Variability Studies of Two Prunus-Infecting Fabaviruses with the Aid of High-Throughput Sequencing

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Sour and Sweet Cherries Host Diverse viruses Belonging to Different Families
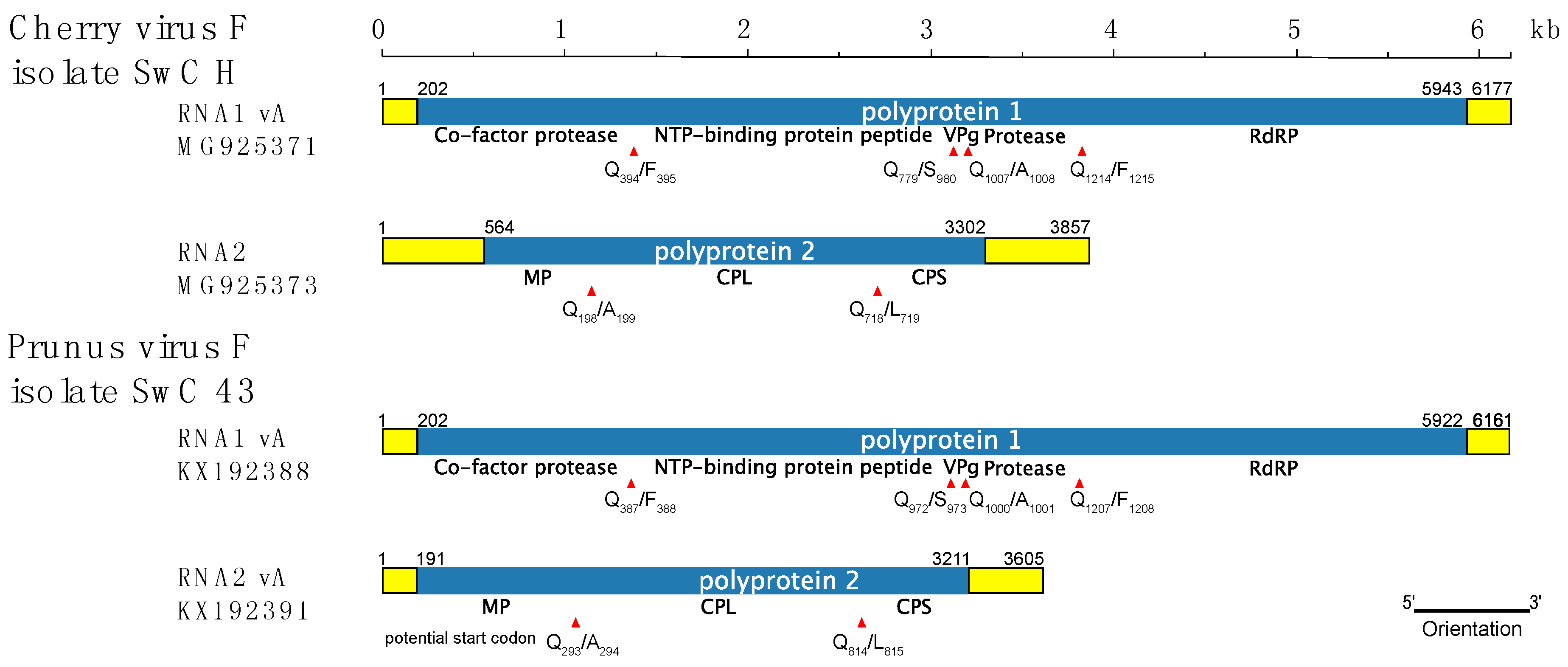
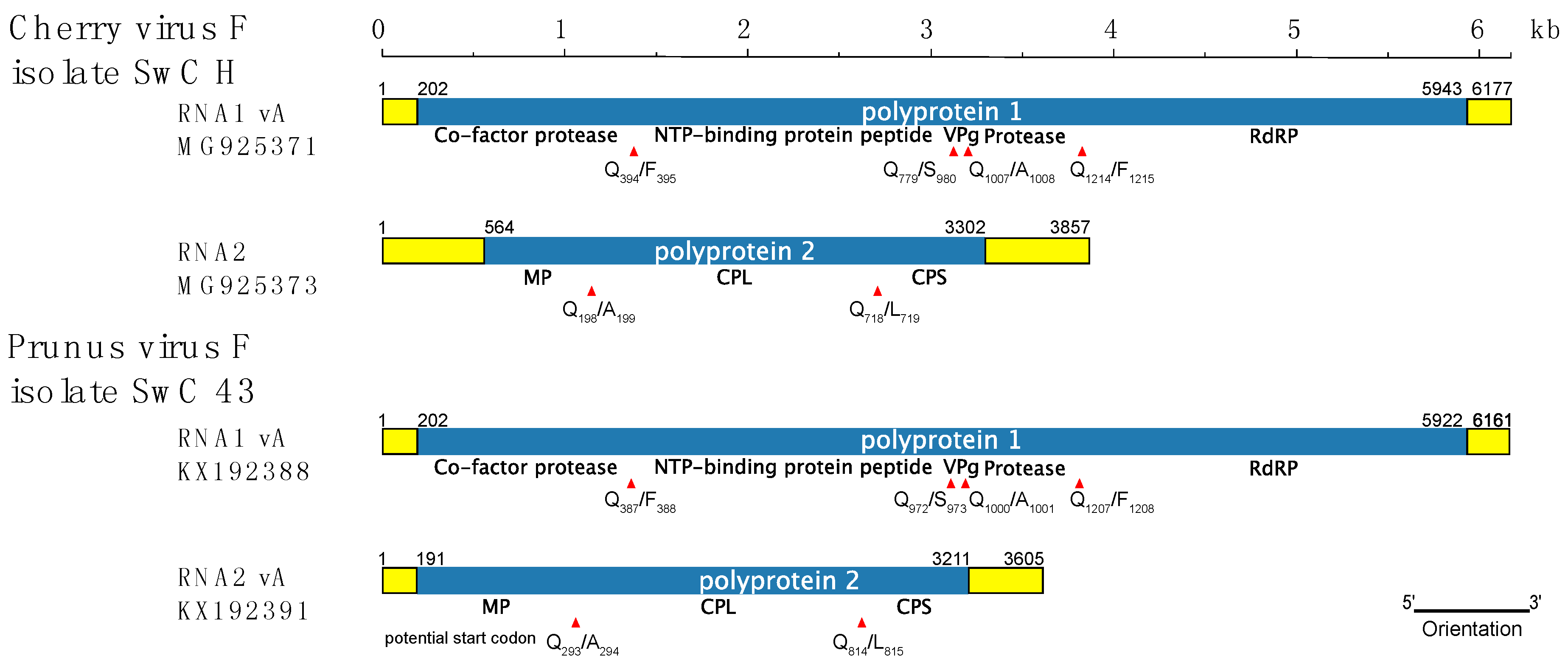
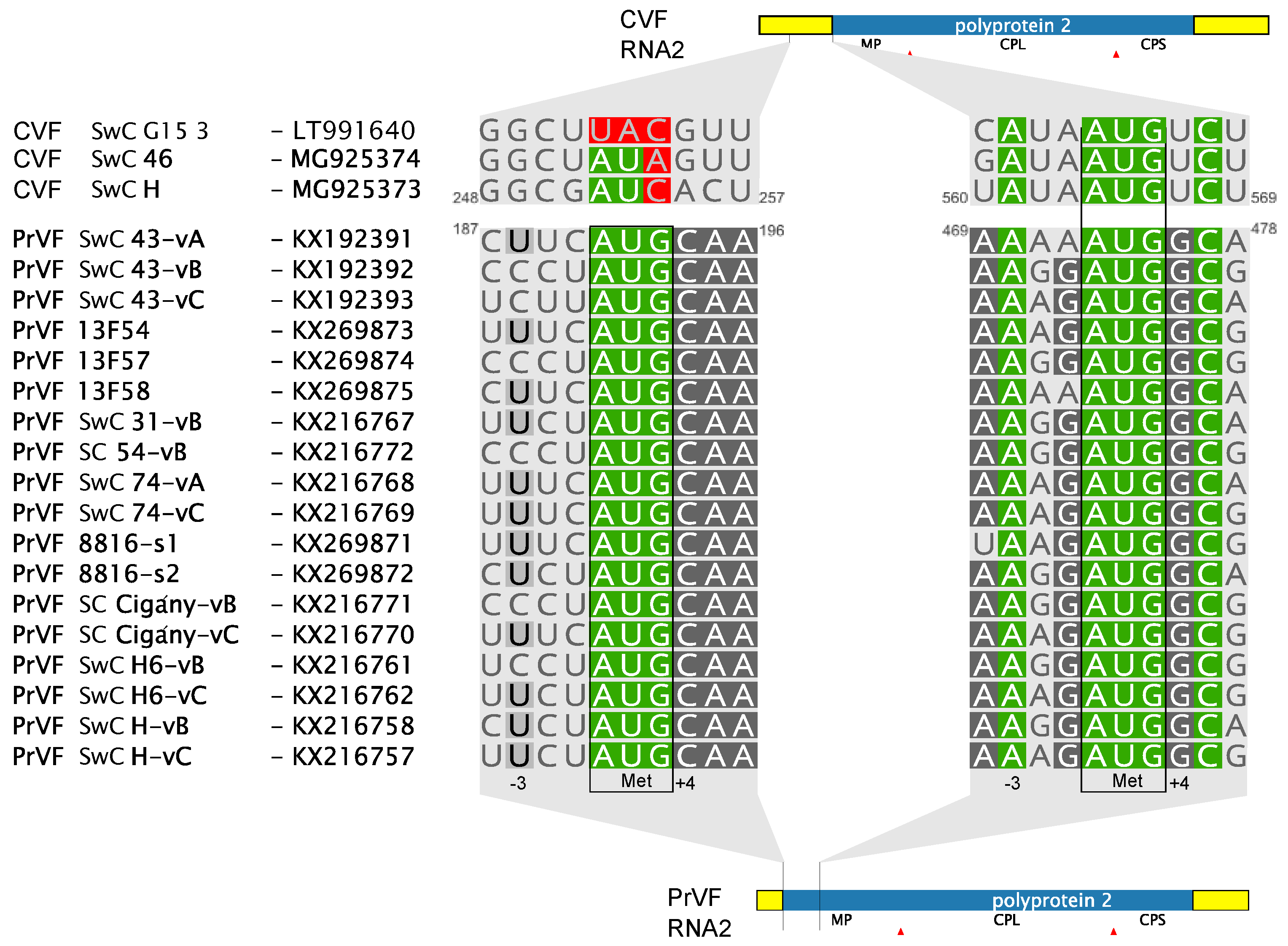
3.2. Comparison of CVF and PrVF Genome Organization
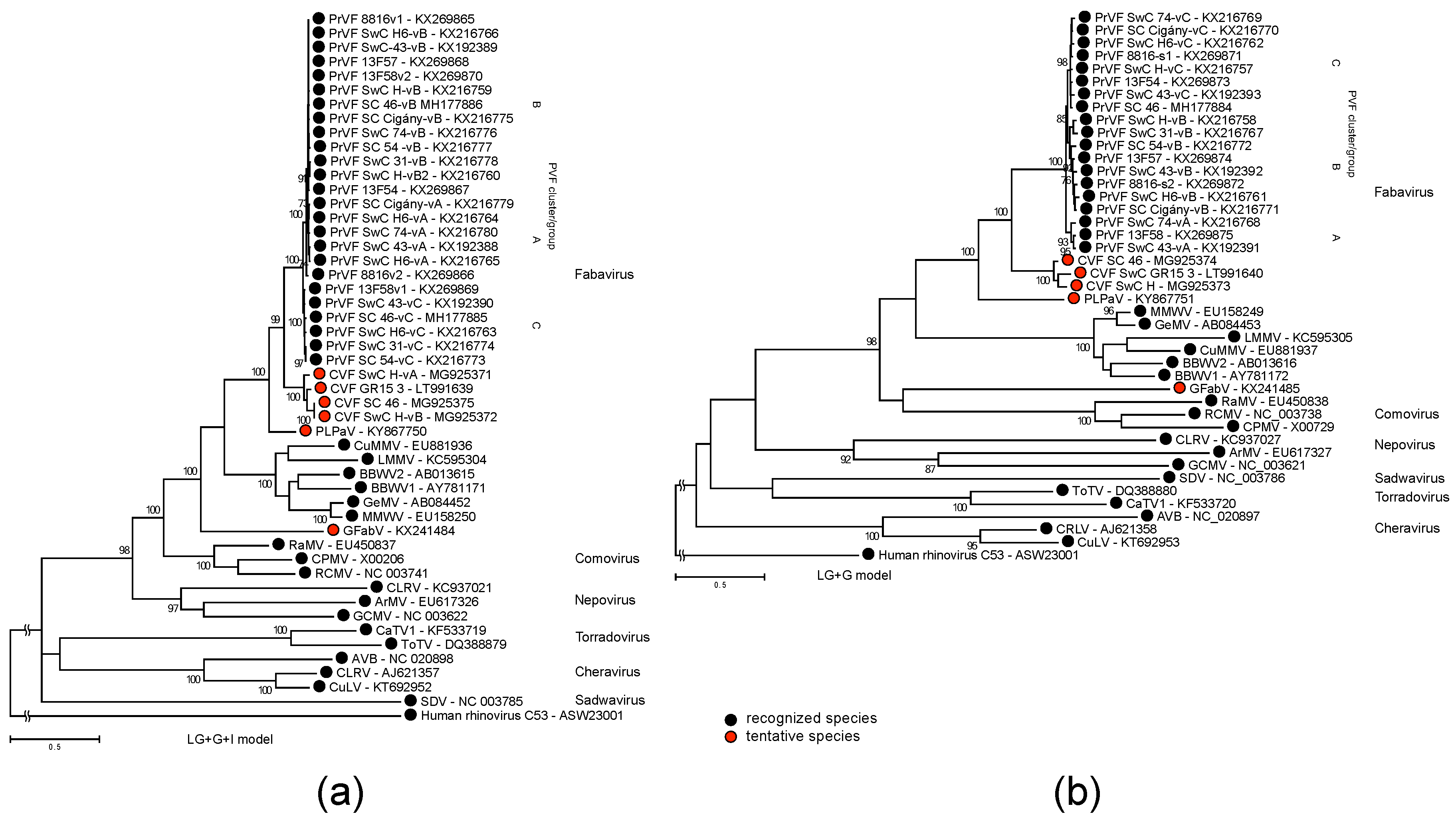
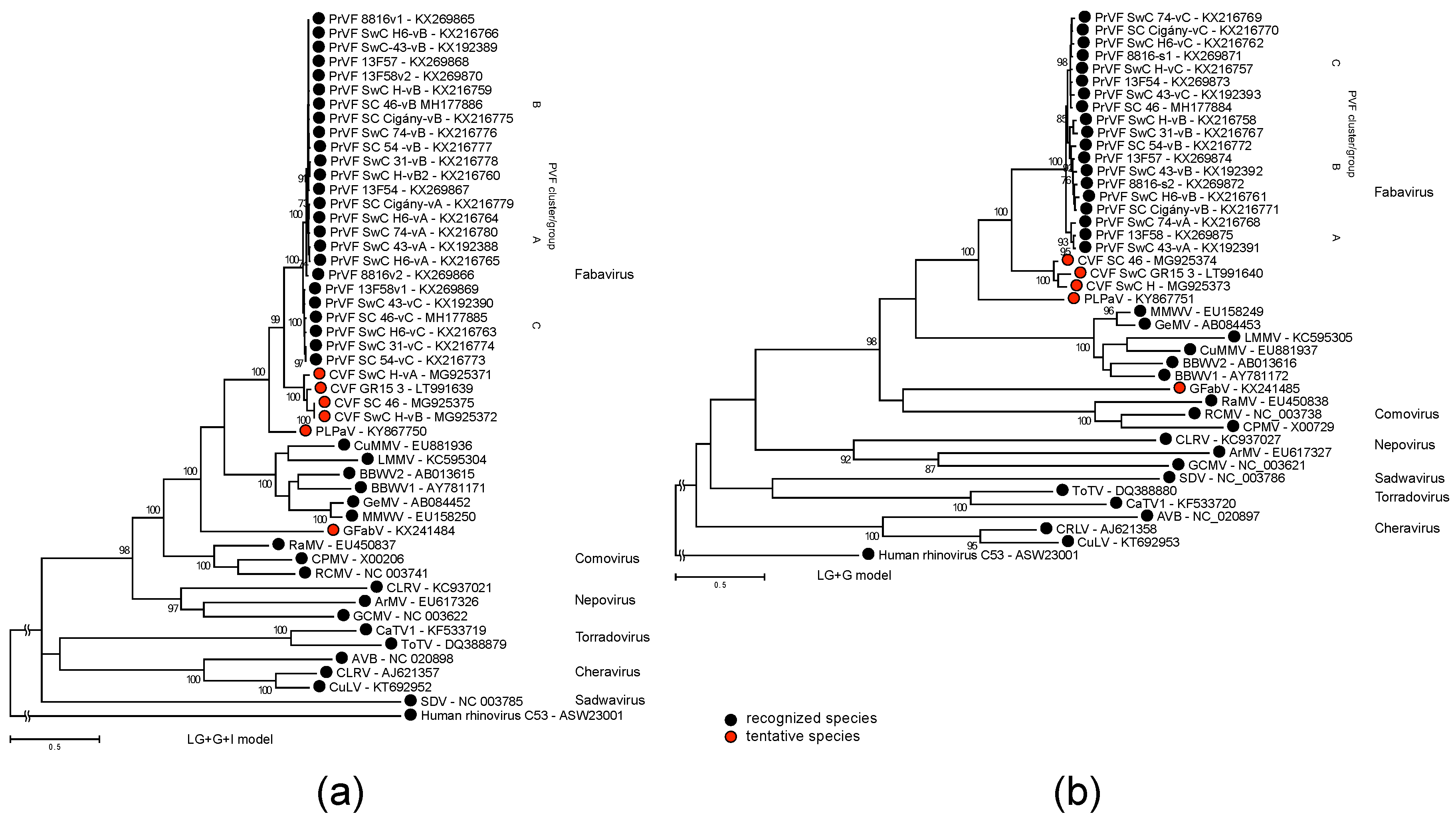
3.3. Phylogenetic Inference
3.4. Analysis of Recombinants
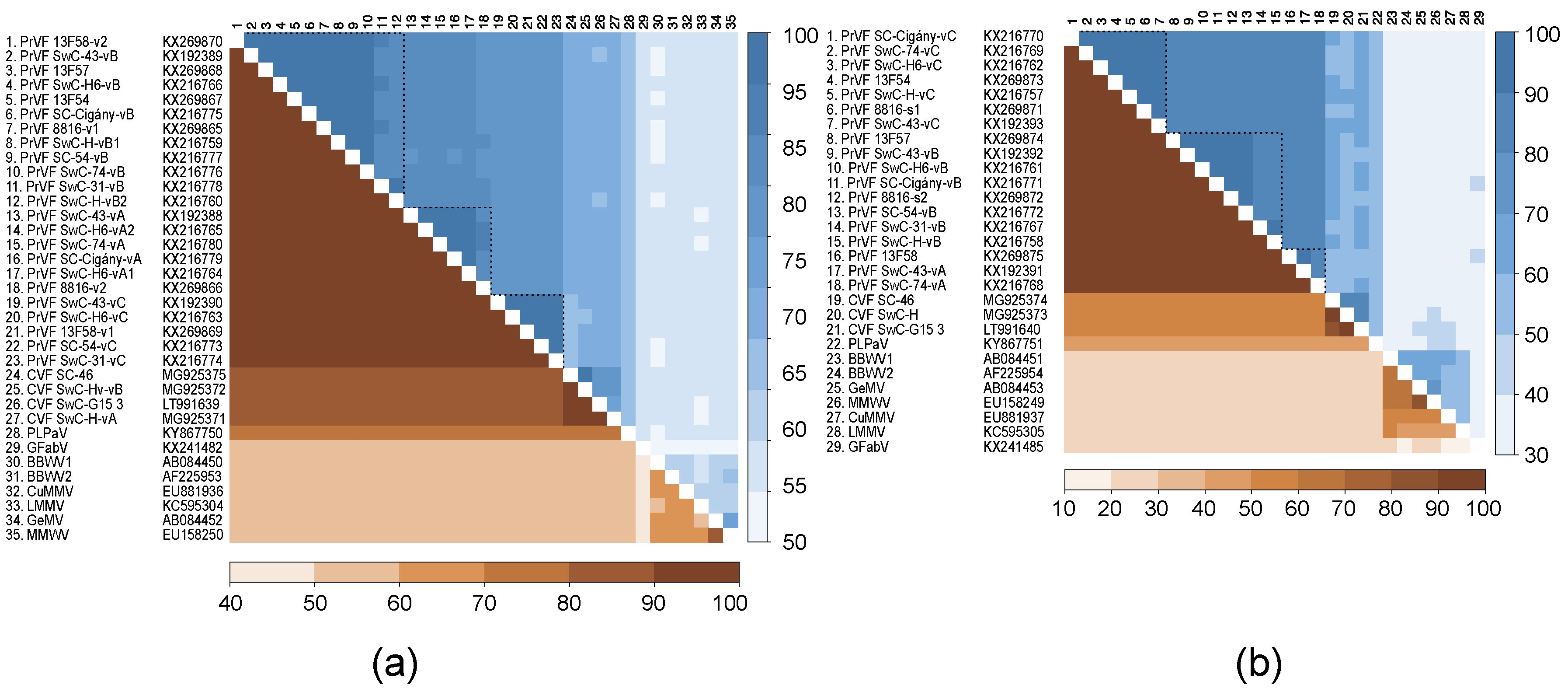
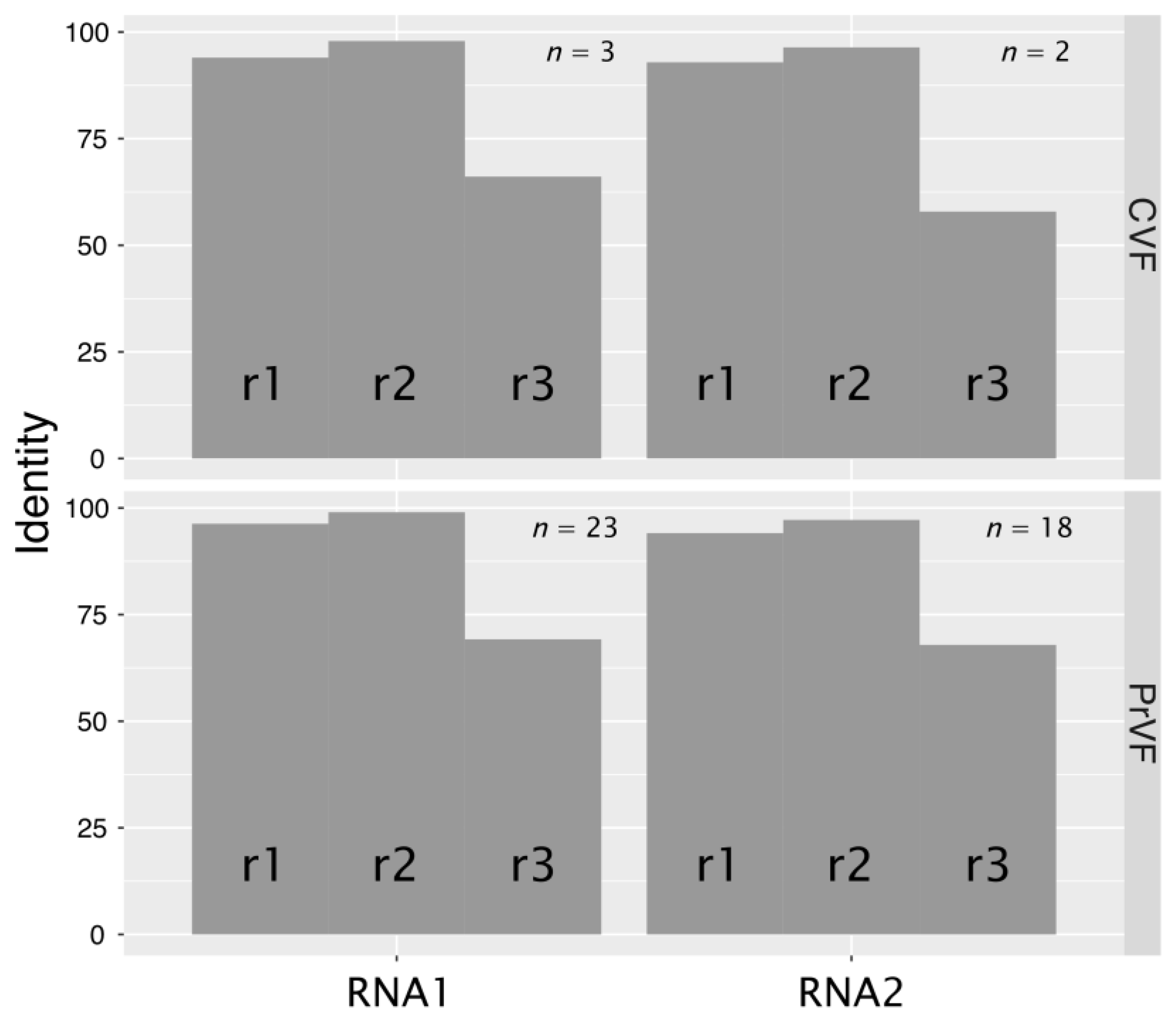
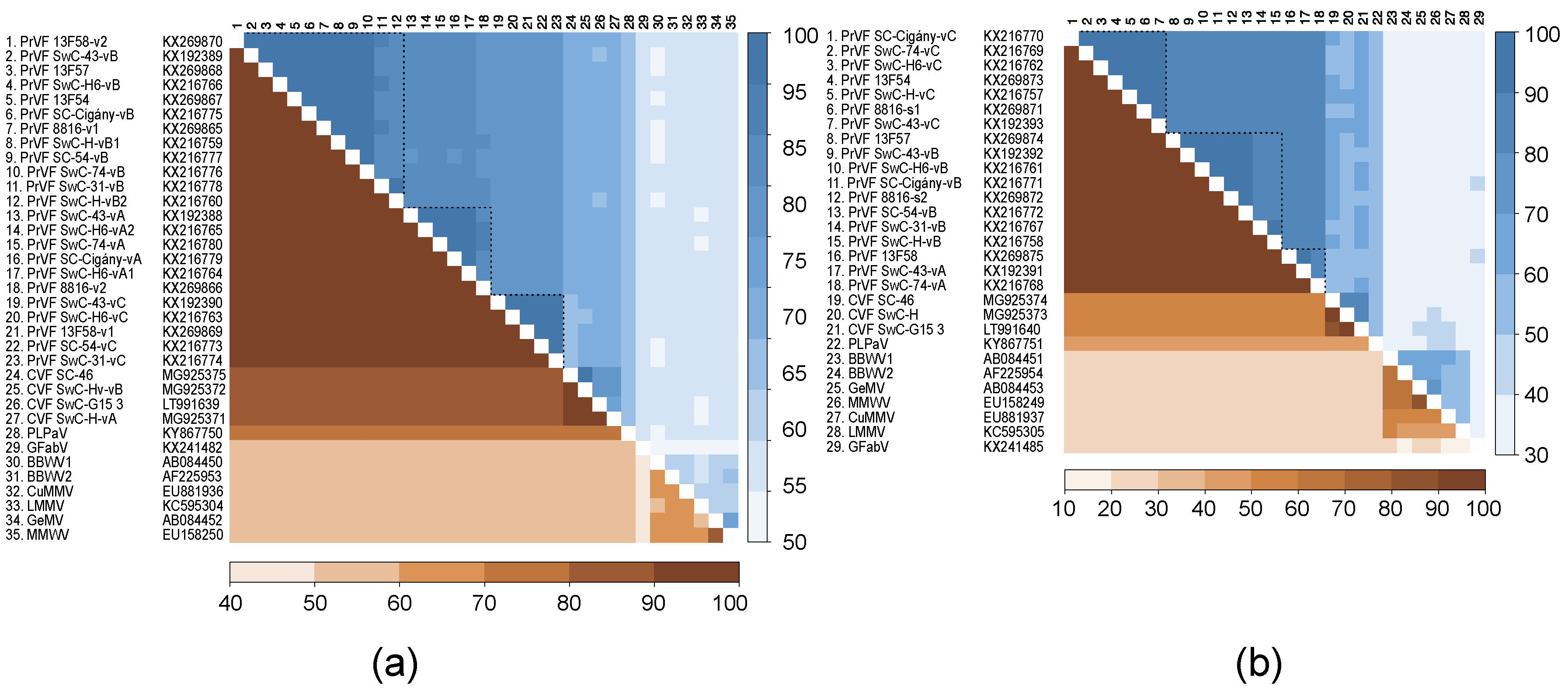
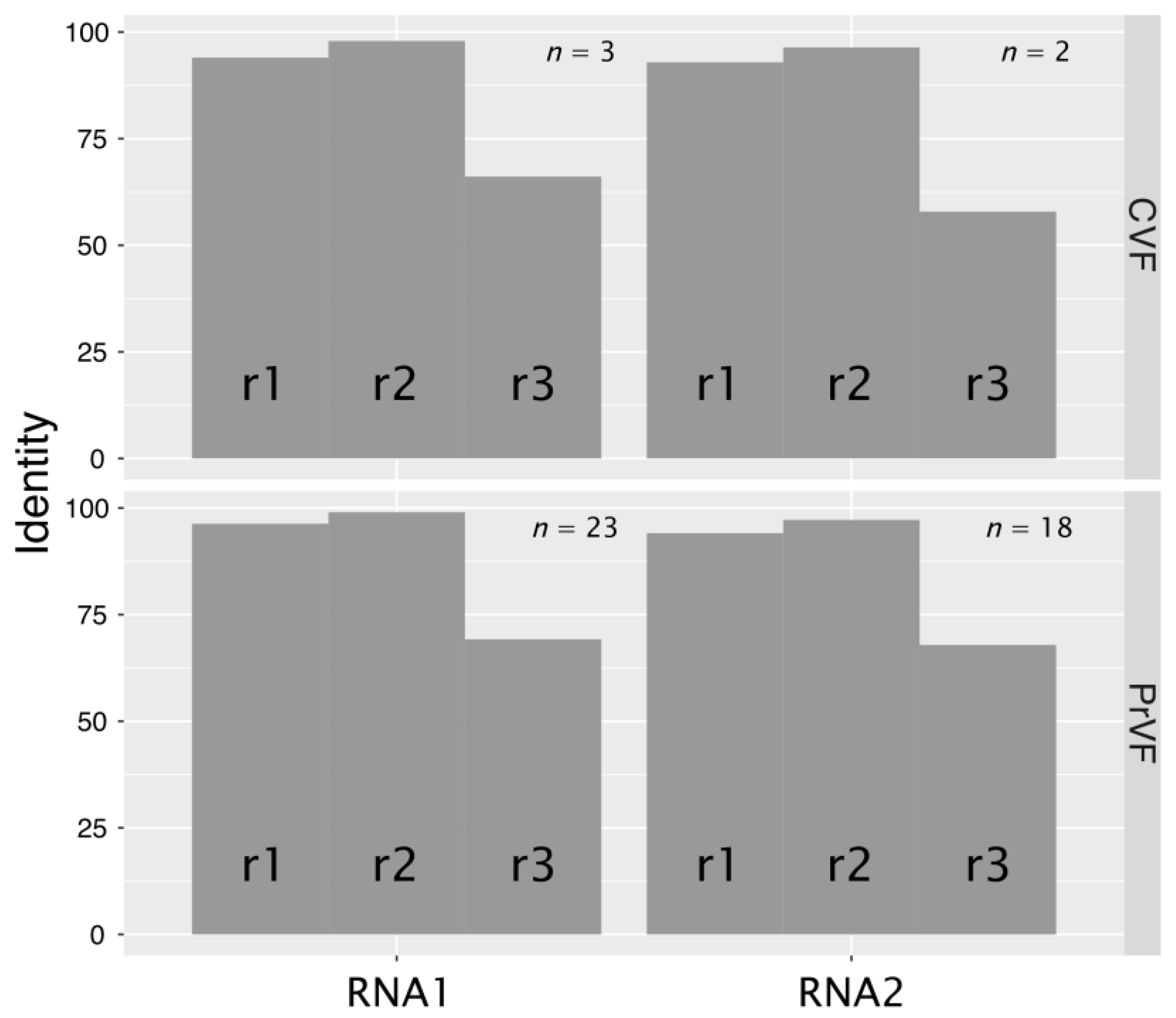
3.5. Genetic Diversity among Different Isolates of PrVF and CVF and Other Fabaviruses
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rubio, M.; Martínez-Gómez, P.; Marais, A.; Sánchez-Navarro, J.A.; Pallás, V.; Candresse, T. Recent advances and prospects in Prunus virology. Ann. Appl. Biol. 2017, 171, 125–138. [Google Scholar] [CrossRef]
- He, Y.; Cai, L.; Zhou, L.; Yang, Z.; Hong, N.; Wang, G.; Li, S.; Xu, W. Deep sequencing reveals the first fabavirus infecting peach. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hadidi, A.; Barba, M.; Candresse, T.; Jelkmann, W. Virus and Virus-like Diseases of Pome and Stone Fruits; Amer Phytopathological Society: Saint Paul, MN, USA, 2011. [Google Scholar]
- Villamor, D.E.V.; Pillai, S.S.; Eastwell, K.C. High throughput sequencing reveals a novel fabavirus infecting sweet cherry. Arch. Virol. 2017, 163, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Lenz, O.; Přibylová, J.; Fránová, J.; Koloniuk, I.; Špak, J. Identification and characterization of a new member of the genus Luteovirus from cherry. Arch. Virol. 2017, 162, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Glasa, M.; Šoltys, K.; Vozárová, Z.; Predajňa, L.; Sihelská, N.; Šubr, Z.; Candresse, T. High intra-host cherry virus a population heterogeneity in cherry trees in Slovakia. J. Plant Pathol. 2017, 99, 745–752. [Google Scholar]
- Giallonardo, F.D.; Töpfer, A.; Rey, M.; Prabhakaran, S.; Duport, Y.; Leemann, C.; Schmutz, S.; Campbell, N.K.; Joos, B.; Lecca, M.R.; et al. Full-length haplotype reconstruction to infer the structure of heterogeneous virus populations. Nucleic Acids Res. 2014, 42, e115. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.R.; Dasgupta, I.; Fuchs, M.; Iwanami, T.; Karasev, A.V.; Petrzik, K.; Sanfaçon, H.; Tzanetakis, I.; van der Vlugt, R.; Wetzel, T.; et al. ICTV Report Consortium ICTV Virus Taxonomy Profile: Secoviridae. J. Gener. Virol. 2017, 98, 529–531. [Google Scholar]
- Šafářová, D.; Faure, C.; Marais, A.; Suchá, J.; Paprštein, F.; Navrátil, M.; Candresse, T. First report of Prunus virus F infecting sour cherry in the Czech Republic. Plant Dis. 2017, 101. [Google Scholar] [CrossRef]
- Morris, T.J.; Dodds, J.A. Isolation and analysis of double-stranded RNA from virus-infected plant and fungal tissue. Phytopathology 1979, 69, 854–858. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, B. HIV signature and sequence variation analysis. In Computational Analysis of HIV Molecular Sequences; Rodrigo, A.G., Learn, J., Gerald, H., Eds.; Springer: Dordrecht, The Netherlands, 2000; pp. 55–72. [Google Scholar]
- Ferriol, I.; Rangel, E.A.; Panno, S.; Davino, S.; Han, C.G.; Olmos, A.; Rubio, L. Rapid detection and discrimination of fabaviruses by flow-through hybridisation with genus- and species-specific riboprobes. Ann. Appl. Biol. 2015, 167, 26–35. [Google Scholar] [CrossRef]
- Kozak, M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 1986, 44, 283–292. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sanfaçon, H.; Wellink, J.; Le Gall, O.; Karasev, A.; van der Vlugt, R.; Wetzel, T. Secoviridae: A proposed family of plant viruses within the order Picornavirales that combines the families Sequiviridae and Comoviridae, the unassigned genera Cheravirus and Sadwavirus, and the proposed genus Torradovirus. Arch. Virol. 2009, 154, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Sanfaçon, H.; Iwanami, T.; Karasev, A.V.; van der Vlugt, R.; Wellink, J.; Wetzel, T.; Yoshikawa, N. Family—Secoviridae. In Virus Taxonomy; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 881–899. [Google Scholar]
- Xia, X. The rate heterogeneity of nonsynonymous substitutions in mammalian mitochondrial genes. Mol. Biol. Evol. 1998, 15, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Wellink, J.; Verver, J.; Van Kammen, A. Mutational analysis of AUG codons of cowpea mosaic virus M RNA. Biochimie 1993, 75, 741–747. [Google Scholar] [CrossRef]
- Lin, J.; Guo, J.; Finer, J.; Dorrance, A.E.; Redinbaugh, M.G.; Qu, F. The bean pod mottle virus RNA2-encoded 58-kilodalton protein P58 is required in cis for RNA2 Accumulation. J. Virol. 2014, 88, 3213–3222. [Google Scholar] [CrossRef] [PubMed]
- Cann, A.J. Principles of Molecular Virology, 4th ed.; Elsevier Academic Press: London, UK, 2005; pp. 1–332. [Google Scholar]
- Jridi, C.; Martin, J.F.; Marie-Jeanne, V.; Labonne, G.; Blanc, S. Distinct viral populations differentiate and evolve independently in a single perennial host plant. J. Virol. 2006, 80, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Jara, P.; Fraile, A.; Canto, T.; García-Arenal, F. The Multiplicity of infection of a plant virus varies during colonization of its eukaryotic host. J. Virol. 2009, 83, 7487–7494. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F.; Bernet, G.P.; Carrasco, J.L. The games plant viruses play. Curr. Opin. Virol. 2014, 8, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.S.; French, R.; Hein, G.L.; Morris, T.J.; Stenger, D.C. Three distinct mechanisms facilitate genetic isolation of sympatric wheat streak mosaic virus lineages. Virology 2001, 282, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Folimonova, S.Y.; Robertson, C.J.; Shilts, T.; Folimonov, A.S.; Hilf, M.E.; Garnsey, S.M.; Dawson, W.O. Infection with strains of Citrus tristeza virus does not exclude superinfection by other strains of the virus. J. Virol. 2010, 84, 1314–1325. [Google Scholar] [CrossRef] [PubMed]
- Folimonova, S.Y.; Harper, S.J.; Leonard, M.T.; Triplett, E.W.; Shilts, T. Superinfection exclusion by Citrus tristeza virus does not correlate with the production of viral small RNAs. Virology 2014, 468–470, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef] [PubMed]
- Morroni, M.; Thompson, J.R.; Tepfer, M. Analysis of recombination between viral RNAs and transgene mRNA under conditions of high selection pressure in favour of recombinants. J. Gen. Virol. 2009, 90, 2798–2807. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Description | Origin | Applied Enrichment | Symptoms | HTS, Length of Reads and Output (Millions) |
|---|---|---|---|---|---|
| SwC–31 | Sweet cherry cv Kisinevskaja, | Chișinău, Moldova | dsRNA | No obvious symptoms | 100b, 24 |
| SwC–43 | Sweet cherry cv Lambert | Canada | dsRNA | Premature leaf fall, dieback, bunches of small leaves and blossoms growing at the old branches | 100b, 25 |
| SC–54 | Sour cherry cv Dneprovka | Chișinău, Moldova | dsRNA | Dwarfism | 100b, 23 |
| SC–46 | Sour cherry cv Rannaja | Chișinău, Moldova | dsRNA | No obvious symptoms | 100b, 24 |
| SwC–74 | Sweet cherry cv Rube | Magdeburg, Germany | dsRNA | Dwarfism | 100b, 25 |
| SC–Cigány | Sour cherry, unknown cultivar | Hungary | dsRNA | Severe mosaic | 100b, 25 |
| SwC–H | Sweet cherry, unknown cultivar | Czech Republic | polyA | No obvious symptoms, old age | 100b, 20 |
| SwC–H6 | Sweet cherry, unknown cultivar | Czech Republic | polyA | No obvious symptoms, old age | 100b, 39 |
| SwC–G15 3 | Sweet cherry cv tragana-edessis | Greece | sRNAs | No obvious symptoms, old age | 21b-24b, 2 |
| Samples | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| SwC–31 | SwC–43 | SC–46 | SC–54 | SwC–74 | SC–Cigány | SwC–H | SwC–H6 | SwC–G15 3 | |
| Betaflexiviridae | |||||||||
| Apple chlorotic leaf spot virus | |||||||||
| Cherry green ring mottle virus | |||||||||
| Cherry necrotic rusty mottle virus | |||||||||
| Cherry virus A | |||||||||
| Bromoviridae | |||||||||
| Prune dwarf virus | |||||||||
| Prunus necrotic ringspot virus | |||||||||
| Closteroviridae | |||||||||
| Little cherry virus 1 | |||||||||
| Little cherry virus 2 | |||||||||
| Luteoviridae | |||||||||
| Cherry–associated luteovirus | |||||||||
| Cherry–associated luteovirus 2 (Thierry Candresse et al. Novel luteovirus infecting sour cherry, submission data preparation) | |||||||||
| Secoviridae | |||||||||
| Cherry leaf roll virus | |||||||||
| Prunus virus F | |||||||||
| Cherry virus F | |||||||||
| Tymovirales | |||||||||
| Grapevine Syrah virus 1-like virus | |||||||||
| Sample | Virus | Variants | |
|---|---|---|---|
| RNA1 | RNA2 | ||
| SwC–31 | PrVF | 2 (B–KX216778, C–KX216774) | 1 (B–KX216767) |
| SwC–43 | PrVF | 3 (A–KX192388, B–KX192389, C–KX192390) | 3 (A–KX192391, B–KX192392, C–KX192393) |
| SC–54 | PrVF | 2 (B–KX216777, C–KX216773) | 1 (B–KX216772) |
| SwC–74 | PrVF | 2 (A–KX216780, B–KX216776) | 2 (A–KX216768, C–KX216769) |
| SC–Cigány | PrVF | 2 (A–KX216779, B–KX216775) | 2 (B–KX216771, C–KX216770) |
| SC–46 | PrVF | 2 (B–MH177886, C–MH177885) | 1 (C–MH177884) |
| CVF | 1 (na–MG925375) | 1 (na–MG925374) | |
| SwC–H | PrVF | 2 (B–KX216759, B–KX216760) | 2 (B–KX216758, C–KX216757) |
| CVF | 2 (A–MG925371, B–MG925372) | 1 (na–MG925373) | |
| SwC–H6 | PrVF | 2 (A–KX216764, A–KX216765, B–KX216766, C–KX216763) | 1 (B–KX216761, C–KX216762) |
| SwC–G15 3 | CVF | 1 (na–LT991639) | 1 (na–LT991640) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koloniuk, I.; Sarkisova, T.; Petrzik, K.; Lenz, O.; Přibylová, J.; Fránová, J.; Špak, J.; Lotos, L.; Beta, C.; Katsiani, A.; et al. Variability Studies of Two Prunus-Infecting Fabaviruses with the Aid of High-Throughput Sequencing. Viruses 2018, 10, 204. https://doi.org/10.3390/v10040204
Koloniuk I, Sarkisova T, Petrzik K, Lenz O, Přibylová J, Fránová J, Špak J, Lotos L, Beta C, Katsiani A, et al. Variability Studies of Two Prunus-Infecting Fabaviruses with the Aid of High-Throughput Sequencing. Viruses. 2018; 10(4):204. https://doi.org/10.3390/v10040204
Chicago/Turabian StyleKoloniuk, Igor, Tatiana Sarkisova, Karel Petrzik, Ondřej Lenz, Jaroslava Přibylová, Jana Fránová, Josef Špak, Leonidas Lotos, Christina Beta, Asimina Katsiani, and et al. 2018. "Variability Studies of Two Prunus-Infecting Fabaviruses with the Aid of High-Throughput Sequencing" Viruses 10, no. 4: 204. https://doi.org/10.3390/v10040204