RNA Replicons - A New Approach for Influenza Virus Immunoprophylaxis


Abstract
:1. Introduction
Potential obstacles with current influenza vaccines
2. RNA Virus Vectors
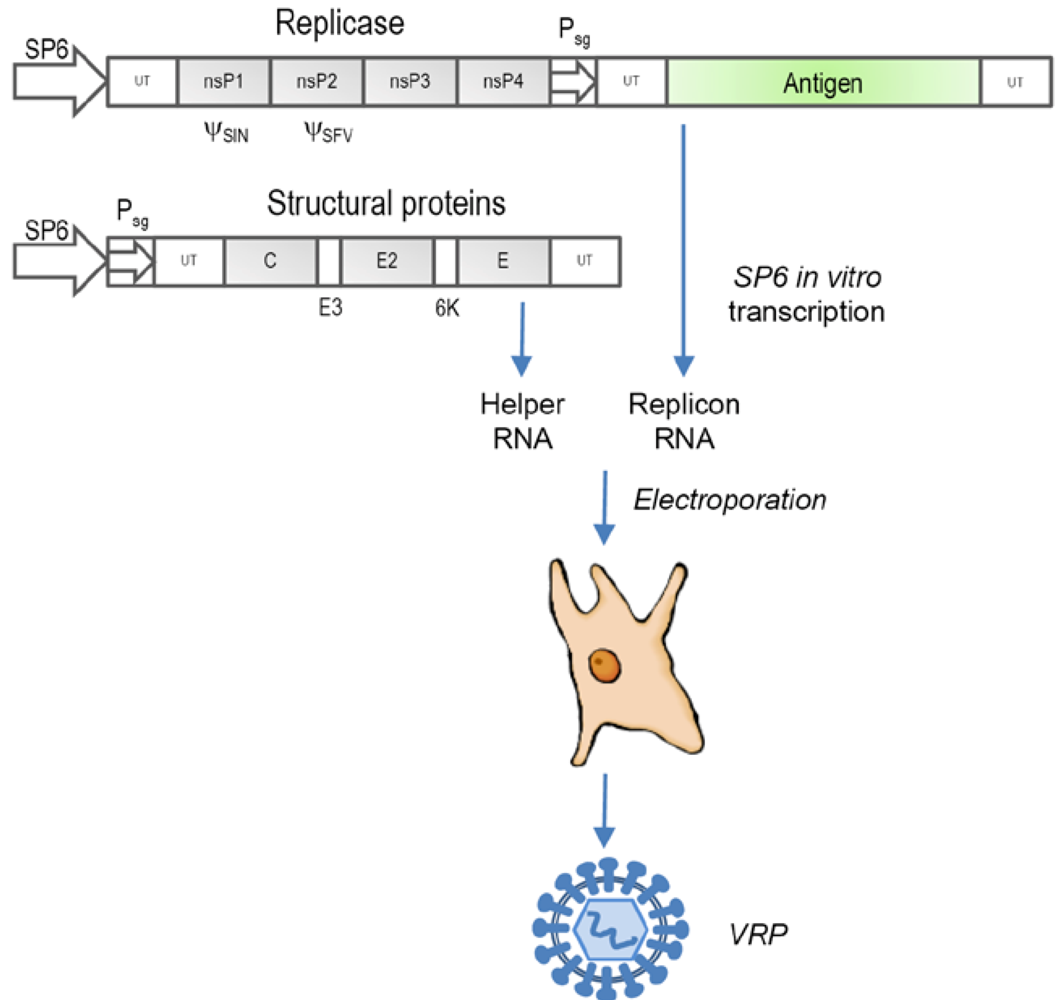
2.1. RNA replicons
2.2. RNA replicons and initiation of immune response development
2.3. RNA replicons in current use for vaccine studies
2.3.1. Positive-strand RNA replicons for vaccine studies

2.3.2. Negative-strand RNA replicons for vaccine studies

2.4. RNA replicon vaccine studies
2.4.1. Alphavirus replicons
2.4.2. VSV replicons
2.5 RNA replicon vaccines for protection of poultry
2.6. Immunological correlates of protection
3. Resume and future perspectives

{kind=link}
{kind=link}
| Parameter | Inactivated subunit vaccine | Live attenuated vaccine | RNA replicon vaccine |
|---|---|---|---|
| Safety | High | Risk of revertants, reassortants | High |
| Induction of humoral immunity | Yes | Yes | Yes |
| Induction of cellular immunity | Low | Yes | Yes |
| Mucosal Immunity | Low | Yes | Not yet tested |
| Booster Immunisation | May be required (dependent on the vaccine) | No | Likely to be required (further work needed) |
| Immune memory | Short-term | Long-term | Long-term |
| Protection against drift virus | Low | Yes | Yes |
| Heterosubtypic protection | No | Yes | Not known |
| Shedding of vaccine virus | No | Yes | No |
| Application route | Parenteral (Intranasal possible, but there are safety queries*) | Parenteral or Intranasal* | Parenteral (Intranasal under investigation*) |
| Adjuvants | Not used** | Not usually required | Not usually required |
| Antigens | HA, NA | Multiple | Maximum number yet to be determined |
| Availability | Low | High | High |
3.1. RNA replicon vaccines for multiple antigen expression
3.2. RNA replicon vaccines for mucosal immunizations
3.3. RNA replicon vaccines for the future
Acknowledgments
References
- Rothberg, M.B.; Haessler, S.D.; Brown, R.B. Complications of viral influenza. Am. J. Med. 2008, 121, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Carrat, F.; Flahault, A. Influenza vaccine: the challenge of antigenic drift. Vaccine 2007, 25, 6852–6862. [Google Scholar] [CrossRef] [PubMed]
- Palache, B. New vaccine approaches for seasonal and pandemic influenza. Vaccine 2008, 26, 6232–6236. [Google Scholar] [CrossRef] [PubMed]
- Gambotto, A.; Barratt-Boyes, S.M.; de Jong, M.D.; Neumann, G.; Kawaoka, Y. Human infection with highly pathogenic H5N1 influenza virus. Lancet 2008, 371, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Webby, R.J.; Webster, R.G.; Richt, J.A. Influenza viruses in animal wildlife populations. Curr. Top. Microbiol. Immunol. 2007, 315, 67–83. [Google Scholar] [PubMed]
- Weingartl, H.M.; Albrecht, R.A.; Lager, K.M.; Babiuk, S.; Marszal, P.; Neufeld, J.; Embury-Hyatt, C.; Lekcharoensuk, P.; Tumpey, T.M.; Garcia-Sastre, A.; Richt, J.A. Experimental infection of pigs with the human 1918 pandemic influenza virus. J. Virol. 2009, 83, 4287–4296. [Google Scholar] [CrossRef] [PubMed]
- Gerdil, C. The annual production cycle for influenza vaccine. Vaccine 2003, 21, 1776–1779. [Google Scholar] [CrossRef] [PubMed]
- Rimmelzwaan, G.F.; Fouchier, R.A.; Osterhaus, A.D. Influenza virus-specific cytotoxic T lymphocytes: a correlate of protection and a basis for vaccine development. Curr. Opin. Biotechnol. 2007, 18, 529–536. [Google Scholar] [CrossRef] [PubMed]
- McMurry, J.A.; Johansson, B.E.; De Groot, A.S. A call to cellular & humoral arms: enlisting cognate T cell help to develop broad-spectrum vaccines against influenza A. Hu. Hum. Vaccin. 2008, 4, 148–157. [Google Scholar] [CrossRef] [PubMed]
- de Jong, J.C.; Beyer, W.E.; Palache, A.M.; Rimmelzwaan, G.F.; Osterhaus, A.D. Mismatch between the 1997/1998 influenza vaccine and the major epidemic A(H3N2) virus strain as the cause of an inadequate vaccine-induced antibody response to this strain in the elderly. J. Med. Virol. 2000, 61, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Kreijtz, J.H.; Osterhaus, A.D.; Rimmelzwaan, G.F. Vaccination strategies and vaccine formulations for epidemic and pandemic influenza control. Hum. Vaccin. 2009, 5, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Wareing, M.D.; Tannock, G.A. Live attenuated vaccines against influenza; an historical review. Vaccine 2001, 19, 3320–3330. [Google Scholar] [CrossRef] [PubMed]
- Beyer, W.E.; Palache, A.M.; de Jong, J.C.; Osterhaus, A.D. Cold-adapted live influenza vaccine versus inactivated vaccine: systemic vaccine reactions, local and systemic antibody response, and vaccine efficacy. A meta-analysis. Vaccine 2002, 20, 1340–1353. [Google Scholar] [CrossRef] [PubMed]
- Powell, T.J.; Strutt, T.; Reome, J.; Hollenbaugh, J.A.; Roberts, A.D.; Woodland, D.L.; Swain, S.L.; Dutton, R.W. Priming with cold-adapted influenza A does not prevent infection but elicits long-lived protection against supralethal challenge with heterosubtypic virus. J. Immunol. 2007, 178, 1030–1038. [Google Scholar] [PubMed]
- Karron, R.A.; Talaat, K.; Luke, C.; Callahan, K.; Thumar, B.; Dilorenzo, S.; McAuliffe, J.; Schappell, E.; Suguitan, A.; Mills, K.; Chen, G.; Lamirande, E.; Coelingh, K.; Jin, H.; Murphy, B.R.; Kemble, G.; Subbarao, K. Evaluation of two live attenuated cold-adapted H5N1 influenza virus vaccines in healthy adults. Vaccine 2009, 27, 4953–4960. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, C.S.; Luke, C.; Coelingh, K. Current status of live attenuated influenza vaccine in the United States for seasonal and pandemic influenza. Influenza Other Respi. Viruses 2008, 2, 193–202. [Google Scholar] [CrossRef]
- Liniger, M.; Zuniga, A.; Naim, H.Y. Use of viral vectors for the development of vaccines. Expert Rev. Vaccines 2007, 6, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Minor, P. Vaccine-derived poliovirus (VDPV): Impact on poliomyelitis eradication. Vaccine 2009, 27, 2649–2652. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, E.; Modlin, J.; Chumakov, K. Future of polio vaccines. Expert Rev. Vaccines 2009, 8, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Dory, D.; Gravier, R.; Jestin, A. Risk assessment in new and conventional vaccines. Dev. Biol. (Basel) 2006, 126 discussion 327, 253–259. [Google Scholar] [PubMed]
- Vyas, J.M.; Van der Veen, A.G.; Ploegh, H.L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 2008, 8, 607–618. [Google Scholar] [CrossRef]
- Nakhaei, P.; Genin, P.; Civas, A.; Hiscott, J. RIG-I-like receptors: Sensing and responding to RNA virus infection. Semin. Immunol. 2009. [Google Scholar]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Powers, C.; DeFilippis, V.; Malouli, D.; Fruh, K. Cytomegalovirus immune evasion. Curr. Top. Microbiol. Immunol. 2008, 325, 333–359. [Google Scholar] [PubMed]
- Seet, B.T.; Johnston, J.B.; Brunetti, C.R.; Barrett, J.W.; Everett, H.; Cameron, C.; Sypula, J.; Nazarian, S.H.; Lucas, A.; McFadden, G. Poxviruses and immune evasion. Annu. Rev. Immunol. 2003, 21, 377–423. [Google Scholar] [CrossRef] [PubMed]
- Rayner, J.O.; Dryga, S.A.; Kamrud, K.I. Alphavirus vectors and vaccination. Rev. Med. Virol. 2002, 12, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Atkins, G.J.; Fleeton, M.N.; Sheahan, B.J. Therapeutic and prophylactic applications of alphavirus vectors. Expert Rev Mol Med 2008, 10, e33. [Google Scholar] [CrossRef] [PubMed]
- Pijlman, G.P.; Suhrbier, A.; Khromykh, A.A. Kunjin virus replicons: an RNA-based, non-cytopathic viral vector system for protein production, vaccine and gene therapy applications. Expert Opin. Biol. Ther. 2006, 6, 135–145. [Google Scholar] [CrossRef]
- Widman, D.G.; Ishikawa, T.; Fayzulin, R.; Bourne, N.; Mason, P.W. Construction and characterization of a second-generation pseudoinfectious West Nile virus vaccine propagated using a new cultivation system. Vaccine 2008, 26, 2762–2771. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Winkelmann, E.R.; Mason, P.W. Construction and characterization of a single-cycle chimeric flavivirus vaccine candidate that protects mice against lethal challenge with dengue virus type 2. J. Virol. 2009, 83, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, R.; Heinz, F.X.; Davis, N.L.; Mandl, C.W. Heterologous gene expression by infectious and replicon vectors derived from tick-borne encephalitis virus and direct comparison of this flavivirus system with an alphavirus replicon. J. Gen. Virol. 2005, 86, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Gerbaud, S.; van der Werf, S.; Escriou, N. Naked RNA immunization with replicons derived from poliovirus and Semliki Forest virus genomes for the generation of a cytotoxic T cell response against the influenza A virus nucleoprotein. J. Gen. Virol. 2001, 82, 1737–1747. [Google Scholar] [PubMed]
- Vignuzzi, M.; Gerbaud, S.; van der Werf, S.; Escriou, N. Expression of a membrane-anchored glycoprotein, the influenza virus hemagglutinin, by dicistronic replicons derived from the poliovirus genome. J. Virol. 2002, 76, 5285–5290. [Google Scholar] [CrossRef] [PubMed]
- Fleeton, M.N.; Chen, M.; Berglund, P.; Rhodes, G.; Parker, S.E.; Murphy, M.; Atkins, G.J.; Liljestrom, P. Self-replicative RNA vaccines elicit protection against influenza A virus, respiratory syncytial virus, and a tickborne encephalitis virus. J. Infect. Dis. 2001, 183, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Driver, D.A.; Latham, E.M.; Polo, J.M.; Belli, B.A.; Banks, T. A.; Chada, S.; Brumm, D.; Chang, S.M.; Mento, S.J.; Jolly, D.J.;et al. Layered amplification of gene expression with a DNA gene delivery system. Ann. NY Acad. Sci. 1995, 772, 261–264. [Google Scholar] [CrossRef]
- Berglund, P.; Smerdou, C.; Fleeton, M.N.; Tubulekas, I.; Liljestrom, P. Enhancing immune responses using suicidal DNA vaccines. Nat. Biotechnol. 1998, 16, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Liljestrom, P.; Garoff, H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Biotechnology (NY) 1991, 9, 1356–1361. [Google Scholar] [CrossRef]
- Bredenbeek, P.J.; Frolov, I.; Rice, C.M.; Schlesinger, S. Sindbis virus expression vectors: packaging of RNA replicons by using defective helper RNAs. J. Virol. 1993, 67, 6439–6446. [Google Scholar] [PubMed]
- Pushko, P.; Parker, M.; Ludwig, G.V.; Davis, N.L.; Johnston, R.E.; Smith, J.F. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology 1997, 239, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Schnell, M.J.; Mebatsion, T.; Conzelmann, K.K. Infectious rabies viruses from cloned cDNA. EMBO J. 1994, 13, 4195–4203. [Google Scholar] [PubMed]
- Li, H.O.; Zhu, Y.F.; Asakawa, M.; Kuma, H.; Hirata, T.; Ueda, Y.; Lee, Y.S.; Fukumura, M.; Iida, A.; Kato, A.; Nagai, Y.; Hasegawa, M. A cytoplasmic RNA vector derived from nontransmissible Sendai virus with efficient gene transfer and expression. J. Virol. 2000, 74, 6564–6569. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Tokusumi, Y.; Ban, H.; Kanaya, T.; Shirakura, M.; Tokusumi, T.; Hirata, T.; Nagai, Y.; Iida, A.; Hasegawa, M. A new Sendai virus vector deficient in the matrix gene does not form virus particles and shows extensive cell-to-cell spreading. J. Virol. 2003, 77, 6419–6429. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Tokusumi, Y.; Ban, H.; Kanaya, T.; Tokusumi, T.; Nagai, Y.; Iida, A.; Hasegawa, M. Nontransmissible virus-like particle formation by F-deficient sendai virus is temperature sensitive and reduced by mutations in M and HN proteins. J. Virol. 2003, 77, 3238–3246. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Tokusumi, Y.; Ban, H.; Shirakura, M.; Kanaya, T.; Yoshizaki, M.; Hironaka, T.; Nagai, Y.; Iida, A.; Hasegawa, M. Recombinant Sendai virus vectors deleted in both the matrix and the fusion genes: efficient gene transfer with preferable properties. J. Gene Med. 2004, 6, 1069–1081. [Google Scholar] [CrossRef]
- Yoshizaki, M.; Hironaka, T.; Iwasaki, H.; Ban, H.; Tokusumi, Y.; Iida, A.; Nagai, Y.; Hasegawa, M.; Inoue, M. Naked Sendai virus vector lacking all of the envelope-related genes: reduced cytopathogenicity and immunogenicity. J. Gene Med. 2006, 8, 1151–1159. [Google Scholar] [CrossRef]
- Bernloehr, C.; Bossow, S.; Ungerechts, G.; Armeanu, S.; Neubert, W.J.; Lauer, U.M.; Bitzer, M. Efficient propagation of single gene deleted recombinant Sendai virus vectors. Virus Res. 2004, 99, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Takada, A.; Robison, C.; Goto, H.; Sanchez, A.; Murti, K. G.; Whitt, M.A.; Kawaoka, Y. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. USA 1997, 94, 14764–14769. [Google Scholar] [CrossRef]
- Roberts, A.; Buonocore, L.; Price, R.; Forman, J.; Rose, J.K. Attenuated vesicular stomatitis viruses as vaccine vectors. J. Virol. 1999, 73, 3723–3732. [Google Scholar] [PubMed]
- Hanika, A.; Larisch, B.; Steinmann, E.; Schwegmann-Wessels, C.; Herrler, G.; Zimmer, G. Use of influenza C virus glycoprotein HEF for generation of vesicular stomatitis virus pseudotypes. J. Gen. Virol. 2005, 86, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Kalhoro, N.H.; Veits, J.; Rautenschlein, S.; Zimmer, G. A recombinant vesicular stomatitis virus replicon vaccine protects chickens from highly pathogenic avian influenza virus (H7N1). Vaccine 2009, 27, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Spann, K.M.; Collins, P.L.; Teng, M.N. Genetic recombination during coinfection of two mutants of human respiratory syncytial virus. J. Virol. 2003, 77, 11201–11211. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Bukreyev, A.; Murphy, B.R. What are the risks--hypothetical and observed--of recombination involving live vaccines and vaccine vectors based on nonsegmented negative-strain RNA viruses? J. Virol. 2008, 82, 9805–9806. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.; Subramaniam, S.V.; Hajjou, M. Genesis of Sindbis virus by in vivo recombination of nonreplicative RNA precursors. J. Virol. 1995, 69, 7391–7401. [Google Scholar] [PubMed]
- Gallei, A.; Pankraz, A.; Thiel, H.J.; Becher, P. RNA recombination in vivo in the absence of viral replication. J. Virol. 2004, 78, 6271–6281. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Berglund, P.; Rhodes, G.; Parker, S.E.; Jondal, M.; Liljestrom, P. Self-replicating Semliki Forest virus RNA as recombinant vaccine. Vaccine 1994, 12, 1510–1514. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Berglund, P.; Zhao, H.; Liljestrom, P.; Jondal, M. Generation of cytotoxic and humoral immune responses by nonreplicative recombinant Semliki Forest virus. Proc. Natl. Acad. Sci. USA 1995, 92, 3009–3013. [Google Scholar] [CrossRef]
- Berglund, P.; Fleeton, M.N.; Smerdou, C.; Liljestrom, P. Immunization with recombinant Semliki Forest virus induces protection against influenza challenge in mice. Vaccine 1999, 17, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.G.; Wo, J.E.; Li, M.W.; Yu, C.B.; Lv, G.L.; Cao, H.C.; Lu, H.F.; Wang, B.H.; Zhu, H.P.; Li, L.J. Construction and packaging of Semliki forest virus replicon particles efficiently expressing Influenza A virus (H5N1) hemagglutinin. Acta Virol. 2009, 53, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Hubby, B.; Talarico, T.; Maughan, M.; Reap, E.A.; Berglund, P.; Kamrud, K.I.; Copp, L.; Lewis, W.; Cecil, C.; Norberg, P.; Wagner, J.; Watson, A.; Negri, S.; Burnett, B.K.; Graham, A.; Smith, J.F.; Chulay, J.D. Development and preclinical evaluation of an alphavirus replicon vaccine for influenza. Vaccine 2007, 25, 8180–8189. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, S.U.; Simon, I.D.; Rose, J.K. SARS vaccine based on a replication-defective recombinant vesicular stomatitis virus is more potent than one based on a replication-competent vector. Virology 2008, 376, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Publicover, J.; Ramsburg, E.; Rose, J.K. A single-cycle vaccine vector based on vesicular stomatitis virus can induce immune responses comparable to those generated by a replication-competent vector. J. Virol. 2005, 79, 13231–13238. [Google Scholar] [CrossRef] [PubMed]
- Majid, A.M.; Ezelle, H.; Shah, S.; Barber, G.N. Evaluating replication-defective vesicular stomatitis virus as a vaccine vehicle. J. Virol. 2006, 80, 6993–7008. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.S.; Roberts, A.; Weibel, C.; Buonocore, L.; Rose, J.K. Replication-competent or attenuated, nonpropagating vesicular stomatitis viruses expressing respiratory syncytial virus (RSV) antigens protect mice against RSV challenge. J. Virol. 2001, 75, 11079–11087. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.A.; Buonocore, L.; Roberts, A.; Suguitan, A.; Kobasa, D.; Kobinger, G.; Feldmann, H.; Subbarao, K.; Rose, J.K. Vesicular stomatitis virus vectors expressing avian influenza H5 HA induce cross-neutralizing antibodies and long-term protection. Virology 2007, 366, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Senne, D.A.; Suarez, D.L. Effect of vaccine use in the evolution of Mexican lineage H5N2 avian influenza virus. J. Virol. 2004, 78, 8372–8381. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Suarez, D.L. Avian influenza virus: prospects for prevention and control by vaccination. Anim. Health Res. Rev. 2005, 6, 1–15. [Google Scholar] [CrossRef]
- Schultz-Cherry, S.; Dybing, J.K.; Davis, N.L.; Williamson, C.; Suarez, D.L.; Johnston, R.; Perdue, M.L. Influenza virus (A/HK/156/97) hemagglutinin expressed by an alphavirus replicon system protects chickens against lethal infection with Hong Kong-origin H5N1 viruses. Virology 2000, 278, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Sylte, M.J.; Hubby, B.; Suarez, D.L. Influenza neuraminidase antibodies provide partial protection for chickens against high pathogenic avian influenza infection. Vaccine 2007, 25, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Becht, H.; Orlich, M. The significance of influenza virus neuraminidase in immunity. J. Gen. Virol. 1974, 22, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, S.P.; Veits, J.; Keil, G.M.; Mettenleiter, T.C.; Fuchs, W. Protection of chickens against H5N1 highly pathogenic avian influenza virus infection by live vaccination with infectious laryngotracheitis virus recombinants expressing H5 hemagglutinin and N1 neuraminidase. Vaccine 2009, 27, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Salomon, R.; Webster, R.G. The influenza virus enigma. Cell 2009, 136, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.C.; McKeon, R.M.; Brackin, M.N.; Miller, L.A.; Keating, R.; Brown, S.A.; Makarova, N.; Perez, D.R.; Macdonald, G.H.; McCullers, J.A. Distinct contributions of vaccine-induced immunoglobulin G1 (IgG1) and IgG2a antibodies to protective immunity against influenza. Clin. Vaccine Immunol. 2006, 13, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.M.; Whitmore, A.C.; Konopka, J.L.; Collier, M.L.; Richmond, E.M.; Davis, N.L.; Staats, H.F.; Johnston, R.E. Mucosal and systemic adjuvant activity of alphavirus replicon particles. Proc. Natl. Acad. Sci. USA 2006, 103, 3722–3727. [Google Scholar] [CrossRef]
- Thompson, J.M.; Whitmore, A.C.; Staats, H.F.; Johnston, R. The contribution of type I interferon signaling to immunity induced by alphavirus replicon vaccines. Vaccine 2008, 26, 4998–5003. [Google Scholar] [CrossRef] [PubMed]
- Maassab, H.F.; Bryant, M.L. The development of live attenuated cold-adapted influenza virus vaccine for humans. Rev. Med. Virol. 1999, 9, 237–244. [Google Scholar] [CrossRef]
- Jameson, J.; Cruz, J.; Ennis, F.A. Human cytotoxic T-lymphocyte repertoire to influenza A viruses. J. Virol. 1998, 72, 8682–8689. [Google Scholar] [PubMed]
- Boon, A.C.; de Mutsert, G.; van Baarle, D.; Smith, D.J.; Lapedes, A.S.; Fouchier, R.A.; Sintnicolaas, K.; Osterhaus, A.D.; Rimmelzwaan, G.F. Recognition of homo- and heterosubtypic variants of influenza A viruses by human CD8+ T lymphocytes. J. Immunol. 2004, 172, 2453–2460. [Google Scholar] [PubMed]
- Roy, S.; Kobinger, G.P.; Lin, J.; Figueredo, J.; Calcedo, R.; Kobasa, D.; Wilson, J.M. Partial protection against H5N1 influenza in mice with a single dose of a chimpanzee adenovirus vector expressing nucleoprotein. Vaccine 2007, 25, 6845–6851. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.P.; Compans, R.W. Virus infection of polarized epithelial cells. Adv. Virus Res. 1993, 42, 187–247. [Google Scholar] [PubMed]
- Fuller, S.; von Bonsdorff, C.H.; Simons, K. Vesicular stomatitis virus infects and matures only through the basolateral surface of the polarized epithelial cell line, MDCK. Cell 1984, 38, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Fuller, S.D.; von Bonsdorff, C.H.; Simons, K. Cell surface influenza haemagglutinin can mediate infection by other animal viruses. EMBO J. 1985, 4, 2475–2485. [Google Scholar] [PubMed]
- Schnell, M.J.; Buonocore, L.; Kretzschmar, E.; Johnson, E.; Rose, J.K. Foreign glycoproteins expressed from recombinant vesicular stomatitis viruses are incorporated efficiently into virus particles. Proc. Natl. Acad. Sci. USA 1996, 93, 11359–11365. [Google Scholar] [CrossRef]
- Ross, T.M.; Xu, Y.; Bright, R.A.; Robinson, H.L. C3d enhancement of antibodies to hemagglutinin accelerates protection against influenza virus challenge. Nat. Immunol. 2000, 1, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Fiers, W.; Neirynck, S.; Deroo, T.; Saelens, X.; Jou, W.M. Soluble recombinant influenza vaccines. Philos. Trans. R Soc. Lond. B Biol. Sci. 2001, 356, 1961–1963. [Google Scholar] [CrossRef] [PubMed]
- Bukreyev, A.; Skiadopoulos, M.H.; McAuliffe, J.; Murphy, B.R.; Collins, P.L.; Schmidt, A.C. More antibody with less antigen: can immunogenicity of attenuated live virus vaccines be improved? Proc. Natl. Acad. Sci. USA 2002, 99, 16987–16991. [Google Scholar] [CrossRef]
- Nishimoto, K.P.; Laust, A. K.; Wang, K.; Kamrud, K.I.; Hubby, B.; Smith, J.F.; Nelson, E.L. Restricted and selective tropism of a Venezuelan equine encephalitis virus-derived replicon vector for human dendritic cells. Viral Immunol. 2007, 20, 88–104. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.P.; Frolov, I.; Perri, S.; Ji, Y.; MacKichan, M.L.; zur Megede, J.; Chen, M.; Belli, B.A.; Driver, D.A.; Sherrill, S.; Greer, C.E.; Otten, G.R.; Barnett, S.W.; Liu, M.A.; Dubensky, T.W.; Polo, J.M. Infection of human dendritic cells by a sindbis virus replicon vector is determined by a single amino acid substitution in the E2 glycoprotein. J. Virol. 2000, 74, 11849–11857. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Zimmer, G. RNA Replicons - A New Approach for Influenza Virus Immunoprophylaxis. Viruses 2010, 2, 413-434. https://doi.org/10.3390/v2020413
Zimmer G. RNA Replicons - A New Approach for Influenza Virus Immunoprophylaxis. Viruses. 2010; 2(2):413-434. https://doi.org/10.3390/v2020413
Chicago/Turabian StyleZimmer, Gert. 2010. "RNA Replicons - A New Approach for Influenza Virus Immunoprophylaxis" Viruses 2, no. 2: 413-434. https://doi.org/10.3390/v2020413