Innate and Adaptive Immune Responses to Herpes Simplex Virus


Abstract
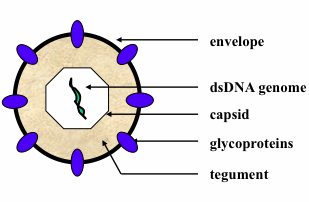
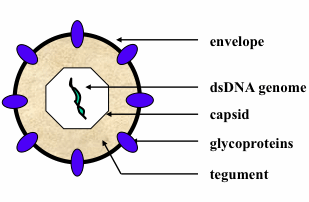
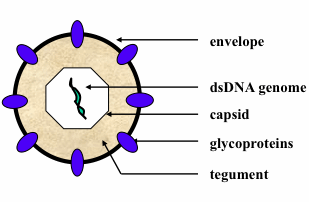
:
{kind=link}
{kind=link}
1. Introduction
2. The role of type I interferons in vivo
3. Recognition of HSV and activation of type I IFN pathways
3.1. Cellular sensors of HSV
3.2. Signal transduction pathways leading to type I IFN production
3.3. Downstream effector pathways
4. Cell type specific contributions to innate immunity against HSV
4.1. NK cells
4.2. pDCs
5. Adaptive immune mechanisms against HSV
5.1. Humoral immunity against HSV
5.2. Cellular immunity against HSV
6. Conclusion
References
- Ellermann-Eriksen, S.; Liberto, M.; Iannello, D.; Mogensen, S. X-linkage of the early in vitro alpha/beta interferon response of mouse peritoneal macrophages to herpes simplex virus type 2. J Gen Virol 1986, 67, 1025–103. [Google Scholar] [CrossRef] [PubMed]
- Gresser, I.; Tovey, M.; Maury, C.; Bandu, M. Role of interferon in the pathogenesis of virus diseases in mice as demonstrated by the use of anti-interferon serum II. Studies with herpes simplex, Moloney sarcoma, vesicular stomatitis, Newcastle disease, and influenza viruses. J Exp Med 1976, 144, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C. Genetics of natural resistance to herpesvirus infections in mice. Nature 1975, 258, 152–153. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, E.; Haahr, S.; Mogensen, S. X-linked resistance of mice to high doses of herpes simplex virus type 2 correlates with early interferon production. Infect Immun 1983, 42, 740–746. [Google Scholar] [PubMed]
- Shupack, J.; Stiller, M.; Davis, I.; Kenny, C.; Jondreau, L. Topical alpha-interferon ointment with dimethyl sulfoxide in the treatment of recurrent genital herpes simplex. Dermatology 1992, 184, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Vollstedt, S.; Arnold, S.; Schwerdel, C.; Franchini, M.; Alber, G.; Di Santo, J.; Ackermann, M.; Suter, M. Interplay between alpha/beta and gamma interferons with B, T, and natural killer cells in the defense against herpes simplex virus type 1. J Virol 2004, 78, 3846–3850. [Google Scholar] [CrossRef] [PubMed]
- Zawatzky, R.; Engler, H.; Kirchner, H. Experimental infection of inbred mice with herpes simplex virus III. Comparison between newborn and adult C57BL/6 mice. J Gen Virol 1982, 60, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Zawatzky, R.; Gresser, I.; DeMaeyer, E.; Kirchner, H. The role of interferon in the resistance of C57BL/6 mice to various doses of herpes simplex virus type 1. J Infect Dis 1982, 146, 405–410. [Google Scholar] [PubMed]
- Zawatzky, R.; Kirchner, H.; DeMaeyer-Guignard, J.; DeMaeyer, E. An X-linked locus influences the amount of circulating interferon induced in the mouse by herpes simplex virus type 1. J Gen Virol 1982, 63, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.; Brossay, L. NK cells and NKT cells in innate defense against viral infections. Curr Opin Immunol 2001, 13, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Cerwenka, A.; Lanier, L. Ligands for natural killer cell receptors: redundancy or specificity. Immunol Rev 2001, 181, 158–169. [Google Scholar] [PubMed]
- Cooper, M.; Fehniger, T.; Caligiuri, M. The biology of human natural killer-cell subsets. Trends Immunol 2001, 22, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Kurago, Z.; Lutz, C.; Smith, K.; Colonna, M. NK cell natural cytotoxicity and IFN-gamma production are not always coordinately regulated: engagement of DX9 KIR+ NK cells by HLA-B7 variants and target cells. J Immunol 1998, 160, 1573–1580. [Google Scholar] [PubMed]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med 2003, 198, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G. N.; Bernstein, D. I. Generation of humoral immune responses against herpes simplex virus type 2 in the murine female genital tract. Virology 1995, 206, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L. A.; Zhu, L.; Thebeau, L. G. Vaccine-induced serum immunoglobin contributes to protection from herpes simplex virus type 2 genital infection in the presence of immune T cells. J Virol 2001, 75, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Seppanen, M.; Meri, S.; Notkola, I.-L.; Seppala, I. J. T.; Hiltunen-Back, E.; Sarvas, H.; Lappalainen, M.; Valimaa, H.; Palikhe, A.; Valtonen, V. V.; Lokki, M.-L. Subtly impaired humoral immunity predisposes to frequently recurring genital herpes simplex virus type 2 infection and herpetic neuralgia. J Infect Dis 2006, 194, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Rouse, B. T.; Gierynska, M. Immunity to herpes simplex virus: a hypothesis. Herpes: the journal of the IHMF 2001, 8, 2A–5A. [Google Scholar] [PubMed]
- Dobbs, M. E.; Strasser, J. E.; Chu, C.-F.; Chalk, C.; Milligan, G. N. Clearance of herpes simplex virus type 2 by CD8+ T cells requires gamma interferon and either perforin- or Fas-mediated cytolytic mechanisms. J Virol 2005, 79, 14546–14554. [Google Scholar] [CrossRef] [PubMed]
- Iijima, N.; Linehan, M. M.; Zamora, M.; Butkus, D.; Dunn, R.; Kehry, M. R.; Laufer, T. M.; Iwasaki, A. Dendritic cells and B cells maximize mucosal Th1 memory response to herpes simplex virus. J Exp Med 2008, 205, 3041–3052. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G. N.; Bernstein, D. I. Interferon-gamma enhances resolution of herpes simplex virus type 2 infection of the murine genital tract. Virology 1997, 229, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G. N.; Dudley-McClain, K. L.; Young, C. G.; Chu, C.-F. T-cell-mediated mechanisms involved in resolution of genital herpes simplex virus type 2 (HSV-2) infection of mice. J Reprod Immunol 2004, 61, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A. J.; Chu, C.-F.; Milligan, G. N. Effector CD4+ T-cell involvement in clearance of infectious herpes simplex virus type 1 from sensory ganglia and spinal cords. J Virol 2008, 82, 9678–9688. [Google Scholar] [CrossRef] [PubMed]
- Ellermann-Eriksen, S.; Justesen, J.; Mogensen, S. Genetically determined difference in the antiviral action of alpha/beta interferon in cells from mice resistant or susceptible to herpes simplex virus type 2. J Gen Virol 67, 1986, 1859–1866. [Google Scholar]
- Stulting, R.; Kindle, J.; Nahmias, A. Patterns of herpes simplex keratitis in inbred mice. Invest Ophthalmol Vis Sci 1985, 26, 1360–1367. [Google Scholar] [PubMed]
- Pepose, J.; Whittum-Hudson, J. An immunogenetic analysis of resistance to herpes simplex virus retinitis in inbred strains of mice. Invest Ophthalmol Vis Sci 1987, 28, 1549–1552. [Google Scholar] [PubMed]
- Abghari, S.; Stulting, R.; Nigida, S.; Downer, D.; Nahmias, A. Comparative replication of HSV-1 in BALB/c and C57BL/6 mouse embryo fibroblasts in vitro. Invest Ophthalmol Vis Sci 1986, 27, 909–914. [Google Scholar] [PubMed]
- Kastrukoff, L.; Lau, A.; Kim, S. Multifocal CNS demyelination following peripheral inoculation with herpes simplex virus type 1. Ann Neurol 1987, 22, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Vahlne, A.; Nilheden, E.; Svennerholm, B. Multiplicity activation of herpes simplex virus in mouse neuroblastoma (C1300) cells. Arch Virol 1981, 70, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Lau, A.; Kim, S.; Osborne, D.; Kastrukoff, L. Variation in resistance to herpes simplex virus type 1 of oligodendrocytes derived from inbred strains of mice. J Gen Virol 1991, 72, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Abghari, S.; Stulting, R.; Zhu, Z.; Schinazi, R.; Kaufman, H. Effect of genetically determined host factors on the efficacy of vidarabine, acyclovir and 5-trifluorothymidine in herpes simplex virus type 1 infection. Ophthalmic Res 1994, 26, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.; Kurimasa, A.; Oshimura, M.; Dynan, W.; Bradbury, E.; Chen, D. Loss of the catalytic subunit of the DNA-dependent protein kinase in DNA double-strand-break-repair mutant mammalian cells. Proc Natl Acad Sci U S A 1995, 92, 3171–3174. [Google Scholar] [CrossRef] [PubMed]
- Halford, W. P.; Balliet, J. W.; Gebhardt, B. M. Re-evaluating natural resistance to herpes simplex virus type 1. J Virol 2004, 78, 10086–10095. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Duerst, R.; Smith, T.; Morrison, L. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J Virol 2003, 77, 9337–9345. [Google Scholar] [CrossRef] [PubMed]
- Gill, N.; Deacon, P.; Lichty, B.; Mossman, K.; Ashkar, A. Induction of innate immunity against herpes simplex virus type 2 infection via local delivery of Toll-like receptor ligands correlates with beta interferon production. J Virol 2006, 80, 9943–9950. [Google Scholar] [CrossRef] [PubMed]
- Svensson, A.; Bellner, L.; Magnusson, M.; Eriksson, K. Role of IFN-alpha/beta signaling in the prevention of genital herpes virus type 2 infection. J Reprod Immunol 2007, 74, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.; Harrison, T.; Laslo, K.; Machalek, M.; Moorman, N.; Virgin, H. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med 1999, 189, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Luker, G.; Prior, J.; Song, J.; Pica, C.; Leib, D. Bioluminescence imaging reveals systemic dissemination of herpes simplex virus type 1 in the absence of interferon receptors. J Virol 2003, 77, 11082–11093. [Google Scholar] [CrossRef] [PubMed]
- Casrouge, A.; Zhang, S.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; Lorenzo, L.; Plancoulaine, S.; Senechal, B.; Geissmann, F.; Tabeta, K.; Hoebe, K.; Du, X.; Miller, R.; Heron, B.; Mignot, C.; de Villemeur, T.; Lebon, P.; Dulac, O.; Rozenberg, F.; Beutler, B.; Tardieu, M.; Abel, L.; Casanova, J. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, S.; Jouanguy, E.; Al-Hajjar, S.; Fieschi, C.; Al-Mohsen, I.; Al-Jumaah, S.; Yang, K.; Chapgier, A.; Eidenschenk, C.; Eid, P.; Al Ghonaium, A.; Tufenkeji, H.; Frayha, H.; Al-Gazlan, S.; Al-Rayes, H.; Schreiber, R.; Gresser, I.; Casanova, J. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet 2003, 33, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Picard, C.; Chapgier, A.; Plancoulaine, S.; Titeux, M.; Cognet, C.; von Bernuth, H.; Ku, C.; Casrouge, A.; Zhang, X.; Barreiro, L.; Leonard, J.; Hamilton, C.; Lebon, P.; Heron, B.; Vallee, L.; Quintana-Murci, L.; Hovnanian, A.; Rozenberg, F.; Vivier, E.; Geissmann, F.; Tardieu, M.; Abel, L.; Casanova, J. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.; Medzhitov, R.; Flavell, R. Recognition of double-stranded RNA and activation of NF-[kappa]B by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E. S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D. R.; Latz, E.; Fitzgerald, K. A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K. J.; Yamaguchi, O.; Otsu, K.; Tsujimura, T.; Koh, C.-S.; Reis e Sousa, C.; Matsuura, Y.; Fujita, T.; Akira, S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Limmon, G. V.; Arredouani, M.; McCann, K. L.; Corn Minor, R. A.; Kobzik, L.; Imani, F. Scavenger receptor class-A is a novel cell surface receptor for double-stranded RNA. FASEB J 2008, 22, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Muruve, D. A.; Pétrilli, V.; Zaiss, A. K.; White, L. R.; Clark, S. A.; Ross, P. J.; Parks, R. J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Aravalli, R. N.; Hu, S.; Rowen, T. N.; Palmquist, J. M.; Lokensgard, J. R. Cutting edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus. J Immunol 2005, 175, 4189–4193. [Google Scholar] [PubMed]
- Kurt-Jones, E. A.; Chan, M.; Zhou, S.; Wang, J.; Reed, G.; Bronson, R.; Arnold, M. M.; Knipe, D. M.; Finberg, R. W. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci USA 2004, 101, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; Akira, S. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, S.; Marq, J.-B.; Tapparel, C.; Kolakofsky, D.; Garcin, D. RIG-I and dsRNA-induced IFNbeta activation. PLoS ONE 2008, 3, e3965. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T. S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M. K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; Ohba, Y.; Taniguchi, T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-H.; Macmillan, J. B.; Chen, Z. J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Choi, M. K.; Ban, T.; Yanai, H.; Negishi, H.; Lu, Y.; Tamura, T.; Takaoka, A.; Nishikura, K.; Taniguchi, T. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci USA 2008, 105, 5477–5482. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S. B.; Jensen, S. B.; Nielsen, C.; Quartin, E.; Kato, H.; Chen, Z. J.; Silverman, R. H.; Akira, S.; Paludan, S. R. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene- like receptors, which synergize to induce type I interferon production. J Gen Virol 2009, 90, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S. B.; Sørensen, L. N.; Malmgaard, L.; Ank, N.; Baines, J. D.; Chen, Z. J.; Paludan, S. R. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J Virol 2007, 81, 13315–13324. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zhong, J.; Chung, J.; Chisari, F. V. Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc Natl Acad Sci USA 2007, 104, 9035–9040. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Luker, G. D.; Barchet, W.; Leib, D. A.; Akira, S.; Colonna, M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 2004, 103, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Linehan, M.; Iijima, N.; Iwasaki, A. Cutting Edge: Plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. J Immunol 2006, 177, 7510–7514. [Google Scholar] [PubMed]
- Mansur, D.; Kroon, E.; Nogueira, M.; Arantes, R.; Rodrigues, S.; Akira, S.; Gazzinelli, R.; Campos, M. Lethal encephalitis in myeloid differentiation factor 88-deficient mice infected with herpes simplex virus 1. Am J Pathol 2005, 166, 1419–1426. [Google Scholar] [PubMed]
- Wuest, T.; Austin, B.; Uematsu, S.; Thapa, M.; Akira, S.; Carr, D. Intact TRL 9 and type I interferon signaling pathways are required to augment HSV-1 induced corneal CXCL9 and CXCL10. J Neuroimmunol 2006, 179, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Ashkar, A.; Rosenthal, K. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol 2003, 77, 10168–10171. [Google Scholar] [CrossRef] [PubMed]
- Ashkar, A.; Yao, X.; Gill, N.; Sajic, D.; Patrick, A.; Rosenthal, K. Toll-like receptor (TLR)-3, but not TLR4, agonist protects against genital herpes infection in the absence of inflammation seen with CpG DNA. J Infect Dis 2004, 190, 1841–1849. [Google Scholar] [CrossRef] [PubMed]
- Harandi, A.; Eriksson, K.; Holmgren, J. A protective role of locally administered immunostimulatory CpG oligodeoxynucleotide in a mouse model of genital herpes infection. J Virol 2003, 77, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Herbst-Kralovetz, M.; Pyles, R. Quantification of poly(I:C)-mediated protection against genital herpes simplex virus type 2 infection. J Virol 2006, 80, 9988–9997. [Google Scholar] [CrossRef] [PubMed]
- Pyles, R.; Higgins, D.; Chalk, C.; Zalar, A.; Eiden, J.; Brown, C.; Van Nest, G.; Stanberry, L. Use of immunostimulatory sequence-containing oligonucleotides as topical therapy for genital herpes simplex virus type 2 infection. J Virol 2002, 76, 11387–11396. [Google Scholar] [CrossRef] [PubMed]
- Sajic, D.; Ashkar, A.; Patrick, A.; McCluskie, M.; Davis, H.; Levine, K.; Holl, R.; Rosenthal, K. Parameters of CpG oligodeoxynucleotide-induced protection against intravaginal HSV-2 challenge. J Med Virol 2003, 71, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Boivin, N.; Sergerie, Y.; Rivest, S.; Boivin, G. Effect of pretreatment with toll-like receptor agonists in a mouse model of herpes simplex virus type 1 encephalitis. J Infect Dis 2008, 198, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.; Pietropaolo, R.; Compton, T. Engagement of the Cellular Receptor for Glycoprotein B of Human Cytomegalovirus Activates the Interferon-Responsive Pathway. Mol. Cell. Biol. 1999, 19, 3607–3613. [Google Scholar] [PubMed]
- Collins, S.; Noyce, R.; Mossman, K. Innate cellular response to virus particle entry requires IRF3 but not virus replication. J Virol 2004, 78, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Netterwald, J.; Jones, T.; Britt, W.; Yang, S.-J.; McCrone, I.; Zhu, H. Postattachment Events Associated with Viral Entry Are Necessary for Induction of Interferon-Stimulated Genes by Human Cytomegalovirus. J. Virol. 2004, 78, 6688–6691. [Google Scholar] [CrossRef] [PubMed]
- Noyce, R. S.; Collins, S. E.; Mossman, K. L. Differential modification of interferon regulatory factor 3 following virus particle entry. J Virol 2009, 83, 4013–4022. [Google Scholar] [CrossRef] [PubMed]
- Paladino, P.; Cummings, D.; Noyce, R.; Mossman, K. The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J Immunol 2006, 177, 8008–8016. [Google Scholar] [PubMed]
- Prescott, J.; Ye, C.; Sen, G.; Hjelle, B. Induction of Innate Immune Response Genes by Sin Nombre Hantavirus Does Not Require Viral Replication. J. Virol. 2005, 79, 15007–15015. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.; Harman, A.; Nicholl, M. Activation of interferon response factor-3 in human cells infected with herpes simplex virus type 1 or human cytomegalovirus. J Virol 2001, 75, 8909–8916. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J. A.; Bergstralh, D. T.; Wang, Y.; Willingham, S. B.; Ye, Z.; Zimmermann, A. G.; Ting, J. P.-Y. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci USA 2007, 104, 8041–8046. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D. S.; Newton, K.; McBride, J.; O'Rourke, K.; Roose-Girma, M.; Lee, W. P.; Weinrauch, Y.; Monack, D. M.; Dixit, V. M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 2007, 14, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.; McWhirter, S.; Faia, K.; Rowe, D.; Latz, E.; Golenbock, D.; Coyle, A.; Liao, S.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; tenOever, B.; Grandvaux, N.; Zhou, G.-P.; Lin, R.; Hiscott, J. Triggering the Interferon Antiviral Response Through an IKK-Related Pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.-Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; Shu, H.-B. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, S. M.; Fitzgerald, K. A.; Rosains, J.; Rowe, D. C.; Golenbock, D. T.; Maniatis, T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci USA 2004, 101, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Servant, M.; tenOever, B.; Sen, G.; Balachandran, S.; Barber, G.; Lin, R.; Hiscott, J. Transcriptional Profiling of Interferon Regulatory Factor 3 Target Genes: Direct Involvement in the Regulation of Interferon-Stimulated Genes. J. Virol. 2002, 76, 5532–5539. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; McBride, K.; Weaver, B.; Dingwall, C.; Reich, N. Regulated Nuclear-Cytoplasmic Localization of Interferon Regulatory Factor 3, a Subunit of Double-Stranded RNA-Activated Factor 1. Mol. Cell. Biol. 2000, 20, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Heylbroeck, C.; Pitha, P.; Hiscott, J. Virus-Dependent Phosphorylation of the IRF-3 Transcription Factor Regulates Nuclear Translocation, Transactivation Potential, and Proteasome-Mediated Degradation. Mol. Cell. Biol. 1998, 18, 2986–2996. [Google Scholar] [PubMed]
- Qin, B.; Liu, C.; Srinath, H.; Lam, S.; Correia, J.; Derynck, R.; Lin, K. Crystal Structure of IRF-3 in Complex with CBP. Structure 2005, 13, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Qin, B. Y.; Liu, C.; Lam, S. S.; Srinath, H.; Delston, R.; Correia, J. J.; Derynck, R.; Lin, K. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol 2003, 10, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Takahasi, K.; Suzuki, N. N.; Horiuchi, M.; Mori, M.; Suhara, W.; Okabe, Y.; Fukuhara, Y.; Terasawa, H.; Akira, S.; Fujita, T.; Inagaki, F. X-ray crystal structure of IRF-3 and its functional implications. Nat Struct Biol 2003, 10, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Noyce, R.; Collins, S.; Everett, R.; Mossman, K. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J Virol 2004, 78, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Melroe, G.; DeLuca, N.; Knipe, D. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J Virol 2004, 78, 8411–8420. [Google Scholar] [CrossRef] [PubMed]
- Melroe, G.; Silva, L.; Schaffer, P.; Knipe, D. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: Potential role in blocking IFN-beta induction. Virology 2007, 360, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; Taniguchi, T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- D'Cunha, J.; Knight, E.; Haas, A.; Truitt, R.; Borden, E. Immunoregulatory properties of ISG15, an interferon-induced cytokine. PNAS 1996, 93, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Peters, K.; Sen, G. Induction of the Human Protein P56 by Interferon, Double-Stranded RNA, or Virus Infection. Virology 2000, 267, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.; Bhasker, C.; Merrick, W.; Sen, G. Viral Stress-inducible Protein p56 Inhibits Translation by Blocking the Interaction of eIF3 with the Ternary Complex eIF2{middle dot}GTP{middle dot}Met-tRNAi. J. Biol. Chem. 2003, 278, 39477–39482. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.; Rebouillat, D.; Ramana, C.; Murakami, J.; Hill, J.; Gudkov, A.; Silverman, R.; Stark, G.; Williams, B. Down-Regulation of p53 by Double-Stranded RNA Modulates the Antiviral Response. J. Virol. 2005, 79, 11105–11114. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Hassel, B.; Silverman, R. Expression cloning of 2-5A-dependent RNAase: A uniquely regulated mediator of interferon action. Cell 1993, 72, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Paladino, P.; Mossman, K. L. Mechanisms employed by herpes simplex virus 1 to inhibit the interferon response. J Interferon Cytokine Res 2009, 29, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Lavau, C.; Marchio, A.; Fagioli, M.; Jansen, J.; Falini, B.; Lebon, P.; Grosveld, F.; Pandolfi, P. P.; Pelicci, P. G.; Dejean, A. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene 1995, 11, 871–876. [Google Scholar] [PubMed]
- Wang, Z. G.; Ruggero, D.; Ronchetti, S.; Zhong, S.; Gaboli, M.; Rivi, R.; Pandolfi, P. P. PML is essential for multiple apoptotic pathways. Nat Genet 1998, 20, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Guo, Y.; Niu, Q.; Levy, D. E.; Dyck, J. A.; Lu, S.; Sheiman, L. A.; Liu, Y. Proto-oncogene PML controls genes devoted to MHC class I antigen presentation. Nature 1998, 396, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Everett, R. D.; Maul, G. G. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J 1994, 13, 5062–5069. [Google Scholar] [PubMed]
- Maul, G. G.; Everett, R. D. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J Gen Virol 1994, 75, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P. J.; Sen, G. C.; Dubois, M. F.; Ratner, L.; Slattery, E.; Lengyel, P. Interferon action: two distinct pathways for inhibition of protein synthesis by double-stranded RNA. Proc Natl Acad Sci USA 1978, 75, 5893–5897. [Google Scholar] [CrossRef] [PubMed]
- Al-Khatib, K.; Williams, B.; Silverman, R.; Halford, W.; Carr, D. Distinctive roles for 2',5'-oligoadenylate synthetases and double-stranded RNA-dependent protein kinase R in the in vivo antiviral effect of an adenoviral vector expressing murine IFN-beta. J Immunol 2004, 172, 5638–5647. [Google Scholar] [PubMed]
- Carr, D.; Tomanek, L.; Silverman, R.; Campbell, I.; Williams, B. RNA-dependent protein kinase is required for alpha-1 interferon transgene-induced resistance to genital herpes simplex virus type 2. J Virol 2005, 79, 9341–9345. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA 1997, 94, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Poppers, J.; Mulvey, M.; Khoo, D.; Mohr, I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J Virol 2000, 74, 11215–11221. [Google Scholar] [CrossRef] [PubMed]
- Duerst, R. J.; Morrison, L. A. Herpes simplex virus 2 virion host shutoff protein interferes with type I interferon production and responsiveness. Virology 2004, 322, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Karlhofer, F.; Ribaudo, R.; Yokoyama, W. MHC class I alloantigen specificity of Ly-49+ IL-2-activated natural killer cells. Nature 1992, 358, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Wagtmann, N.; Biassoni, R.; Cantoni, C.; Verdiani, S.; Malnati, M.; Vitale, M.; Bottino, C.; Moretta, L.; Moretta, A.; Long, E. Molecular clones of the p58 NK cell receptor reveal immunoglobulin-related molecules with diversity in both the extra- and intracellular domains. Immunity 1995, 2, 439–449. [Google Scholar] [CrossRef]
- Mocikat, R.; Braumuller, H.; Gumy, A.; Egeter, O.; Ziegler, H.; Reusch, U.; Bubeck, A.; Louis, J.; Mailhammer, R.; Riethmuller, G.; Koszinowski, U.; Rocken, M. Natural killer cells activated by MHC class I(low) targets prime dendritic cells to induce protective CD8 T cell responses. Immunity 2003, 19, 561–569. [Google Scholar] [CrossRef] [PubMed]
- York, I.; Roop, C.; Andrews, D.; Riddell, S.; Graham, F.; Johnson, D. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 1994, 77, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Rola-Pleszczynski, M. In vitro induction of human cell-mediated cytotoxicity directed against herpes simplex virus-infected cells Kinetics in normal donors and patients with recurrent herpes labialis. J Clin Lab Immunol 1981, 6, 39–43. [Google Scholar] [PubMed]
- Bishop, G.; Glorioso, J.; Schwartz, S. Relationship between expression of herpes simplex virus glycoproteins and susceptibility of target cells to human natural killer activity. J Exp Med 1983, 157, 1544–1561. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, P.; Mendelsohn, M.; Lopez, C. Human natural killer cells limit replication of herpes simplex virus type 1 in vitro. J Immunol 1985, 134, 2666–2672. [Google Scholar] [PubMed]
- Smith, H.; Heusel, J.; Mehta, I.; Kim, S.; Dorner, B.; Naidenko, O.; Iizuka, K.; Furukawa, H.; Beckman, D.; Pingel, J.; Scalzo, A.; Fremont, D.; Yokoyama, W. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U S A 2002, 99, 8826–8831. [Google Scholar] [PubMed]
- Ghiasi, H.; Cai, S.; Perng, G.; Nesburn, A.; Wechsler, S. The role of natural killer cells in protection of mice against death and corneal scarring following ocular HSV-1 infection. Antiviral Res 2000, 45, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Habu, S.; Akamatsu, K.; Tamaoki, N.; Okumura, K. In vivo significance of NK cell on resistance against virus (HSV-1) infections in mice. J Immunol 1984, 133, 2743–2747. [Google Scholar] [PubMed]
- Nandakumar, S.; Woolard, S.; Yuan, D.; Rouse, B.; Kumaraguru, U. Natural killer cells as novel helpers in anti-herpes simplex virus immune response. J Virol 2008, 82, 10820–10831. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Scalzo, A.; Simmons, A. Cutting edge: a NK complex-linked locus governs acute versus latent herpes simplex virus infection of neurons. J Immunol 2001, 166, 5869–5873. [Google Scholar] [PubMed]
- Staats, H.; Oakes, J.; Lausch, R. Anti-glycoprotein D monoclonal antibody protects against herpes simplex virus type 1-induced diseases in mice functionally depleted of selected T-cell subsets or asialo GM1+ cells. J Virol 1991, 65, 6008–6014. [Google Scholar] [PubMed]
- Tanigawa, M.; Bigger, J.; Kanter, M.; Atherton, S. Natural killer cells prevent direct anterior-to-posterior spread of herpes simplex virus type 1 in the eye. Invest Ophthalmol Vis Sci 2000, 41, 132–137. [Google Scholar] [PubMed]
- Thapa, M.; Kuziel, W.; Carr, D. Susceptibility of CCR5-deficient mice to genital herpes simplex virus type 2 is linked to NK cell mobilization. J Virol 2007, 81, 3704–3713. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, J.; Welsh, R. The role of natural killer cells and interferon in resistance to acute infection of mice with herpes simplex virus type 1. J Immunol 1986, 136, 3481–3485. [Google Scholar] [PubMed]
- Chmielarczyk, W.; Engler, H.; Ernst, R.; Opitz, U.; Kirchner, H. Injection of anti-thy-1.2 serum breaks genetic resistance of mice against herpes simplex virus. J Gen Virol 1985, 66, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Kassim, S.; Rajasagi, N.; Ritz, B.; Pruett, S.; Gardner, E.; Chervenak, R.; Jennings, S. Dendritic cells are required for optimal activation of natural killer functions following primary infection with herpes simplex virus type 1. J Virol 2009, 83, 3175–3186. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, A.; Schwarting, G.; Robbins, P. Glycolipids of the mouse peritoneal macrophage. Alterations in amount and surface exposure of specific glycolipid species occur in response to inflammation and tumoricidal activation. J Exp Med 1984, 160, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Schuler, G. The dendritic, Thy-1-positive cell of murine epidermis: a new epidermal cell type of bone marrow origin. J Invest Dermatol 1984, 83, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Suttles, J.; Schwarting, G.; Stout, R. Flow cytometric analysis reveals the presence of asialo GM1 on the surface membrane of alloimmune cytotoxic T lymphocytes. J Immunol 1986, 136, 1586–1591. [Google Scholar] [PubMed]
- Wiltrout, R.; Santoni, A.; Peterson, E.; Knott, D.; Overton, W.; Herberman, R.; Holden, H. Reactivity of anti-asialo GM1 serum with tumoricidal and non-tumoricidal mouse macrophages. J Leukoc Biol 1985, 37, 597–614. [Google Scholar] [PubMed]
- Halford, W. P.; Maender, J. L.; Gebhardt, B. M. Re-evaluating the role of natural killer cells in innate resistance to herpes simplex virus type 1. Virol J 2005, 2, 56. [Google Scholar] [CrossRef] [Green Version]
- Slifka, M.; Pagarigan, R.; Whitton, J. NK markers are expressed on a high percentage of virus-specific CD8+ and CD4+ T cells. J Immunol 2000, 164, 2009–2015. [Google Scholar] [PubMed]
- Rager-Zisman, B.; Quan, P.; Rosner, M.; Moller, J.; Bloom, B. Role of NK cells in protection of mice against herpes simplex virus-1 infection. J Immunol 1987, 138, 884–888. [Google Scholar] [PubMed]
- Adler, H.; Beland, J.; Del-Pan, N.; Kobzik, L.; Sobel, R.; Rimm, I. In the absence of T cells, natural killer cells protect from mortality due to HSV-1 encephalitis. J Neuroimmunol 1999, 93, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.; Klem, J.; Puzanov, I.; Sivakumar, P.; Schatzle, J.; Bennett, M.; Kumar, V. Natural killer cell differentiation: insights from knockout and transgenic mouse models and in vitro systems. Immunol Rev 1998, 165, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Gill, N.; Rosenthal, K.; Ashkar, A. NK and NKT cell-independent contribution of interleukin-15 to innate protection against mucosal viral infection. J Virol 2005, 79, 4470–4478. [Google Scholar] [CrossRef] [PubMed]
- Grubor-Bauk, B.; Simmons, A.; Mayrhofer, G.; Speck, P. Impaired clearance of herpes simplex virus type 1 from mice lacking CD1d or NKT cells expressing the semivariant V alpha 14-J alpha 281 TCR. J Immunol 2003, 170, 1430–1434. [Google Scholar] [PubMed]
- Brandt, C.; Salkowski, C. Activation of NK cells in mice following corneal infection with herpes simplex virus type-1. Invest Ophthalmol Vis Sci 1992, 33, 113–120. [Google Scholar] [PubMed]
- Gill, N.; Ashkar, A. Overexpression of interleukin-15 compromises CD4-dependent adaptive immune responses against herpes simplex virus 2. J Virol 2009, 83, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Tomazin, R.; van Schoot, N.; Goldsmith, K.; Jugovic, P.; Sempe, P.; Fruh, K.; Johnson, D. Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J Virol 1998, 72, 2560–2563. [Google Scholar] [PubMed]
- Ching, C.; Lopez, C. Natural killing of herpes simplex virus type 1-infected target cells: normal human responses and influence of antiviral antibody. Infect Immun 1979, 26, 49–56. [Google Scholar] [PubMed]
- Biron, C.; Byron, K.; Sullivan, J. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med 1989, 320, 1731–1735. [Google Scholar] [PubMed]
- Dalloul, A.; Oksenhendler, E.; Chosidow, O.; Ribaud, P.; Carcelain, G.; Louvet, S.; Massip, P.; Lebon, P.; Autran, B. Severe herpes virus (HSV-2) infection in two patients with myelodysplasia and undetectable NK cells and plasmacytoid dendritic cells in the blood. J Clin Virol 2004, 30, 329–336. [Google Scholar] [CrossRef] [PubMed]
- McKenna, K.; Beignon, A.; Bhardwaj, N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol 2005, 79, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Siegal, F.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Hochrein, H.; Schlatter, B.; O'Keeffe, M.; Wagner, C.; Schmitz, F.; Schiemann, M.; Bauer, S.; Suter, M.; Wagner, H. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A 2004, 101, 11416–11421. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, A.; Wagner, M.; Gunther, S.; Towarowski, A.; Tuma, E.; Moderer, M.; Rothenfusser, S.; Wetzel, S.; Endres, S.; Hartmann, G. Plasmacytoid dendritic cells: a new cutaneous dendritic cell subset with distinct role in inflammatory skin diseases. J Invest Dermatol 2002, 119, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Kittan, N.; Bergua, A.; Haupt, S.; Donhauser, N.; Schuster, P.; Korn, K.; Harrer, T.; Schmidt, B. Impaired plasmacytoid dendritic cell innate immune responses in patients with herpes virus-associated acute retinal necrosis. J Immunol 2007, 179, 4219–4230. [Google Scholar] [PubMed]
- Donaghy, H.; Bosnjak, L.; Harman, A.; Marsden, V.; Tyring, S.; Meng, T.; Cunningham, A. Role for plasmacytoid dendritic cells in the immune control of recurrent human herpes simplex virus infection. J Virol 2009, 83, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z. A.; Selke, S.; Zeh, J.; Kopelman, J.; Maslow, A.; Ashley, R. L.; Watts, D. H.; Berry, S.; Herd, M.; Corey, L. The acquisition of herpes simplex virus during pregnancy. N Engl J Med 1997, 337, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Kuklin, N. A.; Daheshia, M.; Chun, S.; Rouse, B. T. Role of mucosal immunity in herpes simplex virus infection. J Immunol 1998, 160, 5998–6003. [Google Scholar] [PubMed]
- Kohl, S.; Loo, L. S. The relative role of transplacental and milk immune transfer in protection against lethal neonatal herpes simplex virus infection in mice. J Infect Dis 1984, 149, 38–42. [Google Scholar] [PubMed]
- Sherwood, J. K.; Zeitlin, L.; Whaley, K. J.; Cone, R. A.; Saltzman, M. Controlled release of antibodies for long-term topical passive immunoprotection of female mice against genital herpes. Nat Biotechnol 1996, 14, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, L.; Whaley, K. J.; Sanna, P. P.; Moench, T. R.; Bastidas, R.; De Logu, A.; Williamson, R. A.; Burton, D. R.; Cone, R. A. Topically applied human recombinant monoclonal IgG1 antibody and its Fab and F(ab')2 fragments protect mice from vaginal transmission of HSV-2. Virology 1996, 225, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.-F.; Meador, M. G.; Young, C. G.; Strasser, J. E.; Bourne, N.; Milligan, G. N. Antibody-mediated protection against genital herpes simplex virus type 2 disease in mice by Fc gamma receptor-dependent and -independent mechanisms. J Reprod Immunol 2008, 78, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G. N.; Meador, M. G.; Chu, C.-F.; Young, C. G.; Martin, T. L.; Bourne, N. Long-term presence of virus-specific plasma cells in sensory ganglia and spinal cord following intravaginal inoculation of herpes simplex virus type 2. J Virol 2005, 79, 11537–11540. [Google Scholar] [CrossRef] [PubMed]
- Para, M. F.; Baucke, R. B.; Spear, P. G. Immunoglobulin G(Fc)-binding receptors on virions of herpes simplex virus type 1 and transfer of these receptors to the cell surface by infection. J Virol 1980, 34, 512–520. [Google Scholar] [PubMed]
- Baucke, R. B.; Spear, P. G. Membrane proteins specified by herpes simplex viruses V. Identification of an Fc-binding glycoprotein. J Virol 1979, 32, 779–789. [Google Scholar] [PubMed]
- Mitchell, B. M.; Stevens, J. G. Neuroinvasive properties of herpes simplex virus type 1 glycoprotein variants are controlled by the immune response. J Immunol 1996, 156, 246–255. [Google Scholar] [PubMed]
- Deshpande, S. P.; Zheng, M.; Daheshia, M.; Rouse, B. T. Pathogenesis of herpes simplex virus-induced ocular immunoinflammatory lesions in B-cell-deficient mice. J Virol 2000, 74, 3517–3524. [Google Scholar] [CrossRef] [PubMed]
- Dudley, K. L.; Bourne, N.; Milligan, G. N. Immune protection against HSV-2 in B-cell-deficient mice. Virology 2000, 270, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S. N.; Jones, C. M.; Smith, C. M.; Heath, W. R.; Carbone, F. R. Rapid cytotoxic T lymphocyte activation occurs in the draining lymph nodes after cutaneous herpes simplex virus infection as a result of early antigen presentation and not the presence of virus. J Exp Med 2002, 195, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, B. S.; Cherpes, T. L.; Urban, J.; Kalinski, P.; Hendricks, R. L. Reevaluating the CD8 T-cell response to herpes simplex virus type 1: involvement of CD8 T cells reactive to subdominant epitopes. J Virol 2009, 83, 2237–2245. [Google Scholar] [CrossRef] [PubMed]
- Hosken, N.; McGowan, P.; Meier, A.; Koelle, D. M.; Sleath, P.; Wagener, F.; Elliott, M.; Grabstein, K.; Posavad, C.; Corey, L. Diversity of the CD8+ T-cell response to herpes simplex virus type 2 proteins among persons with genital herpes. J Virol 2006, 80, 5509–5515. [Google Scholar] [CrossRef] [PubMed]
- Cantin, E.; Tanamachi, B.; Openshaw, H.; Mann, J.; Clarke, K. Gamma interferon (IFN-gamma) receptor null-mutant mice are more susceptible to herpes simplex virus type 1 infection than IFN-gamma ligand null-mutant mice. J Virol 1999, 73, 5196–5200. [Google Scholar] [PubMed]
- Wakim, L. M.; Jones, C. M.; Gebhardt, T.; Preston, C. M.; Carbone, F. R. CD8(+) T-cell attenuation of cutaneous herpes simplex virus infection reduces the average viral copy number of the ensuing latent infection. Immunol Cell Biol 2008, 86, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Cantin, E.; Hinton, D.; Chen, J.; Openshaw, H. Gamma interferon expression during acute and latent nervous system infection by herpes simplex virus type 1. J Virol 1995, 69, 4898–4905. [Google Scholar] [PubMed]
- Posavad, C. M.; Huang, M. L.; Barcy, S.; Koelle, D. M.; Corey, L. Long term persistence of herpes simplex virus-specific CD8+ CTL in persons with frequently recurring genital herpes. J Immunol 2000, 165, 1146–1152. [Google Scholar] [PubMed]
- Cantin, E.; Tanamachi, B.; Openshaw, H. Role for gamma interferon in control of herpes simplex virus type 1 reactivation. J Virol 1999, 73, 3418–3423. [Google Scholar] [PubMed]
- Milligan, G. N.; Bernstein, D. I.; Bourne, N. T lymphocytes are required for protection of the vaginal mucosae and sensory ganglia of immune mice against reinfection with herpes simplex virus type 2. J Immunol 1998, 160, 6093–6100. [Google Scholar] [PubMed]
- Balish, M.; Abrams, M.; Pumfery, A.; Brandt, C. Enhanced inhibition of herpes simplex virus type 1 growth in human corneal fibroblasts by combinations of interferon-alpha and -gamma. J Infect Dis 1992, 166, 1401–1403. [Google Scholar] [PubMed]
- Chen, S.; Oakes, J.; Lausch, R. Synergistic anti-HSV effect of tumor necrosis factor alpha and interferon gamma in human corneal fibroblasts is associated with interferon beta induction. Antiviral Res 1993, 22, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Czarniecki, C.; Fennie, C.; Powers, D.; Estell, D. Synergistic antiviral and antiproliferative activities of Escherichia coli-derived human alpha, beta, and gamma interferons. J Virol 1984, 49, 490–496. [Google Scholar] [PubMed]
- Neumann-Haefelin, D.; Sundmacher, R.; Frey, H.; Merk, W. Recombinant HuIFN-gamma prevents herpes simplex keratitis in African green monkeys: demonstration of synergism with recombinant HuIFN-alpha 2. Med Microbiol Immunol 1985, 174, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Zerial, A.; Hovanessian, A.; Stefanos, S.; Huygen, K.; Werner, G.; Falcoff, E. Synergistic activities of type I (alpha, beta) and type II (gamma) murine interferons. Antiviral Res 1982, 2, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B.; Halford, W. Alpha/Beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1. J Virol 2002, 76, 11541–11550. [Google Scholar] [CrossRef] [PubMed]
- Chee, A.; Lopez, P.; Pandolfi, P.; Roizman, B. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J Virol 2003, 77, 7101–7105. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.; DeSalvo, J.; Foster, T.; Kosinski, A.; Weller, S.; Halford, W. Beta interferon and gamma interferon synergize to block viral DNA and virion synthesis in herpes simplex virus-infected cells. J Gen Virol 2005, 86, 2421–2432. [Google Scholar] [CrossRef] [PubMed]
- Pasieka, T.; Cilloniz, C.; Lu, B.; Teal, T.; Proll, S.; Katze, M.; Leib, D. Host responses to wild-type and attenuated herpes simplex virus infection in the absence of Stat1. J Virol 2009, 83, 2075–2087. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Durand, V.; Kamphuis, E.; Thompson, C.; Bulfone-Paus, S.; Rossmann, C.; Kalinke, U.; Tough, D. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J Immunol 2006, 176, 4682–4689. [Google Scholar] [PubMed]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D. F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol 2003, 4, 1009–1015. [Google Scholar] [CrossRef]
- Trilling, M.; Le, V. T. K.; Zimmermann, A.; Ludwig, H.; Pfeffer, K.; Sutter, G.; Smith, G. L.; Hengel, H. Gamma interferon-induced interferon regulatory factor 1-dependent antiviral response inhibits vaccinia virus replication in mouse but not human fibroblasts. J Virol 2009, 83, 3684–3695. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Chew, T.; Taylor, K.E.; Mossman, K.L. Innate and Adaptive Immune Responses to Herpes Simplex Virus. Viruses 2009, 1, 979-1002. https://doi.org/10.3390/v1030979
Chew T, Taylor KE, Mossman KL. Innate and Adaptive Immune Responses to Herpes Simplex Virus. Viruses. 2009; 1(3):979-1002. https://doi.org/10.3390/v1030979
Chicago/Turabian StyleChew, Tracy, Kathryne E. Taylor, and Karen L. Mossman. 2009. "Innate and Adaptive Immune Responses to Herpes Simplex Virus" Viruses 1, no. 3: 979-1002. https://doi.org/10.3390/v1030979