Pectobacterium atrosepticum Phage vB_PatP_CB5: A Member of the Proposed Genus ‘Phimunavirus’

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial and Phage Propagation Conditions
2.2. Phage Isolation
2.3. Host Range and General Characterization
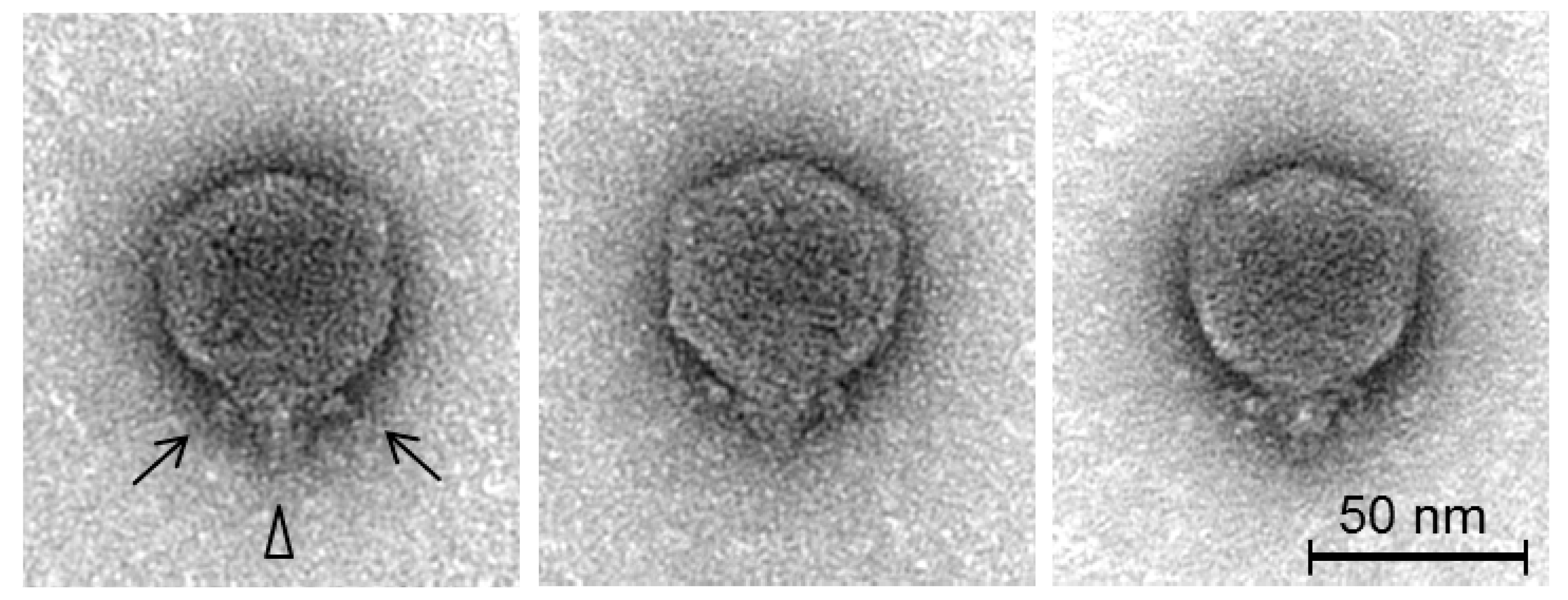
2.4. Transmission Electron Microscopy
2.5. DNA Isolation and Sequencing
2.6. Bioinformatic Analysis
2.7. Comparative Genomics
2.8. Accession Number
3. Results
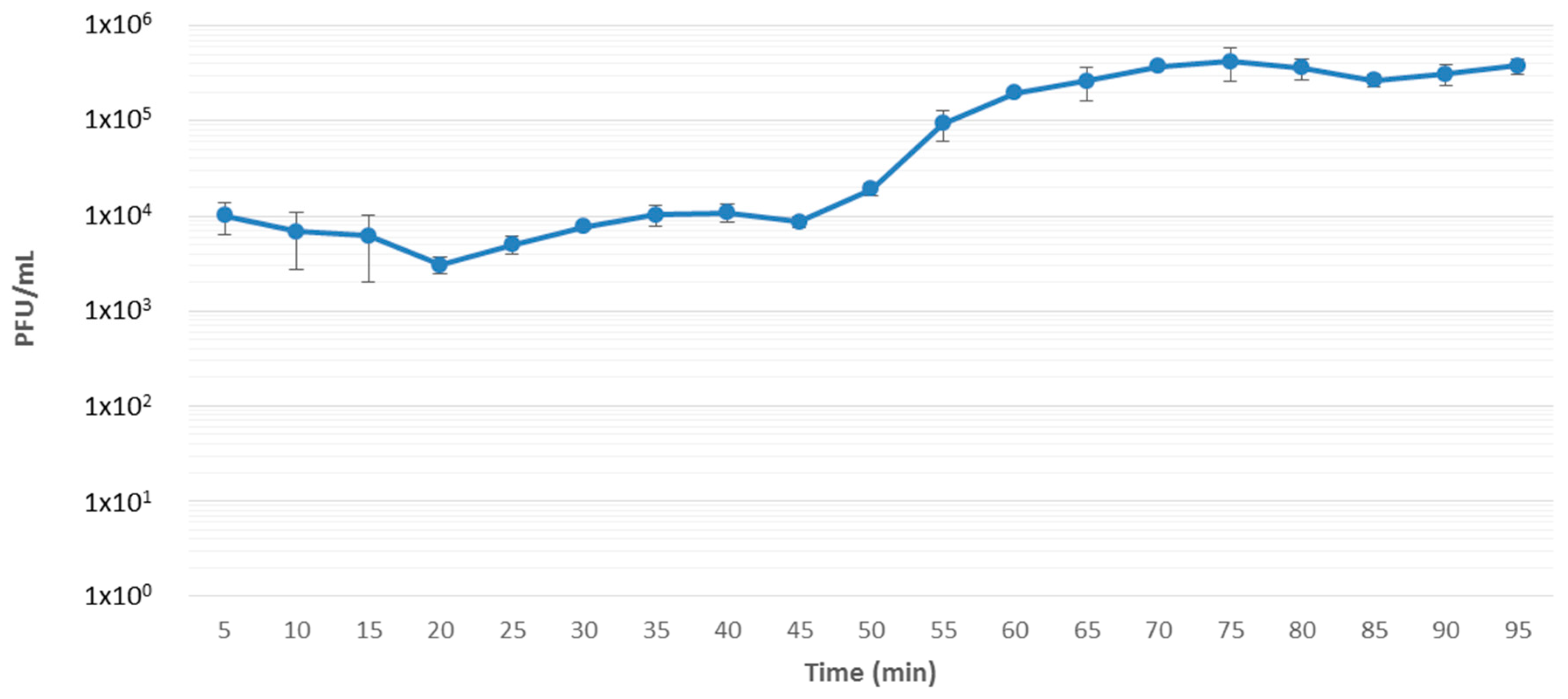
3.1. Isolation, Host Range, Growth Characteristics and Morphology
3.2. General Genome Information of CB5
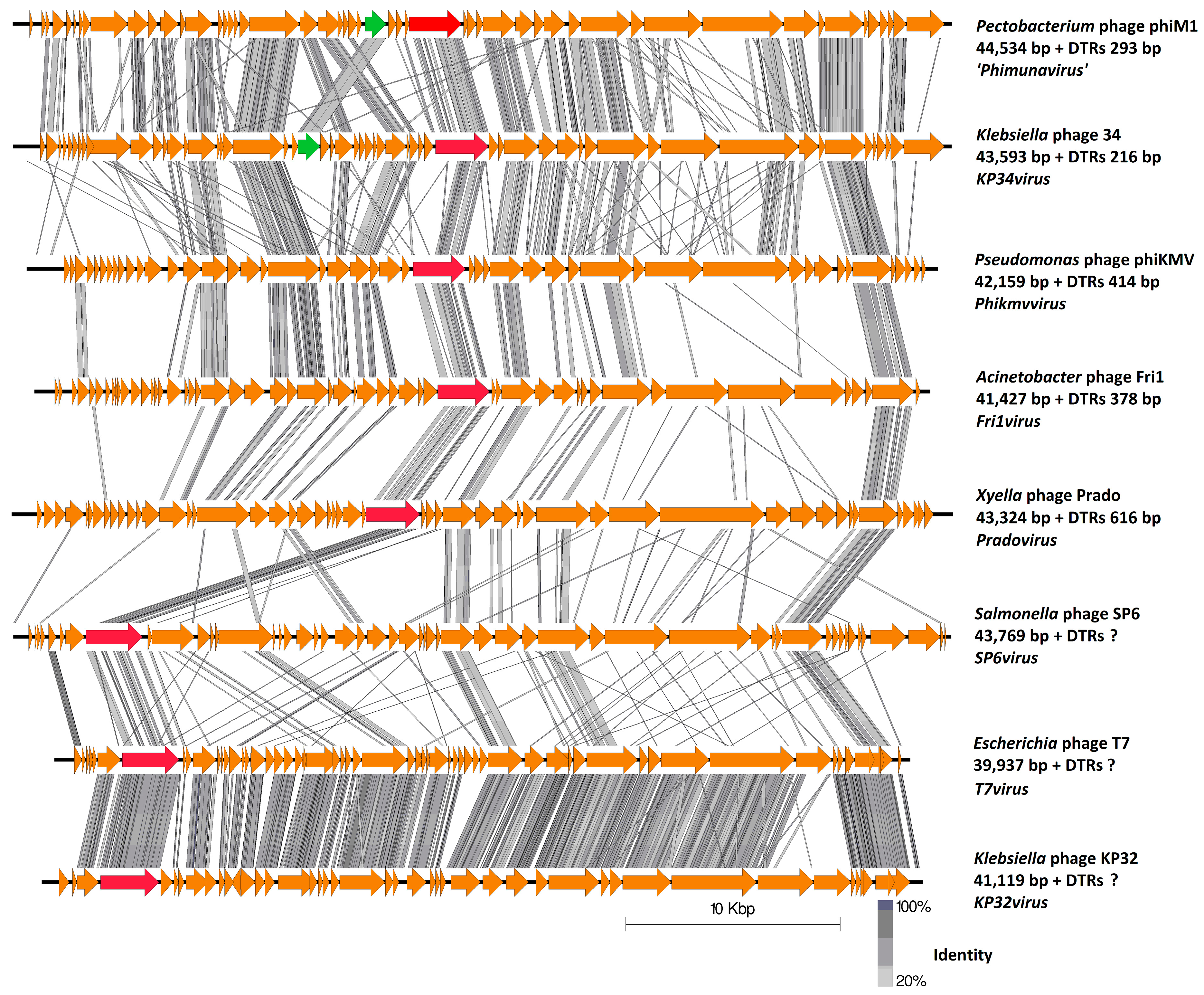
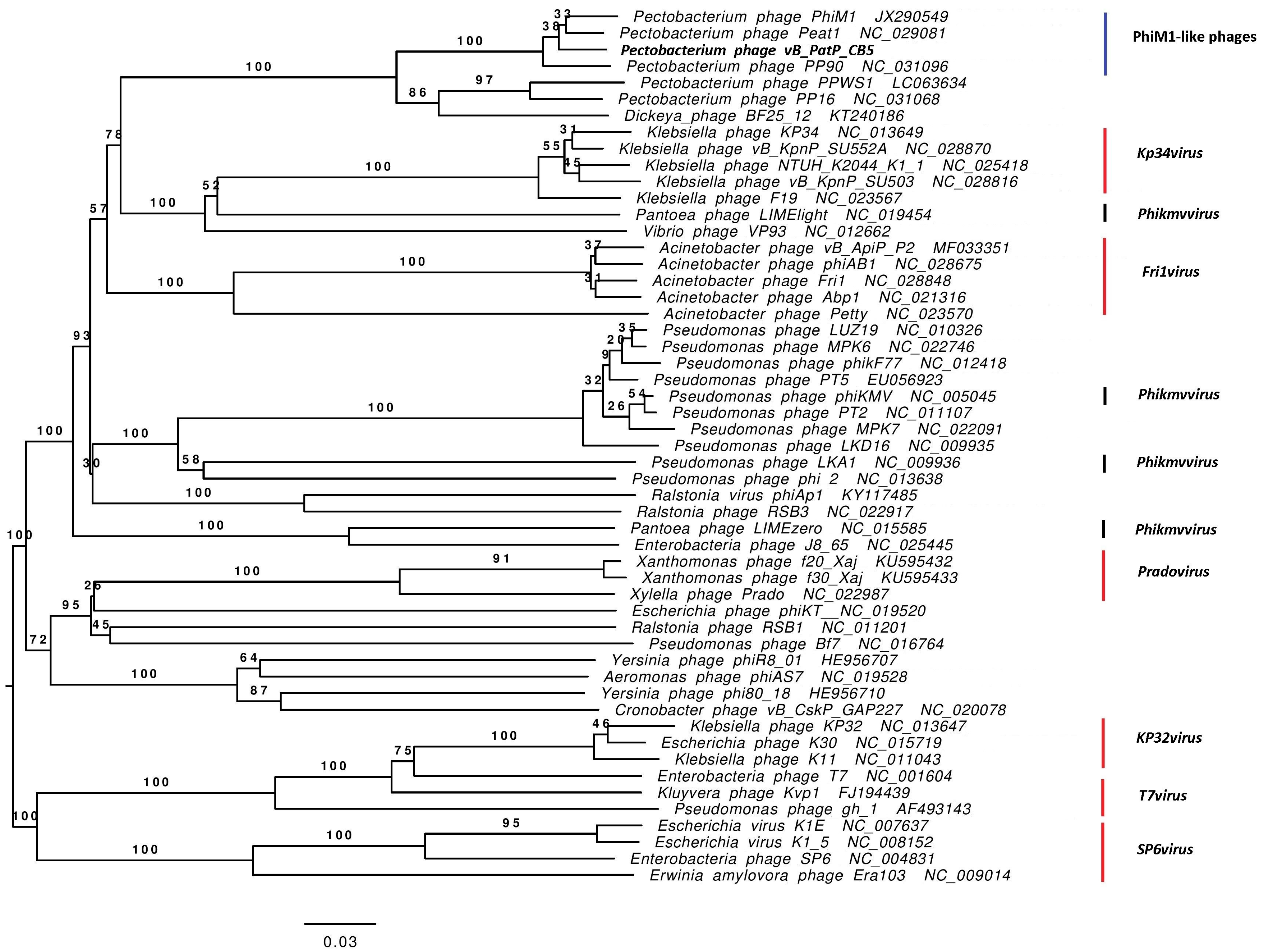
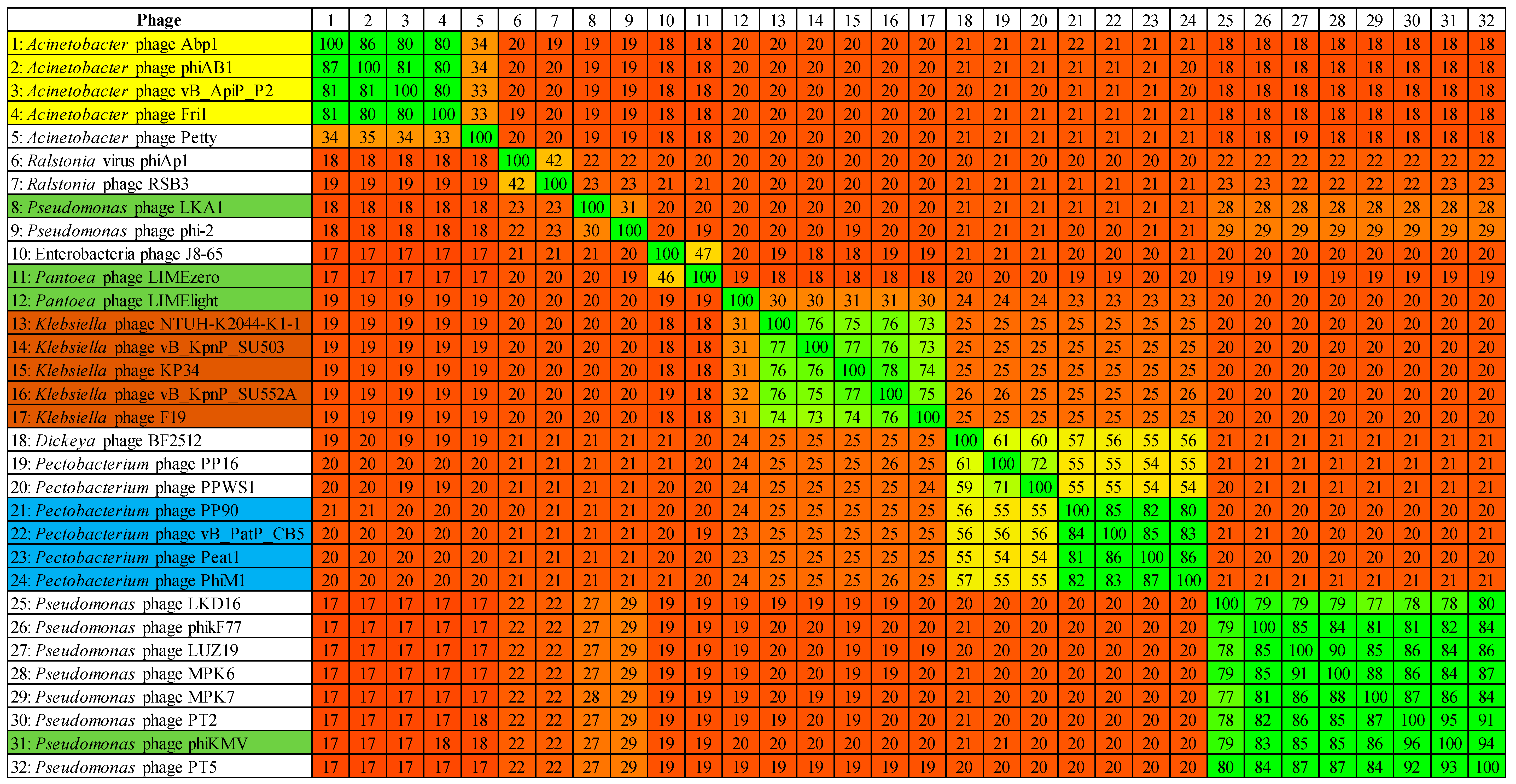
3.3. Comparative Genomics of PhiM1-Like Phages
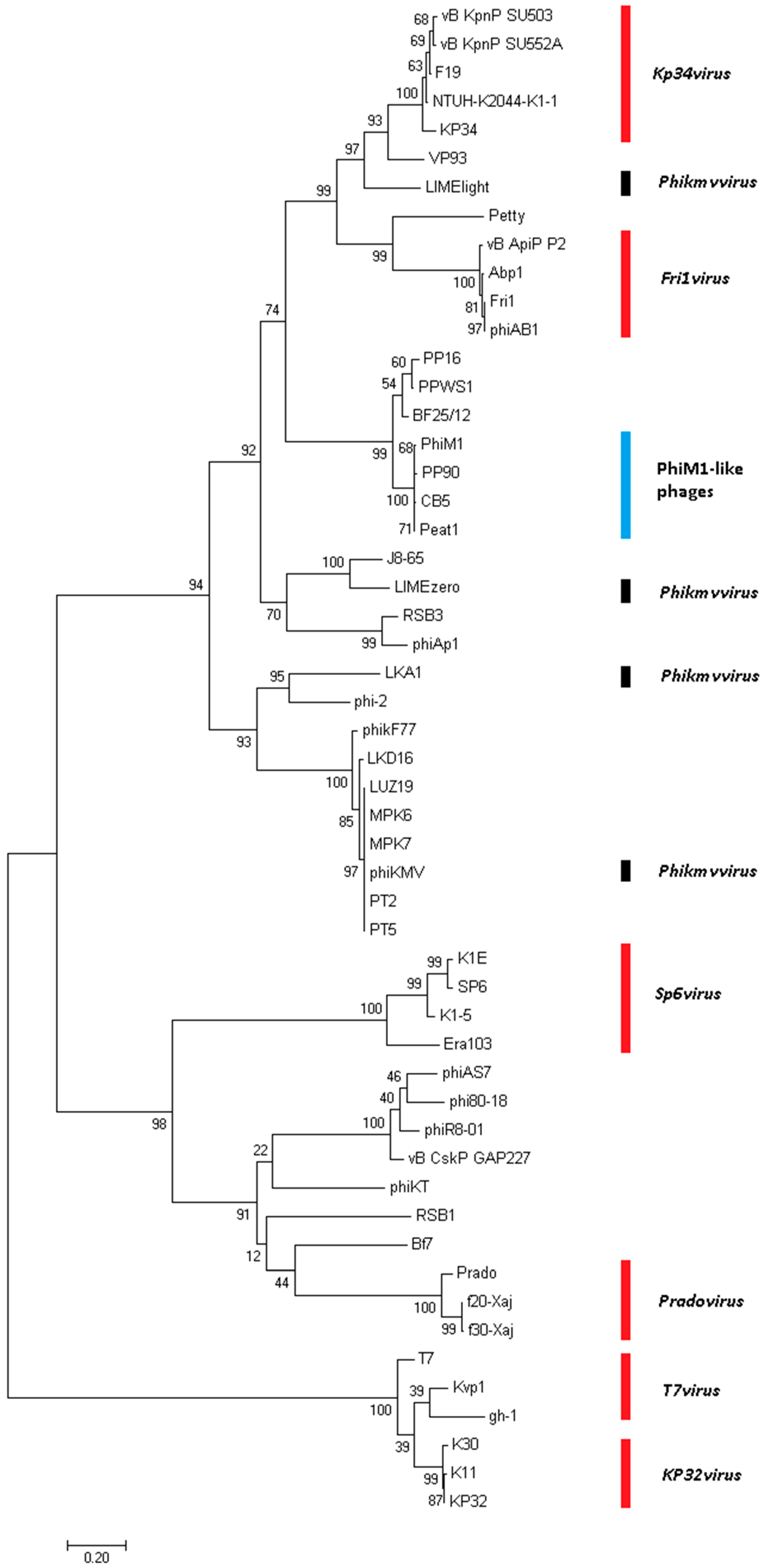
3.4. Phimunavirus Evolutionary Position within the Autographivirinae
3.5. RNAP of the PhiM1-Like Phages
3.6. Early Gene Region
3.7. DNA Replication, Repair, and Related Metabolism
3.8. Structure-Related Genes
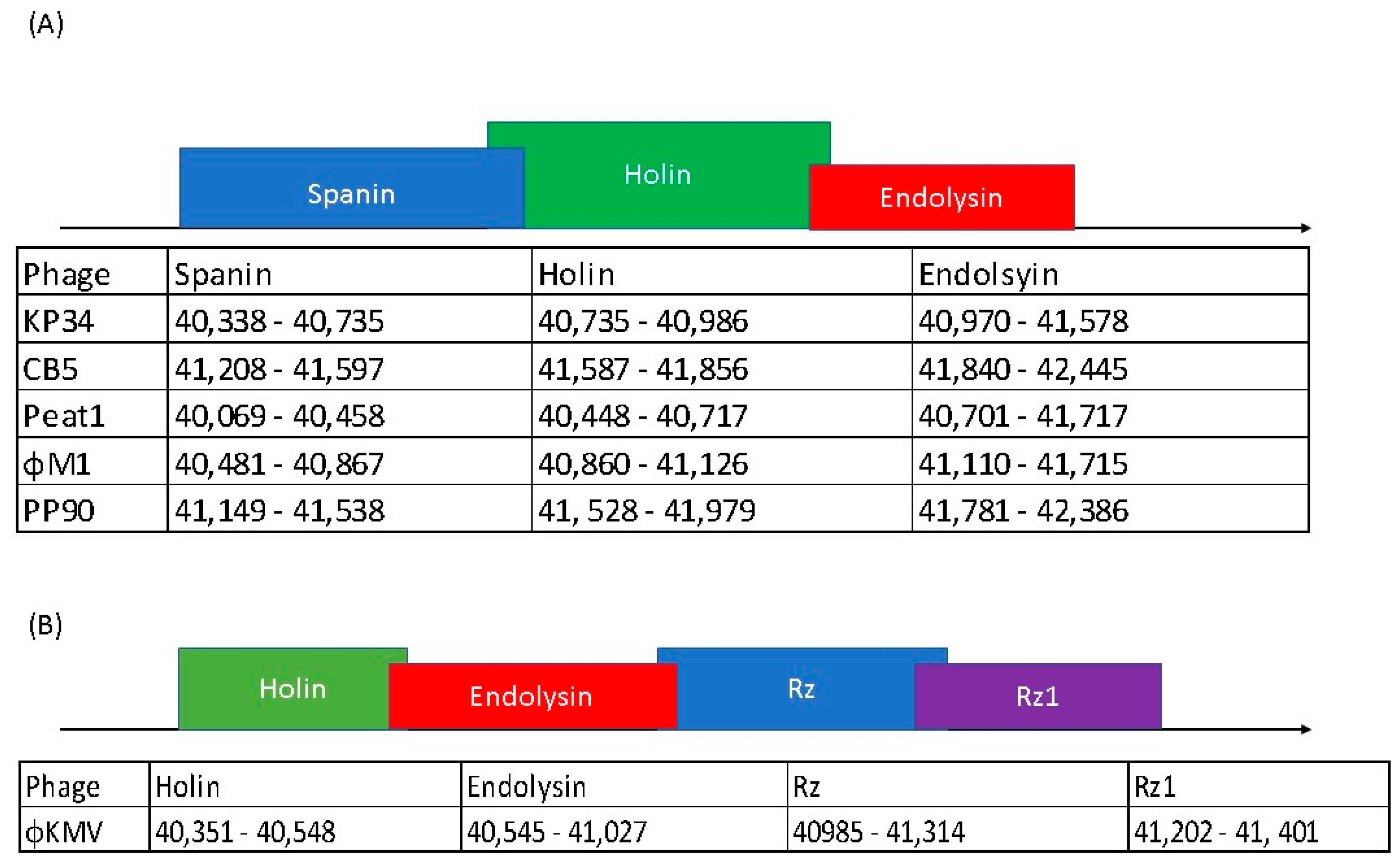
3.9. Lysis Cassette of PhiM1-Like Phages Resembles That of KP34virus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of interest
Disclaimer
References
- Calendar, R. The Bacteriophages; Oxford University Press: Oxford, UK, 2006; ISBN 0195148509. [Google Scholar]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.-W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefknowitz, E.J. (Eds.) Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: New York, NY, USA, 2012; ISBN 9780123846846. [Google Scholar]
- Adriaenssens, E.M.; Clokie, M.R.; Sullivan, M.B.; Gillis, A.; Kuhn, J.H.; Kropinski, A.M. Taxonomy of prokaryotic viruses: 2016 update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2017, 162, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Toth, I.K.; van der Wolf, J.M.; Saddler, G.; Lojkowska, E.; Hélias, V.; Pirhonen, M.; Tsror Lahkim, L.; Elphinstone, J.G. Dickeya species: An emerging problem for potato production in Europe. Plant Pathol. 2011, 60, 385–399. [Google Scholar] [CrossRef]
- Pérombelon, M.C.M. Potato diseases caused by soft rot erwinias: An overview of pathogenesis. Plant Pathol. 2002, 51, 1–12. [Google Scholar] [CrossRef]
- Kalischuk, M.; Hachey, J.; Kawchuk, L. Complete genome sequence of phytopathogenic Pectobacterium atrosepticum bacteriophage Peat1. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Blower, T.R.; Chai, R.; Przybilski, R.; Chindhy, S.; Fang, X.; Kidman, S.E.; Tan, H.; Luisi, B.F.; Fineran, P.C.; Salmond, G.P.C. Evolution of Pectobacterium bacteriophage ΦM1 to escape two bifunctional type III toxin-antitoxin and abortive infection systems through mutations in a single viral gene. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed]
- Toth, I.K.; Mulholland, V.; Cooper, V.; Bentley, S.; Shih, Y.; Perombelon, M.C.M.; Salmond, G.P.C. Generalized transduction in the potato blackleg pathogen Ewinia carotovora subsp. atroseptica by bacteriophage φM1. Microbiology 1997, 143, 2433–2438. [Google Scholar]
- Alič, Š.; Naglič, T.; Tušek-Žnidarič, M.; Ravnikar, M.; Rački, N.; Peterka, M.; Dreo, T. Newly isolated bacteriophages from the Podoviridae, Siphoviridae, and Myoviridae families have variable effects on putative novel Dickeya spp. Front. Microbiol. 2017, 8, 1870. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Kashihara, M.; Horiike, T.; Suzuki, T.; Dohra, H.; Netsu, O.; Tsuyumu, S. Genome sequence of Pectobacterium carotovorum phage PPWS1, isolated from Japanese horseradish [Eutrema japonicum (Miq.) Koidz] showing soft-rot symptoms. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Preparing stocks of bacteriophage Lamda by plate lysis and elution. In Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; Volume 1, p. 2.34. ISBN 0879695773. [Google Scholar]
- Buttimer, C.; Hendrix, H.; Lucid, A.; Neve, H.; Noben, J.-P.; Franz, C.; O’Mahony, J.; Lavigne, R.; Coffey, A. Novel N4-Like bacteriophages of Pectobacterium atrosepticum. Pharmaceuticals 2018, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Picking bacteriophage Lambda plaques. In Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; Volume 1, p. 2.32. ISBN 0879695773. [Google Scholar]
- Sambrook, J.; Russell, D.W. Plating bacteriophage Lambda. In Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; Volume 1, p. 2.25. ISBN 0879698773. [Google Scholar]
- Park, M.; Lee, J.-H.; Shin, H.; Kim, M.; Choi, J.; Kang, D.-H.; Heu, S.; Ryu, S. Characterization and comparative genomic analysis of a novel bacteriophage, SFP10, simultaneously inhibiting both Salmonella enterica and Escherichia coli O157:H7. Appl. Environ. Microbiol. 2012, 78, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liang, L.; Lin, S.; Jia, S. Isolation and characterization of a virulent bacteriophage AB1 of Acinetobacter baumannii. BMC Microbiol. 2010, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Pickard, D.J.J. Preparation of bacteriophage lysates and pure DNA. Methods Mol. Biol. 2009, 502, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2015, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.; Chang, H.-Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2014, 43, D213–D221. [Google Scholar] [CrossRef] [PubMed]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Juncker, A.S.; Willenbrock, H.; von Heijne, G.; Brunak, S.; Nielsen, H.; Krogh, A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003, 12, 1652–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, D.; Reynolds, D.; Seto, D.; Mahadevan, P. CoreGenes3.5: A webserver for the determination of core genes from sets of viral and small bacterial genomes. BMC Res. Notes 2013, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- Rambaut, A. FigTreeFigTree 1.4.3—A Graphical Viewer of Phylogenetic Trees and a Program for Producing Publication-Ready Figures. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 2 May 2018).
- Göker, M.; García-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom. Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Ågren, J.; Sundström, A.; Håfström, T.; Segerman, B. Gegenees: Fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS ONE 2012, 7, e39107. [Google Scholar] [CrossRef] [PubMed]
- Ceyssens, P.-J.; Lavigne, R.; Mattheus, W.; Chibeu, A.; Hertveldt, K.; Mast, J.; Robben, J.; Volckaert, G. Genomic analysis of Pseudomonas aeruginosa phages LKD16 and LKA1: Establishment of the ϕKMV subgroup within the T7 supergroup. J. Bacteriol. 2006, 188, 6924–6931. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, H.; Maciejewska, B.; Latka, A.; Majkowska-Skrobek, G.; Hellstrand, M.; Melefors, Ö.; Wang, J.T.; Kropinski, A.M.; Drulis-Kawa, Z.; Nilsson, A.S. A suggested new bacteriophage genus, “Kp34likevirus”, within the Autographivirinae subfamily of Podoviridae. Viruses 2015, 7, 1804–1822. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. Frequency of morphological phage descriptions in the year 2000. Arch. Virol. 2001, 146, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Burkal’tseva, M.V.; Robben, J.; Sykilinda, N.N.; Kurochkina, L.P.; Grymonprez, B.; Jonckx, B.; Krylov, V.N.; Mesyanzhinov, V.V.; Volckaert, G. The genome of bacteriophage phiKMV, a T7-like virus infecting Pseudomonas aeruginosa. Virology 2003, 312, 49–59. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Prangishvili, D.; Lavigne, R. Position paper: The creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ. Microbiol. 2009, 11, 2775–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Fan, H.; An, X.; Fan, H.; Jiang, H.; Chen, Y.; Tong, Y. Scrutinizing virus genome termini by high-throughput sequencing. PLoS ONE 2014, 9, e85806. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E.; Klumpp, J.; Bishop-Lilly, K.A.; Rajavel, M.; Willner, K.M.; Butani, A.; Henry, M.; Biswas, B.; Li, M.; Albert, M.; et al. Whole genome sequencing and comparative genomic analyses of two Vibrio cholerae O139 Bengal-specific Podoviruses to other N4-like phages reveal extensive genetic diversity. Virol. J. 2013, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Buttimer, C.; Hendrix, H.; Oliveira, H.; Casey, A.; Neve, H.; McAuliffe, O.; Ross, R.P.; Hill, C.; Noben, J.-P.; O’Mahony, J.; et al. Things are getting hairy: Enterobacteria bacteriophage vB_PcaM_CBB. Front. Microbiol. 2017, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, K.S.; Sebaihia, M.; Pritchard, L.; Holden, M.T.G.; Hyman, L.J.; Holeva, M.C.; Thomson, N.R.; Bentley, S.D.; Churcher, L.J.C.; Mungall, K.; et al. Genome sequence of the enterobacterial phytopathogen Erwinia carotovora subsp. atroseptica and characterization of virulence factors. Proc. Natl. Acad. Sci. USA 2004, 101, 11105–11110. [Google Scholar] [CrossRef] [PubMed]
- Nikolaichik, Y.; Gorshkov, V.; Gogolev, Y.; Valentovich, L.; Evtushenkov, A. Genome sequence of Pectobacterium atrosepticum strain 21A. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Marín, A.; Xia, X. GC skew in protein-coding genes between the leading and lagging strands in bacterial genomes: New substitution models incorporating strand bias. J. Theor. Biol. 2008, 253, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Roucourt, B.; Lavigne, R. The role of interactions between phage and bacterial proteins within the infected cell: A diverse and puzzling interactome. Environ. Microbiol. 2009, 11, 2789–2805. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, J.; Mayans, O.; Pickersgill, R. Structure and evolution of parallel β-helix proteins. J. Struct. Biol. 1998, 122, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Andres, D.; Baxa, U.; Hanke, C.; Seckler, R.; Barbirz, S. Carbohydrate binding of Salmonella phage P22 tailspike protein and its role during host cell infection. Biochem. Soc. Trans. 2010, 38, 1386–1389. [Google Scholar] [CrossRef] [PubMed]
- Moak, M.; Molineux, I.J. Role of the Gp16 lytic transglycosylase motif in bacteriophage T7 virions at the initiation of infection. Mol. Microbiol. 2000, 37, 345–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briers, Y.; Peeters, L.M.; Volckaert, G.; Lavigne, R. The lysis cassette of bacteriophage ϕKMV encodes a signal-arrest-release endolysin and a pinholin. Bacteriophage 2011, 1, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.; Savva, C.G.; Fleming, K.G.; Struck, D.K.; Young, R. Structure of the lethal phage pinhole. Proc. Natl. Acad. Sci. USA 2009, 106, 18966–18971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, J.D.; Rajaure, M.; Young, R. Spanin function requires subunit homodimerization through intermolecular disulfide bonds. Mol. Microbiol. 2013, 88, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.-A.; Heu, S.; Park, J.; Roh, E. Genomic characterization of bacteriophage vB_PcaP_PP2 infecting Pectobacterium carotovorum subsp. carotovorum, a new member of a proposed genus in the subfamily Autographivirinae. Arch. Virol. 2017, 162, 2441–2444. [Google Scholar] [CrossRef] [PubMed]
- Abbasifar, R.; Kropinski, A.M.; Sabour, P.M.; Ackermann, H.-W.; Alanis Villa, A.; Abbasifar, A.; Griffiths, M.W. The genome of Cronobacter sakazakii bacteriophage vB_CsaP_GAP227 suggests a new genus within the Autographivirinae. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, G.; Walkowiak-Nowicka, K.; Zemleduch-Barylska, A.; Mleczko, A.; Frąckowiak, P.; Nowaczyk, N.; Kozdrowska, E.; Barylski, J. Complete genome sequences of two novel autographiviruses infecting a bacterium from the Pseudomonas fluorescens group. Arch. Virol. 2017, 162, 2907–2911. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Sensitivity |
|---|---|---|
| Pectobacterium atrosepticum | DSM 18077 (type strain) | ++ |
| DSM 30184 | + | |
| DSM 30185 | + | |
| DSM 30186 | ++ * | |
| CB BL1-1 | + | |
| CB BL2-1 | + | |
| CB BL3-1 | + | |
| CB BL4-1 | + | |
| CB BL5-1 | + | |
| CB BL7-1 | + | |
| CB BL9-1 | + | |
| CB BL11-1 | + | |
| CB BL12-2 | ++ | |
| CB BL13-1 | + | |
| CB BL14-1 | + | |
| CB BL15-1 | − | |
| CB BL16-1 | + | |
| CB BL18-1 | + | |
| CB BL19-1 | + | |
| Pectobacterium carotovorum subsp. carotovorum | DSM 30168 (type strain) | − |
| DSM 30169 | − | |
| DSM 30170 | − | |
| CB BL19-1-37 | − | |
| Dickeya chrysanthemi bv chrysanthemi | LMG 2804 | − |
| Dickeya dianthicola | PD 482 | − |
| PD 2174 | − | |
| GBBC 1538 | − | |
| Dickeya solani | sp. PRI 2222 (D36) | − |
| LMG 25865 (D10) | − | |
| GBBC 1502 | − | |
| GBBC 1586 | − |
| Phage | Genome Size (bp) | DTRs (bp) | G + C Content, % | ORFs | tRNA | DNA Identity, % * | Homologous Proteins, % ** |
|---|---|---|---|---|---|---|---|
| φM1 | 43,534 | 293 | 49.18 | 52 | 1 | 100 | 100 |
| CB5 | 44,262 | 287 | 48.98 | 60 | 0 | 84 | 73 |
| Peat1 | 45,633 | NA | 48.86 | 61 | 0 | 86 | 87 |
| PP90 | 44,570 | NA | 48.89 | 56 | 0 | 86 | 80 |
| No. | Product | φM1 Accession No. | Φm1 Locus Tag | pVOG |
|---|---|---|---|---|
| 1 | hypothetical protein | AFQ22488.1 | PhiM1_03 | VOG6006 |
| 2 | hypothetical protein | AFQ22489.1 | PhiM1_04 | VOG1073 |
| 3 | hypothetical protein | AFQ22493.1 | PhiM1_08 | VOG5528 |
| 4 | hypothetical protein | AFQ22494.1 | PhiM1_09 | - |
| 5 | putative peptidase | AFQ22495.1 | PhiM1_10 | VOG5332 |
| 6 | hypothetical protein | AFQ22496.1 | PhiM1_11 | VOG5704 |
| 7 | putative DNA primase | AFQ22497.1 | PhiM1_12 | VOG4551 |
| 8 | putative DNA helicase | AFQ22499.1 | PhiM1_14 | VOG0025 |
| 9 | hypothetical protein | AFQ22501.1 | PhiM1_16 | - |
| 10 | hypothetical protein | AFQ22503.1 | PhiM1_18 | - |
| 11 | DNA polymerase | AFQ22505.1 | PhiM1_20 | VOG0026 |
| 12 | hypothetical protein | AFQ22506.1 | PhiM1_21 | VOG1076 |
| 13 | DNA exonuclease | AFQ22507.1 | PhiM1_22 | VOG0028 |
| 14 | hypothetical protein | AFQ22508.1 | PhiM1_23 | - |
| 15 | DNA endonuclease VII | AFQ22510.1 | PhiM1_25 | VOG8238 |
| 16 | putative metallophosphoesterase | AFQ22512.1 | PhiM1_27 | VOG1606 |
| 17 | hypothetical protein | AFQ22514.1 | PhiM1_29 | VOG1254 |
| 18 | hypothetical protein | AFQ22515.1 | PhiM1_30 | VOG9679 |
| 19 | putative RNA polymerase | AFQ22516.1 | PhiM1_31 | VOG0019 |
| 20 | hypothetical protein | AFQ22517.1 | PhiM1_32 | VOG1406 |
| 21 | hypothetical protein | AFQ22518.1 | PhiM1_33 | VOG9202 |
| 22 | putative structural protein | AFQ22519.1 | PhiM1_34 | VOG8332 |
| 23 | putative head–tail connector protein | AFQ22520.1 | PhiM1_35 | VOG0030 |
| 24 | putative scaffolding protein | AFQ22521.1 | PhiM1_36 | VOG0031 |
| 25 | putative endonuclease | AFQ22522.1 | PhiM1_37 | - |
| 26 | putative capsid protein | AFQ22523.1 | PhiM1_38 | VOG4572 |
| 27 | putative tail tubular protein A | AFQ22524.1 | PhiM1_39 | VOG4592 |
| 28 | putative tail tubular protein B | AFQ22525.1 | PhiM1_40 | VOG0034 |
| 29 | putative internal core protein A | AFQ22526.1 | PhiM1_41 | VOG1080 |
| 30 | putative internal core protein B | AFQ22527.1 | PhiM1_42 | VOG3794 |
| 31 | putative internal core protein C | AFQ22528.1 | PhiM1_43 | VOG0038 |
| 32 | putative tail fiber protein | AFQ22529.1 | PhiM1_44 | - |
| 33 | putative DNA maturase A | AFQ22530.1 | PhiM1_45 | VOG0041 |
| 34 | putative DNA maturase B | AFQ22531.1 | PhiM1_46 | VOG4544 |
| 35 | hypothetical protein | AFQ22532.1 | PhiM1_47 | - |
| 36 | putative Rz1A protein | AFQ22534.1 | PhiM1_49 | VOG1082 |
| 37 | putative holin | AFQ22535.1 | PhiM1_50 | VOG0765 |
| 38 | endolysin | AFQ22536.1 | PhiM1_51 | VOG4565 |
| 39 | phage tail spike protein | AFQ22537.1 | PhiM1_52 | VOG4640 |
| Phage | Recognition Loop | Specificity Loop |
|---|---|---|
| φKMV | HQEAKAAGPAAKL | EEVRVRLRAEAVEYVTLYEAK-DE |
| KP34 | MRNVKAPGIGGKY | EEVRVRIDCMNLSAVLVHNRDFKT |
| Fri1 | VKKQKIRGVGGKY | VTKTVAIRSMGINNIAYRYPD-NQ |
| φM1 | ICSKGTRGVGGKY | SITRVSLKALGVALNMRVFDD-HS |
| CB5 | ICSKGTRGVGGKY | SITRVSLKALGVALNMRVFDD-HS |
| Peat1 | ICSKGTRGVGGKY | SITRVSLKALGVALNMRVFDD-HS |
| PP90 | ICSKGTRGVGGKY | SITRVSLKALGVALNMRVFDD-HS |
| BF25/12 | MCSTGSRGLGGKY | DSTRINLNALGTQLVMRTFND-HL |
| PP16 | ICTTGNRGLNGKY | DSTRIELRSLGIKLVMRTFDD-TQ |
| PPWS1 | ICTTGNRGLNGKY | DSTRIELRSLGIKLVMRTFDD-TQ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buttimer, C.; Lucid, A.; Neve, H.; Franz, C.M.A.P.; O’Mahony, J.; Turner, D.; Lavigne, R.; Coffey, A. Pectobacterium atrosepticum Phage vB_PatP_CB5: A Member of the Proposed Genus ‘Phimunavirus’. Viruses 2018, 10, 394. https://doi.org/10.3390/v10080394
Buttimer C, Lucid A, Neve H, Franz CMAP, O’Mahony J, Turner D, Lavigne R, Coffey A. Pectobacterium atrosepticum Phage vB_PatP_CB5: A Member of the Proposed Genus ‘Phimunavirus’. Viruses. 2018; 10(8):394. https://doi.org/10.3390/v10080394
Chicago/Turabian StyleButtimer, Colin, Alan Lucid, Horst Neve, Charles M. A. P. Franz, Jim O’Mahony, Dann Turner, Rob Lavigne, and Aidan Coffey. 2018. "Pectobacterium atrosepticum Phage vB_PatP_CB5: A Member of the Proposed Genus ‘Phimunavirus’" Viruses 10, no. 8: 394. https://doi.org/10.3390/v10080394