3560. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Dao Thi, V.L.; Fresquet, J.; Guerin, M.; Julia, Z.; Verney, G.; Durantel, D.; Zoulim, F.; Lavillette, D.; Cosset, F.L.; Bartosch, B. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog. 2009, 5, e1000310. [Google Scholar] [CrossRef] [PubMed]
- Helle, F.; Dubuisson, J. Hepatitis C virus entry into host cells. Cell. Mol. Life Sci. 2008, 65, 100–112. [Google Scholar] [CrossRef] [PubMed]
- von Hahn, T.; Rice, C.M. Hepatitis C virus entry. J. Biol. Chem. 2008, 283, 3689–3693. [Google Scholar] [CrossRef] [PubMed]
- Lavillette, D.; Morice, Y.; Germanidis, G.; Donot, P.; Soulier, A.; Pagkalos, E.; Sakellariou, G.; Intrator, L.; Bartosch, B.; Pawlotsky, J.M.; Cosset, F.L. Human serum facilitates hepatitis C virus infection, and neutralizing responses inversely correlate with viral replication kinetics at the acute phase of hepatitis C virus infection. J. Virol. 2005, 79, 6023–6034. [Google Scholar] [CrossRef] [PubMed]
- Pestka, J.M.; Zeisel, M.B.; Blaser, E.; Schurmann, P.; Bartosch, B.; Cosset, F.L.; Patel, A.H.; Meisel, H.; Baumert, J.; Viazov, S.; Rispeter, K.; Blum, H.E.; Roggendorf, M.; Baumert, T.F. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. U S A 2007, 104, 6025–6030. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.A.; Netski, D.M.; Wang, X.H.; Cox, A.L.; Ray, S.C. Selection Pressure From Neutralizing Antibodies Drives Sequence Evolution During Acute Infection With Hepatitis C Virus. Gastroenterology 2009, 136, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Cosset, F.L.; Baumert, T.F. Host neutralizing responses and pathogenesis of hepatitis C virus infection. Hepatology 2008, 48, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Gastaminza, P.; Chung, J.; Stamataki, Z.; Isogawa, M.; Cheng, G.; McKeating, J.A.; Chisari, F.V. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J. Virol. 2006, 80, 11082–11093. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.Y.; Li, S.H.; Xia, J.; von Hahn, T.; Balfe, P.; McKeating, J.A.; Witteveldt, J.; Patel, A.H.; Alter, H.; Rice, C.M.; Foung, S.K. Mutations in HCV E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. J. Virol. 2009, 83, 6149–6160. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Doyle, M.L.; Casper, D.J.; Cicala, C.; Leavitt, S.A.; Majeed, S.; Steenbeke, T.D.; Venturi, M.; Chaiken, I.; Fung, M.; Katinger, H.; Parren, P.W.; Robinson, J.; Van Ryk, D.; Wang, L.; Burton, D.R.; Freire, E.; Wyatt, R.; Sodroski, J.; Hendrickson, W.A.; Arthos, J. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 2002, 420, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Gal-Tanamy, M.; Keck, Z.Y.; Yi, M.; McKeating, J.A.; Patel, A.H.; Foung, S.K.; Lemon, S.M. In vitro selection of a neutralization-resistant hepatitis C virus escape mutant. Proc. Natl. Acad. Sci. U S A 2008, 105, 19450–19455. [Google Scholar] [CrossRef] [PubMed]
- Diepolder, H.M.; Zachoval, R.; Hoffmann, R.M.; Wierenga, E.A.; Santantonio, T.; Jung, M.C.; Eichenlaub, D.; Pape, G.R. Possible mechanism involving T-lymphocyte response to non-structural protein 3 in viral clearance in acute hepatitis C virus infection. Lancet 1995, 346, 1006–1007. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Oldach, D.; Chang, K.M.; Steiger, C.; Ray, S.C.; Chisari, F.V. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- von Hahn, T.; Yoon, J.C.; Alter, H.; Rice, C.M.; Rehermann, B.; Balfe, P.; McKeating, J.A. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 2007, 132, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wu, C.G.; Mihalik, K.; Virata-Theimer, M.L.; Yu, M.Y.; Alter, H.J.; Feinstone, S.M. Hepatitis C virus epitope-specific neutralizing antibodies in Igs prepared from human plasma. Proc. Natl. Acad. Sci. U S A 2007, 104, 8449–8454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhong, L.; Struble, E.B.; Watanabe, H.; Kachko, A.; Mihalik, K.; Virata-Theimer, M.L.; Alter, H.J.; Feinstone, S.; Major, M. Depletion of interfering antibodies in chronic hepatitis C patients and vaccinated chimpanzees reveals broad cross-genotype neutralizing activity. Proc. Natl. Acad. Sci. U S A 2009, 106, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.G.; Shoukry, N.H.; Grakoui, A.; Fuller, M.J.; Cawthon, A.G.; Dong, C.; Hasselschwert, D.L.; Brasky, K.M.; Freeman, G.J.; Seth, N.P.; Wucherpfennig, K.W.; Houghton, M.; Walker, C.M. Variable patterns of programmed death-1 expression on fully functional memory T cells after spontaneous resolution of hepatitis C virus infection. J. Virol. 2008, 82, 5109–5114. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.G.; Walker, C.M. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 2005, 436, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, H.; He, X.S.; Nascimbeni, M.; Davis, A.R.; Greenberg, H.B.; Hoofnagle, J.H.; Liang, T.J.; Alter, H.; Rehermann, B. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J. Immunol. 2002, 169, 3447–3458. [Google Scholar] [PubMed]
- Schulze zur Wiesch, J.; Lauer, G.M.; Day, C.L.; Kim, A.Y.; Ouchi, K.; Duncan, J.E.; Wurcel, A.G.; Timm, J.; Jones, A.M.; Mothe, B.; Allen, T.M.; McGovern, B.; Lewis-Ximenez, L.; Sidney, J.; Sette, A.; Chung, R.T.; Walker, B.D. Broad repertoire of the CD4+ Th cell response in spontaneously controlled hepatitis C virus infection includes dominant and highly promiscuous epitopes. J. Immunol. 2005, 175, 3603–3613. [Google Scholar] [PubMed]
- Smyk-Pearson, S.; Tester, I.A.; Lezotte, D.; Sasaki, A.W.; Lewinsohn, D.M.; Rosen, H.R. Differential antigenic hierarchy associated with spontaneous recovery from hepatitis C virus infection: implications for vaccine design. J. Infect. Dis. 2006, 194, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, J.T.; Diepolder, H.M.; Jung, M.C.; Gruener, N.H.; Schraut, W.W.; Zachoval, R.; Hoffmann, R.; Schirren, C.A.; Santantonio, T.; Pape, G.R. Recurrence of hepatitis C virus after loss of virus-specific CD4(+) T- cell response in acute hepatitis C. Gastroenterology 1999, 117, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, C.; Valli, A.; Galati, L.; Penna, A.; Scaccaglia, P.; Giuberti, T.; Schianchi, C.; Missale, G.; Marin, M.G.; Fiaccadori, F. T-cell response to structural and nonstructural hepatitis C virus antigens in persistent and self-limited hepatitis C virus infections. Hepatology 1994, 19, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Wertheimer, A.M.; Miner, C.; Lewinsohn, D.M.; Sasaki, A.W.; Kaufman, E.; Rosen, H.R. Novel CD4+ and CD8+ T-cell determinants within the NS3 protein in subjects with spontaneously resolved HCV infection. Hepatology 2003, 37, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Aberle, J.H.; Formann, E.; Steindl-Munda, P.; Weseslindtner, L.; Gurguta, C.; Perstinger, G.; Grilnberger, E.; Laferl, H.; Dienes, H.P.; Popow-Kraupp, T.; Ferenci, P.; Holzmann, H. Prospective study of viral clearance and CD4(+) T-cell response in acute hepatitis C primary infection and reinfection. J. Clin. Virol. 2006, 36, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.L.; Mosbruger, T.; Lauer, G.M.; Pardoll, D.; Thomas, D.L.; Ray, S.C. Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology 2005, 42, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Smyk-Pearson, S.; Tester, I.A.; Klarquist, J.; Palmer, B.E.; Pawlotsky, J.M.; Golden-Mason, L.; Rosen, H.R. Spontaneous recovery in acute human hepatitis C virus infection: functional T-cell thresholds and relative importance of CD4 help. J. Virol. 2008, 82, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Folgori, A.; Spada, E.; Pezzanera, M.; Ruggeri, L.; Mele, A.; Garbuglia, A.R.; Perrone, M.P.; Del Porto, P.; Piccolella, E.; Cortese, R.; Nicosia, A.; Vitelli, A. Early impairment of hepatitis C virus specific T cell proliferation during acute infection leads to failure of viral clearance. Gut 2006, 55, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Urbani, S.; Amadei, B.; Fisicaro, P.; Tola, D.; Orlandini, A.; Sacchelli, L.; Mori, C.; Missale, G.; Ferrari, C. Outcome of acute hepatitis C is related to virus-specific CD4 function and maturation of antiviral memory CD8 responses. Hepatology 2006, 44, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Grakoui, A.; Shoukry, N.H.; Woollard, D.J.; Han, J.H.; Hanson, H.L.; Ghrayeb, J.; Murthy, K.K.; Rice, C.M.; Walker, C.M. HCV persistence and immune evasion in the absence of memory T cell help. Science 2003, 302, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191–1198. [Google Scholar] [CrossRef]
- Chang, K.M.; Thimme, R.; Melpolder, J.J.; Oldach, D.; Pemberton, J.; Moorhead-Loudis, J.; McHutchison, J.G.; Alter, H.J.; Chisari, F.V. Differential CD4(+) and CD8(+) T-cell responsiveness in hepatitis C virus infection. Hepatology 2001, 33, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Weiner, A.J.; Paliard, X.; Selby, M.J.; Medina-Selby, A.; Coit, D.; Nguyen, S.; Kansopon, J.; Arian, C.L.; Ng, P.; Tucker, J.; Lee, C.T.; Polakos, N.K.; Han, J.; Wong, S.; Lu, H.H.; Rosenberg, S.; Brasky, K.M.; Chien, D.; Kuo, G.; Houghton, M. Intrahepatic genetic inoculation of hepatitis C virus RNA confers cross-protective immunity. J. Virol. 2001, 75, 7142–7148. [Google Scholar] [CrossRef] [PubMed]
- Shoukry, N.H.; Grakoui, A.; Houghton, M.; Chien, D.Y.; Ghrayeb, J.; Reimann, K.A.; Walker, C.M. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J. Exp. Med. 2003, 197, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, M.; Mizukoshi, E.; Bosmann, M.; Major, M.E.; Mihalik, K.; Rice, C.M.; Feinstone, S.M.; Rehermann, B. Kinetics of CD4+ and CD8+ memory T-cell responses during hepatitis C virus rechallenge of previously recovered chimpanzees. J. Virol. 2003, 77, 4781–4793. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Guerra, B.; Chavez, D.; Bigger, C.; Brasky, K.M.; Wang, X.H.; Ray, S.C.; Thomas, D.L. Cross-genotype immunity to hepatitis C virus. J. Virol. 2004, 78, 1575–1581. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J.; Thimme, R.; Meunier, J.C.; Faulk, K.; Spangenberg, H.C.; Chang, K.M.; Satterfield, W.; Chisari, F.V.; Purcell, R.H. Previously infected chimpanzees are not consistently protected against reinfection or persistent infection after reexposure to the identical hepatitis C virus strain. J. Virol. 2008, 82, 8183–8195. [Google Scholar] [CrossRef] [PubMed]
- Bassett, S.E.; Thomas, D.L.; Brasky, K.M.; Lanford, R.E. Viral persistence, antibody to E1 and E2, and hypervariable region 1 sequence stability in hepatitis C virus-inoculated chimpanzees. J. Virol. 1999, 73, 1118–1126. [Google Scholar] [PubMed]
- Thimme, R.; Lohmann, V.; Weber, F. A target on the move: innate and adaptive immune escape strategies of hepatitis C virus. Antiviral. Res. 2006, 69, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.E.; Sugimoto, K.; Newton, K.; Valiga, M.E.; Ikeda, F.; Aytaman, A.; Nunes, F.A.; Lucey, M.R.; Vance, B.A.; Vonderheide, R.H.; Reddy, K.R.; McKeating, J.A.; Chang, K.M. Discordant role of CD4 T-cell response relative to neutralizing antibody and CD8 T-cell responses in acute hepatitis C. Gastroenterology 2007, 132, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Welsh, R.M.; Kim, S.K.; Cornberg, M.; Clute, S.C.; Selin, L.K.; Naumov, Y.N. The privacy of T cell memory to viruses. Curr. Top. Microbiol. Immunol. 2006, 311, 117–153. [Google Scholar] [PubMed]
- Prlic, M.; Lefrancois, L.; Jameson, S.C. Multiple choices: regulation of memory CD8 T cell generation and homeostasis by interleukin (IL)-7 and IL-15. J. Exp. Med. 2002, 195, F49–F52. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Williams, M.A.; Bevan, M.J. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat. Immunol. 2004, 5, 927–933. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, E.M.; de Bree, G.J.; Remmerswaal, E.B.; Yong, S.L.; Tesselaar, K.; ten Berge, I.J.; van Lier, R.A. IL-7 receptor alpha chain expression distinguishes functional subsets of virus-specific human CD8+ T cells. Blood 2005, 106, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Boettler, T.; Panther, E.; Bengsch, B.; Nazarova, N.; Spangenberg, H.C.; Blum, H.E.; Thimme, R. Expression of the interleukin-7 receptor alpha chain (CD127) on virus-specific CD8+ T cells identifies functionally and phenotypically defined memory T cells during acute resolving hepatitis B virus infection. J. Virol. 2006, 80, 3532–3540. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, M.H.; Hardy, M.P.; Rooney, J.; Labrecque, N. IL-7 receptor expression levels do not identify CD8+ memory T lymphocyte precursors following peptide immunization. J. Immunol. 2005, 175, 4400–4407. [Google Scholar] [PubMed]
- Klonowski, K.D.; Williams, K.J.; Marzo, A.L.; Lefrancois, L. Cutting edge: IL-7-independent regulation of IL-7 receptor alpha expression and memory CD8 T cell development. J. Immunol. 2006, 177, 4247–4251. [Google Scholar] [PubMed]
- Goldrath, A.W.; Sivakumar, P.V.; Glaccum, M.; Kennedy, M.K.; Bevan, M.J.; Benoist, C.; Mathis, D.; Butz, E.A. Cytokine requirements for acute and Basal homeostatic proliferation of naive and memory CD8+ T cells. J. Exp. Med. 2002, 195, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.; Ernst, B.; Kieper, W.C.; LeRoy, E.; Sprent, J.; Surh, C.D. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J. Exp. Med. 2002, 195, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Kieper, W.C.; Tan, J.T.; Bondi-Boyd, B.; Gapin, L.; Sprent, J.; Ceredig, R.; Surh, C.D. Overexpression of interleukin (IL)-7 leads to IL-15-independent generation of memory phenotype CD8+ T cells. J. Exp. Med. 2002, 195, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.C.; Wherry, E.J.; Boone, D.; Murali-Krishna, K.; Antia, R.; Ma, A.; Ahmed, R. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J. Exp. Med. 2002, 195, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Schluns, K.S.; Williams, K.; Ma, A.; Zheng, X.X.; Lefrancois, L. Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J. Immunol. 2002, 168, 4827–4831. [Google Scholar] [PubMed]
- Frohlich, A.; Kisielow, J.; Schmitz, I.; Freigang, S.; Shamshiev, A.T.; Weber, J.; Marsland, B.J.; Oxenius, A.; Kopf, M. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science 2009, 324, 1576–1580. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.S.; Du, M.; Zajac, A.J. A vital role for interleukin-21 in the control of a chronic viral infection. Science 2009, 324, 1572–1576. [Google Scholar] [CrossRef] [PubMed]
- Elsaesser, H.; Sauer, K.; Brooks, D.G. IL-21 is required to control chronic viral infection. Science 2009, 324, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Burton, J.R.; Castelblanco, N.; Klarquist, J.; Benlloch, S.; Wang, C.; Rosen, H.R. Loss of IL-7 receptor alpha-chain (CD127) expression in acute HCV infection associated with viral persistence. Hepatology 2006, 44, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Radziewicz, H.; Ibegbu, C.C.; Fernandez, M.L.; Workowski, K.A.; Obideen, K.; Wehbi, M.; Hanson, H.L.; Steinberg, J.P.; Masopust, D.; Wherry, E.J.; Altman, J.D.; Rouse, B.T.; Freeman, G.J.; Ahmed, R.; Grakoui, A. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J. Virol. 2007, 81, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Spangenberg, H.C.; Kersting, N.; Neumann-Haefelin, C.; Panther, E.; von Weizsacker, F.; Blum, H.E.; Pircher, H.; Thimme, R. Analysis of CD127 and KLRG1 expression on hepatitis C virus-specific CD8+ T cells reveals the existence of different memory T-cell subsets in the peripheral blood and liver. J. Virol. 2007, 81, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Urbani, S.; Boni, C.; Missale, G.; Elia, G.; Cavallo, C.; Massari, M.; Raimondo, G.; Ferrari, C. Virus-specific CD8+ lymphocytes share the same effector-memory phenotype but exhibit functional differences in acute hepatitis B and C. J. Virol. 2002, 76, 12423–12434. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, H.C.; Viazov, S.; Kersting, N.; Neumann-Haefelin, C.; McKinney, D.; Roggendorf, M.; von Weizsacker, F.; Blum, H.E.; Thimme, R. Intrahepatic CD8+ T-cell failure during chronic hepatitis C virus infection. Hepatology 2005, 42, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Castellino, F.; Germain, R.N. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu. Rev. Immunol. 2006, 24, 519–540. [Google Scholar] [CrossRef] [PubMed]
- Ulsenheimer, A.; Gerlach, J.T.; Gruener, N.H.; Jung, M.C.; Schirren, C.A.; Schraut, W.; Zachoval, R.; Pape, G.R.; Diepolder, H.M. Detection of functionally altered hepatitis C virus-specific CD4 T cells in acute and chronic hepatitis C. Hepatology 2003, 37, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; Mncube, Z.; Duraiswamy, J.; Zhu, B.; Eichbaum, Q.; Altfeld, M.; Wherry, E.J.; Coovadia, H.M.; Goulder, P.J.; Klenerman, P.; Ahmed, R.; Freeman, G.J.; Walker, B.D. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Urbani, S.; Amadei, B.; Tola, D.; Massari, M.; Schivazappa, S.; Missale, G.; Ferrari, C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J. Virol. 2006, 80, 11398–11403. [Google Scholar] [CrossRef] [PubMed]
- Rutebemberwa, A.; Ray, S.C.; Astemborski, J.; Levine, J.; Liu, L.; Dowd, K.A.; Clute, S.; Wang, C.; Korman, A.; Sette, A.; Sidney, J.; Pardoll, D.M.; Cox, A.L. High-programmed death-1 levels on hepatitis C virus-specific T cells during acute infection are associated with viral persistence and require preservation of cognate antigen during chronic infection. J. Immunol. 2008, 181, 8215–8225. [Google Scholar] [PubMed]
- Shevach, E.M. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 2009, 30, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Ikeda, F.; Stadanlick, J.; Nunes, F.A.; Alter, H.J.; Chang, K.M. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology 2003, 38, 1437–1448. [Google Scholar] [PubMed]
- Cabrera, R.; Tu, Z.; Xu, Y.; Firpi, R.J.; Rosen, H.R.; Liu, C.; Nelson, D.R. An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology 2004, 40, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Boettler, T.; Spangenberg, H.C.; Neumann-Haefelin, C.; Panther, E.; Urbani, S.; Ferrari, C.; Blum, H.E.; von Weizsacker, F.; Thimme, R. T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J. Virol. 2005, 79, 7860–7867. [Google Scholar] [CrossRef] [PubMed]
- Rushbrook, S.M.; Ward, S.M.; Unitt, E.; Vowler, S.L.; Lucas, M.; Klenerman, P.; Alexander, G.J. Regulatory T cells suppress in vitro proliferation of virus-specific CD8+ T cells during persistent hepatitis C virus infection. J. Virol. 2005, 79, 7852–7859. [Google Scholar] [CrossRef] [PubMed]
- Manigold, T.; Shin, E.C.; Mizukoshi, E.; Mihalik, K.; Murthy, K.K.; Rice, C.M.; Piccirillo, C.A.; Rehermann, B. Foxp3+CD4+CD25+ T cells control virus-specific memory T cells in chimpanzees that recovered from hepatitis C. Blood 2006, 107, 4424–4432. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.M.; Rehermann, B.; McHutchison, J.G.; Pasquinelli, C.; Southwood, S.; Sette, A.; Chisari, F.V. Immunological significance of cytotoxic T lymphocyte epitope variants in patients chronically infected by the hepatitis C virus. J. Clin. Invest. 1997, 100, 2376–2385. [Google Scholar] [CrossRef] [PubMed]
- Weiner, A.; Erickson, A.L.; Kansopon, J.; Crawford, K.; Muchmore, E.; Hughes, A.L.; Houghton, M.; Walker, C.M. Persistent hepatitis C virus infection in a chimpanzee is associated with emergence of a cytotoxic T lymphocyte escape variant. Proc. Natl. Acad. Sci. U S A 1995, 92, 2755–2759. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.L.; Kimura, Y.; Igarashi, S.; Eichelberger, J.; Houghton, M.; Sidney, J.; McKinney, D.; Sette, A.; Hughes, A.L.; Walker, C.M. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity 2001, 15, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.L.; Mosbruger, T.; Mao, Q.; Liu, Z.; Wang, X.H.; Yang, H.C.; Sidney, J.; Sette, A.; Pardoll, D.; Thomas, D.L.; Ray, S.C. Cellular immune selection with hepatitis C virus persistence in humans. J. Exp. Med. 2005, 201, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Tester, I.; Smyk-Pearson, S.; Wang, P.; Wertheimer, A.; Yao, E.; Lewinsohn, D.M.; Tavis, J.E.; Rosen, H.R. Immune evasion versus recovery after acute hepatitis C virus infection from a shared source. J. Exp. Med. 2005, 201, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Timm, J.; Lauer, G.M.; Kavanagh, D.G.; Sheridan, I.; Kim, A.Y.; Lucas, M.; Pillay, T.; Ouchi, K.; Reyor, L.L.; Schulze zur Wiesch, J.; Gandhi, R.T.; Chung, R.T.; Bhardwaj, N.; Klenerman, P.; Walker, B.D.; Allen, T.M. CD8 epitope escape and reversion in acute HCV infection. J. Exp. Med. 2004, 200, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.C.; Fanning, L.; Wang, X.H.; Netski, D.M.; Kenny-Walsh, E.; Thomas, D.L. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J. Exp. Med. 2005, 201, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Haefelin, C.; McKiernan, S.; Ward, S.; Viazov, S.; Spangenberg, H.C.; Killinger, T.; Baumert, T.F.; Nazarova, N.; Sheridan, I.; Pybus, O.; von Weizsacker, F.; Roggendorf, M.; Kelleher, D.; Klenerman, P.; Blum, H.E.; Thimme, R. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology 2006, 43, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Uebelhoer, L.; Han, J.H.; Callendret, B.; Mateu, G.; Shoukry, N.H.; Hanson, H.L.; Rice, C.M.; Walker, C.M.; Grakoui, A. Stable cytotoxic T cell escape mutation in hepatitis C virus is linked to maintenance of viral fitness. PLoS Pathog. 2008, 4, e1000143. [Google Scholar] [CrossRef] [PubMed]
- Timm, J.; Li, B.; Daniels, M.G.; Bhattacharya, T.; Reyor, L.L.; Allgaier, R.; Kuntzen, T.; Fischer, W.; Nolan, B.E.; Duncan, J.; Schulze zur Wiesch, J.; Kim, A.Y.; Frahm, N.; Brander, C.; Chung, R.T.; Lauer, G.M.; Korber, B.T.; Allen, T.M. Human leukocyte antigen-associated sequence polymorphisms in hepatitis C virus reveal reproducible immune responses and constraints on viral evolution. Hepatology 2007, 46, 339–349. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Zeisel, M.B.; Fafi-Kremer, S.; Robinet, E.; Habersetzer, F.; Baumert, T.F.; Stoll-Keller, F. Adaptive Immunity to Hepatitis C Virus. Viruses 2009, 1, 276-297. https://doi.org/10.3390/v1020276
Zeisel MB, Fafi-Kremer S, Robinet E, Habersetzer F, Baumert TF, Stoll-Keller F. Adaptive Immunity to Hepatitis C Virus. Viruses. 2009; 1(2):276-297. https://doi.org/10.3390/v1020276
Chicago/Turabian StyleZeisel, Mirjam B., Samira Fafi-Kremer, Eric Robinet, François Habersetzer, Thomas F. Baumert, and Françoise Stoll-Keller. 2009. "Adaptive Immunity to Hepatitis C Virus" Viruses 1, no. 2: 276-297. https://doi.org/10.3390/v1020276