Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. HIV-1 RT Function in Viral Replication
3. A Historical Perspective of Three-Dimensional Structures of HIV-1 RT
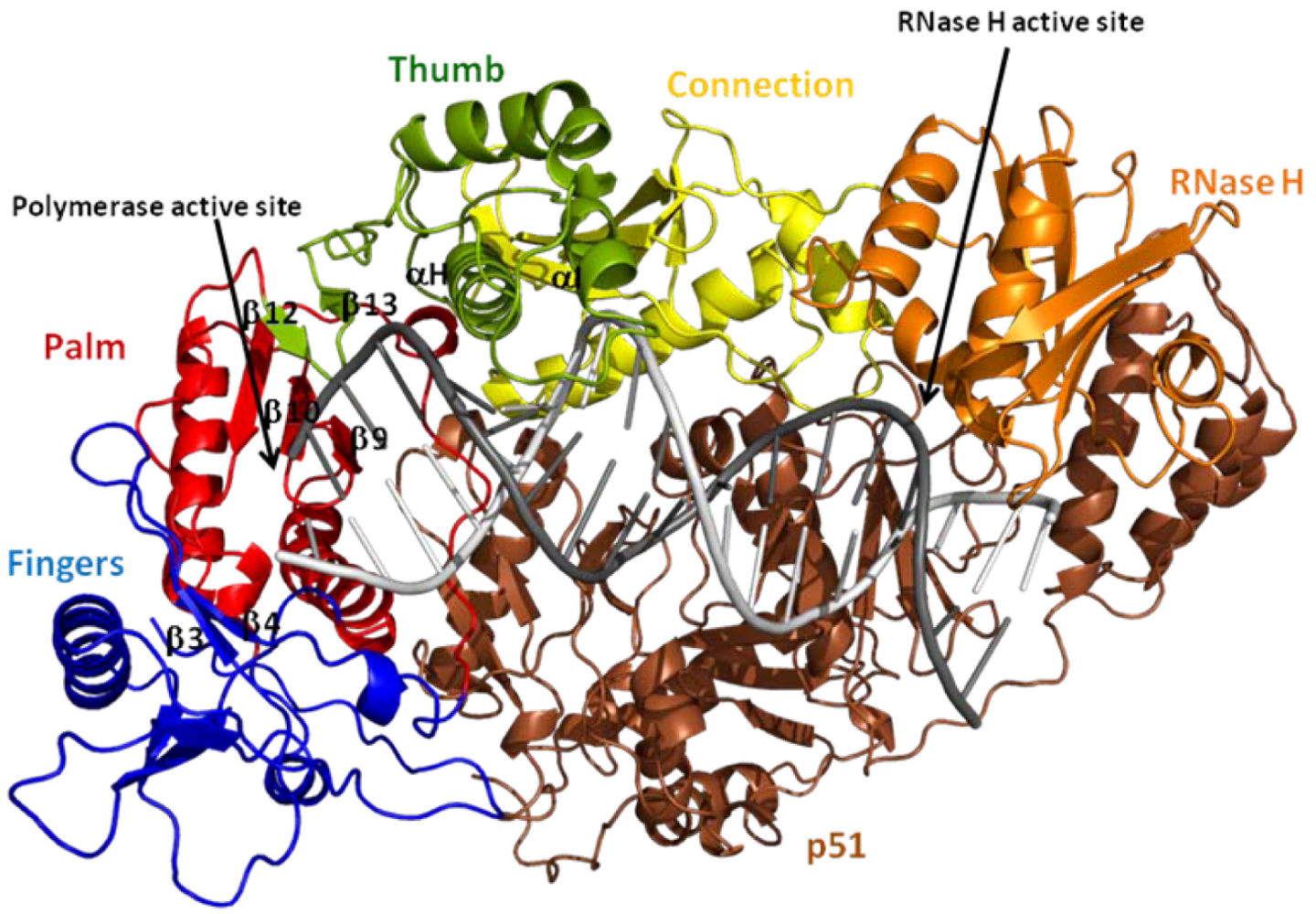
4. Structural Features of HIV-1 RT
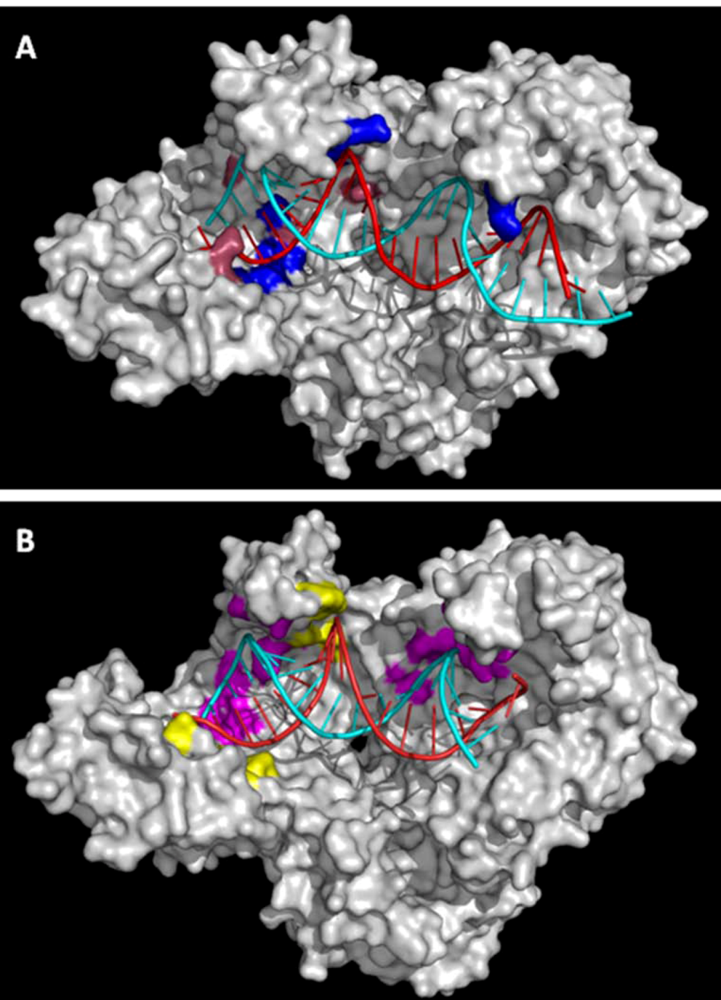
4.1. Nucleic Acid Binding Cleft

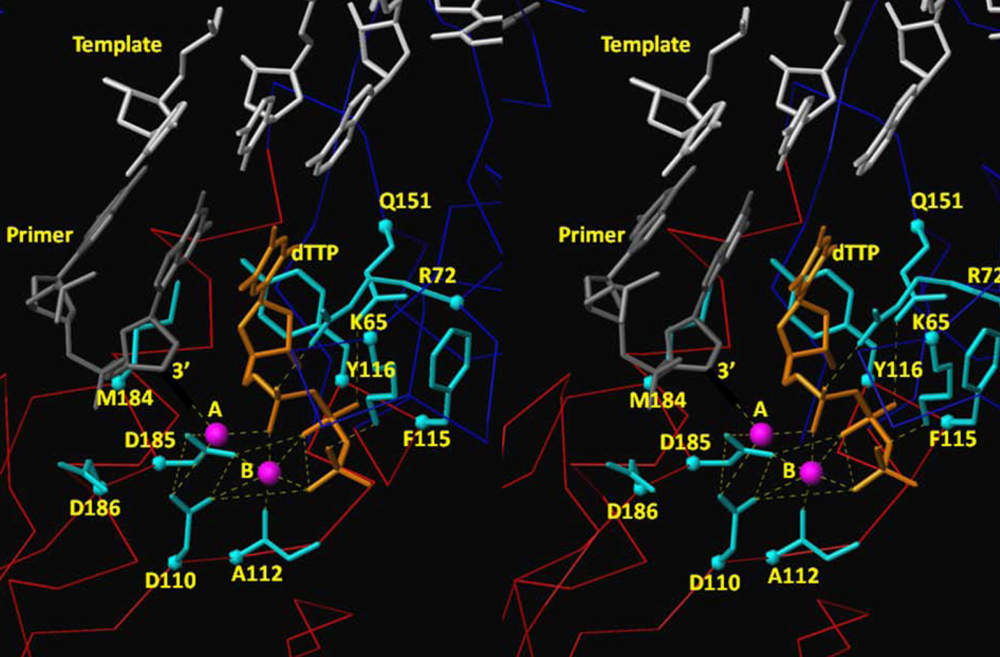
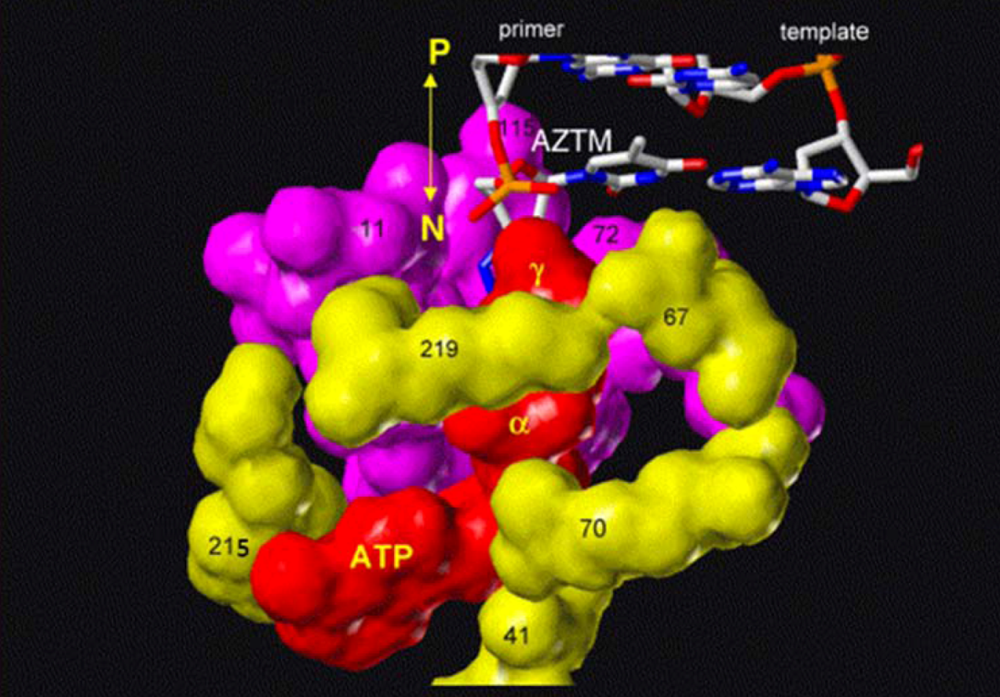
4.2. dNTP Binding Site



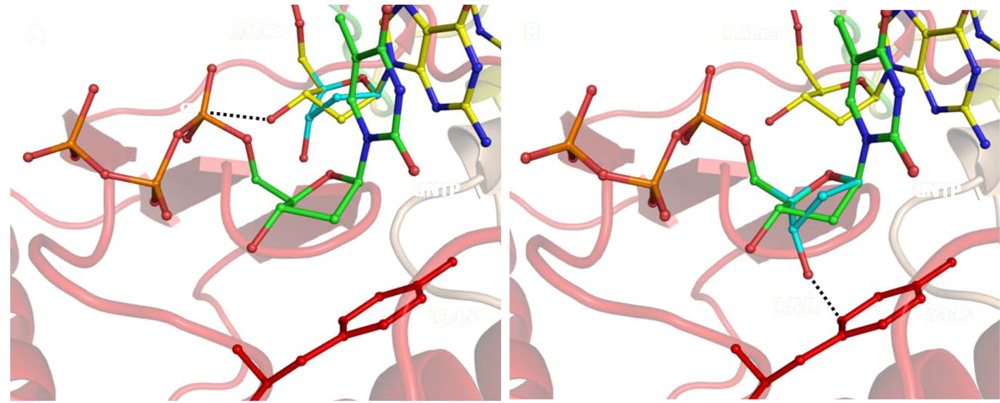
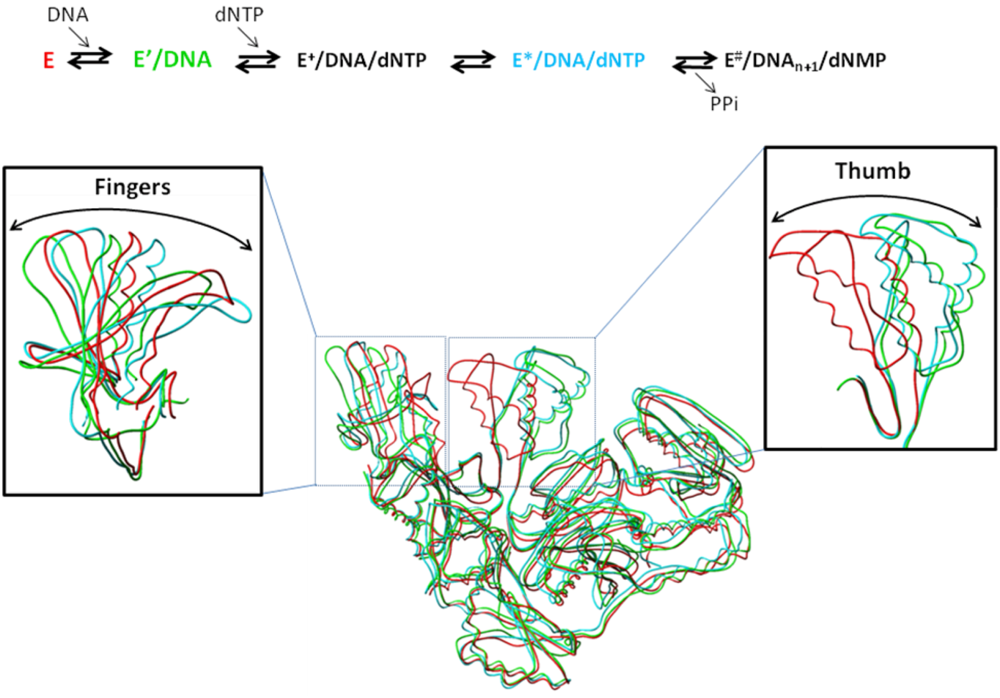
5. Structural Aspects of the Mechanism of DNA Synthesis by HIV-1 RT
6. Structural Aspects of HIV-1 RT Inhibition Mechanisms
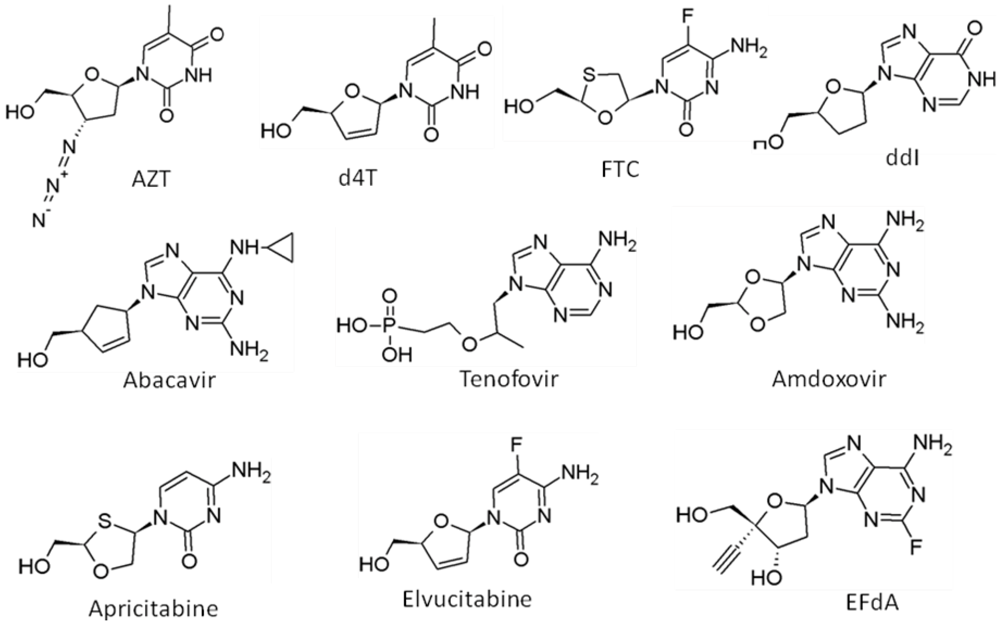
6.1. Nucleoside Reverse Transcriptase Inhibitors (NRTIs)
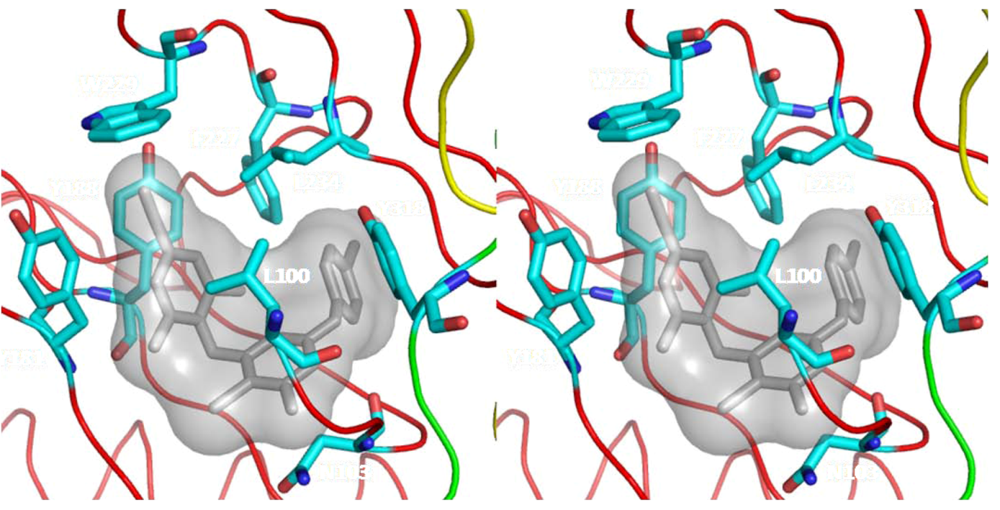
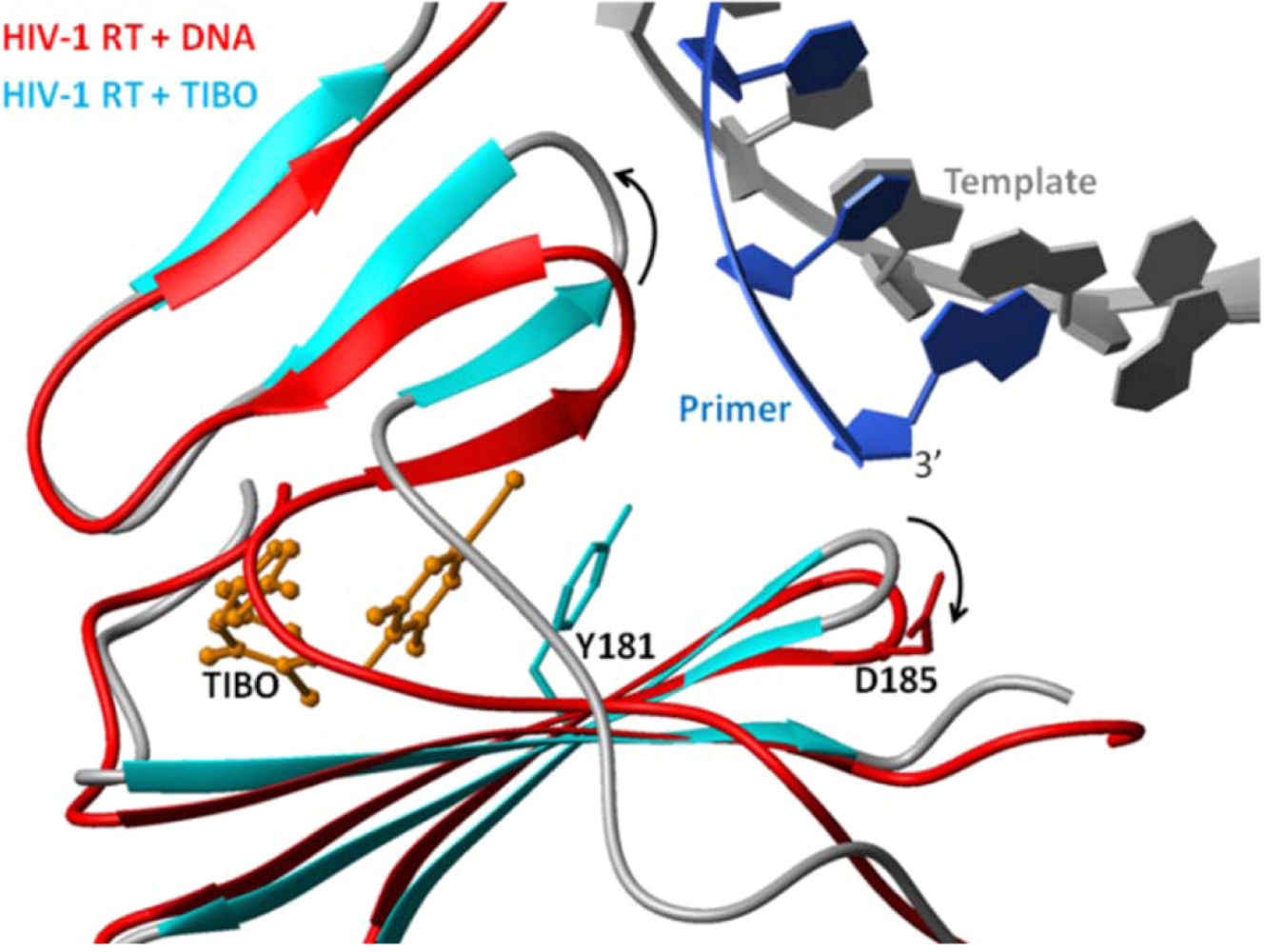
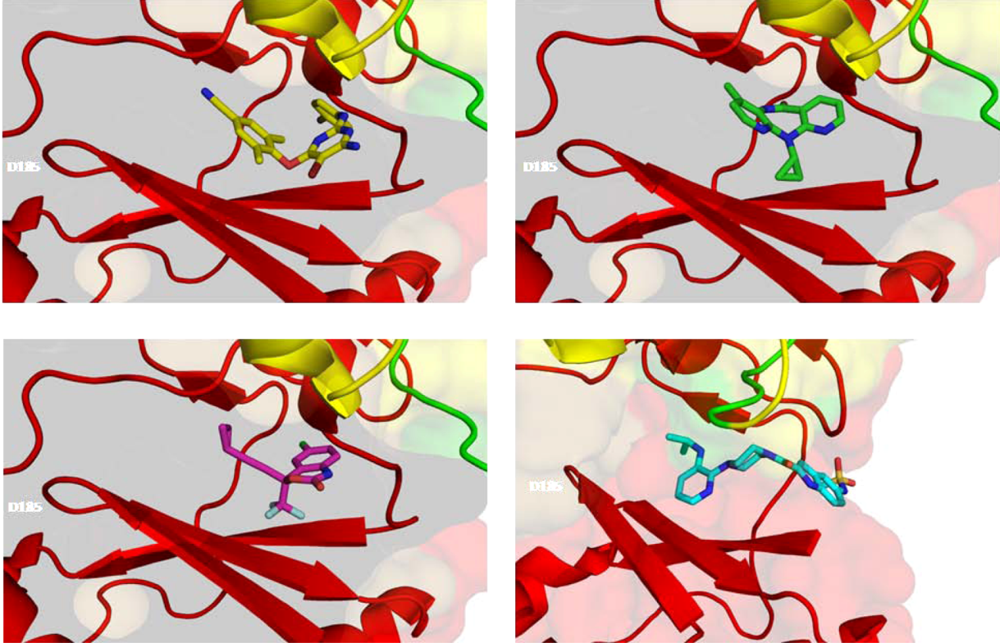
6.2. Nonnucleoside Reverse Transcriptase Inhibitors (NNRTIs)


7. Molecular Mechanisms of Resistance
7.1. Resistance to NRTIs
7.2. Interference with the Incorporation of NRTIs
7.3. Excision of Incorporated NRTIs

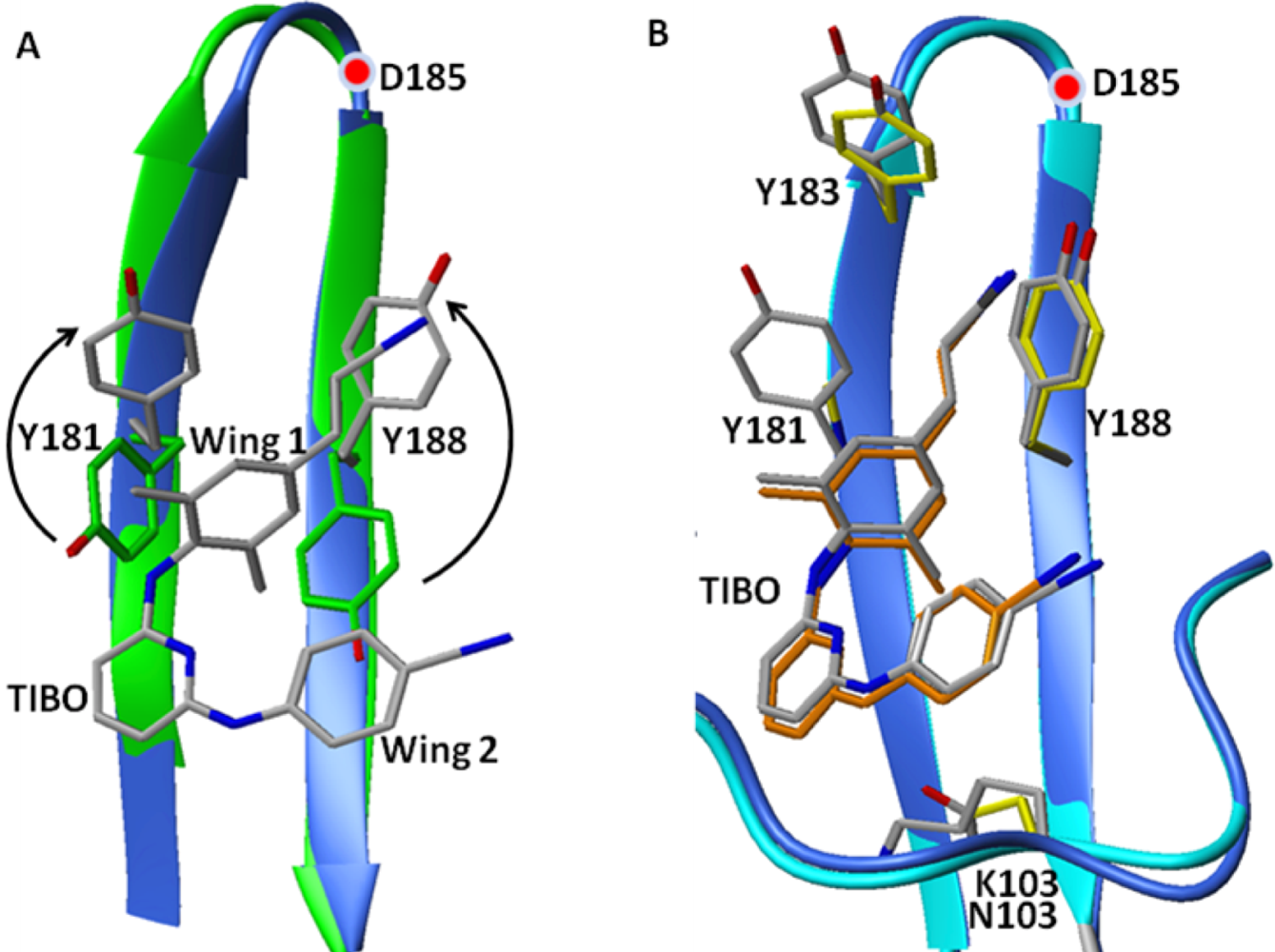
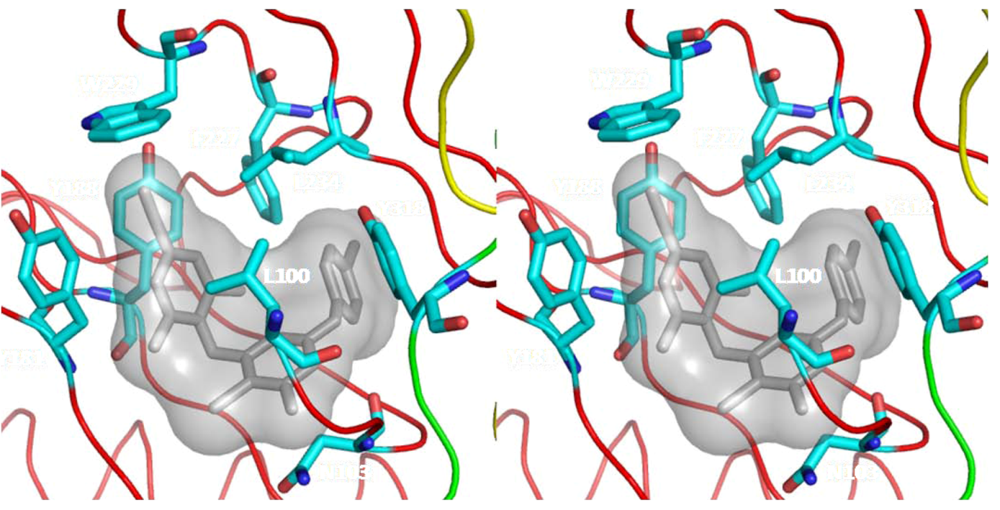
8. Resistance to NNRTIs

8.1. Loss/Change of Interactions at the NNRTI Binding Pocket

8.2. Steric Hindrance
8.3. Access to NNRTI binding pocket
9. Conclusions
Acknowledgments
References
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. Cold Spring Harbor Laboratory Press: New York, NY, USA, 1997.
- Gilboa, E.; Mitra, S.W.; Goff, S.; Baltimore, D. A detailed model of reverse transcription and tests of crucial aspects. Cell 1979, 18, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. Cold Spring Harbor Laboratory Press: New York, NY, USA, 1993.
- DeLano, W.L. The PyMol Molecular Graphics System. 2002. Available online: http://www.pymol.org.
- Esnouf, R.; Ren, J.; Ross, C.; Jones, Y.; Stammers, D.; Stuart, D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat. Struct. Biol. 1995, 2, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Hsiou, Y.; Ding, J.; Das, K.; Clark Jr., A.D.; Hughes, S.H.; Arnold, E. Structure of unliganded HIV-1 reverse transcriptase at 2.7 Ã resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure 1996, 4, 853–860. [Google Scholar] [CrossRef]
- Rodgers, D.W.; Gamblin, S.J.; Harris, B.A.; Ray, S.; Culp, J.S.; Hellmig, B.; Woolf, D.J.; Debouck, C.; Harrison, S.C. The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1995, 92, 1222–1226. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Das, K.; Tantillo, C.; Clark Jr., A.D.; Ding, J.; Whitcomb, J.M.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001, 20, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Das, K.; Hsiou, Y.; Sarafianos, S.G.; Clark Jr., A.D.; Jacobo-Molina, A.; Tantillo, C.; Hughes, S.H.; Arnold, E. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution. J. Mol. Biol. 1998, 284, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Das, K.; Clark Jr., A.D.; Ding, J.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with beta-branched amino acids. Proc. Natl. Acad. Sci. USA 1999, 96, 10027–10032. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Clark Jr., A.D.; Das, K.; Tuske, S.; Birktoft, J.J.; Ilankumaran, P.; Ramesha, A.R.; Sayer, J.M.; Jerina, D.M.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA. EMBO J. 2002, 21, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Tuske, S.; Sarafianos, S.G.; Clark Jr., A.D.; Ding, J.; Naeger, L.K.; White, K.L.; Miller, M.D.; Gibbs, C.S.; Boyer, P.L.; Clark Jr., P.; Wang, G.; Gaffney, B.L.; Jones, R.A.; Jerina, D.M.; Hughes, S.H.; Arnold, E. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 2004, 11, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Das, K.; Moereels, H.; Koymans, L.; Andries, K.; Janssen, P.A.; Hughes, S.H.; Arnold, E. Structure of HIV-1 RT/TIBO R 86183 complex reveals similarity in the binding of diverse nonnucleoside inhibitors. Nat. Struct. Biol. 1995, 2, 407–415. [Google Scholar] [CrossRef]
- Esnouf, R.M.; Ren, J.; Hopkins, A.L.; Ross, C.K.; Jones, E.Y.; Stammers, D.K.; Stuart, D.I. Unique features in the structure of the complex between HIV-1 reverse transcriptase and the bis(heteroaryl)piperazine (BHAP) U-90152 explain resistance mutations for this nonnucleoside inhibitor. Proc. Natl. Acad. Sci. USA 1997, 94, 3984–3989. [Google Scholar] [CrossRef]
- Hsiou, Y.; Ding, J.; Das, K.; Clark Jr., A.D.; Boyer, P.L.; Lewi, P.; Janssen, P.A.; Kleim, J.P.; Rosner, M.; Hughes, S.H.; Arnold, E. The Lys103Asn mutation of HIV-1 RT: a novel mechanism of drug resistance. J. Mol. Biol. 2001, 309, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar] [PubMed]
- Arnold, E.; Jacobo-Molina, A.; Nanni, R.G.; Williams, R.L.; Lu, X.; Ding, J.; Clark Jr., A.D.; Zhang, A.; Ferris, A. L.; Clark Jr., P. Structure of HIV-1 reverse transcriptase/DNA complex at 7 A resolution showing active site locations. Nature 1992, 357, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Ollis, D.L.; Brick, P.; Hamlin, R.; Xuong, N.G.; Steitz, T.A. Structure of large fragment of Escherichia coli DNA polymerase I complexed with dTMP. Nature 1985, 313, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Jacobo-Molina, A.; Ding, J.; Nanni, R.G.; Clark Jr., A.D.; Lu, X.; Tantillo, C.; Williams, R.L.; Kamer, G.; Ferris, A.L.; Clark Jr., P. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6320–6324. [Google Scholar] [CrossRef]
- Ren, J.; Esnouf, R.; Garman, E.; Somers, D.; Ross, C.; Kirby, I.; Keeling, J.; Darby, G.; Jones, Y.; Stuart, D. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat. Struct. Biol. 1995, 2, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Unge, T.; Knight, S.; Bhikhabhai, R.; Lovgren, S.; Dauter, Z.; Wilson, K.; Strandberg, B. 2.2 A resolution structure of the amino-terminal half of HIV-1 reverse transcriptase (fingers and palm subdomains. Structure 1994, 2, 953–961. [Google Scholar] [CrossRef]
- Das, K.; Bandwar, R.P.; White, K.L.; Feng, J.Y.; Sarafianos, S.G.; Tuske, S.; Tu, X.; Clark Jr., A.D.; Boyer, P.L.; Hou, X.; Gaffney, B.L.; Jones, R.A.; Miller, M.D.; Hughes, S.H.; Arnold, E. Structural basis for the role of the K65R mutation in HIV-1 reverse transcriptase polymerization, excision antagonism, and tenofovir resistance. J. Biol. Chem. 2009, 284, 35092–35100. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Milton, J.; Weaver, K.L.; Short, S.A.; Stuart, D.I.; Stammers, D.K. Structural basis for the resilience of efavirenz (DMP-266) to drug resistance mutations in HIV-1 reverse transcriptase. Structure 2000, 8, 1089–1094. [Google Scholar] [CrossRef]
- Das, K.; Bauman, J.D.; Clark Jr., A.D.; Frenkel, Y.V.; Lewi, P.J.; Shatkin, A.J.; Hughes, S.H.; Arnold, E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc. Natl. Acad. Sci. USA 2008, 105, 1466–1471. [Google Scholar] [CrossRef]
- Das, K.; Ding, J.; Hsiou, Y.; Clark Jr., A.D.; Moereels, H.; Koymans, L.; Andries, K.; Pauwels, R.; Janssen, P.A.; Boyer, P.L.; Clark Jr., P.; Smith Jr., R.H.; Kroeger Smith, M.B.; Michejda, C.J.; Hughes, S.H.; Arnold, E. Crystal structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1 RT and 8-Cl TIBO complexed with the Tyr181Cys HIV-1 RT drug-resistant mutant. J. Mol. Biol. 1996, 264, 1085–1100. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Sarafianos, S. G.; Clark Jr., A.D.; Boyer, P. L.; Hughes, S. H.; Arnold, E. Crystal structures of clinically relevant Lys103Asn/Tyr181Cys double mutant HIV-1 reverse transcriptase in complexes with ATP and non-nucleoside inhibitor HBY 097. J. Mol. Biol. 2007, 365, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Das, K.; Tantillo, C.; Zhang, W.; Clark Jr., A.D.; Jessen, S.; Lu, X.; Hsiou, Y.; Jacobo-Molina, A.; Andries, K. Structure of HIV-1 reverse transcriptase in a complex with the non-nucleoside inhibitor alpha-APA R 95845 at 2.8 A resolution. Structure 1995, 3, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Hsiou, Y.; Das, K.; Ding, J.; Clark Jr., A.D.; Kleim, J.P.; Rosner, M.; Winkler, I.; Riess, G.; Hughes, S.H.; Arnold, E. Structures of Tyr188Leu mutant and wild-type HIV-1 reverse transcriptase complexed with the non-nucleoside inhibitor HBY 097: inhibitor flexibility is a useful design feature for reducing drug resistance. J. Mol. Biol. 1998, 284, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.; Sigurdsson, S.; Lowgren, S.; Andersson, H.O.; Sahlberg, C.; Noreen, R.; Fridborg, K.; Zhang, H.; Unge, T. Structural basis for the inhibitory efficacy of efavirenz (DMP-266), MSC194 and PNU142721 towards the HIV-1 RT K103N mutant. Eur. J. Biochem. 2002, 269, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Pata, J. D.; Stirtan, W. G.; Goldstein, S. W.; Steitz, T. A. Structure of HIV-1 reverse transcriptase bound to an inhibitor active against mutant reverse transcriptases resistant to other nonnucleoside inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 10548–10553. [Google Scholar] [CrossRef]
- Ren, J.; Nichols, C.; Bird, L.E.; Fujiwara, T.; Sugimoto, H.; Stuart, D.I.; Stammers, D.K. Binding of the second generation non-nucleoside inhibitor S-1153 to HIV-1 reverse transcriptase involves extensive main chain hydrogen bonding. J. Biol. Chem. 2000, 275, 14316–14320. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Nichols, C.E.; Chamberlain, P.P.; Weaver, K.L.; Short, S.A.; Stammers, D.K. Crystal structures of HIV-1 reverse transcriptases mutated at codons 100, 106 and 108 and mechanisms of resistance to non-nucleoside inhibitors. J. Mol. Biol. 2004, 336, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Nichols, C.E.; Stamp, A.; Chamberlain, P.P.; Ferris, R.; Weaver, K.L.; Short, S.A.; Stammers, D. K. Structural insights into mechanisms of non-nucleoside drug resistance for HIV-1 reverse transcriptases mutated at codons 101 or 138. FEBS J. 2006, 273, 3850–3860. [Google Scholar] [CrossRef] [PubMed]
- Beilhartz, G.L.; Wendeler, M.; Baichoo, N.; Rausch, J.; Le Grice, S.; Gotte, M. HIV-1 reverse transcriptase can simultaneously engage its DNA/RNA substrate at both DNA polymerase and RNase H active sites: implications for RNase H inhibition. J. Mol. Biol. 2009, 388, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Delarue, M.; Poch, O.; Tordo, N.; Moras, D.; Argos, P. An attempt to unify the structure of polymerases. Protein Eng. 1990, 3, 461–467. [Google Scholar] [CrossRef]
- Poch, O.; Sauvaget, I.; Delarue, M.; Tordo, N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989, 8, 3867–3874. [Google Scholar] [PubMed]
- Johnson, M.S.; McClure, M.A.; Feng, D.F.; Gray, J.; Doolittle, R.F. Computer analysis of retroviral pol genes: assignment of enzymatic functions to specific sequences and homologies with nonviral enzymes. Proc. Natl. Acad. Sci. USA 1986, 83, 7648–7652. [Google Scholar] [CrossRef]
- Xiong, Y.; Eickbush, T.H. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990, 9, 3353–3362. [Google Scholar] [PubMed]
- Bebenek, K.; Beard, W.A.; Darden, T.A.; Li, L.; Prasad, R.; Luton, B.A.; Gorenstein, D.G.; Wilson, S.H.; Kunkel, T.A. A minor groove binding track in reverse transcriptase. Nat. Struct. Biol. 1997, 4, 194–197. [Google Scholar] [CrossRef]
- Hamburgh, M.E.; Curr, K.A.; Monaghan, M.; Rao, V.R.; Tripathi, S.; Preston, B.D.; Sarafianos, S.; Arnold, E.; Darden, T.; Prasad, V.R. Structural determinants of slippage-mediated mutations by human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 2006, 281, 7421–7428. [Google Scholar] [CrossRef] [PubMed]
- Powell, M.D.; Ghosh, M.; Jacques, P.S.; Howard, K.J.; Le Grice, S.F.; Levin, J.G. Alanine-scanning mutations in the "primer grip" of p66 HIV-1 reverse transcriptase result in selective loss of RNA priming activity. J. Biol. Chem. 1997, 272, 13262–13269. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Jacques, P.S.; Rodgers, D.W.; Ottman, M.; Darlix, J.L.; Le Grice, S.F. Alterations to the primer grip of p66 HIV-1 reverse transcriptase and their consequences for template-primer utilization. Biochemistry 1996, 35, 8553–8562. [Google Scholar] [CrossRef] [PubMed]
- Julias, J.G.; McWilliams, M.J.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Mutations in the RNase H domain of HIV-1 reverse transcriptase affect the initiation of DNA synthesis and the specificity of RNase H cleavage in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 9515–9520. [Google Scholar] [CrossRef]
- Wisniewski, M.; Palaniappan, C.; Fu, Z.; Le Grice, S.F.; Fay, P.; Bambara, R.A. Mutations in the primer grip region of HIV reverse transcriptase can increase replication fidelity. J. Biol. Chem. 1999, 274, 28175–28184. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Williams, J.; Powell, M.D.; Levin, J.G.; Le Grice, S.F. Mutating a conserved motif of the HIV-1 reverse transcriptase palm subdomain alters primer utilization. Biochemistry 1997, 36, 5758–5768. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.S.; Wohrl, B.M.; Ottmann, M.; Darlix, J.L.; Le Grice, S.F. Mutating the "primer grip" of p66 HIV-1 reverse transcriptase implicates tryptophan-229 in template-primer utilization. J. Biol. Chem. 1994, 269, 26472–26478. [Google Scholar] [PubMed]
- Doublie, S.; Tabor, S.; Long, A.M.; Richardson, C.C.; Ellenberger, T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature 1998, 391, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Korolev, S.; Waksman, G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 1998, 17, 7514–7525. [Google Scholar] [CrossRef] [PubMed]
- Eom, S.H.; Wang, J.; Steitz, T.A. Structure of Taq polymerase with DNA at the polymerase active site. Nature 1996, 382, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Sarafianos, S.G.; Nicklaus, M.C.; Russ, P.; Siddiqui, M.A.; Ford Jr., H.; Mitsuya, H.; Le, R.; Kodama, E.; Meier, C.; Knispel, T.; Anderson, L.; Barchi Jr., J.J.; Marquez, V.E. Interactions of conformationally biased north and south 2'-fluoro-2', 3'-dideoxynucleoside 5'-triphosphates with the active site of HIV-1 reverse transcriptase. Biochemistry 2000, 39, 11205–11215. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Orlova, M.; Georgiadis, M.M.; Hendrickson, W.A.; Goff, S.P. Conferring RNA polymerase activity to a DNA polymerase: a single residue in reverse transcriptase controls substrate selection. Proc. Natl. Acad. Sci. USA 1997, 94, 407–411. [Google Scholar] [CrossRef]
- Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Analysis of mutations at positions 115 and 116 in the dNTP binding site of HIV-1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 2000, 97, 3056–3061. [Google Scholar] [CrossRef]
- Martin-Hernandez, A.M.; Domingo, E.; Menendez-Arias, L. Human immunodeficiency virus type 1 reverse transcriptase: role of Tyr115 in deoxynucleotide binding and misinsertion fidelity of DNA synthesis. EMBO J. 1996, 15, 4434–4442. [Google Scholar] [PubMed]
- Emsley, P.; Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, M. A.; Salomon, H.; Gu, Z.; Montaner, J. S.; Cooley, T. P.; McCaffrey, R.; Ruedy, J.; Hirst, H.M.; Cammack, N.; Cameron, J. Development of HIV-1 resistance to (-)2'-deoxy-3'-thiacytidine in patients with AIDS or advanced AIDS-related complex. AIDS 1995, 9, 351–357. [Google Scholar] [PubMed]
- Gu, Z.; Gao, Q.; Li, X.; Parniak, M.A.; Wainberg, M.A. Novel mutation in the human immunodeficiency virus type 1 reverse transcriptase gene that encodes cross-resistance to 2',3'-dideoxyinosine and 2',3'-dideoxycytidine. J. Virol. 1992, 66, 7128–7135. [Google Scholar] [PubMed]
- Koradi, R.; Billeter, M.; Wuthrich, K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Kuchta, R.D.; Mizrahi, V.; Benkovic, P.A.; Johnson, K.A.; Benkovic, S. J. Kinetic mechanism of DNA polymerase I (Klenow). Biochemistry 1987, 26, 8410–8417. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.; Patel, S.S.; Johnson, K.A. An induced-fit kinetic mechanism for DNA replication fidelity: direct measurement by single-turnover kinetics. Biochemistry 1991, 30, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Wong, I.; Johnson, K.A. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry 1991, 30, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Kati, W.M.; Johnson, K.A.; Jerva, L.F.; Anderson, K.S. Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 1992, 267, 25988–25997. [Google Scholar] [PubMed]
- Wohrl, B.M.; Krebs, R.; Goody, R.S.; Restle, T. Refined model for primer/template binding by HIV-1 reverse transcriptase: pre-steady-state kinetic analyses of primer/template binding and nucleotide incorporation events distinguish between different binding modes depending on the nature of the nucleic acid substrate. J. Mol. Biol. 1999, 292, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y. W.; Steitz, T. A. The structural mechanism of translocation and helicase activity in T7 RNA polymerase. Cell 2004, 116, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Gotte, M. Effects of nucleotides and nucleotide analogue inhibitors of HIV-1 reverse transcriptase in a ratchet model of polymerase translocation. Current Pharm. Des. 2006, 12, 1867–1877. [Google Scholar] [CrossRef]
- Marchand B.; Tchesnokov E.P.; Götte M. The pyrophosphate analogue foscarnet traps the pre-translocational state of HIV-1 reverse transcriptase in a Brownian ratchet model of polymerase translocation. J. Biol. Chem. 2007, 282, 3337–3346. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A.; Steitz, J.A. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6498–6502. [Google Scholar] [CrossRef]
- Steitz, T.A. A mechanism for all polymerases. Nature 1998, 391, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, H.; Sawaya, M.R.; Kumar, A.; Wilson, S.H.; Kraut, J. Structures of ternary complexes of rat DNA polymerase beta, a DNA template-primer, and ddCTP. Science 1994, 264, 1891–1903. [Google Scholar] [PubMed]
- Sawaya, M.R.; Pelletier, H.; Kumar, A.; Wilson, S.H.; Kraut, J. Crystal structure of rat DNA polymerase beta: evidence for a common polymerase mechanism. Science 1994, 264, 1930–1935. [Google Scholar] [PubMed]
- De Clercq, E. Antiviral drugs in current clinical use. J. Clin. Virol. 2004, 30, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Parniak, M.A.; Sluis-Cremer, N. Inhibitors of HIV-1 reverse transcriptase. Adv. Pharmacol. 2000, 49, 67–109. [Google Scholar] [PubMed]
- Perno, C.F.; Yarchoan, R.; Cooney, D.A.; Hartman, N.R.; Gartner, S.; Popovic, M.; Hao, Z.; Gerrard, T.L.; Wilson, Y.A.; Johns, D. G. Inhibition of human immunodeficiency virus (HIV-1/HTLV-IIIBa-L) replication in fresh and cultured human peripheral blood monocytes/ macrophages by azidothymidine and related 2',3'-dideoxynucleosides. J. Exp. Med. 1988, 168, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Sarfati, R.; Deville-Bonne, D.; Véron, M. Role of nucleoside diphosphate kinase in the activation of anti-HIV nucleoside analogs. J. Bioenerg. Biomembr. 2000, 36, 317–324. [Google Scholar] [CrossRef]
- Gallois-Montbrun, S.; Schneider, B.; Chen, Y.; Giacomoni-Fernandes, V.; Mulard, L.; Morera, S.; Janin, J.; Deville-Bonne, D.; Véron, M. Improving nucleoside diphosphate kinase for antiviral nucleotide analogs activation. J. Biol. Chem. 2002, 277, 39953–39959. [Google Scholar] [CrossRef] [PubMed]
- Kerr, S.G.; Anderson, K.S. Pre-steady-state kinetic characterization of wild type and 3'-azido-3'-deoxythymidine (AZT) resistant human immunodeficiency virus type 1 reverse transcriptase: implication of RNA directed DNA polymerization in the mechanism of AZT resistance. Biochemistry 1997, 36, 14064–14070. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Y.; Anderson, K.S. Mechanistic studies comparing the incorporation of (+) and (-) isomers of 3TCTP by HIV-1 reverse transcriptase. Biochemistry 1999, 38, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, A.; Kodama, E.; Sarafianos, S.G.; Sakagami, Y.; Kohgo, S.; Kitano, K.; Ashida, N.; Iwai, Y.; Hayakawa, H.; Nakata, H.; Mitsuya, H.; Arnold, E.; Matsuoka, M. 2'-deoxy-4'-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int. J. Biochem. Cell Biol. 2008, 40, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, E.; Marchand, B.; Kodama, E.N.; Singh, K.; Matsuoka, M.; Kirby, K.A.; Ryan, E.M.; Sawani, A.M.; Nagy, E.; Ashida, N.; Mitsuya, H.; Parniak, M.A.; Sarafianos, S.G. Mechanism of inhibition of HIV-1 reverse transcriptase by 4'-ethynyl-2-fluoro-2'-deoxyadenosine triphosphate, a translocation defective reverse transcriptase inhibitor. J. Biol. Chem. 2009, 284, 35681–35691. [Google Scholar] [CrossRef] [PubMed]
- Tantillo, C.; Ding, J.; Jacobo-Molina, A.; Nanni, R.G.; Boyer, P.L.; Hughes, S.H.; Pauwels, R.; Andries, K.; Janssen, P.A.; Arnold, E. Locations of anti-AIDS drug binding sites and resistance mutations in the three-dimensional structure of HIV-1 reverse transcriptase. Implications for mechanisms of drug inhibition and resistance. J. Mol. Biol. 1994, 243, 369–387. [Google Scholar] [CrossRef] [PubMed]
- Tachedjian, G.; Orlova, M.; Sarafianos, S.G.; Arnold, E.; Goff, S.P. Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 2001, 98, 7188–7193. [Google Scholar] [CrossRef]
- Spessard, G.O. ACD Labs/LogP dB 3.5 and ChemSketch 3.5. J. Chem. Inf. Comput. Sci. 1998, 38, 1250–1253. [Google Scholar] [CrossRef]
- Rittinger, K.; Divita, G.; Goody, R.S. Human immunodeficiency virus reverse transcriptase substrate-induced conformational changes and the mechanism of inhibition by nonnucleoside inhibitors. Proc. Natl. Acad. Sci. USA 1995, 92, 8046–8049. [Google Scholar] [CrossRef]
- Spence, R.A.; Kati, W.M.; Anderson, K.S.; Johnson, K.A. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science 1995, 267, 988–993. [Google Scholar] [PubMed]
- Menendez-Arias, L. Targeting HIV: antiretroviral therapy and development of drug resistance. Trends Pharmacol. Sci. 2002, 23, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Arias, L. Mechanisms of resistance to nucleoside analogue inhibitors of HIV-1 reverse transcriptase. Virus Res. 2008, 134, 124–146. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.; Kemp, S.D.; Parry, N.R.; Larder, B.A. Rapid in vitro selection of human immunodeficiency virus type 1 resistant to 3'-thiacytidine inhibitors due to a mutation in the YMDD region of reverse transcriptase. Proc. Natl. Acad. Sci. USA 1993, 90, 5653–5656. [Google Scholar] [CrossRef]
- Back, N.K.; Nijhuis, M.; Keulen, W.; Boucher, C.A.; Oude Essink, B.O.; van Kuilenburg, A.B.; van Gennip, A.H.; Berkhout, B. Reduced replication of 3TC-resistant HIV-1 variants in primary cells due to a processivity defect of the reverse transcriptase enzyme. EMBO J. 1996, 15, 4040–4049. [Google Scholar] [PubMed]
- Wei, X.; Liang, C.; Gotte, M.; Wainberg, M.A. Negative effect of the M184V mutation in HIV-1 reverse transcriptase on initiation of viral DNA synthesis. Virology 2003, 311, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Winters, M.A.; Shafer, R.W.; Jellinger, R.A.; Mamtora, G.; Gingeras, T.; Merigan, T.C. Human immunodeficiency virus type 1 reverse transcriptase genotype and drug susceptibility changes in infected individuals receiving dideoxyinosine monotherapy for 1 to 2 years. Antimicrob Agents Chemother 1997, 41, 757–762. [Google Scholar] [PubMed]
- Quan, Y.; Gu, Z.; Li, X.; Li, Z.; Morrow, C.D.; Wainberg, M.A. Endogenous reverse transcription assays reveal high-level resistance to the triphosphate of (-)2'-dideoxy-3'-thiacytidine by mutated M184V human immunodeficiency virus type 1. J. Virol. 1996, 70, 5642–5645. [Google Scholar] [PubMed]
- Harrigan, P.R.; Stone, C.; Griffin, P.; Najera, I.; Bloor, S.; Kemp, S.; Tisdale, M.; Larder, B. Resistance profile of the human immunodeficiency virus type 1 reverse transcriptase inhibitor abacavir (1592U89) after monotherapy and combination therapy. CNA2001 Investigative Group. J. Infect. Dis. 2000, 181, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.; Ait-Khaled, M.; Stone, C.; Griffin, P.; Mesogiti, D.; Cutrell, A.; Harrigan, R.; Staszewski, S.; Katlama, C.; Pearce, G.; Tisdale, M. HIV-1 reverse transcriptase (RT) genotype and susceptibility to RT inhibitors during abacavir monotherapy and combination therapy. AIDS 2000, 14, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Hughes, S.H.; Arnold, E. Designing anti-AIDS drugs targeting the major mechanism of HIV-1 RT resistance to nucleoside analog drugs. Int. J. Biochem. Cell. Biol. 2004, 36, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Zala, C.; Ochoa, C.; Tharnish, P.; Fromentin, E.; Asil, G.; UHurwitz, S.J.; Kivel, N.M.; Schinazi, R.F. Pharmacokinetics and potent anti-HIV Activity of Amdoxovir plus Zidovudine in a randomized double-blind placebo-controlled study. 15th CROI Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 3â6 February 2008. Available online: http://www.natap.org/2008/CROI/croi_96.htm.
- De Clercq, E. Emerging anti-HIV drugs. Expert Opin. Emerg. Drugs 2005, 10, 241–273. [Google Scholar] [CrossRef] [PubMed]
- Dunkle, L.M.; Gathe, J.C.; Pedevillano, D.E.; Robison, H.G.; Rice, W.G.; Pottage, J.C.J.; Team, A.S. Elvucitabine: Potent Antiviral activity demonstrated in multi-drug resistant HIV infection. Antivir. Ther. 2003, 8, S5. [Google Scholar]
- Bethell, R.C.; Lie, Y.S.; Parkin, N.T. In vitro activity of SPD754, a new deoxycytidine nucleoside reverse transcriptase inhibitor (NRTI), against 215 HIV-1 isolates resistant to other NRTIs. Antivir. Chem. Chemother. 2005, 16, 295–302. [Google Scholar] [PubMed]
- Ueno, T.; Mitsuya, H. Comparative enzymatic study of HIV-1 reverse transcriptase resistant to 2',3'-dideoxynucleotide analogs using the single-nucleotide incorporation assay. Biochemistry 1997, 36, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Shirasaka, T.; Mitsuya, H. Enzymatic characterization of human immunodeficiency virus type 1 reverse transcriptase resistant to multiple 2',3'-dideoxynucleoside 5'-triphosphates. J. Biol. Chem. 1995, 270, 23605–23611. [Google Scholar] [CrossRef] [PubMed]
- Shirasaka, T.; Kavlick, M.F; Ueno, T.; Gao, W.Y.; Kojima, E.; Alcaide, M.L.; Chokekijchai, S.; Roy, B.M.; Arnold, E.; Yarchoan, R. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc. Natl. Acad. Sci. USA 1995, 92, 2398–2402. [Google Scholar] [CrossRef]
- Iversen, A.K.; Shafer, R.W.; Wehrly, K.; Winters, M.A.; Mullins, J.I.; Chesebro, B.; Merigan, T.C. Multidrug-resistant human immunodeficiency virus type 1 strains resulting from combination antiretroviral therapy. J. Virol. 1996, 70, 1086–1090. [Google Scholar] [PubMed]
- Deval, J.; Selmi, B.; Boretto, J.; Egloff, M.P.; Guerreiro, C.; Sarfati, S.; Canard, B. The molecular mechanism of multidrug resistance by the Q151M human immunodeficiency virus type 1 reverse transcriptase and its suppression using alpha-boranophosphate nucleotide analogues. J. Biol. Chem. 2002, 277, 42097–42104. [Google Scholar] [CrossRef] [PubMed]
- St Clair, M.H.; Martin, J.L.; Tudor-Williams, G.; Bach, M.C.; Vavro, C.L.; King, D.M.; Kellam, P.; Kemp, S.D.; Larder, B.A. Resistance to ddI and sensitivity to AZT induced by a mutation in HIV-1 reverse transcriptase. Science 1991, 253, 1557–1559. [Google Scholar] [PubMed]
- Martin, J.L.; Wilson, J.E.; Haynes, R.L.; Furman, P.A. Mechanism of resistance of human immunodeficiency virus type 1 to 2',3'-dideoxyinosine. Proc. Natl. Acad. Sci. USA 1993, 90, 6135–6139. [Google Scholar] [CrossRef]
- Deval, J.; Navarro, J.M.; Selmi, B.; Courcambeck, J.; Boretto, J.; Halfon, P.; Garrido-Urbani, S.; Sire, J.; Canard, B. A loss of viral replicative capacity correlates with altered DNA polymerization kinetics by the human immunodeficiency virus reverse transcriptase bearing the K65R and L74V dideoxynucleoside resistance substitutions. J. Biol. Chem. 2004, 279, 25489–25496. [Google Scholar] [CrossRef] [PubMed]
- Lacey, S.F.; Larder, B.A. Novel mutation (V75T) in human immunodeficiency virus type 1 reverse transcriptase confers resistance to 2',3'-didehydro-2',3'-dideoxythymidine in cell culture. Antimicrob. Agents Chemother. 1994, 38, 1428–1432. [Google Scholar] [PubMed]
- Matamoros, T.; Kim, B.; Menendez-Arias, L. Mechanistic insights into the role of Val75 of HIV-1 reverse transcriptase in misinsertion and mispair extension fidelity of DNA synthesis. J. Mol. Biol. 2008, 375, 1234–1248. [Google Scholar] [CrossRef] [PubMed]
- Selmi, B.; Boretto, J.; Navarro, J.M.; Sire, J.; Longhi, S.; Guerreiro, C.; Mulard, L.; Sarfati, S.; Canard, B. The valine-to-threonine 75 substitution in human immunodeficiency virus type 1 reverse transcriptase and its relation with stavudine resistance. J. Biol. Chem. 2001, 276, 13965–13974. [Google Scholar] [PubMed]
- Margot, N.A.; Waters, J.M.; Miller, M.D. In vitro human immunodeficiency virus type 1 resistance selections with combinations of tenofovir and emtricitabine or abacavir and lamivudine. Antimicrob. Agents Chemother. 2006, 50, 4087–4095. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, M.A.; Miller, M.D.; Quan, Y.; Salomon, H.; Mulato, A.S.; Lamy, P.D.; Margot, N.A.; Anton, K.E.; Cherrington, J.M. In vitro selection and characterization of HIV-1 with reduced susceptibility to PMPA. Antivir. Ther. 1999, 4, 87–94. [Google Scholar] [PubMed]
- Jeffrey, J.L.; Feng, J.Y.; Qi, C.C.; Anderson, K.S.; Furman, P.A. Dioxolane guanosine 5'-triphosphate, an alternative substrate inhibitor of wild-type and mutant HIV-1 reverse transcriptase. Steady state and pre-steady state kinetic analyses. J. Biol. Chem. 2003, 278, 18971–18979. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Margot, N.A.; Wrin, T.; Petropoulos, C.J.; Miller, M.D.; Naeger, L.K. Molecular mechanisms of resistance to human immunodeficiency virus type 1 with reverse transcriptase mutations K65R and K65R+M184V and their effects on enzyme function and viral replication capacity. Antimicrob. Agents Chemother. 2002, 46, 3437–3446. [Google Scholar] [CrossRef] [PubMed]
- Selmi, B.; Boretto, J.; Sarfati, S.R.; Guerreiro, C.; Canard, B. Mechanism-based suppression of dideoxynucleotide resistance by K65R human immunodeficiency virus reverse transcriptase using an alpha-boranophosphate nucleoside analogue. J. Biol. Chem. 2001, 276, 48466–48472. [Google Scholar] [PubMed]
- Parikh, U.M.; Zelina, S.; Sluis-Cremer, N.; Mellors, J.W. Molecular mechanisms of bidirectional antagonism between K65R and thymidine analog mutations in HIV-1 reverse transcriptase. AIDS 2007, 21, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Sluis-Cremer, N.; Sheen, C.W.; Zelina, S.; Torres, P.S.; Parikh, U.M.; Mellors, J.W. Molecular mechanism by which the K70E mutation in human immunodeficiency virus type 1 reverse transcriptase confers resistance to nucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2007, 51, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Deval, J.; White, K.L.; Miller, M.D.; Parkin, N.T.; Courcambeck, J.; Halfon, P.; Selmi, B.; Boretto, J.; Canard, B. Mechanistic basis for reduced viral and enzymatic fitness of HIV-1 reverse transcriptase containing both K65R and M184V mutations. J. Biol. Chem. 2004, 279, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Arion, D.; Kaushik, N.; McCormick, S.; Borkow, G.; Parniak, M.A. Phenotypic mechanism of HIV-1 resistance to 3'-azido-3'-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry 1998, 37, 15908–15917. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.R.; Matsuura, S.E.; So, A.G.; Scott, W.A. Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. USA 1998, 95, 13471–13476. [Google Scholar] [CrossRef]
- Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Selective excision of AZTMP by drug-resistant human immunodeficiency virus reverse transcriptase. J. Virol. 2001, 75, 4832–4842. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.R.; Matsuura, S. E.; Mian, A.M.; So, A.G.; Scott, W.A. A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell. 1999, 4, 35–43. [Google Scholar] [CrossRef]
- Meyer, P.R.; Matsuura, S.E.; Schinazi, R.F.; So, A.G.; Scott, W.A. Differential removal of thymidine nucleotide analogues from blocked DNA chains by human immunodeficiency virus reverse transcriptase in the presence of physiological concentrations of 2'-deoxynucleoside triphosphates. Antimicrob Agents Chemother 2000, 44, 3465–3472. [Google Scholar] [CrossRef] [PubMed]
- Dharmasena, S.; Pongracz, Z.; Arnold, E.; Sarafianos, S.G.; Parniak, M.A. 3'-Azido-3'-deoxythymidine-(5')-tetraphospho-(5')-adenosine, the product of ATP-mediated excision of chain-terminating AZTMP, is a potent chain-terminating substrate for HIV-1 reverse transcriptase. Biochemistry 2007, 46, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Lacey, S.F.; Reardon, J.E.; Furfine, E.S.; Kunkel, T.A.; Bebenek, K.; Eckert, K.A.; Kemp, S.D.; Larder, B.A. Biochemical studies on the reverse transcriptase and RNase H activities from human immunodeficiency virus strains resistant to 3'-azido-3'-deoxythymidine. J. Biol. Chem. 1992, 267, 15789–15794. [Google Scholar] [PubMed]
- Tu, X. Structural studies of HIV-1 reverse transcriptase: resistanceto azt via atp-mediated excision. Ph.D Dissertation, Rutgers The State University Of New Jersey-New Brunswick: New Jersey, USA, 2008. Available online: http://mss3.libraries.rutgers.edu/dlr/TMP/rutgers-lib_24491-PDF-1.pdf (accessed on 9 November 2009).
- Marchand, B.; Gotte, M. Site-specific footprinting reveals differences in the translocation status of HIV-1 reverse transcriptase. Implications for polymerase translocation and drug resistance. J. Biol. Chem. 2003, 278, 35362–35372. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Clark Jr., A.D.; Tuske, S.; Squire, C.J.; Das, K.; Sheng, D.; Ilankumaran, P.; Ramesha, A.R.; Kroth, H.; Sayer, J.M.; Jerina, D.M.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Trapping HIV-1 reverse transcriptase before and after translocation on DNA. J. Biol. Chem. 2003, 278, 16280–16288. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.R.; Rutvisuttinunt, W.; Matsuura, S.E.; So, A.G.; Scott, W.A. Stable complexes formed by HIV-1 reverse transcriptase at distinct positions on the primer-template controlled by binding deoxynucleoside triphosphates or foscarnet. J. Mol. Biol. 2007, 369, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Lu, C.D.; Sharma, S.K.; Matsuura, S.; So, A.G.; Scott, W.A. Nucleotide-induced stable complex formation by HIV-1 reverse transcriptase. Biochemistry 1997, 36, 5749–5757. [Google Scholar] [CrossRef] [PubMed]
- Marchand, B.; White, K.L.; Ly, J.K.; Margot, N.A.; Wang, R.; McDermott, M.; Miller, M.D.; Götte, M. Effects of the translocation status of human immunodeficiency virus type 1 reverse transcriptase on the efficiency of excision of tenofovir. Antimicrob. Agents Chemother. 2007, 51, 2911–2919. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. The M184V mutation reduces the selective excision of zidovudine 5'-monophosphate (AZTMP) by the reverse transcriptase of human immunodeficiency virus type 1. J. Virol. 2002, 76, 3248–3256. [Google Scholar] [CrossRef] [PubMed]
- Gotte, M.; Arion, D.; Parniak, M.A.; Wainberg, M.A. The M184V mutation in the reverse transcriptase of human immunodeficiency virus type 1 impairs rescue of chain-terminated DNA synthesis. J. Virol. 2000, 74, 3579–3585. [Google Scholar] [CrossRef] [PubMed]
- Sluis-Cremer, N.; Arion, D.; Parniak, M.A. Molecular mechanisms of HIV-1 resistance to nucleoside reverse transcriptase inhibitors (NRTIs). Cell Mol. Life Sci. 2000, 57, 1408–1422. [Google Scholar] [CrossRef] [PubMed]
- Larder, B.A.; Kemp, S.D.; Harrigan, P.R. Potential mechanism for sustained antiretroviral efficacy of AZT-3TC combination therapy. Science 1995, 269, 696–699. [Google Scholar] [PubMed]
- Miranda, L.R.; Gotte, M.; Liang, F.; Kuritzkes, D.R. The L74V mutation in human immunodeficiency virus type 1 reverse transcriptase counteracts enhanced excision of zidovudine monophosphate associated with thymidine analog resistance mutations. Antimicrob. Agents Chemother. 2005, 49, 2648–2656. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Margot, N.A.; Ly, J.K.; Chen, J.M.; Ray, A.S.; Pavelko, M.; Wang, R.; McDermott, M.; Swaminathan, S.; Miller, M.D. A combination of decreased NRTI incorporation and decreased excision determines the resistance profile of HIV-1 K65R RT. AIDS 2005, 19, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Frankel, F.A.; Marchand, B.; Turner, D.; Gotte, M.; Wainberg, M.A. Impaired rescue of chain-terminated DNA synthesis associated with the L74V mutation in human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 2005, 49, 2657–2664. [Google Scholar] [CrossRef] [PubMed]
- Larder, B. A. 3'-Azido-3'-deoxythymidine resistance suppressed by a mutation conferring human immunodeficiency virus type 1 resistance to nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 1992, 36, 2664–2669. [Google Scholar] [PubMed]
- Byrnes, V.W.; Emini, E.A.; Schleif, W.A.; Condra, J.H.; Schneider, C.L.; Long, W.J.; Wolfgang, J.A.; Graham, D.J.; Gotlib, L.; Schlabach, A.J. Susceptibilities of human immunodeficiency virus type 1 enzyme and viral variants expressing multiple resistance-engendering amino acid substitutions to reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 1994, 38, 1404–1407. [Google Scholar] [PubMed]
- Selmi, B.; Deval, J.; Alvarez, K.; Boretto, J.; Sarfati, S.; Guerreiro, C.; Canard, B. The Y181C substitution in 3'-azido-3'-deoxythymidine-resistant human immunodeficiency virus, type 1, reverse transcriptase suppresses the ATP-mediated repair of the 3'-azido-3'-deoxythymidine 5'-monophosphate-terminated primer. J. Biol. Chem. 2003, 278, 40464–40472. [Google Scholar] [CrossRef] [PubMed]
- Tachedjian, G.; Mellors, J.; Bazmi, H.; Birch, C.; Mills, J. Zidovudine resistance is suppressed by mutations conferring resistance of human immunodeficiency virus type 1 to foscarnet. J. Virol. 1996, 70, 7171–7181. [Google Scholar] [PubMed]
- De Antoni, A.; Foli, A.; Lisziewicz, J.; Lori, F. Mutations in the pol gene of human immunodeficiency virus type 1 in infected patients receiving didanosine and hydroxyurea combination therapy. J. Infect. Dis. 1997, 176, 899–903. [Google Scholar] [PubMed]
- Winters, M.A.; Coolley, K.L.; Girard, Y.A.; Levee, D.J.; Hamdan, H.; Shafer, R.W.; Katzenstein, D.A.; Merigan, T.C. A 6-basepair insert in the reverse transcriptase gene of human immunodeficiency virus type 1 confers resistance to multiple nucleoside inhibitors. J. Clin. Invest. 1998, 102, 1769–1775. [Google Scholar] [CrossRef] [PubMed]
- Mas, A.; Parera, M.; Briones, C.; Soriano, V.; Martinez, M.A.; Domingo, E.; Menendez-Arias, L. Role of a dipeptide insertion between codons 69 and 70 of HIV-1 reverse transcriptase in the mechanism of AZT resistance. EMBO J. 2000, 19, 5752–5761. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Chen, J.M.; Margot, N.A.; Wrin, T.; Petropoulos, C.J.; Naeger, L.K.; Swaminathan, S.; Miller, M.D. Molecular mechanisms of tenofovir resistance conferred by human immunodeficiency virus type 1 reverse transcriptase containing a diserine insertion after residue 69 and multiple thymidine analog-associated mutations. Antimicrob. Agents Chemother. 2004, 48, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Nucleoside analog resistance caused by insertions in the fingers of human immunodeficiency virus type 1 reverse transcriptase involves ATP-mediated excision. J. Virol. 2002, 76, 9143–9151. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.R.; Lennerstrand, J.; Matsuura, S.E.; Larder, B.A.; Scott, W.A. Effects of dipeptide insertions between codons 69 and 70 of human immunodeficiency virus type 1 reverse transcriptase on primer unblocking, deoxynucleoside triphosphate inhibition, and DNA chain elongation. J. Virol. 2003, 77, 3871–3877. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, T.; Franco, S.; Vazquez-Alvarez, B.M.; Mas, A.; Martinez, M.A.; Menendez-Arias, L. Molecular determinants of multi-nucleoside analogue resistance in HIV-1 reverse transcriptases containing a dipeptide insertion in the fingers subdomain: effect of mutations D67N and T215Y on removal of thymidine nucleotide analogues from blocked DNA primers. J. Biol. Chem. 2004, 279, 24569–24577. [Google Scholar] [CrossRef] [PubMed]
- Hertogs, K.; Bloor, S.; De Vroey, V.; van Den Eynde, C.; Dehertogh, P.; van Cauwenberge, A.; Sturmer, M.; Alcorn, T.; Wegner, S.; van Houtte, M.; Miller, V.; Larder, B.A. A novel human immunodeficiency virus type 1 reverse transcriptase mutational pattern confers phenotypic lamivudine resistance in the absence of mutation 184V. Antimicrob. Agents Chemother. 2000, 44, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Girouard, M.; Diallo, K.; Marchand, B.; McCormick, S.; Gotte, M. Mutations E44D and V118I in the reverse transcriptase of HIV-1 play distinct mechanistic roles in dual resistance to AZT and 3TC. J. Biol. Chem. 2003, 278, 34403–34410. [Google Scholar] [CrossRef] [PubMed]
- Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Barr, R.; Pathak, V.K. Mutations in human immunodeficiency virus type 1 RNase H primer grip enhance 3'-azido-3'-deoxythymidine resistance. J. Virol. 2007, 81, 6837–6845. [Google Scholar] [CrossRef] [PubMed]
- Nikolenko, G.N.; Delviks-Frankenberry, K.A.; Palmer, S.; Maldarelli, F.; Fivash Jr., M.J.; Coffin, J.M.; Pathak, V.K. Mutations in the connection domain of HIV-1 reverse transcriptase increase 3'-azido-3'-deoxythymidine resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 317–322. [Google Scholar] [CrossRef]
- Hachiya, A.; Kodama, E.N.; Sarafianos, S.G.; Schuckmann, M.M.; Sakagami, Y.; Matsuoka, M.; Takiguchi, M.; Gatanaga, H.; Oka, S. Amino acid mutation N348I in the connection subdomain of human immunodeficiency virus type 1 reverse transcriptase confers multiclass resistance to nucleoside and nonnucleoside reverse transcriptase inhibitors. J. Virol. 2008, 82, 3261–3270. [Google Scholar] [CrossRef] [PubMed]
- Yap, S.H.; Sheen, C.W.; Fahey, J.; Zanin, M.; Tyssen, D.; Lima, V.D.; Wynhoven, B.; Kuiper, M.; Sluis-Cremer, N.; Harrigan, P. R.; Tachedjian, G. N348I in the connection domain of HIV-1 reverse transcriptase confers zidovudine and nevirapine resistance. PLoS Med. 2007, 4, e335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehteshami, M.; Beilhartz, G.L.; Scarth, B.J.; Tchesnokov, E.P.; McCormick, S.; Wynhoven, B.; Harrigan, P.R.; Gotte, M. Connection domain mutations N348I and A360V in HIV-1 reverse transcriptase enhance resistance to 3'-azido-3'-deoxythymidine through both RNase H-dependent and -independent mechanisms. J. Biol. Chem. 2008, 283, 22222–22232. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Sarafianos, S.G.; Clark Jr., P.K.; Arnold, E.; Hughes, S.H. Why do HIV-1 and HIV-2 use different pathways to develop AZT resistance? PLoS Pathog. 2006, 2, e10. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bird, L.E.; Chamberlain, P.P.; Stewart-Jones, G.B.; Stuart, D.I.; Stammers, D.K. Structure of HIV-2 reverse transcriptase at 2.35-A resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc. Natl. Acad. Sci. USA 2002, 99, 14410–14415. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Das, K.; Hughes, S.H.; Arnold, E. Taking aim at a moving target: designing drugs to inhibit drug-resistant HIV-1 reverse transcriptases. Curr. Opin. Struct. Biol. 2004, 14, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Nichols, C.; Bird, L.; Chamberlain, P.; Weaver, K.; Short, S.; Stuart, D.I.; Stammers, D.K. Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation non-nucleoside inhibitors. J. Mol. Biol. 2001, 312, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Stammers, D.K. Structural basis for drug resistance mechanisms for non-nucleoside inhibitors of HIV reverse transcriptase. Virus Res. 2008, 134, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Geitmann, M.; Unge, T; Danielson, U.H. Interaction kinetic characterization of HIV-1 reverse transcriptase non-nucleoside inhibitor resistance. J. Med. Chem. 2006, 49, 2375–2387. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Clark Jr., A.D.; Lewi, P.J.; Heeres, J.; De Jonge, M.R.; Koymans, L.M.; Vinkers, H.M.; Daeyaert, F.; Ludovici, D. W.; Kukla, M. J.; De Corte, B.; Kavash, R. W.; Ho, C. Y.; Ye, H.; Lichtenstein, M.A.; Andries, K.; Pauwels, R.; De Bethune, M.P.; Boyer, P.L.; Clark Jr., P.; Hughes, S.H.; Janssen, P. A.; Arnold, E. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J. Med. Chem. 2004, 47, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Singh, K.; Marchand, B.; Kirby, K.A.; Michailidis, E.; Sarafianos, S.G. Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase. Viruses 2010, 2, 606-638. https://doi.org/10.3390/v2020606
Singh K, Marchand B, Kirby KA, Michailidis E, Sarafianos SG. Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase. Viruses. 2010; 2(2):606-638. https://doi.org/10.3390/v2020606
Chicago/Turabian StyleSingh, Kamalendra, Bruno Marchand, Karen A. Kirby, Eleftherios Michailidis, and Stefan G. Sarafianos. 2010. "Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase" Viruses 2, no. 2: 606-638. https://doi.org/10.3390/v2020606