Retroviral Integration Site Selection


Institute of Microbiology, University Hospital Center and University of Lausanne, Bugnon 48, CH-1011 Lausanne, Switzerland
*
Author to whom correspondence should be addressed.
Viruses 2010, 2(1), 111-130; https://doi.org/10.3390/v2010111
Submission received: 8 October 2009
/
Revised: 21 December 2009
/
Accepted: 5 January 2010
/
Published: 12 January 2010
(This article belongs to the Special Issue Retroviral Enzymes)
Abstract
:The stable insertion of a copy of their genome into the host cell genome is an essential step of the life cycle of retroviruses. The site of viral DNA integration, mediated by the viral-encoded integrase enzyme, has important consequences for both the virus and the host cell. The analysis of retroviral integration site distribution was facilitated by the availability of the human genome sequence, revealing the non-random feature of integration site selection and identifying different favored and disfavored genomic locations for individual retroviruses. This review will summarize the current knowledge about retroviral differences in their integration site preferences as well as the mechanisms involved in this process.
Keywords:
retrovirus; integration; site selection; integrase; HIV; LEDGF; LEDGF/p75; PSIP1; lentivirus; transcription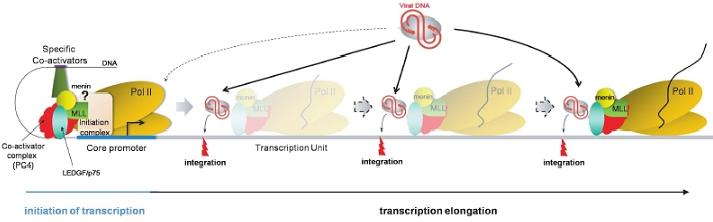
1. Introduction
The principal feature of retroviruses is that upon entry and release of their viral RNA genome into the cytoplasm of the host cell, it is reverse transcribed by the viral reverse transcriptase into a linear double stranded cDNA copy (Figure 1A) (for reviews, see [1-4]). This viral DNA is not naked, but is associated with viral and cellular proteins in a nucleoprotein complex called the preintegration complex (PIC). Depending on the retrovirus, the PIC is subsequently translocated into the nucleus, either actively through nuclear pores, or upon nuclear membrane disruption occurring during mitosis. There, it is, either integrated into the genome of the host cell, or remains unintegrated for a certain time, or is degraded (Figure 1) (for reviews, see [3,5]).
The insertion of the viral DNA into the host cell genome is catalyzed by the virally encoded integrase (IN) enzyme (for reviews, see [6-9]). Retroviral INs typically range between 280 and 450 amino acids (HIV-1 IN: 288 amino acids, 32 kDa), and are characterized by three functional domains: (i) the N-terminal domain, containing an HHCC zinc-binding motif, (ii) the catalytic core domain (residues 50-212 of HIV-1), containing the critical magnesium-binding D-D-35-E motif that constitutes the active site, and (iii) the C-terminal domain. The three domains of IN appear to be involved in DNA binding and multimerization. Indeed, the full concerted integration seems to require an IN tetramer, i.e., one IN dimer at each viral end [10-14].
Although unintegrated viral DNA can be used as template for viral transcription [15-17], integration is required for productive viral replication. However, the efficiency of integration is quite low, and depends on restrictions occurring during the early steps of infection. By infecting human osteosarcoma (HOS) cells with VSV-G pseudotyped HIV particles, Thomas et al. measured that only 5% of HIV viruses successfully entered the cell and initiated reverse transcription [18]. Of these, 28% (i.e., 1.5% total) completed reverse transcription, translocated to the nucleus (with an efficiency ranging around 2-3% according to data using IN-eGFP fusion proteins from Cereseto and collaborators [19,20]) and finally only ~13% of viruses that initiated reverse transcription achieved insertion in the host cell genome, which represented 0.41% of the virus input [18].
The site of viral DNA insertion is critical for the virus, as it can influence the rate of viral transcription. Indeed, integration into transcriptionally active regions may favor viral gene expression, thus facilitating productive infectious progeny particles, while integration into transcriptionally repressed chromatin may disfavor viral gene expression, thus possibly facilitating viral latency [21-24].
The ability of retroviruses to integrate has also important consequences for the host as it can affect the expression of genes surrounding the proviral DNA. Indeed, viral DNA disruptive insertion into a gene may alter its expression (reduced gene expression), thereby potentially affecting cellular physiology. More importantly, the activity of inserted viral promoters or enhancers near cellular genes may also affect their physiological expression (increased gene expression), potentially leading to tumorigenesis when these genes are proto-oncogenes [25,26]. This process, known as insertional mutagenesis, raised justified critical issues regarding the safety of retroviral-based vectors used in gene therapy (for reviews, see [27-32]). However, not all retroviruses display the same genotoxic potential, as gammaretroviruses for example appeared to be more prone to insertional mutagenesis than lentiviruses [33-37]. This phenomenon can be partly explained by their preferred genomic site for proviral DNA insertion, i.e., into promoter regions for gammaretroviruses and along transcription units for lentiviruses.
It is now clear that the chromosomal site of viral DNA integration is not random, but in contrast that retroviruses display specific preferences at distinct genomic positions. This review will summarize the current knowledge about retrovirus-specific favored integration sites, as well as the current models explaining these preferences.
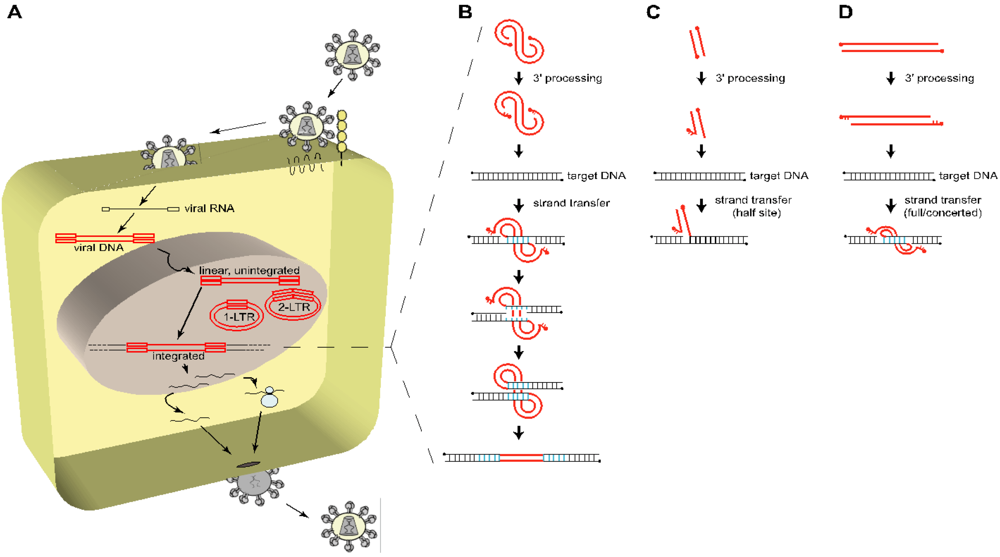
Figure 1.
Overview of the early steps of HIV-1 life cycle. (A) To enter a target cell, HIV-1 gp120 binds to specific cellular receptors, i.e., CD4 and a chemokine coreceptor (CCR5 or CXCR4), triggering the gp41-mediated fusion between the viral and the cellular membrane, and releasing the viral core in the cytoplasm of the host cell. The viral single stranded, positive, RNA genome (black line, flanked by open black squares depicting R-U5 and U3-R in its 5’ and 3’ termini respectively) is reverse transcribed into a linear double stranded cDNA copy (red line, flanked by open red squares representing the LTR = U3-R-U5), which is a component of the preintegration complex (PIC), also containing the viral integrase (IN), as well as other viral and cellular proteins. The PIC is translocated to the nucleus and the viral cDNA is either integrated through the action of IN or remains unintegrated (linear, 1-LTR circles, 2-LTR circles). From this point on, the cellular machinery of the host is recruited to transcribe the viral genome in order to produce all the components required to generate newly infectious particles. (B) The integration process is divided into three major steps: the 3’ processing and the strand transfer reaction, both catalyzed by IN, and the repair of the integrated viral DNA by the DNA repair machinery of the host cell. The PIC-containing viral DNA (red line, with 5’ ends depicted by filled circles) is first processed by the IN-mediated removal of a dinucleotide (GT) at each 3’ end of the viral DNA, leaving a protruding (AC) dinucleotide at the 5’ ends. IN then catalyzes the stable insertion of the processed viral DNA into a target DNA (black line), by simultaneously and asymmetrically breaking the target DNA 5 bp apart (blue bonds) (4 to 6 bp depending on the retrovirus) and joining it to the 3’ recessed ends of the viral DNA, leaving an integration intermediate with unpaired bases at each viral-target DNA junction. The DNA repair machinery of the host cell fills in the five nucleotide gap at each side of the viral DNA and removes the two 5’ overhang nucleotides from the viral DNA, resulting in the duplication of 5 bp of the target DNA at both sides of the proviral DNA. (C and D) Schematic concepts of in vitro integration assays showing half-site integration (C) and concerted or full-site integration (D).
Figure 1.
Overview of the early steps of HIV-1 life cycle. (A) To enter a target cell, HIV-1 gp120 binds to specific cellular receptors, i.e., CD4 and a chemokine coreceptor (CCR5 or CXCR4), triggering the gp41-mediated fusion between the viral and the cellular membrane, and releasing the viral core in the cytoplasm of the host cell. The viral single stranded, positive, RNA genome (black line, flanked by open black squares depicting R-U5 and U3-R in its 5’ and 3’ termini respectively) is reverse transcribed into a linear double stranded cDNA copy (red line, flanked by open red squares representing the LTR = U3-R-U5), which is a component of the preintegration complex (PIC), also containing the viral integrase (IN), as well as other viral and cellular proteins. The PIC is translocated to the nucleus and the viral cDNA is either integrated through the action of IN or remains unintegrated (linear, 1-LTR circles, 2-LTR circles). From this point on, the cellular machinery of the host is recruited to transcribe the viral genome in order to produce all the components required to generate newly infectious particles. (B) The integration process is divided into three major steps: the 3’ processing and the strand transfer reaction, both catalyzed by IN, and the repair of the integrated viral DNA by the DNA repair machinery of the host cell. The PIC-containing viral DNA (red line, with 5’ ends depicted by filled circles) is first processed by the IN-mediated removal of a dinucleotide (GT) at each 3’ end of the viral DNA, leaving a protruding (AC) dinucleotide at the 5’ ends. IN then catalyzes the stable insertion of the processed viral DNA into a target DNA (black line), by simultaneously and asymmetrically breaking the target DNA 5 bp apart (blue bonds) (4 to 6 bp depending on the retrovirus) and joining it to the 3’ recessed ends of the viral DNA, leaving an integration intermediate with unpaired bases at each viral-target DNA junction. The DNA repair machinery of the host cell fills in the five nucleotide gap at each side of the viral DNA and removes the two 5’ overhang nucleotides from the viral DNA, resulting in the duplication of 5 bp of the target DNA at both sides of the proviral DNA. (C and D) Schematic concepts of in vitro integration assays showing half-site integration (C) and concerted or full-site integration (D).

2. Integration targeting in vitro
In vitro, IN is sufficient to carry out the first two steps of the integration reaction, i.e., 3’ processing and strand transfer reactions, resulting in the covalent attachment of the viral DNA on virtually any DNA target (random integration) (Figure 1B) [38]. To succeed, three principal components are minimally required: (i) purified viral integrase, (ii) a donor DNA mimicking a viral DNA terminal sequence to be recognized by IN, and (iii) an acceptor DNA in which the donor DNA will be inserted (for more details, see [7]).
In the first in vitro assays, the donor DNA consisted in short oligonucleotide duplexes (21 bp minimum) containing the terminal LTR sequence (either U3 or U5), allowing to reproduce the 3’ processing efficiently, as well as the strand transfer reaction (albeit with lower efficiency). However, these were only half-site integration as only one donor DNA (e.g. one viral LTR) was inserted in the acceptor DNA (Figure 1C) [39], and not both in a concerted motion. This gave rise to the development of new assays, full-site or concerted integration assays, which use a longer donor DNA containing both terminal sequences (Figure 1D) [40,41].
These studies showed that in vitro, HIV IN displayed only a weak preference for the primary DNA sequence [42-49], slightly favoring the palindromic TNNGT(A/T)ACNNA DNA sequence (bold nucleotides indicate the asymmetrical insertion points, resulting in the final 5 bp duplication flanking the proviral DNA, depicted in blue in Figure 1B). Furthermore, the addition of nucleosomes on the target DNA improved the in vitro efficiency of integration, and favored integration on distorted DNA and outwardly-facing major grooves sites of the nucleosomal DNA [44,46,47,50-53].
In order to investigate whether simple tethering of IN to a specific DNA site could confer integration preferences in vitro, fusions of IN to specific DNA binding proteins were engineered [54]. The fusion of HIV IN to the DNA binding domain of λ repressor (λR) lead to increased integration targeting at sites surrounding the predetermined λR DNA binding sites (λ operator sites) [54]. Fusion of IN with other DNA binding proteins such as LexA [55] or the polydactyl Zinc finger protein E2C [56] reached similar results. These studies provided proof-of-concept that integration site selection in vitro could be modified and redirected more preferentially to specific DNA sites.
3. Integration targeting in vivo
The availability of the human genome sequence and other vertebrate genomes made possible to interrogate where in the host cell genome retroviruses integrated, and more precisely what were the chromosomal features (according to current genomic annotations) that were favored for retroviral integration. To achieve this, host DNA regions flanking the proviral DNAs were amplified, sequenced, and finally aligned to the host genome sequence (method overview reviewed in [32,57,58]).
Schroder et al., in 2002, revealed for the first time that HIV favored integration in transcription units and disfavored Alu repeats [59]. One year later, Wu et al. showed that murine leukemia virus (MLV) had distinct preferences, favoring integration at transcription start sites and CpG islands [60]. Since then, multiple genome-wide studies confirmed these preferences and revealed the integration site preferences for almost all retroviral genera, with the exception of epsilonretroviruses (Tables 1 and 2).

{kind=link}
{kind=link}
{kind=link}
Table 1 .
Major genome-wide studies of retroviral integration distribution.
| Retroviridae genera | Specimen a | Host cell type b | Approx. Nb. of sites investigated c | References |
|---|---|---|---|---|
| lentiviruses | HIV-1 | human | 59869 | [23,36,59-76] |
| other | 2421 | [37,77-81] | ||
| HIV-2 | human | 202 | [82] | |
| SIV | human | 148 | [83] | |
| simian | 501 | [84] | ||
| EIAV | human | 1241 | [69,81] | |
| other | 70 | [81] | ||
| FIV | human | 226 | [85] | |
| alpharetroviruses | ASLV | human | 695 | [62,86] |
| avian | 658 | [77] | ||
| betaretroviruses | MMTV | human | 298 | [87] |
| murine | 170 | [87] | ||
| gammaretroviruses | MLV | human | 4005 | [60,66,70,73,88] |
| murine | 189 | [37] | ||
| other | 953 | [78,79,84] | ||
| MSCV | murine | 259 | [89] | |
| PERV | human | 1962 | [90,91] | |
| XMRV | human | 472 | [92] | |
| deltaretroviruses | HTLV-I | human | 1235 | [93-95] |
| epsilonretroviruses | Not investigated | |||
| spumaviruses | FV | human | 3457 | [65,70] |
| other | 263 | [78] | ||
| Endogenous retroviruses | HERV-K | human | 1565 | [88] |
a. HIV: human immunodeficiency virus; SIV: simian immunodeficiency virus; EIAV: equine infectious anemia virus; FIV: feline immunodeficiency virus; ASLV: avian sarcoma leukosis virus; MMTV: mouse mammary tumor virus; MLV: murine leukemia virus; MSCV: murine stem cell virus; PERV: porcine endogenous retrovirus; XMRV: xenotropic murine leukemia virus-related virus; HTLV: human T-cell lymphotropic virus; FV: foamy virus; HERV: human endogenous retrovirus.
b. Host cell type includes human, simian, murine, canine and avian cells. Are indicated the human cells and the host cell type specific to each specimen. Other: non human and non species-specific host cell type.
c. Number of integration sites analyzed in untreated/control cells according to the original publication.
These studies demonstrated that in vivo the site of retroviral integration was not random, and that integration site preferences were retrovirus-specific (Table 1 ): lentiviruses favor integration in active transcription units, with no preference along the transcript, nor for introns or exons; gammaretroviruses, spumaviruses and endogenous retroviruses (HERV class II) integrate preferentially around transcription start sites and CpG islands, features associated with host gene promoters; alpharetroviruses and deltaretroviruses displayed only weak preferences for integrating in transcription units and CpG islands; and finally betaretroviruses show no integration site preferences, displaying a random distribution of integration sites in the host genome.
The integration site preferences are not host-specific as the same distribution of integration sites can be observed in different host vertebrate cells, including human, simian, murine, avian and canine cells (Table 1 ). Furthermore, integration targeting is independent of the route of viral entry, as HIV-based vectors using a natural CCR5-tropic HIV envelope or a VSV-G pseudotype envelope displayed the same integration site distribution [67].
Table 1 .
Chromosomal features associated with preferential retroviral integration sites.
| Retroviridae genera | in Transcription Unitsa | ± 2kb Transcription Start Sitesa | ± 2 kb CpG Islandsa |
|---|---|---|---|
| Lentiviruses | +/++ | 0 | -/0 |
| Alpharetroviruses | + | 0 | + |
| Betaretroviruses | 0 | 0 | 0 |
| Gammaretroviruses | + | ++ | ++ |
| Deltaretroviruses | + | + | + |
| Epsilonretroviruses | NA | NA | NA |
| Spumaviruses | 0 | ++ | ++ |
| HERV-class II | + | ++ | ++ |
a: ratio between the proportion of the chromosomal feature over the random proportion in the human genome, according to RefSeq databases and with values from [69,87,88,90,94,95];
0: no statistical difference over random;
+/++: statistically favored feature over random with ++ for ratio greater than 2 and + for a ratio less than 2;
-: statistically disfavored feature over random;
NA: not available.
Three models, which are not mutually exclusive, have been proposed to date to explain integration site selection: (i) chromatin accessibility, (ii) cell cycle effects, and (iii) tethering mechanism. However, while the first two models can globally influence integration site targeting, only the last one - integration by a tethering mechanism - can provide a logical explanation to the observed differences of integration targeting preferences among retroviruses.
3.1. The chromatin accessibility model
According to this model, the structure of the chromatin, either relaxed or condensed, may influence the accessibility of target DNA sequences to preintegration complexes, thereby affecting integration.
In vivo, retroviral integration displayed a weak preference for the primary DNA sequence, similar to the one observed in vitro [60,69,70,72,90,94,96-99]. Furthermore, HIV integration in vivo also favors major grooves facing outwards from the nucleosome core, as predicted by nucleosome positioning [72,100]. These data indicated that local chromatin structure, such as A/T-rich distorted DNA and outwardly-facing major grooves of the nucleosomal DNA, may facilitate integration, however this cannot fully explain the observed differences in retroviral integration site distribution.
The retroviral differences in favored integration target site selection observed in vivo argued against chromatin accessibility being the principal determinant explaining integration targeting (Table 1 ). Indeed, if the accessibility of chromatin was the key player, all retroviruses would display the same integration site distribution pattern, favoring reachable chromatin.
Additional evidence against this model playing a major role in integration targeting came from the correlation analysis of mapped HIV and MLV integration sites with mapped DNase I hypersensitive sites. DNase I cleavage sites are used as a surrogate marker for accessible chromatin, and are enriched in the 5’ ends of transcription units and CpG islands [66,101]. This study revealed that MLV integrated preferentially in 2-kb intervals surrounding DNase I hypersensitive sites, compatible with favored MLV integration sites in promoter regions. In contrast, HIV integration did not display such a preference, consistent with favored integration in transcription units and not promoter regions. Therefore, although chromatin accessibility may influence MLV integration site preferences, it does not seem to affect significantly HIV integration site distribution.
In conclusion, even though chromatin structure can facilitate integration, chromatin accessibility cannot solely explain the differences observed in integration site preferences between HIV and MLV.
3.2. The cell cycle model
This model implies that the phase of the cell cycle may influence integration site selection. Indeed, lentiviruses can infect and successfully integrate regardless of the cell cycle stage (dividing or non-dividing) thanks to the active nuclear import of the PIC, while gammaretroviruses can integrate only into dividing cells as they require the disruption of the nuclear membrane occurring during mitosis to contact the host genome. Thus, it is possible that this difference in cell cycling status during viral infection might affect integration site distribution.
To test this hypothesis, HIV integration site distribution was compared between dividing IMR-90 primary lung fibroblasts and non-dividing G1-arrested IMR-90 cells [64]. HIV integration in active transcription units was favored in both dividing and non-dividing cells, with even stronger preferences in non-dividing cells. Similarly, analysis of HIV integration site distribution in non-dividing differentiated human macrophages also revealed a marked preference for transcription units [67,74]. Comparison between quiescent CD4+ T cells and activated CD4+ T cells revealed a similar integration site distribution with favored integration in transcription units and other chromosomal features (gene density, GC-rich regions, DNase I sites), although to a lower extent for resting cells [75,76].
All together, these data argued against a major positive influence of cycling cells in guiding HIV integration in transcription units and cannot explain the integration site selection differences between HIV and MLV.
3.3. The tethering protein model
This model implicates that a cellular protein, specific for each retroviral genera, would act as a tethering factor, binding both to specific chromatin sites and to the retroviral preintegration complex.
In principle, any PIC component could serve as the docking point between the PIC and the integration site, thereby dictating the integration target site preferences. PIC candidates include both viral and cellular proteins.
3.3.3. Epigenetic modifications and integration targeting
The Encyclopedia of DNA Elements (ENCODE) contains ~1% of the genome that is extensively annotated, allowing comparisons of epigenetic marks with retroviral integration site preferences [126,127]. Consistent with previous findings on favored insertion in active transcription units, lentiviral integration sites associated significantly with the epigenetic marks H3K4me, H3K36me, H3K9/K14Ac and H4Ac, histone modifications usually associated with transcriptionally active chromatin. In contrast, lentiviral integration sites were disfavored in regions containing DNA CpG methylation as well as H3K9me2/3, H3K27me2/3 and H3K79me3, epigenetic marks usually associated with repressed chromatin [72,76]. A similar association pattern was observed for alpharetroviruses, gammaretroviruses and HERV-K [88].
Recently, LEDGF/p75 was identified as a cellular partner of the menin/MLL complex [128]. The menin/MLL histone methyltransferase complex promotes specific trimethylation of histone 3 on lysine 4 (H3K4me3), an epigenetic mark associated with active transcription. This histone modification is also associated with HIV integration sites, coherent with a global picture in which, LEDGF/p75, epigenetic marks associated with transcriptional activity and HIV integration sites converge to similar genomic locations.
4. Conclusions
LEDGF/p75 was initially identified in a complex co-immunoprecipating with positive cofactor 4 (PC4), a general coactivator of transcription [113,129]. PC4 has been involved in many transcription steps: (i) PC4, by interacting with upstream activators and the general transcriptional machinery, can enhance the efficiency of pre-initiation complex assembly, thereby promoting transcription initiation, in cooperation with TBP-associated factors (TAFs) [130]; (ii) PC4 improves transcription activation by stimulating promoter escape [131]; (iii) Sub1, the yeast PC4 homolog, facilitates transcription elongation and may also prevent premature transcription termination [132]. Thus, being associated to a PC4-containing complex, itself associated with the transcription machinery, it is reasonable to hypothesize that LEDGF/p75 might be associated with the RNA polymerase II complex during elongation.
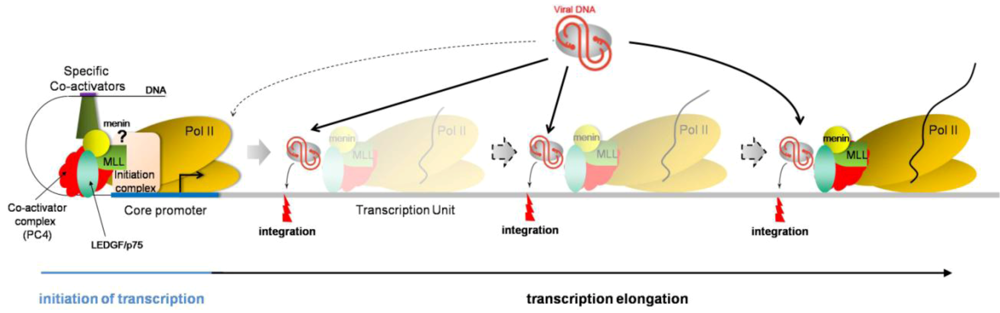
Therefore, based on the current knowledge about HIV integration site selection, i.e., preferentially integrating into active transcription units, it is tempting to speculate a dynamic, more than a static, tethering model, in which LEDGF/p75 would be associated with PC4 and the RNA polymerase II elongation complex (Figure 2). In this model, LEDGF/p75 would recruit HIV preintegration complex while transcribing genes or at pausing sites, thereby explaining integration sites all along the transcription units.
Figure 2.
Dynamic model depicting the mechanism of LEDGF/p75-mediated HIV integration. LEDGF/p75 (green oval) associates with PC4 (red protein) and the RNA polymerase II machinery (yellow ovals) at promoter regions, but steric hindrance may prevent successful recruitment of preintegration complexes (gray oval with viral DNA in red). In this proposed model, LEDGF/p75 remains associated with the RNA pol II transcription elongation complex, potentially interacting with PC4 and menin/MLL complex. While this complex displaces nucleosomes (not depicted) and unwinds DNA to allow RNA polymerization, LEDGF/p75 may recruit HIV PIC and promote integration. This model is consistent with LEDGF/p75-captured DNA sequences and HIV integration sites being present throughout the transcription unit, without specific DNA binding consensus motif.
Figure 2.
Dynamic model depicting the mechanism of LEDGF/p75-mediated HIV integration. LEDGF/p75 (green oval) associates with PC4 (red protein) and the RNA polymerase II machinery (yellow ovals) at promoter regions, but steric hindrance may prevent successful recruitment of preintegration complexes (gray oval with viral DNA in red). In this proposed model, LEDGF/p75 remains associated with the RNA pol II transcription elongation complex, potentially interacting with PC4 and menin/MLL complex. While this complex displaces nucleosomes (not depicted) and unwinds DNA to allow RNA polymerization, LEDGF/p75 may recruit HIV PIC and promote integration. This model is consistent with LEDGF/p75-captured DNA sequences and HIV integration sites being present throughout the transcription unit, without specific DNA binding consensus motif.

Consistent with these results are (i) integration in active transcription units, with no preference along the transcription unit, neither for exons, nor for introns, (ii) LEDGF/p75 initially characterized as a transcriptional coactivator, associated with the transcription machinery (via PC4-containing complex), (iii) LEDGF/p75 interaction with the menin/MLL histone methyltransferase, involved in H3K4me3 histone modification, a mark associated with active transcription, and (iv) no sequence consensus for LEDGF/p75-binding DNA sites, suggesting that the N-terminal domain of LEDGF/p75 may serve as a hook to anchor the protein to the chromatin but that the location specificity is given by an additional chromosome-bound protein, yet to be identified.
Further studies on LEDGF/p75 should help refining the detailed mechanism of LEDGF/p75-mediated HIV integration.
Acknowledgments
This work was supported by the Swiss National Science Foundation (3100A0-120553) and the Novartis Foundation.
Many thanks to Margalida Rotger for critical reading of this manuscript, to Amalio Telenti and Frederic Bushman for mentorship and continuous support, and to colleagues, in particular Raquel Martinez, Julia di Iulio and Miguel Munoz, for helpful discussions.
The authors declare having no potential competing financial interests.
References
- Peterlin, B.M.; Trono, D. Hide, shield and strike back: how HIV-infected cells avoid immune eradication. Nat. Rev. Immunol. 2003, 3, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.P. Host factors exploited by retroviruses. Nat. Rev. Microbiol. 2007, 5, 253–263. [Google Scholar] [CrossRef]
- Suzuki, Y.; Craigie, R. The road to chromatin - nuclear entry of retroviruses. Nat. Rev. Microbiol. 2007, 5, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Flint, S.J.; Lynn W. Enquist, L.W.; Racaniello, V.R.; Skalka, A.M. Principles of Virology, 3rd edAmerican Society for Microbiology Press: Washington, DC, USA, 2009. [Google Scholar]
- Smith, J.A.; Daniel, R. Following the path of the virus: the exploitation of host DNA repair mechanisms by retroviruses. ACS Chem. Biol. 2006, 1, 217–226. [Google Scholar] [CrossRef]
- Vandegraaff, N.; Engelman, A. Molecular mechanisms of HIV integration and therapeutic intervention. Expert Rev. Mol. Med. 2007, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Delelis, O.; Carayon, K.; Saib, A.; Deprez, E.; Mouscadet, J.F. Integrase and integration: biochemical activities of HIV-1 integrase. Retrovirology 2008, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Jaskolski, M.; Alexandratos, J.N.; Bujacz, G.; Wlodawer, A. Piecing together the structure of retroviral integrase, an important target in AIDS therapy. FEBS J. 2009, 276, 2926–2946. [Google Scholar] [CrossRef] [PubMed]
- Ceccherini-Silberstein, F.; Malet, I.; D'Arrigo, R.; Antinori, A.; Marcelin, A.G.; Perno, C.F. Characterization and structural analysis of HIV-1 integrase conservation. AIDS Rev. 2009, 11, 17–29. [Google Scholar] [PubMed]
- McKee, C.J.; Kessl, J.J.; Shkriabai, N.; Dar, M.J.; Engelman, A.; Kvaratskhelia, M. Dynamic modulation of HIV-1 integrase structure and function by cellular lens epithelium-derived growth factor (LEDGF) protein. J. Biol. Chem. 2008, 283, 31802–31812. [Google Scholar] [CrossRef] [PubMed]
- Lesbats, P.; Metifiot, M.; Calmels, C.; Baranova, S.; Nevinsky, G.; Andreola, M.L.; Parissi, V. In vitro initial attachment of HIV-1 integrase to viral ends: control of the DNA specific interaction by the oligomerization state. Nucleic Acids Res. 2008, 36, 7043–7058. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Di Nunzio, F.; Labeja, A.; Wang, J.; Engelman, A.; Cherepanov, P. Structural basis for functional tetramerization of lentiviral integrase. PLoS Pathog. 2009, 5, e1000515:1–e1000515:15. [Google Scholar]
- Michel, F.; Crucifix, C.; Granger, F.; Eiler, S.; Mouscadet, J.F.; Korolev, S.; Agapkina, J.; Ziganshin, R.; Gottikh, M.; Nazabal, A.; Emiliani, S.; Benarous, R.; Moras, D.; Schultz, P.; Ruff, M. Structural basis for HIV-1 DNA integration in the human genome, role of the LEDGF/P75 cofactor. Embo J. 2009, 28, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Cherepanov, P.; Wang, J. Application of general formulas for the correction of a lattice-translocation defect in crystals of a lentiviral integrase in complex with LEDGF. Acta Crystallogr. D. Biol. Crystallogr. 2009, 65, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y. HIV-1 gene expression: lessons from provirus and non-integrated DNA. Retrovirology 2004, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y. The second chance story of HIV-1 DNA: Unintegrated? Not a problem! Retrovirology 2008, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.R.; Yu, D.; Biancotto, A.; Margolis, L.B.; Wu, Y. Measurement of human immunodeficiency virus type 1 preintegration transcription by using Rev-dependent Rev-CEM cells reveals a sizable transcribing DNA population comparable to that from proviral templates. J. Virol. 2009, 83, 8662–8673. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Ott, D.E.; Gorelick, R.J. Efficiency of human immunodeficiency virus type 1 postentry infection processes: evidence against disproportionate numbers of defective virions. J. Virol. 2007, 81, 4367–4370. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Arosio, D.; Terreni, M.; Cereseto, A. HIV-1 pre-integration complexes selectively target decondensed chromatin in the nuclear periphery. PLoS ONE 2008, 3, e2413:1–e2413:9. [Google Scholar] [CrossRef]
- Christ, F.; Thys, W.; De Rijck, J.; Gijsbers, R.; Albanese, A.; Arosio, D.; Emiliani, S.; Rain, J.C.; Benarous, R.; Cereseto, A.; Debyser, Z. Transportin-SR2 imports HIV into the nucleus. Curr. Biol. 2008, 18, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.; Defechereux, P.; Verdin, E. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. Embo J. 2001, 20, 1726–1738. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. Embo J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, M.K.; Bisgrove, D.; Shinn, P.; Chen, H.; Hoffmann, C.; Hannenhalli, S.; Verdin, E.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J. Virol. 2005, 79, 6610–6619. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Lin, Y.B.; An, W.; Xu, J.; Yang, H.C.; O'Connell, K.; Dordai, D.; Boeke, J.D.; Siliciano, J.D.; Siliciano, R.F. Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe 2008, 4, 134–146. [Google Scholar] [CrossRef]
- Bushman, F.D. Lateral DNA Transfer: Mechanisms and Consequences. 2001; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA. [Google Scholar]
- Voigt, K.; Izsvak, Z.; Ivics, Z. Targeted gene insertion for molecular medicine. J. Mol. Med. 2008, 86, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Nienhuis, A.W.; Dunbar, C.E.; Sorrentino, B.P. Genotoxicity of retroviral integration in hematopoietic cells. Mol. Ther. 2006, 13, 1031–1049. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D. Retroviral integration and human gene therapy. J. Clin. Invest. 2007, 117, 2083–2086. [Google Scholar] [CrossRef] [PubMed]
- Pike-Overzet, K.; van der Burg, M.; Wagemaker, G.; van Dongen, J.J.; Staal, F.J. New insights and unresolved issues regarding insertional mutagenesis in X-linked SCID gene therapy. Mol. Ther. 2007, 15, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Fischer, A. Gene therapy for severe combined immunodeficiency: are we there yet? J. Clin. Invest. 2007, 117, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, J. The ethics of human gene transfer. Nat. Rev. Genet. 2008, 9, 239–244. [Google Scholar] [CrossRef]
- Ciuffi, A. Mechanisms governing lentivirus integration site selection. Curr. Gene Ther. 2008, 8, 419–429. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; Fischer, A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; Asnafi, V.; MacIntyre, E.; Dal Cortivo, L.; Radford, I.; Brousse, N.; Sigaux, F.; Moshous, D.; Hauer, J.; Borkhardt, A.; Belohradsky, B.H.; Wintergerst, U.; Velez, M.C.; Leiva, L.; Sorensen, R.; Wulffraat, N.; Blanche, S.; Bushman, F.D.; Fischer, A.; Cavazzana-Calvo, M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.P.; Garrigue, A.; Ciuffi, A.; Ronen, K.; Leipzig, J.; Berry, C.; Lagresle-Peyrou, C.; Benjelloun, F.; Hacein-Bey-Abina, S.; Fischer, A.; Cavazzana-Calvo, M.; Bushman, F.D. DNA bar coding and pyrosequencing to analyze adverse events in therapeutic gene transfer. Nucleic Acids Res. 2008, 36, e49:1–e49:12. [Google Scholar]
- Wang, G.P.; Levine, B.L.; Binder, G.K.; Berry, C.C.; Malani, N.; McGarrity, G.; Tebas, P.; June, C.H.; Bushman, F.D. Analysis of Lentiviral Vector Integration in HIV+ Study Subjects Receiving Autologous Infusions of Gene Modified CD4+ T Cells. Mol. Ther. 2009, 17, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; Doglioni, C.; Di Serio, C.; von Kalle, C.; Naldini, L. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Invest. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D.; Fujiwara, T.; Craigie, R. Retroviral DNA integration directed by HIV integration protein in vitro. Science 1990, 249, 1555–1558. [Google Scholar] [PubMed]
- Chow, S.A. In vitro assays for activities of retroviral integrase. Methods 1997, 12, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Pursley, M.H.; Grandgenett, D.P. Efficient concerted integration by recombinant human immunodeficiency virus type 1 integrase without cellular or viral cofactors. J. Virol. 2002, 76, 3105–3113. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Grandgenett, D.P. Recombinant human immunodeficiency virus type 1 integrase exhibits a capacity for full-site integration in vitro that is comparable to that of purified preintegration complexes from virus-infected cells. J. Virol. 2005, 79, 8208–8216. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D.; Craigie, R. Sequence requirements for integration of Moloney murine leukemia virus DNA in vitro. J. Virol. 1990, 64, 5645–5648. [Google Scholar] [PubMed]
- Leavitt, A.D.; Rose, R.B.; Varmus, H.E. Both substrate and target oligonucleotide sequences affect in vitro integration mediated by human immunodeficiency virus type 1 integrase protein produced in Saccharomyces cerevisiae. J. Virol. 1992, 66, 2359–2368. [Google Scholar] [PubMed]
- Pryciak, P.M.; Muller, H.P.; Varmus, H.E. Simian virus 40 minichromosomes as targets for retroviral integration in vivo. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 9237–9241. [Google Scholar] [CrossRef] [PubMed]
- Pryciak, P.M.; Sil, A.; Varmus, H.E. Retroviral integration into minichromosomes in vitro. Embo J. 1992, 11, 291–303. [Google Scholar] [PubMed]
- Pryciak, P.M.; Varmus, H.E. Nucleosomes, DNA-binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell 1992, 69, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Bor, Y.C.; Bushman, F.D.; Orgel, L.E. In vitro integration of human immunodeficiency virus type 1 cDNA into targets containing protein-induced bends. Proc. Natl. Acad. Sci. USA 1995, 92, 10334–10338. [Google Scholar] [CrossRef]
- Stevens, S.W.; Griffith, J.D. Sequence analysis of the human DNA flanking sites of human immunodeficiency virus type 1 integration. J. Virol. 1996, 70, 6459–6462. [Google Scholar] [PubMed]
- Bor, Y.C.; Miller, M.D.; Bushman, F.D.; Orgel, L.E. Target-sequence preferences of HIV-1 integration complexes in vitro. Virology 1996, 222, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.P.; Varmus, H.E. DNA bending creates favored sites for retroviral integration: an explanation for preferred insertion sites in nucleosomes. Embo J. 1994, 13, 4704–4714. [Google Scholar] [PubMed]
- Pruss, D.; Bushman, F.D.; Wolffe, A.P. Human immunodeficiency virus integrase directs integration to sites of severe DNA distortion within the nucleosome core. Proc. Natl. Acad. Sci. USA 1994, 91, 5913–5917. [Google Scholar] [CrossRef]
- Pruss, D.; Reeves, R.; Bushman, F.D.; Wolffe, A.P. The influence of DNA and nucleosome structure on integration events directed by HIV integrase. J. Biol. Chem. 1994, 269, 25031–25041. [Google Scholar] [PubMed]
- Botbol, Y.; Raghavendra, N.K.; Rahman, S.; Engelman, A.; Lavigne, M. Chromatinized templates reveal the requirement for the LEDGF/p75 PWWP domain during HIV-1 integration in vitro. Nucleic Acids Res. 2008, 36, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D. Tethering human immunodeficiency virus 1 integrase to a DNA site directs integration to nearby sequences. Proc. Natl. Acad. Sci. USA 1994, 91, 9233–9237. [Google Scholar] [CrossRef]
- Goulaouic, H.; Chow, S.A. Directed integration of viral DNA mediated by fusion proteins consisting of human immunodeficiency virus type 1 integrase and Escherichia coli LexA protein. J. Virol. 1996, 70, 37–46. [Google Scholar] [PubMed]
- Tan, W.; Zhu, K.; Segal, D.J.; Barbas 3rd, C.F.; Chow, S.A. Fusion proteins consisting of human immunodeficiency virus type 1 integrase and the designed polydactyl zinc finger protein E2C direct integration of viral DNA into specific sites. J. Virol. 2004, 78, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.; Lewinski, M.; Ciuffi, A.; Barr, S.; Leipzig, J.; Hannenhalli, S.; Hoffmann, C. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 2005, 3, 848–858. [Google Scholar] [CrossRef]
- Ciuffi, A.; Ronen, K.; Brady, T.; Malani, N.; Wang, G.; Berry, C.C.; Bushman, F.D. Methods for integration site distribution analyses in animal cell genomes. Methods 2009, 47, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Craigie, R.; Fujiwara, T.; Bushman, F. The IN protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell 1990, 62, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, e234:1127–e234:1137. [Google Scholar] [CrossRef] [Green Version]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A.; Mitchell, R.S.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F.D. Integration site selection by HIV-based vectors in dividing and growth-arrested IMR-90 lung fibroblasts. Mol. Ther. 2006, 13, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar] [CrossRef]
- Lewinski, M.K.; Yamashita, M.; Emerman, M.; Ciuffi, A.; Marshall, H.; Crawford, G.; Collins, F.; Shinn, P.; Leipzig, J.; Hannenhalli, S.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog. 2006, 2, e60:611–e60:622. [Google Scholar] [CrossRef]
- Barr, S.D.; Ciuffi, A.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F.D. HIV integration site selection: targeting in macrophages and the effects of different routes of viral entry. Mol. Ther. 2006, 14, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.L.; Humeau, L.M.; Boyer, J.; MacGregor, R.R.; Rebello, T.; Lu, X.; Binder, G.K.; Slepushkin, V.; Lemiale, F.; Mascola, J.R.; Bushman, F.D.; Dropulic, B.; June, C.H. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc. Natl. Acad. Sci. USA 2006, 103, 17372–17377. [Google Scholar] [CrossRef]
- Hacker, C.V.; Vink, C.A.; Wardell, T.W.; Lee, S.; Treasure, P.; Kingsman, S.M.; Mitrophanous, K.A.; Miskin, J.E. The integration profile of EIAV-based vectors. Mol. Ther. 2006, 14, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Nowrouzi, A.; Dittrich, M.; Klanke, C.; Heinkelein, M.; Rammling, M.; Dandekar, T.; von Kalle, C.; Rethwilm, A. Genome-wide mapping of foamy virus vector integrations into a human cell line. J. Gen. Virol. 2006, 87, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, Y.; Liang, T.; Sinsheimer, J.S.; Chow, S.A. A high-throughput method for cloning and sequencing human immunodeficiency virus type 1 integration sites. J. Virol. 2006, 80, 11313–11321. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.P.; Ciuffi, A.; Leipzig, J.; Berry, C.C.; Bushman, F.D. HIV integration site selection: Analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007, 17, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Felice, B.; Cattoglio, C.; Cittaro, D.; Testa, A.; Miccio, A.; Ferrari, G.; Luzi, L.; Recchia, A.; Mavilio, F. Transcription factor binding sites are genetic determinants of retroviral integration in the human genome. PLoS ONE 2009, 4, e4571:1–e4571:16. [Google Scholar] [CrossRef]
- Wellensiek, B.P.; Ramakrishnan, R.; Sundaravaradan, V.; Mehta, R.; Harris, D.T.; Ahmad, N. Differential HIV-1 integration targets more actively transcribed host genes in neonatal than adult blood mononuclear cells. Virology 2009, 385, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Vatakis, D.N.; Kim, S.; Kim, N.; Chow, S.A.; Zack, J.A. Human immunodeficiency virus integration efficiency and site selection in quiescent CD4+ T cells. J. Virol. 2009, 83, 6222–6233. [Google Scholar] [CrossRef] [PubMed]
- Brady, T.; Agosto, L.M.; Malani, N.; Berry, C.C.; O'Doherty, U.; Bushman, F. HIV integration site distributions in resting and activated CD4+ T cells infected in culture. Aids 2009, 23, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Barr, S.D.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F.D. Integration targeting by avian sarcoma-leukosis virus and human immunodeficiency virus in the chicken genome. J. Virol. 2005, 79, 12035–12044. [Google Scholar] [CrossRef] [PubMed]
- Beard, B.C.; Keyser, K.A.; Trobridge, G.D.; Peterson, L.J.; Miller, D.G.; Jacobs, M.; Kaul, R.; Kiem, H.P. Unique integration profiles in a canine model of long-term repopulating cells transduced with gammaretrovirus, lentivirus, or foamy virus. Hum. Gene Ther. 2007, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Beard, B.C.; Dickerson, D.; Beebe, K.; Gooch, C.; Fletcher, J.; Okbinoglu, T.; Miller, D.G.; Jacobs, M.A.; Kaul, R.; Kiem, H.P.; Trobridge, G.D. Comparison of HIV-derived Lentiviral and MLV-based Gammaretroviral Vector Integration Sites in Primate Repopulating Cells. Mol. Ther. 2007, 15, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Shun, M.C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.M.; Ronen, K.; Berry, C.; Llano, M.; Sutherland, H.; Saenz, D.; Bickmore, W.; Poeschla, E.; Bushman, F.D. Role of PSIP1/LEDGF/p75 in Lentiviral Infectivity and Integration Targeting. PLoS ONE 2007, 2, e1340:1–e1340:13. [Google Scholar]
- MacNeil, A.; Sankale, J.L.; Meloni, S.T.; Sarr, A.D.; Mboup, S.; Kanki, P. Genomic sites of human immunodeficiency virus type 2 (HIV-2) integration: similarities to HIV-1 in vitro and possible differences in vivo. J. Virol. 2006, 80, 7316–7321. [Google Scholar] [CrossRef] [PubMed]
- Crise, B.; Li, Y.; Yuan, C.; Morcock, D.R.; Whitby, D.; Munroe, D.J.; Arthur, L.O.; Wu, X. Simian immunodeficiency virus integration preference is similar to that of human immunodeficiency virus type 1. J. Virol. 2005, 79, 12199–12204. [Google Scholar] [CrossRef] [PubMed]
- Hematti, P.; Hong, B.K.; Ferguson, C.; Adler, R.; Hanawa, H.; Sellers, S.; Holt, I.E.; Eckfeldt, C.E.; Sharma, Y.; Schmidt, M.; von Kalle, C.; Persons, D.A.; Billings, E.M.; Verfaillie, C.M.; Nienhuis, A.W.; Wolfsberg, T.G.; Dunbar, C.E.; Calmels, B. Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol. 2004, 2, e423:1–e423:8. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Moressi, C.J.; Scheetz, T.E.; Xie, L.; Tran, D.T.; Casavant, T.L.; Ak, P.; Benham, C.J.; Davidson, B.L.; McCray Jr. Jr, P.B. Integration site choice of a feline immunodeficiency virus vector. J. Virol. 2006, 80, 8820–8823. [Google Scholar] [CrossRef] [PubMed]
- Narezkina, A.; Taganov, K.D.; Litwin, S.; Stoyanova, R.; Hayashi, J.; Seeger, C.; Skalka, A.M.; Katz, R.A. Genome-wide analyses of avian sarcoma virus integration sites. J. Virol. 2004, 78, 11656–11663. [Google Scholar] [CrossRef] [PubMed]
- Faschinger, A.; Rouault, F.; Sollner, J.; Lukas, A.; Salmons, B.; Gunzburg, W.H.; Indik, S. Mouse Mammary Tumor Virus Integration Site Selection in Human and Mouse Genomes. J. Virol. 2008, 83, 1360–1367. [Google Scholar] [CrossRef]
- Brady, T.; Lee, Y.N.; Ronen, K.; Malani, N.; Berry, C.C.; Bieniasz, P.D.; Bushman, F.D. Integration target site selection by a resurrected human endogenous retrovirus. Genes Dev. 2009, 23, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Aker, M.; Tubb, J.; Miller, D.G.; Stamatoyannopoulos, G.; Emery, D.W. Integration bias of gammaretrovirus vectors following transduction and growth of primary mouse hematopoietic progenitor cells with and without selection. Mol. Ther. 2006, 14, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Moalic, Y.; Blanchard, Y.; Felix, H.; Jestin, A. Porcine endogenous retrovirus integration sites in the human genome: features in common with those of murine leukemia virus. J. Virol. 2006, 80, 10980–10988. [Google Scholar] [CrossRef] [PubMed]
- Moalic, Y.; Felix, H.; Takeuchi, Y.; Jestin, A.; Blanchard, Y. Genome areas with high gene density and CpG island neighborhood strongly attract porcine endogenous retrovirus for integration and favor the formation of hot spots. J. Virol. 2009, 83, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, N.; Dong, B.; Boren, D.; Lee, S.A.; Das Gupta, J.; Gaughan, C.; Klein, E.A.; Lee, C.; Silverman, R.H.; Chow, S.A. Integration site preference of xenotropic murine leukemia virus-related virus, a new human retrovirus associated with prostate cancer. J. Virol. 2008, 82, 9964–9977. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Wu, X.; Taniguchi, Y.; Yasunaga, J.; Satou, Y.; Okayama, A.; Nosaka, K.; Matsuoka, M. Preferential selection of human T-cell leukemia virus type I provirus integration sites in leukemic versus carrier states. Blood 2005, 106, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Derse, D.; Crise, B.; Li, Y.; Princler, G.; Lum, N.; Stewart, C.; McGrath, C.F.; Hughes, S.H.; Munroe, D.J.; Wu, X. Human T-cell leukemia virus type 1 integration target sites in the human genome: comparison with those of other retroviruses. J. Virol. 2007, 81, 6731–6741. [Google Scholar] [CrossRef] [PubMed]
- Meekings, K.N.; Leipzig, J.; Bushman, F.D.; Taylor, G.P.; Bangham, C.R. HTLV-1 Integration into Transcriptionally Active Genomic Regions Is Associated with Proviral Expression and with HAM/TSP. PLoS Pathog. 2008, 4, e1000027:1–e1000027:10. [Google Scholar] [CrossRef]
- Carteau, S.; Hoffmann, C.; Bushman, F. Chromosome structure and human immunodeficiency virus type 1 cDNA integration: centromeric alphoid repeats are a disfavored target. J. Virol. 1998, 72, 4005–4014. [Google Scholar] [PubMed]
- Holman, A.G.; Coffin, J.M. Symmetrical base preferences surrounding HIV-1, avian sarcoma/leukosis virus, and murine leukemia virus integration sites. Proc. Natl. Acad. Sci. USA 2005, 102, 6103–6107. [Google Scholar] [CrossRef]
- Berry, C.; Hannenhalli, S.; Leipzig, J.; Bushman, F.D. Selection of target sites for mobile DNA integration in the human genome. PLoS Comput. Biol. 2006, 2, e157:1450–e157:1462. [Google Scholar] [CrossRef]
- Shun, M.C.; Daigle, J.E.; Vandegraaff, N.; Engelman, A. Wild-type levels of human immunodeficiency virus type 1 infectivity in the absence of cellular emerin protein. J. Virol. 2007, 81, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Segal, E.; Fondufe-Mittendorf, Y.; Chen, L.; Thastrom, A.; Field, Y.; Moore, I.K.; Wang, J.P.; Widom, J. A genomic code for nucleosome positioning. Nature 2006, 442, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Crawford, G.E.; Holt, I.E.; Whittle, J.; Webb, B.D.; Tai, D.; Davis, S.; Margulies, E.H.; Chen, Y.; Bernat, J.A.; Ginsburg, D.; Zhou, D.; Luo, S.; Vasicek, T.J.; Daly, M.J.; Wolfsberg, T.G.; Collins, F.S. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 2006, 16, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Tobaly-Tapiero, J.; Bittoun, P.; Lehmann-Che, J.; Delelis, O.; Giron, M.L.; de The, H.; Saib, A. Chromatin tethering of incoming foamy virus by the structural Gag protein. Traffic 2008, 9, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D. Integration site selection by lentiviruses: biology and possible control. Curr. Top. Microbiol. Immunol. 2002, 261, 165–177. [Google Scholar] [PubMed]
- Engelman, A. The roles of cellular factors in retroviral integration. Curr. Top. Microbiol. Immunol. 2003, 281, 209–238. [Google Scholar] [PubMed]
- Van Maele, B.; Debyser, Z. HIV-1 integration: an interplay between HIV-1 integrase, cellular and viral proteins. AIDS Rev. 2005, 7, 26–43. [Google Scholar] [PubMed]
- Van Maele, B.; Busschots, K.; Vandekerckhove, L.; Christ, F.; Debyser, Z. Cellular co-factors of HIV-1 integration. Trends Biochem. Sci. 2006, 31, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawsawi, L.Q.; Neamati, N. Blocking interactions between HIV-1 integrase and cellular cofactors: an emerging anti-retroviral strategy. Trends Pharmacol. Sci. 2007, 28, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D.; Malani, N.; Fernandes, J.; D'Orso, I.; Cagney, G.; Diamond, T.L.; Zhou, H.; Hazuda, D.J.; Espeseth, A.S.; Konig, R.; Bandyopadhyay, S.; Ideker, T.; Goff, S.P.; Krogan, N.J.; Frankel, A.D.; Young, J.A.; Chanda, S.K. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 2009, 5, e1000437:1–e1000437:12. [Google Scholar] [CrossRef]
- Ciuffi, A.; Bushman, F.D. Retroviral DNA integration: HIV and the role of LEDGF/p75. Trends Genet. 2006, 22, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The Lentiviral Integrase Binding Protein LEDGF/p75 and HIV-1 Replication. PLoS Pathog. 2008, 4, e1000046:1–e1000046:9. [Google Scholar] [CrossRef]
- Poeschla, E.M. Integrase, LEDGF/p75 and HIV replication. Cell. Mol. Life Sci. 2008, 65, 1403–1424. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Ambrosio, A.L.; Rahman, S.; Ellenberger, T.; Engelman, A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. USA 2005, 102, 17308–17313. [Google Scholar] [CrossRef]
- Ge, H.; Si, Y.; Roeder, R.G. Isolation of cDNAs encoding novel transcription coactivators p52 and p75 reveals an alternate regulatory mechanism of transcriptional activation. Embo J. 1998, 17, 6723–6729. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Daniels, T.; Casiano, C.A. LEDGF/p75: a novel nuclear autoantigen at the crossroads of cell survival and apoptosis. Autoimmun. Rev. 2003, 2, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, T.; Singh, D.P.; Fatma, N. LEDGF, a survival factor, activates stress-related genes. Prog. Retin. Eye Res. 2002, 21, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.G.; Newton, K.; Brownstein, D.G.; Holmes, M.C.; Kress, C.; Semple, C.A.; Bickmore, W.A. Disruption of Ledgf/Psip1 results in perinatal mortality and homeotic skeletal transformations. Mol. Cell. Biol. 2006, 26, 7201–7210. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Saenz, D.T.; Meehan, A.; Wongthida, P.; Peretz, M.; Walker, W.H.; Teo, W.; Poeschla, E.M. An essential role for LEDGF/p75 in HIV integration. Science 2006, 314, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, L.; Christ, F.; Van Maele, B.; De Rijck, J.; Gijsbers, R.; Van den Haute, C.; Witvrouw, M.; Debyser, Z. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 2006, 80, 1886–1896. [Google Scholar] [CrossRef] [PubMed]
- De Rijck, J. and Debyser. KU Leuven, Leuven, Belgium.Personal communication. 2009. [Google Scholar]
- Desfarges, S.; Munoz, M.; Leleu, M.; Lefebvre, G.; Rougemont, J.; Xenarios, I.; Ciuffi, A. Analysis of LEDGF/p75-bound sequences by ChIP-Seq (to be submitted for publication). Genome Res.
- Ciuffi, A.; Diamond, T.L.; Hwang, Y.; Marshall, H.M.; Bushman, F.D. Modulating target site selection during human immunodeficiency virus DNA integration in vitro with an engineered tethering factor. Hum. Gene Ther. 2006, 17, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Meehan, A.M.; Saenz, D.T.; Morrison, J.H.; Garcia-Rivera, J.A.; Peretz, M.; Llano, M.; Poeschla, E.M. LEDGF/p75 proteins with alternative chromatin tethers are functional HIV-1 cofactors. PLoS Pathog. 2009, 5, e1000522:1–e1000522:18. [Google Scholar] [CrossRef]
- Silvers, R.; Smith, J.A.; Schowalter, M.; Litwin, S.; Liang, Z.; Geary, K.; Daniel, R. Modification of Integration Site Preferences of an HIV-1-based Vector by expression of a novel synthetic protein. Hum. Gene Ther. 2009. [Google Scholar]
- Gijsbers, R.; Ronen, K.; Vets, S.; Malani, N.; De Rijck, J.; McNeely, M.; Bushman, F.D.; Debyser, Z. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol. Ther. 2010, In Press. [Google Scholar]
- Studamire, B.; Goff, S.P. Host proteins interacting with the Moloney murine leukemia virus integrase: multiple transcriptional regulators and chromatin binding factors. Retrovirology 2008, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.E.P. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.E.P. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Roeder, R.G. Purification, cloning, and characterization of a human coactivator, PC4, that mediates transcriptional activation of class II genes. Cell 1994, 78, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Roeder, R.G. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem. Sci. 1996, 21, 327–335. [Google Scholar] [PubMed]
- Fukuda, A.; Nakadai, T.; Shimada, M.; Tsukui, T.; Matsumoto, M.; Nogi, Y.; Meisterernst, M.; Hisatake, K. Transcriptional coactivator PC4 stimulates promoter escape and facilitates transcriptional synergy by GAL4-VP16. Mol. Cell. Biol. 2004, 24, 6525–6535. [Google Scholar] [CrossRef] [PubMed]
- Calvo, O.; Manley, J.L. The transcriptional coactivator PC4/Sub1 has multiple functions in RNA polymerase II transcription. Embo J. 2005, 24, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
MDPI and ACS Style
Desfarges, S.; Ciuffi, A. Retroviral Integration Site Selection. Viruses 2010, 2, 111-130. https://doi.org/10.3390/v2010111
AMA Style
Desfarges S, Ciuffi A. Retroviral Integration Site Selection. Viruses. 2010; 2(1):111-130. https://doi.org/10.3390/v2010111
Chicago/Turabian StyleDesfarges, Sébastien, and Angela Ciuffi. 2010. "Retroviral Integration Site Selection" Viruses 2, no. 1: 111-130. https://doi.org/10.3390/v2010111