Antiviral Effects of ABMA against Herpes Simplex Virus Type 2 In Vitro and In Vivo

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus, Compounds and Mice
2.2. Cytotoxicity Assay
2.3. Antiviral Activity Assay of ABMA against HSV-2 In Vitro
2.4. Western Blotting
2.5. Quantitative Polymerase Chain Reaction (qPCR) Assay
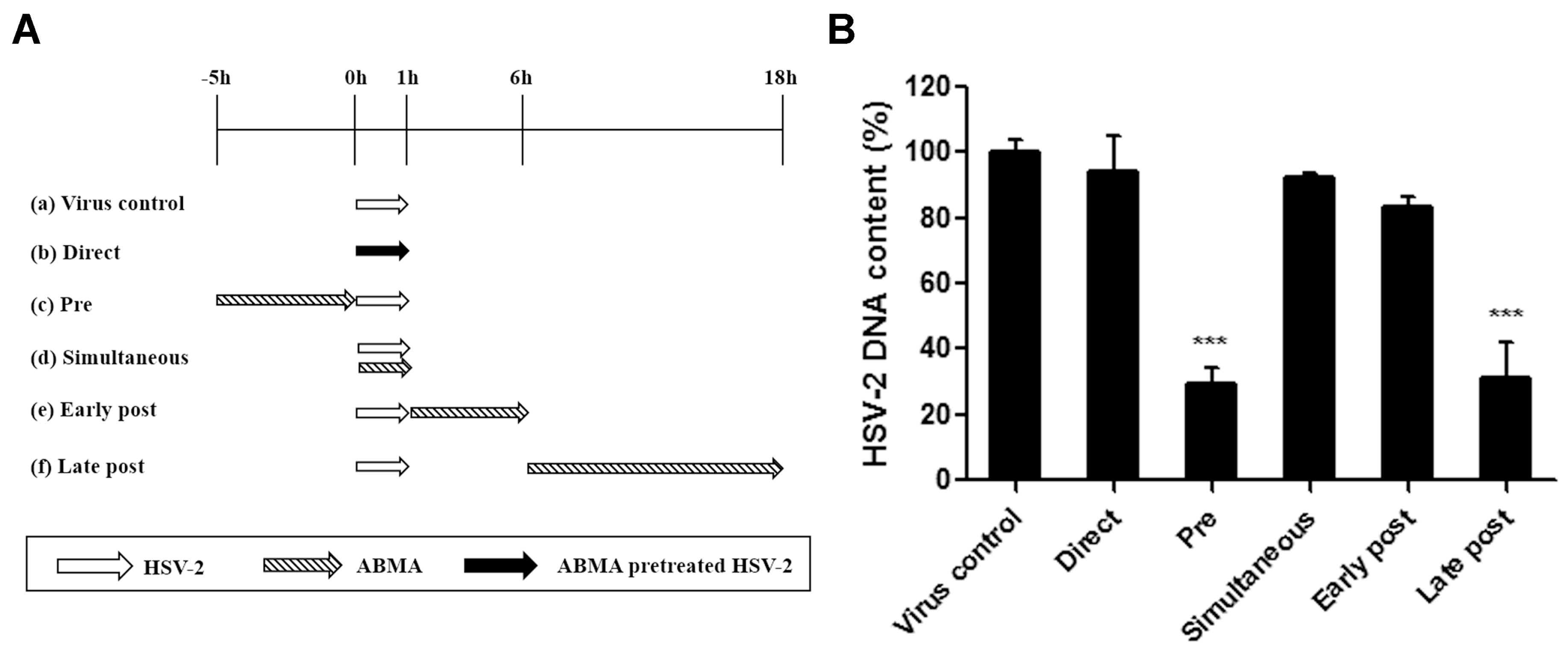
2.6. Time of ABMA Addition Assay
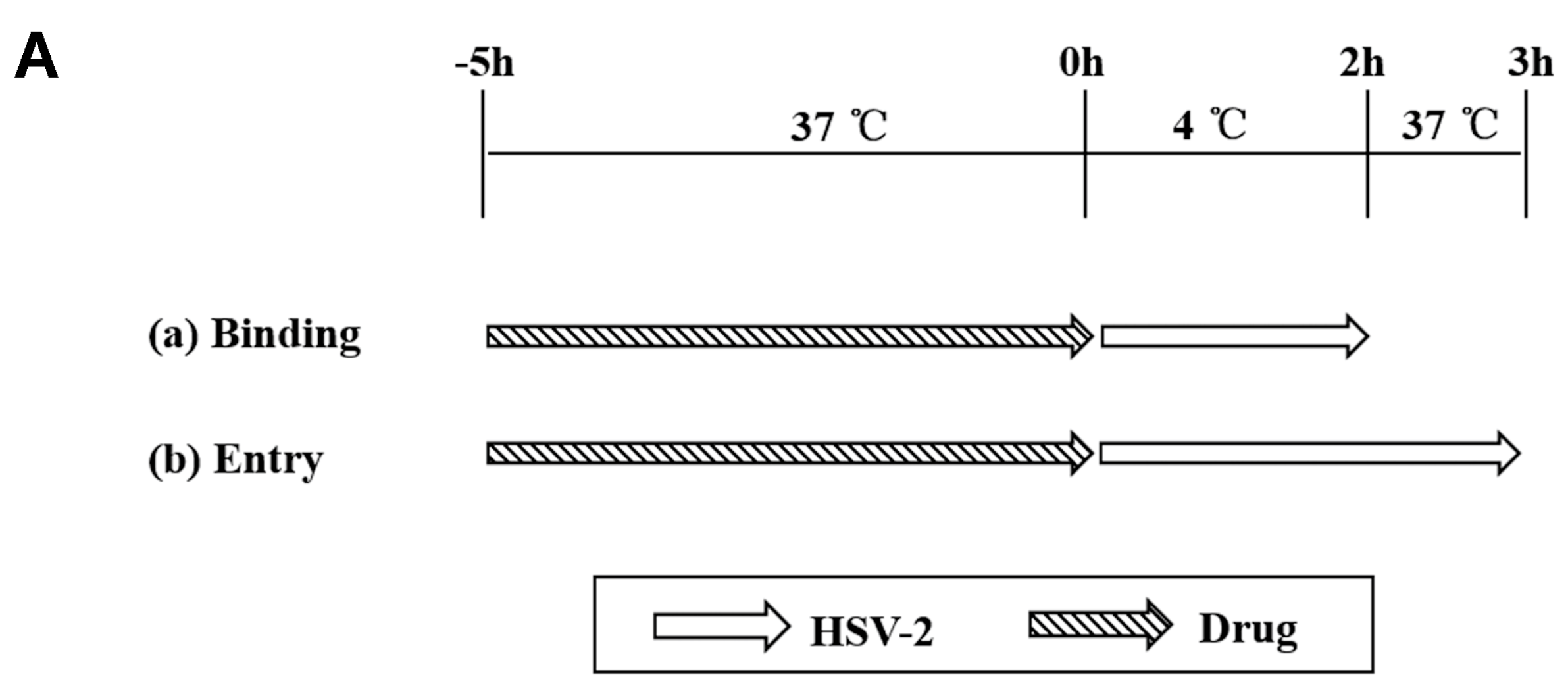
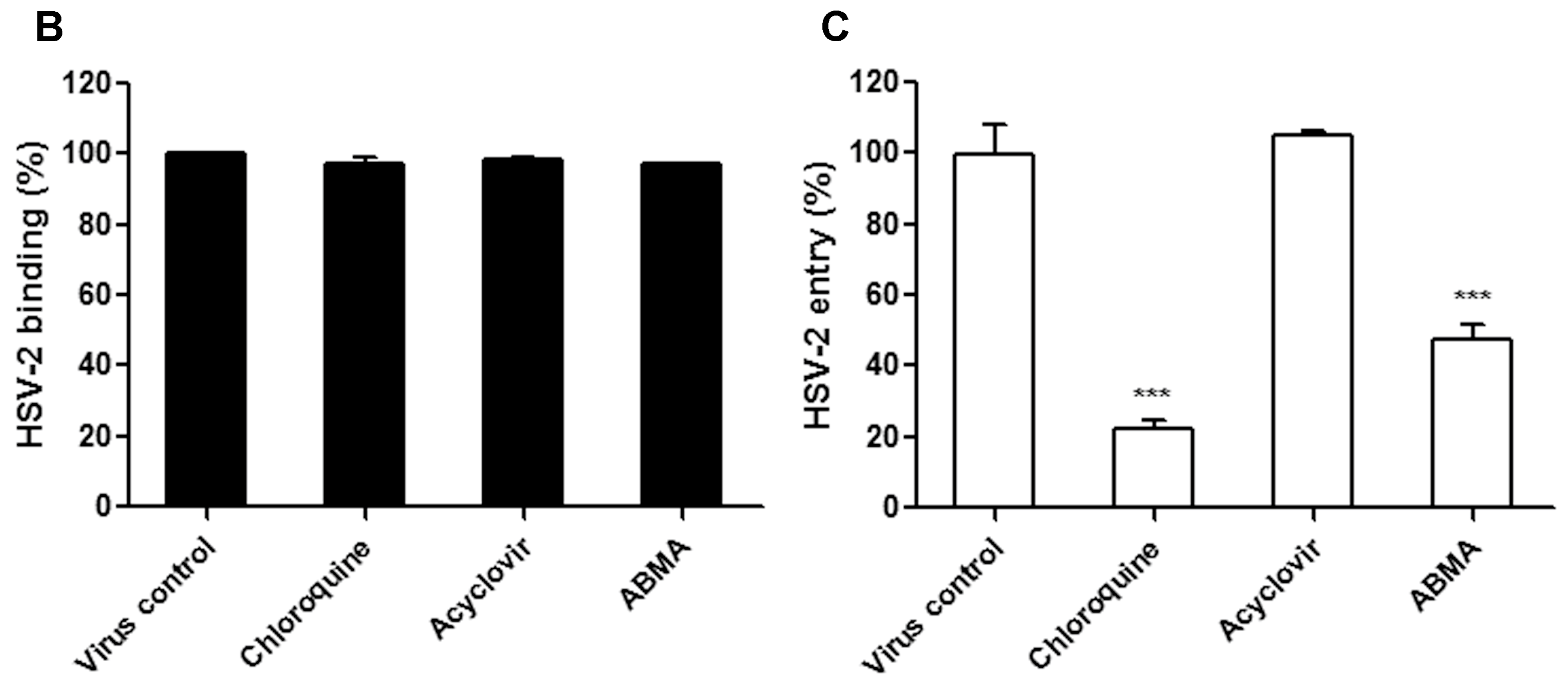
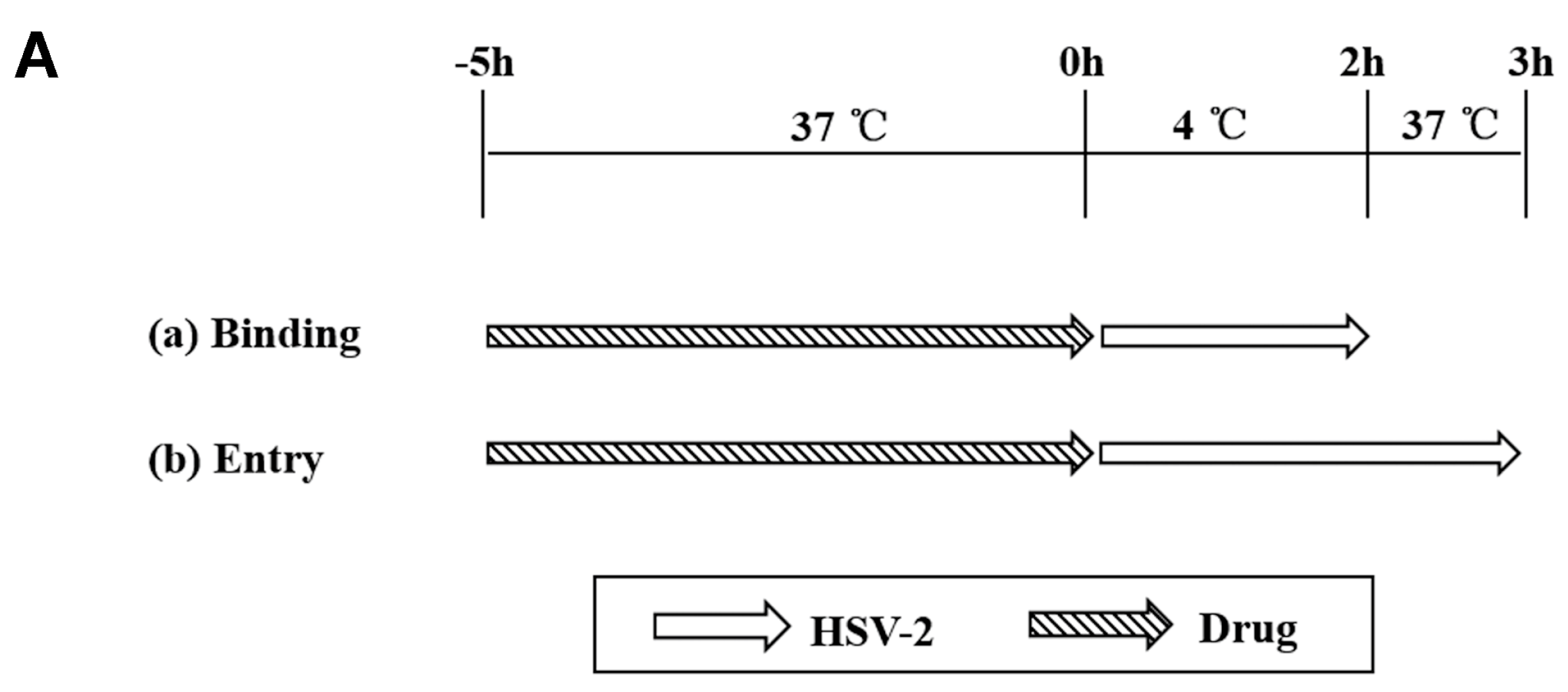
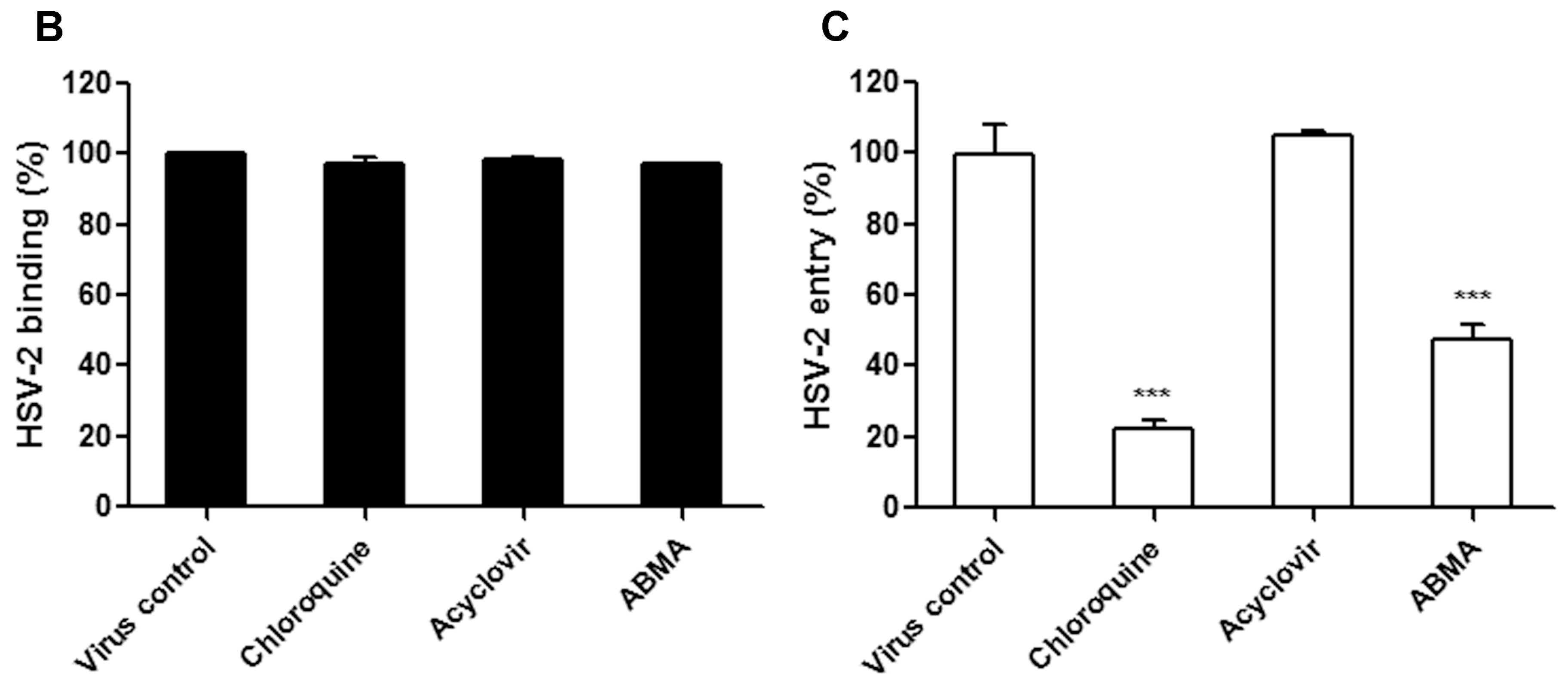
2.7. Binding and Entry Assays
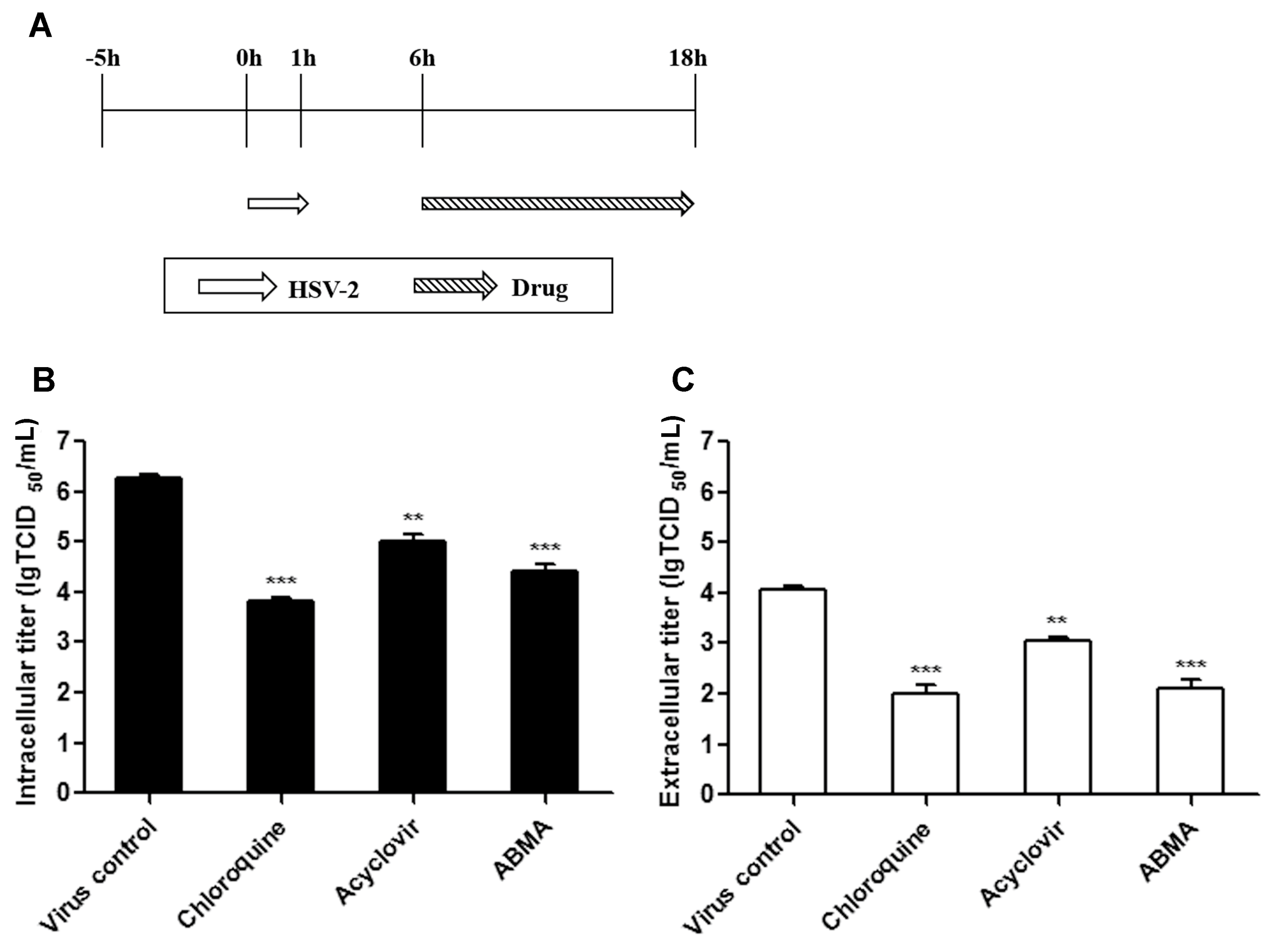
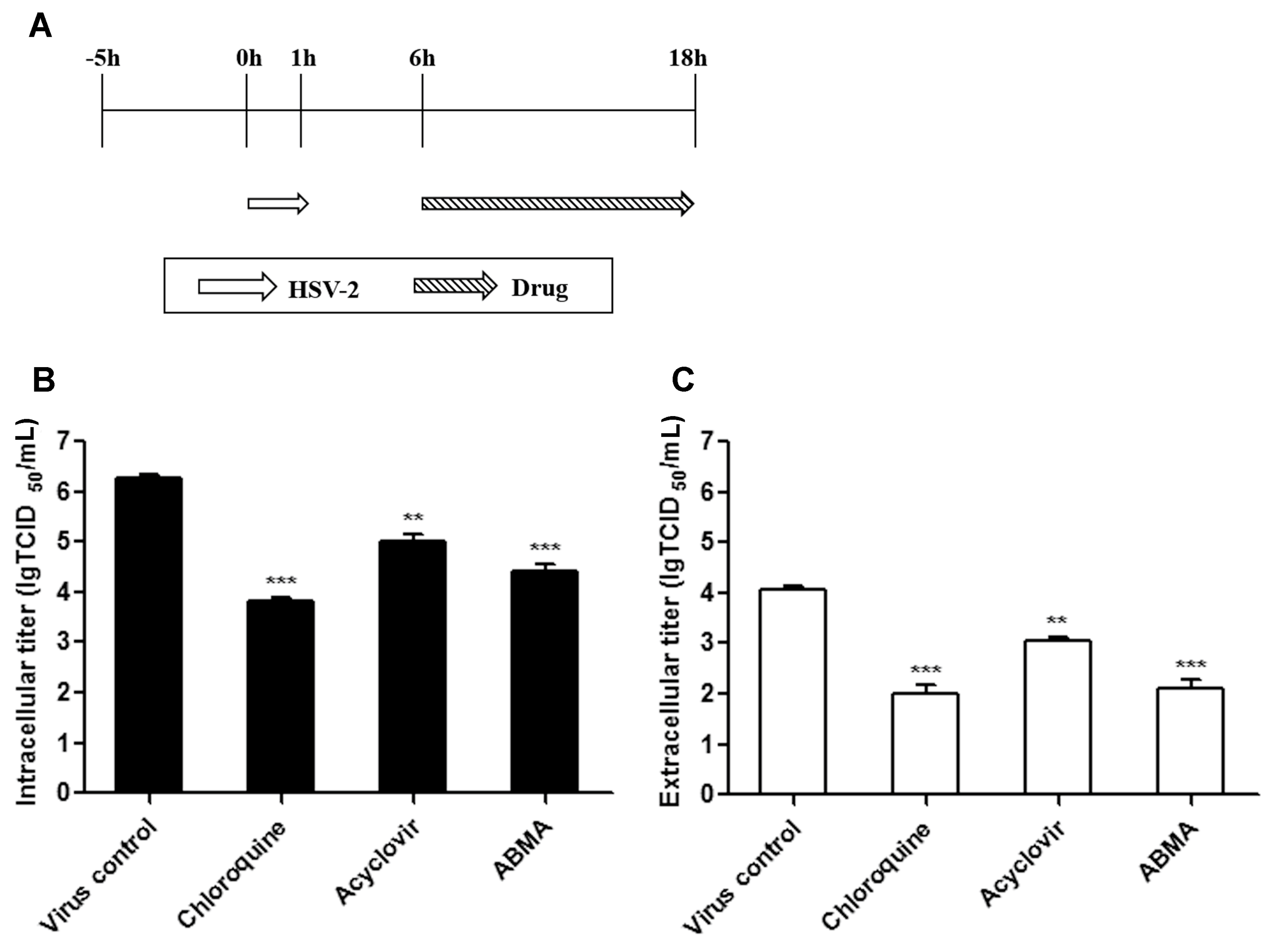
2.8. Late Stage Infection Assay
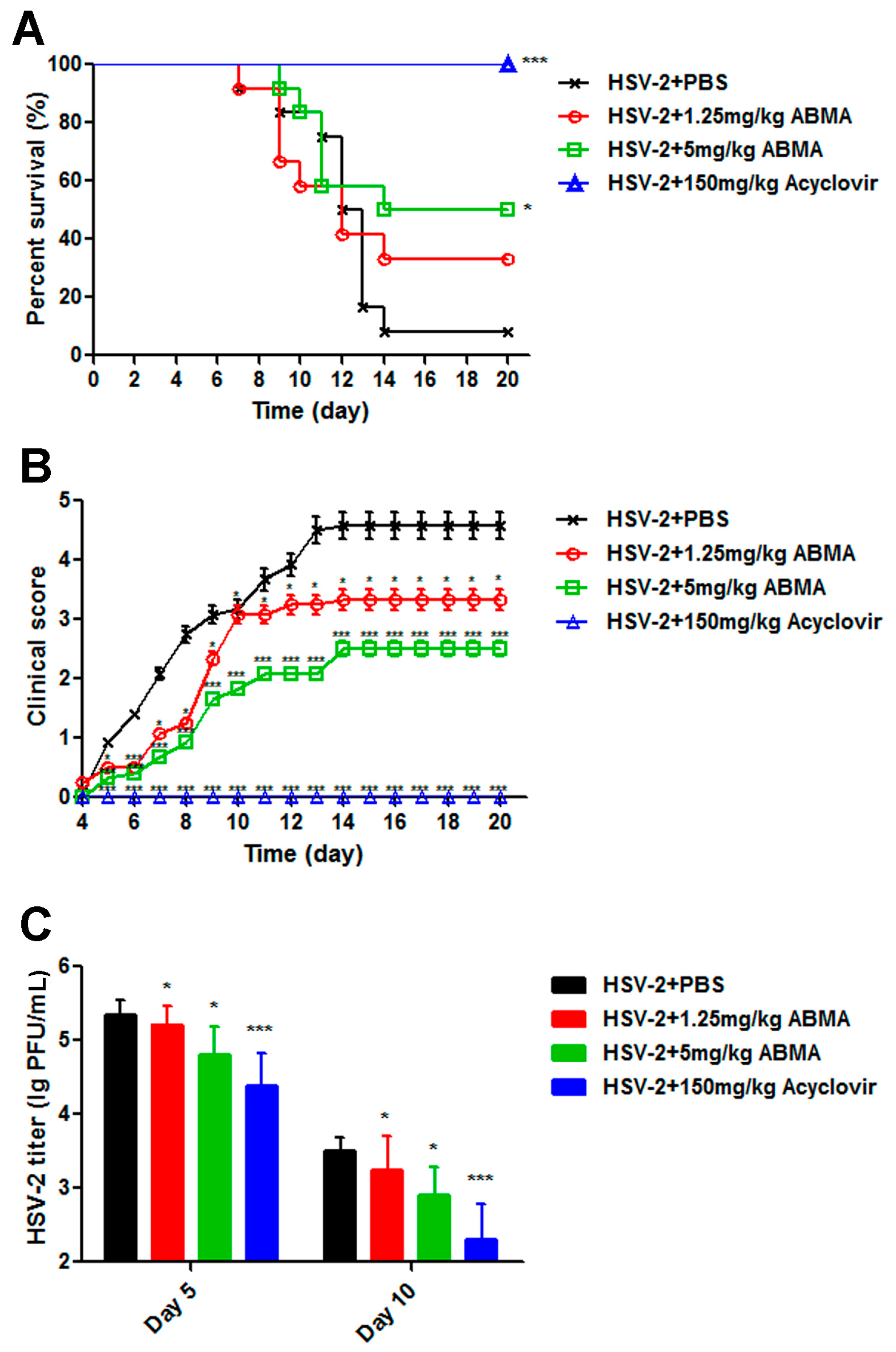
2.9. Antiviral Efficacy Assay of ABMA against HSV-2 In Vivo
2.10. Statistical Analysis
3. Results
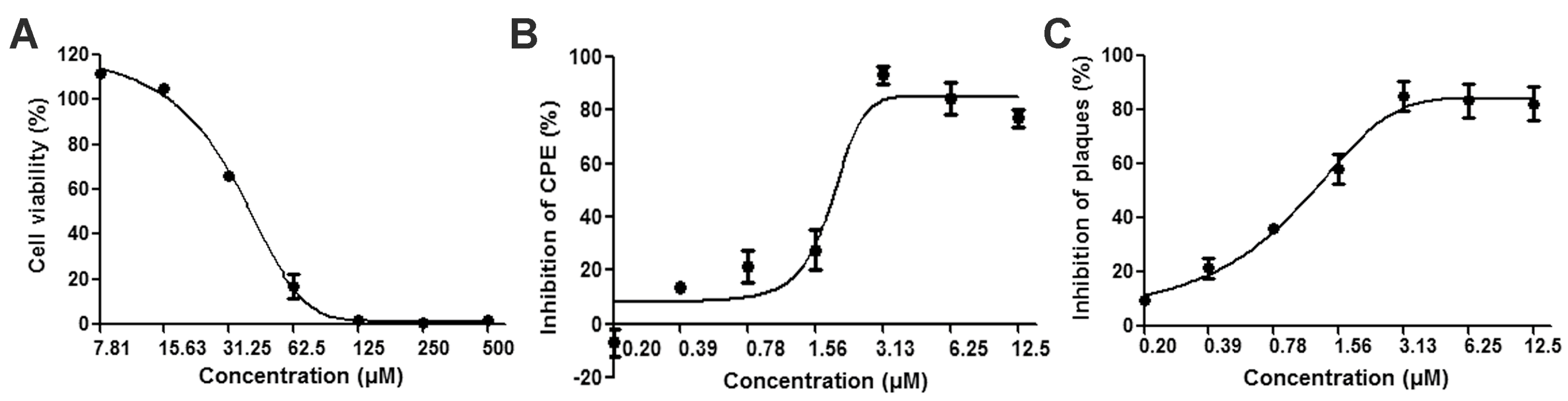
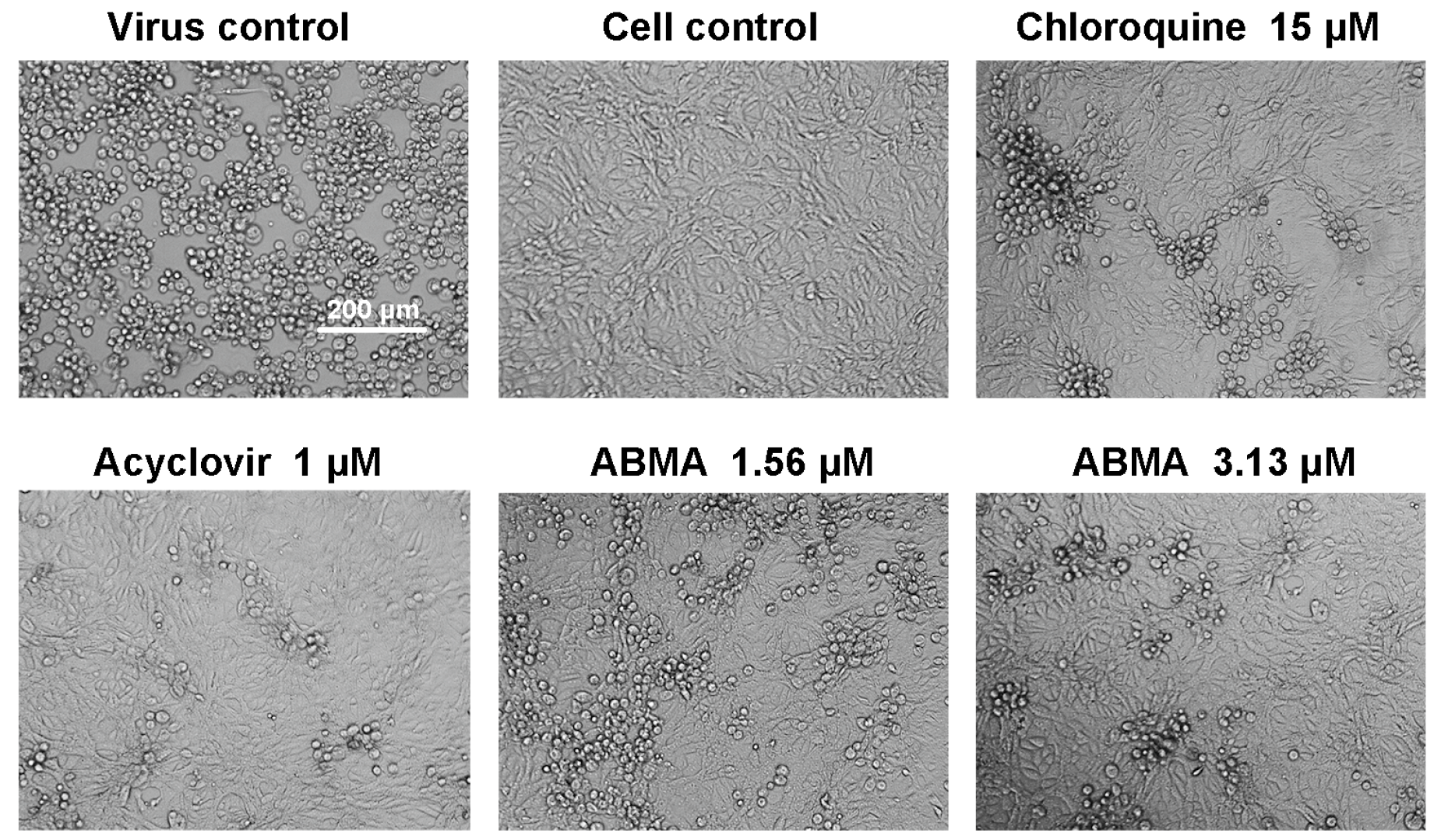
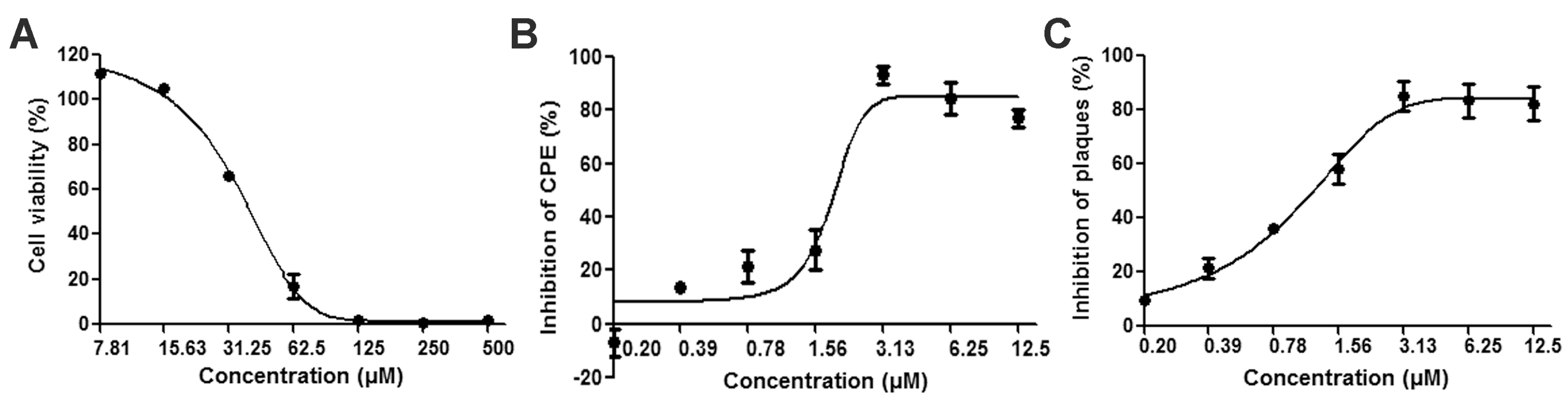
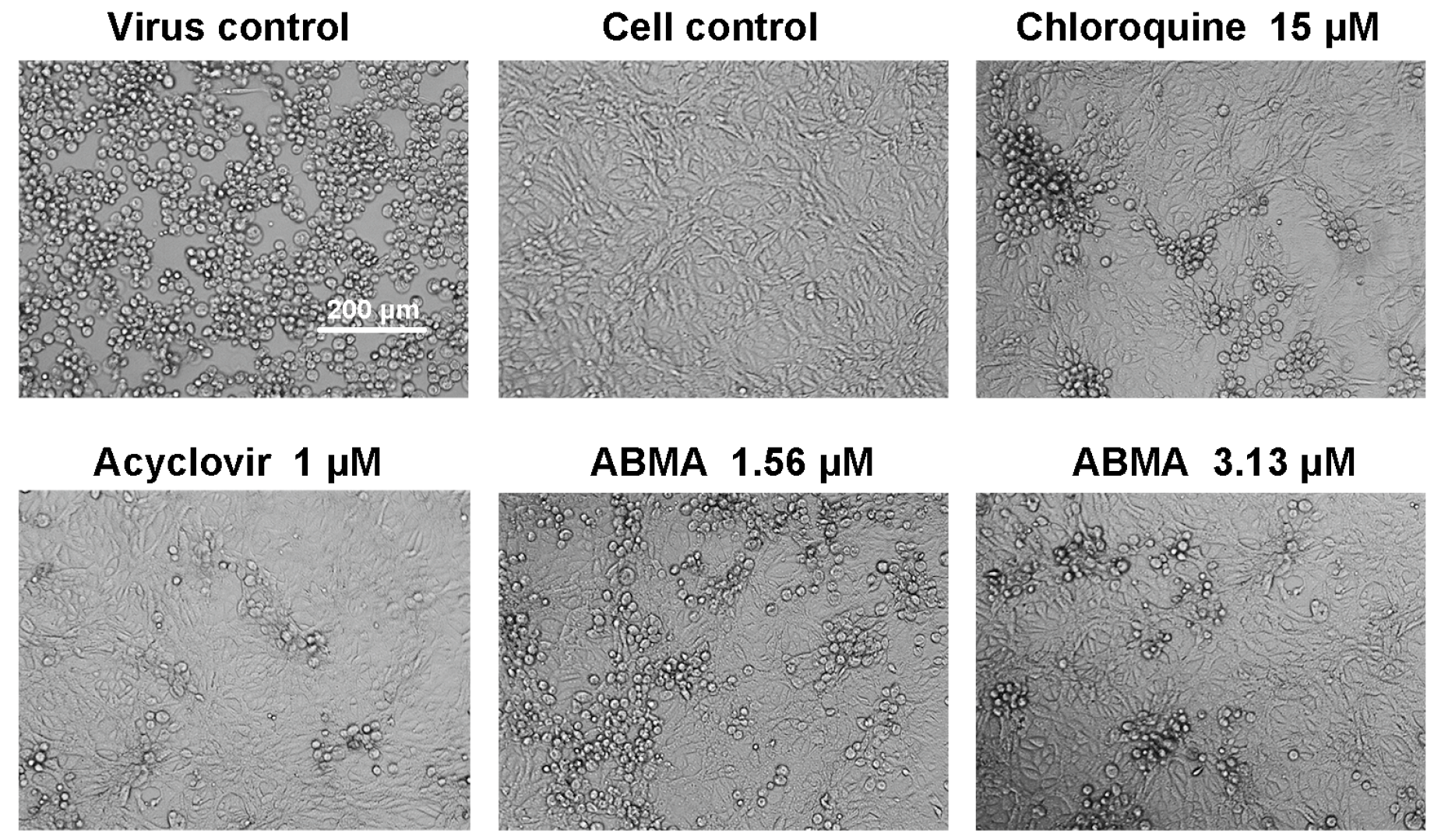
3.1. Reductions of HSV-2-Induced Cytopathic Effects and Plaque Formation Were Detected in ABMA-Treated Cells
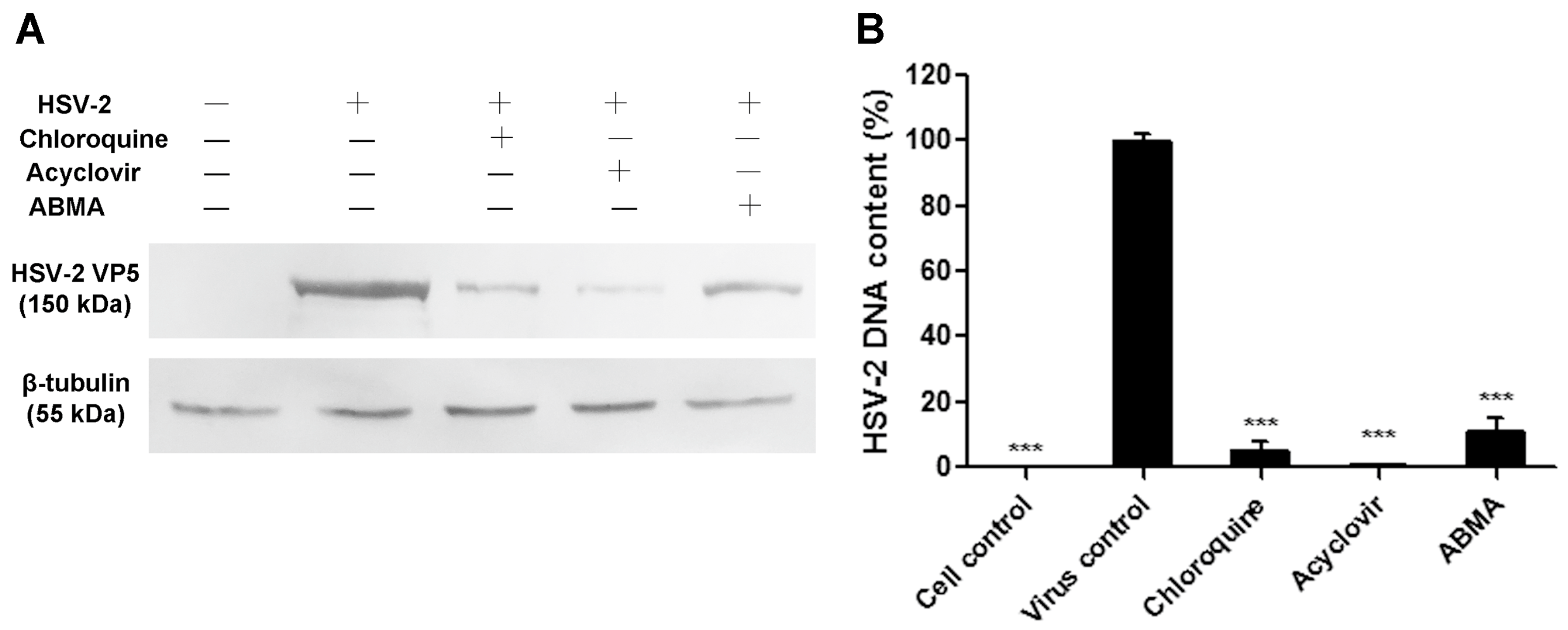
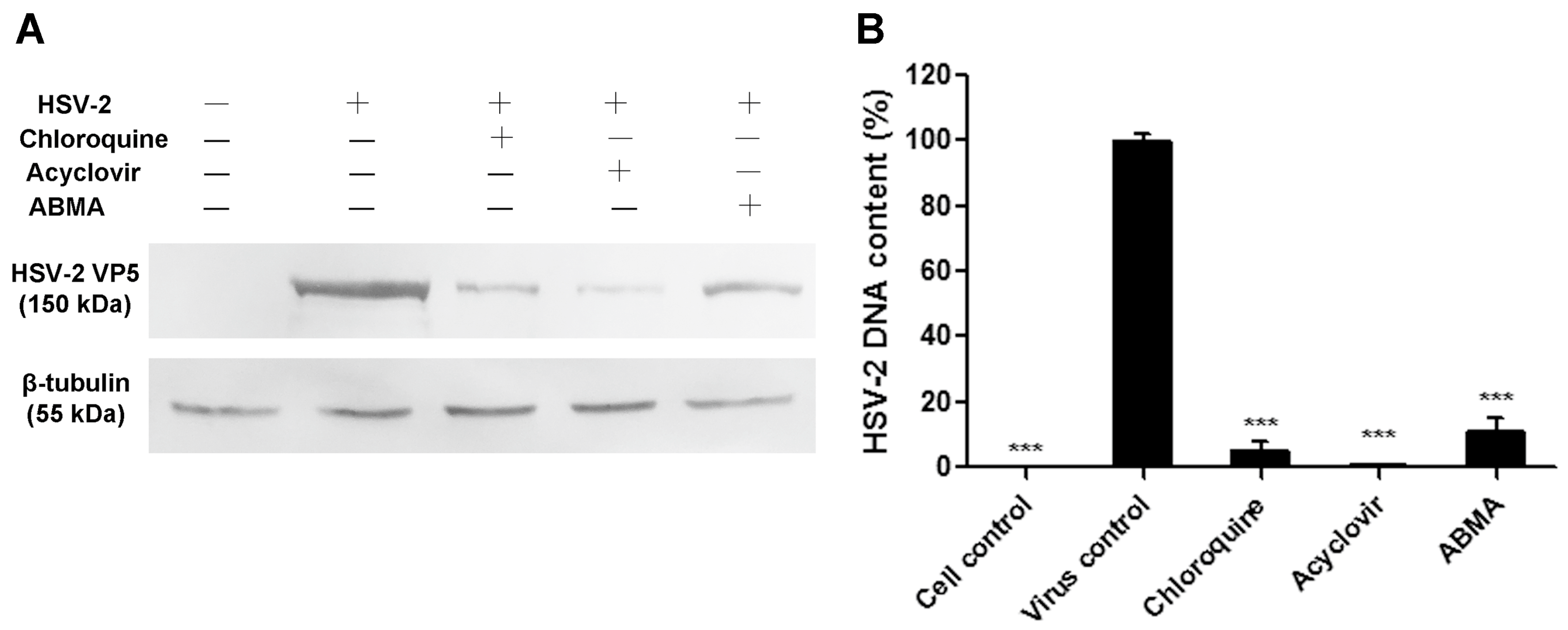
3.2. Reductions of HSV-2 Protein and DNA Content Were Detected in ABMA-Treated Cells
3.3. ABMA Blocks HSV-2 Entry into Cells
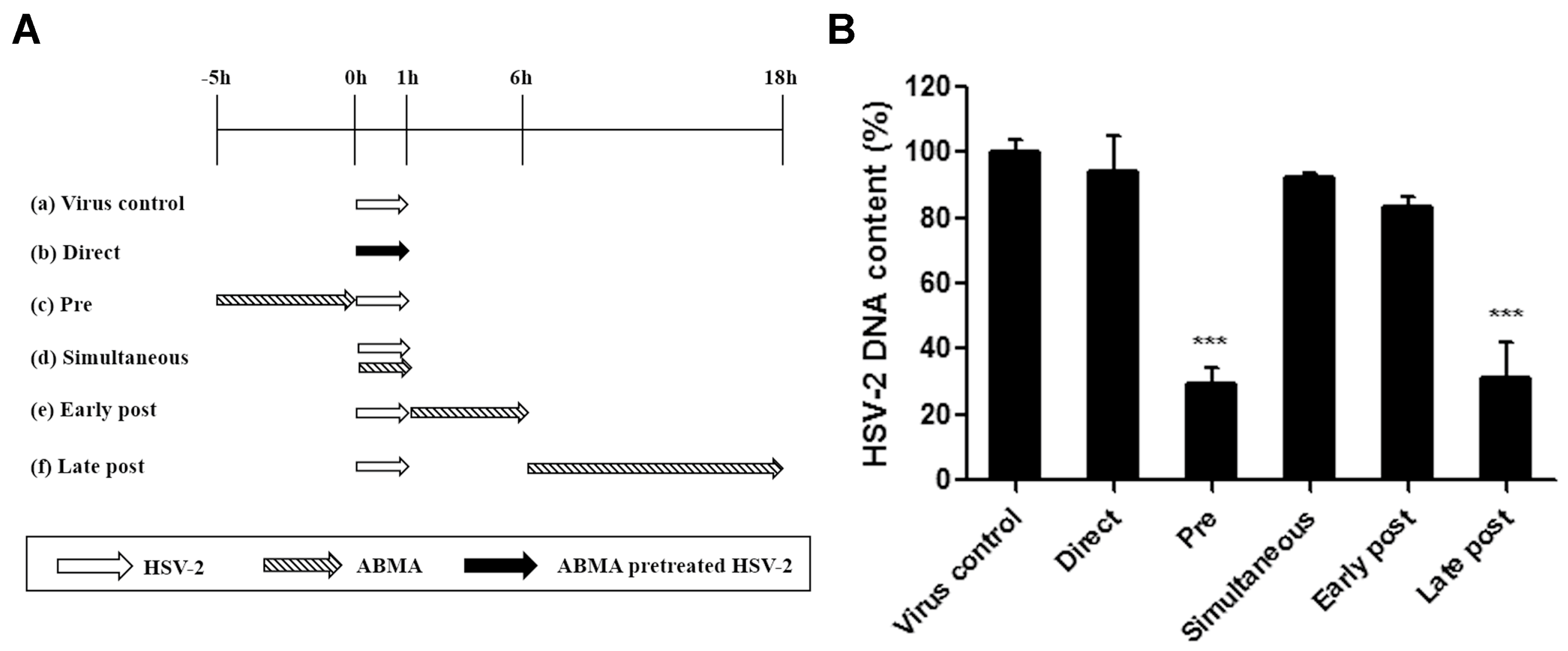
3.4. ABMA Inhibits the Late Stages of the HSV-2 Lifecycle
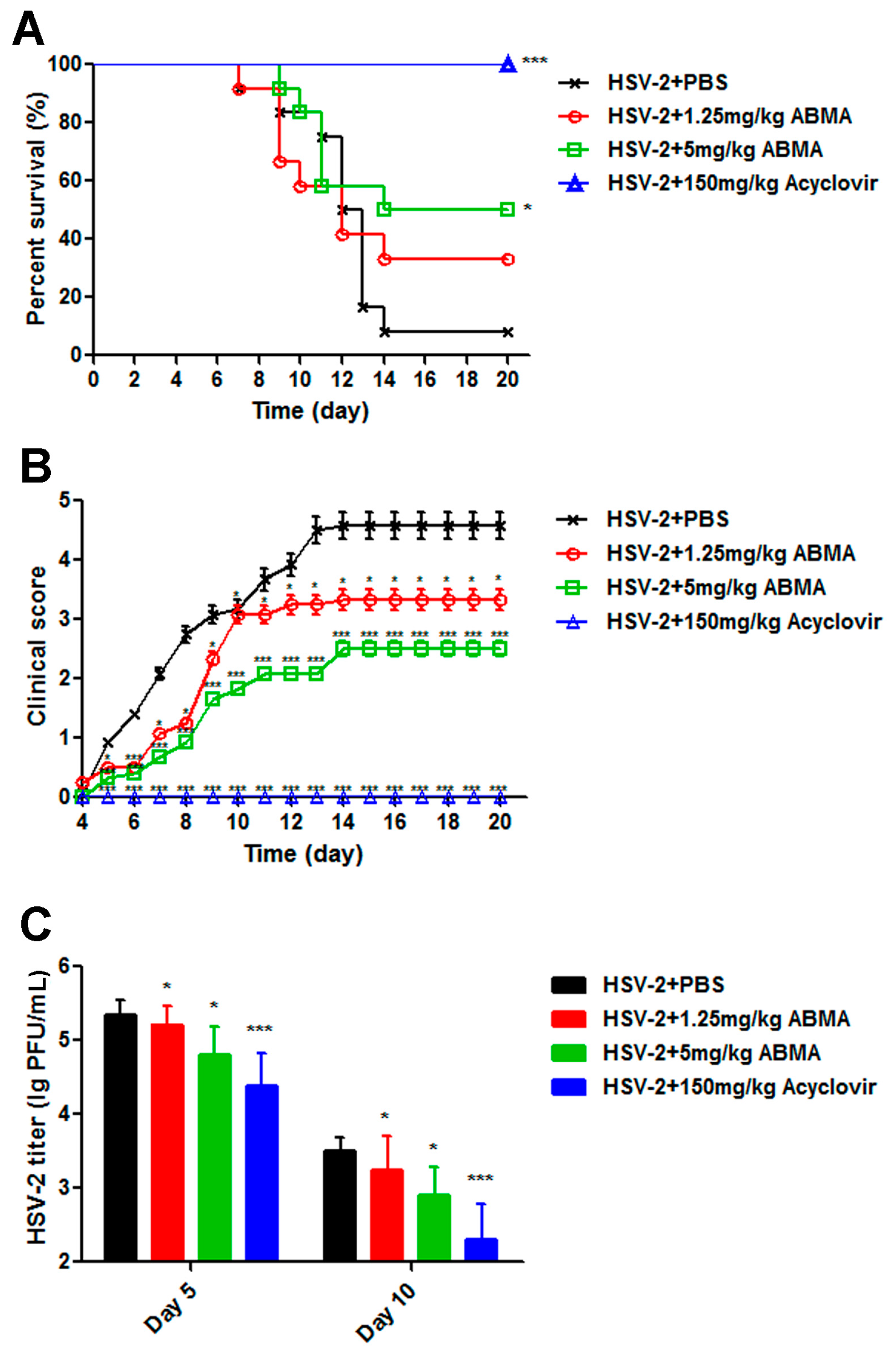
3.5. ABMA Protects BALB/c Mice from Intravaginal HSV-2 Challenge
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kawana, T. Sexually transmitted diseases of alpha herpes virus in women. Nihon Rinsho Jpn. J. Clin. Med. 2000, 58, 883–889. [Google Scholar]
- Corey, L.; Adams, H.G.; Brown, Z.A.; Holmes, K.K. Genital herpes simplex virus infections: Clinical manifestations, course, and complications. Ann. Intern. Med. 1983, 98, 958–972. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Warren, T.; Wald, A. Genital herpes. Lancet 2007, 370, 2127–2137. [Google Scholar] [CrossRef]
- Lehtinen, M.; Koskela, P.; Jellum, E.; Bloigu, A.; Anttila, T.; Hallmans, G.; Luukkaala, T.; Thoresen, S.; Youngman, L.; Dillner, J.; et al. Herpes simplex virus and risk of cervical cancer: A longitudinal, nested case-control study in the Nordic countries. Am. J. Epidemiol. 2002, 156, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.; Link, K. Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: A meta-analysis. J. Infect. Dis. 2002, 185, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Celum, C.L. The interaction between herpes simplex virus and human immunodeficiency virus. Herpes J. IHMF 2004, 11 (Suppl. 1), 36A–45A. [Google Scholar]
- Looker, K.; Magaret, A.; Turner, K.; Vickerman, P.; Gottlieb, S.; Newman, L. Global estimates of prevalent and incident herpes simplex virus type 2 infections in 2012. PLoS ONE 2015, 10, e114989. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.; Koelle, D.M.; Wald, A. HSV-2: In pursuit of a vaccine. J. Clin. Investig. 2011, 121, 4600–4609. [Google Scholar] [CrossRef] [PubMed]
- White, P.J.; Garnett, G.P. Use of antiviral treatment and prophylaxis is unlikely to have a major impact on the prevalence of herpes simplex virus type 2. Sex. Trans. Infect. 1999, 75, 49–54. [Google Scholar] [CrossRef]
- Bacon, T.H.; Levin, M.J.; Leary, J.J.; Sarisky, R.T.; Sutton, D. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin. Microbiol. Rev. 2003, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Limon, L.; Trikha, G.; Wall, H. Acute renal failure and neurotoxicity following oral acyclovir. Ann. Pharm. 1994, 28, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.E.; Franey, R.J. Acyclovir- and ganciclovir-induced neurotoxicity. Ann. Pharm. 1998, 32, 111–113. [Google Scholar] [CrossRef] [PubMed]
- De, S.K.; Hart, J.C.L.; Breuer, J. Herpes simplex virus and varicella zoster virus: Recent advances in therapy. Curr. Opin. Infect. Dis. 2015, 28, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Khurana, R.N.; Charonis, A.; Samuel, M.A.; Gupta, A.; Tawansy, K.A. Intravenous foscarnet in the management of acyclovir-resistant herpes simplex virus type 2 in acute retinal necrosis in children. Med. Sci. Monit. 2005, 11, CS75–CS78. [Google Scholar] [PubMed]
- Wu, Y.; Pons, V.; Goudet, A.; Panigai, L.; Fischer, A.; Herweg, J.-A.; Kali, S.; Davey, R.A.; Laporte, J.; Bouclier, C.; et al. ABMA, a small molecule that inhibits intracellular toxins and pathogens by interfering with late endosomal compartments. Sci. Rep. 2017, 7, 15567. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; van Deurs, B. Delivery into cells: Lessons learned from plant and bacterial toxins. Gene Ther. 2005, 12, 865–872. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Whittaker, G.R. Fusion of Enveloped Viruses in Endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [PubMed]
- Lievin-Le Moal, V.; Loiseau, P.M. Leishmania hijacking of the macrophage intracellular compartments. Febs J. 2016, 283, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.C.; Bernstein, D.I. Treatment of herpes simplex virus infections. Antivir. Res. 2004, 61, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Boelaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases? Lancet Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef]
- Storey, N.; Latchman, D.; Bevan, S. Selective internalization of sodium channels in rat dorsal root ganglion neurons infected with herpes simplex virus-1. J. Cell Biol. 2002, 158, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.A.; Dasgupta, A.; Wilson, D.W. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: Role for organelle acidification in assembly of infectious particles. J. Virol. 2001, 75, 1236–1251. [Google Scholar] [CrossRef] [PubMed]
- McClain, L.; Zhi, Y.; Cheng, H.; Ghosh, A.; Piazza, P.; Yee, M.B.; Kumar, S.; Milosevic, J.; Bloom, D.C.; Arav-Boger, R.; et al. Broad-spectrum non-nucleoside inhibitors of human herpesviruses. Antivir. Res. 2015, 121, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9-mediated recognition of herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Crouch, S.P.; Kozlowski, R.; Slater, K.J.; Fletcher, J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 1993, 160, 81–88. [Google Scholar] [CrossRef]
- Weislow, O.S.; Kiser, R.; Fine, D.L.; Bader, J.; Shoemaker, R.H.; Boyd, M.R. New soluble-formazan assay for HIV-1 cytopathic effects: Application to high-flux screening of synthetic and natural products for AIDS-antiviral activity. J. Natl. Cancer Inst. 1989, 81, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.C.; Newcomb, W.W. Herpesvirus capsid assembly: Insights from structural analysis. Curr. Opin. Virol. 2011, 1, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Eriksson, E. Inhibition of herpes simplex virus type 2 (HSV-2) viral replication by the dominant negative mutant polypeptide of HSV-1 origin binding protein. Antivir. Res. 2002, 53, 127–133. [Google Scholar] [CrossRef]
- Argenta, D.F.; Silva, I.T.; Bassani, V.L.; Koester, L.S.; Teixeira, H.F.; Simoes, C.M.O. Antiherpes evaluation of soybean isoflavonoids. Arch. Virol. 2015, 160, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Mues, M.B.; Cheshenko, N.; Wilson, D.W.; Gunther-Cummins, L.; Herold, B.C. Dynasore Disrupts Trafficking of Herpes Simplex Virus Proteins. J. Virol. 2015, 89, 6673–6684. [Google Scholar] [CrossRef] [PubMed]
- Reefschlager, J.; Wutzler, P.; Thiel, K.D.; Herrmann, G. Efficacy of 5-vinyl-1-beta-d-arabinofuranosyluracil (VaraU) against herpes simplex virus type 2 strains in cell cultures and against experimental herpes encephalitis in mice: Comparison with acyclovir and foscarnet. Pharm. Res. 1987, 4, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Shestakov, A.; Jenssen, H.; Nordstrom, I.; Eriksson, K. Lactoferricin but not lactoferrin inhibit herpes simplex virus type 2 infection in mice. Antivir. Res. 2012, 93, 340–345. [Google Scholar] [PubMed]
- Hong, L.; Xu, X.; Chen, L.; Li, B.; Wu, D.; Hu, M.; Sun, Q.; Zhu, X.; Wu, W.; Hong, S.; et al. The anti-HSV-2 effect of alumen: In vitro and in vivo experimental studies. J. Huazhong Univ. Sci. Technol.-Med. Sci. 2011, 31, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-Y.; Yang, C.-M.; Lin, T.-C.; Lin, L.-T.; Chiang, L.-C.; Lin, C.-C. Excoecarianin, Isolated from Phyllanthus urinaria Linnea, Inhibits Herpes Simplex Virus Type 2 Infection through Inactivation of Viral Particles. Evid.-Based Complement. Altern. Med. 2011, 2011, 259103. [Google Scholar] [CrossRef] [PubMed]
- Hadigal, S.; Shukla, D. Exploiting Herpes Simplex Virus Entry for Novel Therapeutics. Viruses 2013, 5, 1447–1465. [Google Scholar] [PubMed]
- Koyama, A.H.; Uchida, T. Quantitative studies on the maturation process of herpes simplex virus type 1 in Vero cells. Virus Res. 1988, 10, 281–285. [Google Scholar] [CrossRef]
- Parr, M.B.; Kepple, L.; McDermott, M.R.; Drew, M.D.; Bozzola, J.J.; Parr, E.L. A mouse model for studies of mucosal immunity to vaginal infection by herpes simplex virus type 2. Lab. Investig. J. Tech. Methods Pathol. 1994, 70, 369–380. [Google Scholar]
- Yin, S.; Li, Y.; Xia, H.; Zhao, J.; Zhang, Z.; Tang, S.; Kou, Z.; Chen, J.; Yu, J.; Fan, Z.; et al. An improved system for the evaluation of antiviral compounds against herpes simplex virus type 2. J. Virol. Methods 2013, 189, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Drannik, A.G.; Nag, K.; Sallenave, J.-M.; Rosenthal, K.L. Antiviral Activity of Trappin-2 and Elafin In Vitro and In Vivo against Genital Herpes. J. Virol. 2013, 87, 7526–7538. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Shimizu, M.; Hiyama, Y.; Itoh, K.; Hashimoto, Y.; Nakayama, M.; Horie, T.; Morita, N. Antiviral activity of natural occurring flavonoids in vitro. Chem. Pharm. Bull. 1985, 33, 3881–3886. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.H.; Matthews, B.; Cheung, D.; Tam, T.; Gadawski, I.; Leung, D.; Holan, G.; Raff, J.; Sacks, S. Evidence of dual sites of action of dendrimers: SPL-2999 inhibits both virus entry and late stages of herpes simplex virus replication. Antivir. Res. 2002, 55, 319–329. [Google Scholar] [CrossRef]
- Harden, E.A.; Falshaw, R.; Carnachan, S.M.; Kern, E.R.; Prichard, M.N. Virucidal activity of polysaccharide extracts from four algal species against herpes simplex virus. Antivir. Res. 2009, 83, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Agelidis, A.M.; Shukla, D. Cell entry mechanisms of HSV: What we have learned in recent years. Future Virol. 2015, 10, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. Febs J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef] [PubMed]
- Dan, K.; Miyashita, K.; Seto, Y.; Fujita, H.; Yamase, T. The memory effect of heteropolyoxotungstate (PM-19) pretreatment on infection by herpes simplex virus at the penetration stage. Pharmacol. Res. 2002, 46, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Kalu, N.N.; Desai, P.J.; Shirley, C.M.; Gibson, W.; Dennis, P.A.; Ambinder, R.F. Nelfinavir Inhibits Maturation and Export of Herpes Simplex Virus 1. J. Virol. 2014, 88, 5455–5461. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [PubMed]
- Mingo, R.M.; Han, J.; Newcomb, W.W.; Brown, J.C. Replication of Herpes Simplex Virus: Egress of Progeny Virus at Specialized Cell Membrane Sites. J. Virol. 2012, 86, 7084–7097. [Google Scholar] [CrossRef] [PubMed]
- Granzow, H.; Klupp, B.G.; Fuchs, W.; Veits, J.; Osterrieder, N.; Mettenleiter, T.C. Egress of alphaherpesviruses: Comparative ultrastructural study. J. Virol. 2001, 75, 3675–3684. [Google Scholar] [CrossRef] [PubMed]
- Toews, M.L.; Bylund, D.B. Pharmacologic principles for combination therapy. Proc. Am. Thorac. Soc. 2005, 2, 282–289; discussion 290–291. [Google Scholar] [CrossRef] [PubMed]
- Andrei, G.; Snoeck, R. Herpes simplex virus drug-resistance: New mutations and insights. Curr. Opin. Infect. Dis. 2013, 26, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Bekerman, E.; Einav, S. Combating emerging viral threats. Science 2015, 348, 282–283. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytotoxicity | Antiviral Activity | |||
|---|---|---|---|---|
| Compounds | CC50 (μM) | EC50 a (μM) | EC50 b (μM) | SI c |
| ABMA | 34.75 ± 0.28 | 1.66 ± 0.14 | 1.08 ± 0.25 | 20.93 |
| Chloroquine | 15.67 ± 0.47 | 1.86 ± 0.20 | 1.38 ± 0.03 | 8.42 |
| Acyclovir | >1000 | 0.82 ± 0.06 | 0.81 ± 0.07 | 1219.51 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, W.; Wu, Y.; Bi, J.; Wang, S.; Li, F.; Kong, W.; Barbier, J.; Cintrat, J.-C.; Gao, F.; Gillet, D.; et al. Antiviral Effects of ABMA against Herpes Simplex Virus Type 2 In Vitro and In Vivo. Viruses 2018, 10, 119. https://doi.org/10.3390/v10030119
Dai W, Wu Y, Bi J, Wang S, Li F, Kong W, Barbier J, Cintrat J-C, Gao F, Gillet D, et al. Antiviral Effects of ABMA against Herpes Simplex Virus Type 2 In Vitro and In Vivo. Viruses. 2018; 10(3):119. https://doi.org/10.3390/v10030119
Chicago/Turabian StyleDai, Wenwen, Yu Wu, Jinpeng Bi, Shuai Wang, Fang Li, Wei Kong, Julien Barbier, Jean-Christophe Cintrat, Feng Gao, Daniel Gillet, and et al. 2018. "Antiviral Effects of ABMA against Herpes Simplex Virus Type 2 In Vitro and In Vivo" Viruses 10, no. 3: 119. https://doi.org/10.3390/v10030119