How Flaviviruses Activate and Suppress the Interferon Response

Abstract
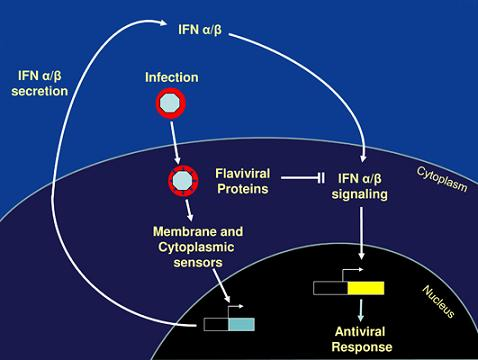
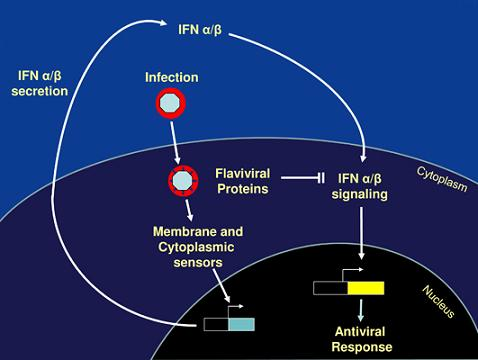
:
{kind=link}
{kind=link}
{kind=link}
Abbreviations
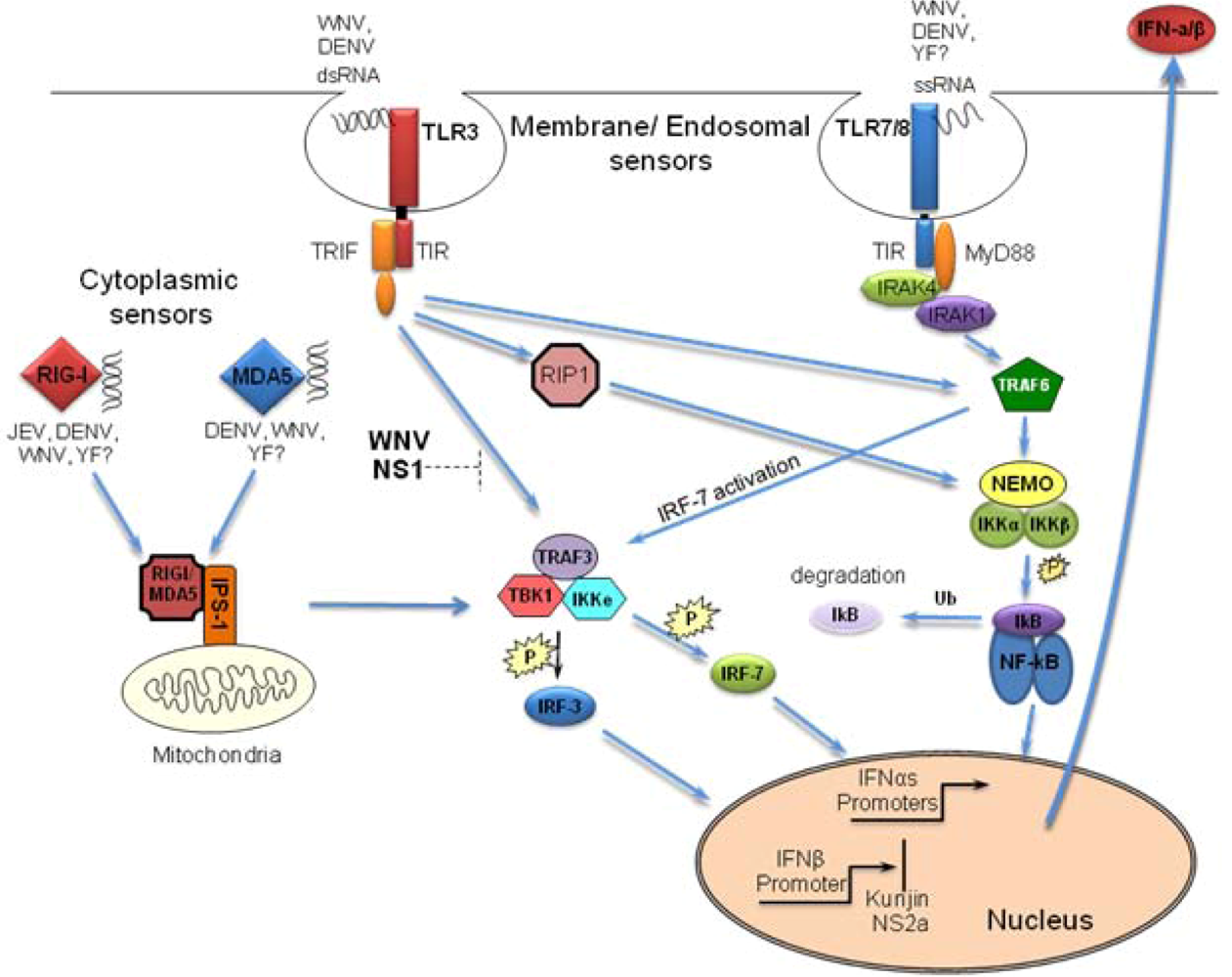
1. Detection of Flaviviruses by the Host Cell
2. Activation of RLR by Flaviviruses

3. Activation of TLRs
4. Evasion of the Host Recognition
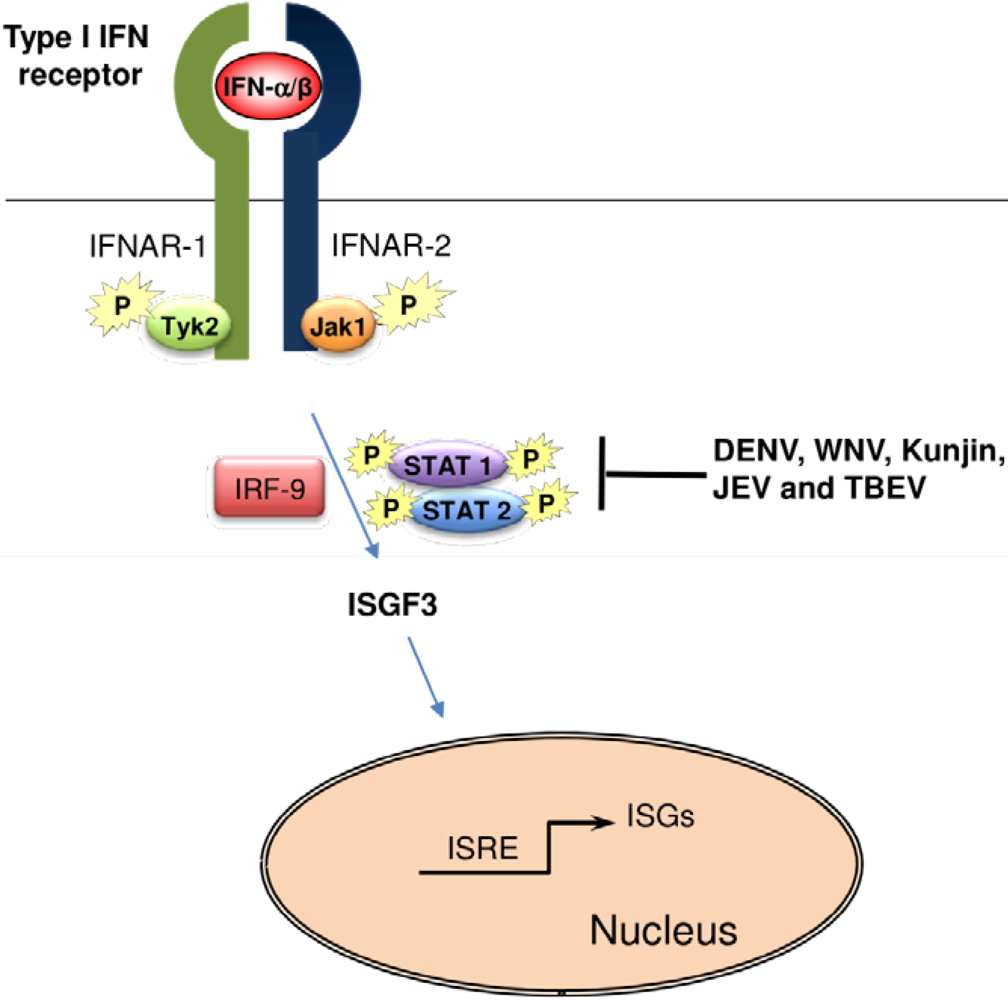
5. Suppression of the IFN-α/β Signaling by Flaviviruses

6. Conclusions
Acknowledgments
References
- Bowie, A.G.; Haga, I.R. The role of Toll-like receptors in the host response to viruses. Mol. Immunol. 2005, 42, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale Jr., M.; Akira, S. Shared and unique functions of the Dexd/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity . J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, J.; Childs, K.S.; Young, D.F.; Carlos, T.S.; Stock, N.; Goodbourn, S.; Randall, R.E. The V proteins of Paramyxoviruses bind the IFN-inducible RNA helicase, MDA-5, and inhibit its activation of the IFN-Beta promoter. Proc. Natl. Acad. Sci. USA 2004, 101, 17264–17269. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I Helicases in the recognition of RNA viruses . Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Eisenacher, K.; Kirchhofer, A.; Brzozka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.K.; Krug, A.; Hopfner, K.P. The C-terminal regulatory domain is the RNA 5'-triphosphate sensor of RIG-I. Mol. Cell 2008, 29, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Takahasi, K.; Yoneyama, M.; Nishihori, T.; Hirai, R.; Kumeta, H.; Narita, R.; Gale Jr., M.; Inagaki, F.; Fujita, T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses . Mol. Cell 2008, 29, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5'-Triphosphate RNA is the ligand for RIG-I . Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, M.; Pichlmair, A.; Martinez-Sobrido, L.; Cros, J.; Garcia-Sastre, A.; Haller, O.; Weber, F. Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol. 2005, 79, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.H.; Liao, C.L.; Lin, Y.L. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-Kappab activation. Microbes Infect. 2006, 8, 157–171. [Google Scholar] [CrossRef]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity . J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Fredericksen, B.L.; Gale Jr., M. West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling . J. Virol. 2006, 80, 291–2923. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Keller, B.C.; Fornek, J.; Katze, M.G.; Gale Jr., M. Establishment and maintenance of the innate antiviral response to West Nile virus involves both RIG-I and MDA5 signaling through IPS-1 . J. Virol. 2008, 82, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Querec, T.D.; Akondy, R.S.; Lee, E.K.; Cao, W.; Nakaya, H.I.; Teuwen, D.; Pirani, A.; Gernert, K.; Deng, J.; Marzolf, B.; et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans . Nat. Immunol. 2009, 10, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Tsunobuchi, H.; Nishimura, H.; Goshima, F.; Daikoku, T.; Suzuki, H.; Nakashima, I.; Nishiyama, Y.; Yoshikai, Y. A protective role of Interleukin-15 in a mouse model for systemic infection with Herpes Simplex virus. Virology 2000, 275, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Akira, S. Toll-like receptors and type I interferons. J. Biol. Chem. 2007, 282, 15319–15323. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis E Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef]
- Wang, J.P.; Liu, P.; Latz, E.; Golenbock, D.T.; Finberg, R.W.; Libraty, D.H. Flavivirus activation of plasmacytoid dendritic cells delineates key elements of TLR7 signaling beyond endosomal recognition. J. Immunol. 2006, 177, 7114–7121. [Google Scholar] [PubMed]
- Severa, M.; Fitzgerald, K.A. TLR-mediated activation of type I IFN during antiviral immune responses: fighting the battle to win the war. Curr. Top. Microbiol. Immunol. 2007, 316, 167–192. [Google Scholar] [PubMed]
- Tsai, Y.T.; Chang, S.Y.; Lee, C.N.; Kao, C.L. Human TLR3 recognizes Dengue virus and modulates viral replication In Vitro. Cell. Microbiol. 2009, 11, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Aleyas, A.G.; George, J.A.; Han, Y.W.; Kim, H.K.; Kim, S.J.; Yoon, H.A.; Eo, S.K. Flaviviruses induce pro-inflammatory and anti-inflammatory cytokines from murine dendritic cells through Myd88-dependent pathway. Immune Network 2007, 66, 66–74. [Google Scholar]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; Flavell, R.A. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Gale Jr., M.; Diamond, M.S. Toll-like receptor 3 has a protective role against West Nile virus infection . J. Virol. 2008, 82, 10349–10358. [Google Scholar] [CrossRef] [PubMed]
- Welte, T.; Reagan, K.; Fang, H.; Machain-Williams, C.; Zheng, X.; Mendell, N.; Chang, G.J.; Wu, P.; Blair, C.D.; Wang, T. Toll-like receptor 7 induced immune response to cutaneous West Nile Virus infection. J. Gen. Virol. 2009, 90, 2660–2668. [Google Scholar] [CrossRef] [PubMed]
- Querec, T.; Bennouna, S.; Alkan, S. Yellow fever vaccine YF-17D Activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J. Exp. Med. 2006, 203, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Elco, C.P.; Guenther, J.M.; Williams, B.R.; Sen, G.C. Analysis of genes induced by Sendai virus infection of mutant cell lines reveals essential roles of interferon regulatory factor 3, NF-kappaB, and interferon but not Toll-like receptor 3. J. Virol. 2005, 79, 3920–3929. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.P.; Kleinman, S.H.; Jackson, B.; Stramer, S.L.; Hewlett, I.; Preston, S. Committee Report Nucleic acid amplification testing of blood donors for transfusion-transmitted infectious diseases: report of the interorganizational task force on nucleic acid amplification testing of blood donors. Transfusion 2000, 40, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Shieh, W.J.; Jung, S.M.; Hsueh, C.; Kuo, T.T.; Mounts, A.; Parashar, U.; Yang, C.F.; Guarner, J.; Ksiazek, T.G.; Dawson, J.; et al. Pathologic studies of fatal cases in outbreak of hand, foot, and mouth disease, taiwan . Emerg. Infect. Dis. 2001, 7, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Suhara, W.; Fukuhara, Y.; Fukuda, M.; Nishida, E.; Fujita, T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/P300. Embo J. 1998, 17, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- tenOever, B.R.; Sharma, S; Zou, W.; Sun, Q.; Grandvaux, N.; Julkunen, I.; Hemmi, H.; Yamamoto, M.; Akira, S.; Yeh, W.C.; et al. Activation of TBK1 and ikkvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity . J. Virol. 2004, 78, 10636–10649. [Google Scholar] [CrossRef] [PubMed]
- Fredericksen, B.; Akkaraju, G.R.; Foy, E.; Wang, C.; Pflugheber, J.; Chen, Z.J.; Gale. M., Jr. Activation of the interferon-beta promoter during Hepatitis C virus RNA replication. Viral Immunol. 2002, 15, 29–40. [Google Scholar] [PubMed]
- Daffis, S.; Samuel, M.A.; Keller, B.C.; Gale Jr., M.; Diamond, M.S. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms . Plos Pathog. 2007, 3, 1005–1015. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Smith, M.; Katze, M.G.; Shi; P.Y.; Gale Jr., M. The host response to West Nile Virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway . J. Virol. 2004, 78, 7737–7747. [Google Scholar] [CrossRef] [PubMed]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef]
- Takaoka, A.; Yanai, H. Interferon signalling network in innate defence. Cell. Microbiol. 2006, 8, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Harris, E. Interferon inhibits Dengue Virus infection by preventing translation of viral RNA through a PKR-independent mechanism. Virology 2001, 289, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Scholle, F.; Mason, P.W. West Nile virus replication interferes with both Poly(I:C)-induced interferon gene transcription and response to interferon treatment. Virology 2005, 342, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.C.; Fredericksen, B.L.; Samuel, M.A.; Mock, R.E.; Mason, P.W.; Diamond, M.S.; Gale Jr., M. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence . J. Virol. 2006, 80, 9424–9434. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.T.; Hayashi, J.; Seeger, C. West Nile Virus inhibits the signal transduction pathway of alpha interferon. J. Virol. 2005, 79, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Pantelic, L.; Sivakumaran, H.; Urosevic, N. Differential induction of antiviral effects against West Nile Virus in primary mouse macrophages derived from flavivirus-susceptible and congenic resistant mice by alpha/beta interferon and Poly(I-C). J. Virol. 2005, 79, 1753–1764. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.A.; Diamond, M.S. Alpha/Beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef] [PubMed]
- Lobigs, M.; Mullbacher, A.; Wang, Y.; Pavy, M.; Lee, E. Role of type I and Type II interferon responses in recovery from infection with an encephalitic flavivirus. J. Gen. Virol. 2003, 84, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.J.; Roehrig, J.T. New Mouse Model For Dengue Virus Vaccine Testing. J. Virol. 1999, 73, 783–786. [Google Scholar] [PubMed]
- Shresta, S.; Sharar, K.L.; Prigozhin, D.M.; Beatty, P.R.; Harris, E. Murine model for dengue virus-induced lethal disease with increased vascular permeability. J. Virol. 2006, 80, 10208–10217. [Google Scholar] [CrossRef] [PubMed]
- Shresta, S.; Sharar, K.L.; Prigozhin, D.M.; Snider, H.M.; Beatty, P.R.; Harris, E. Critical roles for both STAT1-dependent and STAT1-independent pathways in the control of primary Dengue virus infection in mice. J. Immunol. 2005, 175, 3946–3954. [Google Scholar] [PubMed]
- Musso, T.; Gusella, G.L.; Brooks, A.; Longo, D.L.; Varesio, L. Interleukin-4 inhibits indoleamine 2,3-dioxygenase expression in human monocytes. Blood 1994, 83, 1408–1411. [Google Scholar] [PubMed]
- Warke, R.V.; Xhaja, K.; Martin, K.J.; Fournier, M.F.; Shaw, S.K.; Brizuela, N.; De Bosch, N.; Lapointe, D.; Ennis, F.A.; Rothman, A.L.; et al. Dengue virus induces novel changes in gene expression of human umbilical vein endothelial cells . J. Virol. 2003, 77, 11822–11832. [Google Scholar] [CrossRef] [PubMed]
- Warke, R.V.; Becerra, A.; Zawadzka, A.; Schmidt, D.J.; Martin, K.J.; Giaya, K.; Dinsmore, J.H.; Woda, M.; Hendricks, G.; Levine, T.; et al. Efficient Dengue virus (DENV) Infection of human muscle satellite cells upregulates type I interferon response genes and differentially modulates MHC I expression on bystander and DENV-infected cells . J. Gen. Virol. 2008, 89, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Scherbik, S.V.; Stockman, B.M.; Brinton, M.A. Differential expression of interferon (IFN-) regulatory factors and IFN-stimulated genes at early times after West Nile virus infection of mouse embryo fibroblasts. J. Virol. 2007, 81, 12005–12018. [Google Scholar] [CrossRef] [PubMed]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Keller, B.C.; Gale, M.; Diamond, M.S. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection . J. Virol. 2008, 82, 8465–8475. [Google Scholar] [CrossRef] [PubMed]
- Sariol, C.A.; Munoz-Jordan, J.L.; Abel, K.; Rosado, L.C.; Pantoja, P.; Giavedoni, L.; Rodriguez, I.V.; White, L.J.; Martinez, M.; Arana, T.; et al. Transcriptional activation of interferon-stimulated genes but not of cytokine genes after primary infection of rhesus macaques with Dengue virus type 1 . Clin. Vaccine Immunol. 2007, 14, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.; Gu, F.; Ling, L.; Tolfvenstam, T.; Olfat, F.; Chin, K.C.; Aw, P.; George, J.; Kuznetsov, V.A.; Schreiber, M.; et al. Host gene expression profiling of Dengue virus infection in cell lines and patients . Plos Negl. Trop. Dis. 2007, 1, 1–11. [Google Scholar] [CrossRef]
- Ubol, S.; Masrinoul, P.; Chaijaruwanich, J.; Kalayanarooj, S.; Charoensirisuthikul, T.; Kasisith, J. Differences in global gene expression in peripheral blood mononuclear cells indicate a significant role of the innate responses in progression of Dengue fever but not Dengue hemorrhagic fever. J. Infect. Dis. 2008, 197, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Libraty, D.H.; Young, P.R.; Pickering, D.; Endy, T.P.; Kalayanarooj, S.; Green, S.; Vaughn, D.W.; Nisalak, A.; Ennis, F.A.; Rothman, A.L. High circulating levels of the Dengue Virus nonstructural protein NS1 early in Dengue illness correlate with the development of Dengue Hemorrhagic fever. J. Infect. Dis. 2002, 186, 1165–1168. [Google Scholar] [CrossRef] [PubMed]
- Libraty, D.H.; Pichyangkul, S.; Ajariyakhajorn, C.; Endy, T.P.; Ennis, F.A. Human dendritic cells are activated by Dengue virus infection: enhancement by gamma interferon and implications for disease pathogenesis. J. Virol. 2001, 75, 3501–3508. [Google Scholar] [CrossRef] [PubMed]
- Pichyangkul, S.; Endy, T.P.; Kalayanarooj, S.; Nisalak, A.; Yongvanitchit, K.; Green, S.; Rothman, A.L.; Ennis, F.A.; Libraty, D.H. A blunted blood plasmacytoid dendritic cell response to an acute systemic viral infection is associated with increased disease severity. J. Immunol. 2003, 171, 5571–5578. [Google Scholar] [PubMed]
- Munoz-Jordan, J.L.; Sanchez-Burgos, G.G.; Laurent-Rolle, M.; Garcia-Sastre, A. Inhibition of interferon signaling by Dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar] [CrossRef]
- Munoz-Jordan, J.L.; Laurent-Rolle, M.; Ashour, J.; Martinez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; Garcia-Sastre, A. hibition of alpha/beta interferon signaling by the NS4B protein of Flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef] [PubMed]
- Lundin, M.; Monne, M.; Widell, A.; Von Heijne, G.; Persson, M.A. Topology of the membrane-associated Hepatitis C virus protein NS4B. J. Virol. 2003, 77, 5428–5438. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Mcmullan, L.K.; Rice, C.M. Isolation and characterization of noncytopathic pestivirus mutants reveals a role for nonstructural protein NS4B in viral cytopathogenicity. J. Virol. 2001, 75, 10651–10662. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Wang, X.J.; Mokhonov, V.V.; Shi, P.Y.; Randall, R; Khromykh, A.A. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile Virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins . J. Virol. 2005, 79, 1934–1942. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Davidson, A.; Hibbert, L.; Gruenwald, P.; Schlaak, J.; Ball, S.; Foster, G.R.; Jacobs, M. Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J. Virol. 2005, 79, 5414–5420. [Google Scholar] [CrossRef] [PubMed]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; Garcia-Sastre, A. NS5 of Dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Jones, M.; Davidson, A.; Chain, B.; Jacobs, M. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J. Infect. Dis. 2009, 200, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.J.; Hung, L.F.; Weng, C.Y.; Wu, W.L.; Chou, P.; Lin, Y.L.; Chang, D.M.; Tai, T.Y.; Lai, J.H. Dengue virus type 2 antagonizes IFN-alpha but not IFN-gamma antiviral effect via down-regulating Tyk2-STAT signaling in the human dendritic cell. J. Immunol. 2005, 174, 8163–8172. [Google Scholar] [PubMed]
- Lin, R.J.; Liao, C.L.; Lin, E.; Lin, Y.L. Blocking of the alpha interferon-induced Jak-Stat signaling pathway by japanese encephalitis virus infection. J. Virol. 2004, 78, 9285–9294. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M.; Morris, K.L.; Shannon, J.G.; Robertson, S.J.; Mitzel, D.N.; Park, G.S.; Boer, E.; Wolfinbarger, J.B.; Bloom, M.E. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne Flavivirus and identification of NS5 as an interferon antagonist. J. Virol. 2005, 79, 12828–12839. [Google Scholar] [CrossRef] [PubMed]
- Khromykh, A.A.; Sedlak, P.L.; Guyatt, K.J.; Hall, R.A.; Westaway, E.G. Efficient trans-complementation of the Flavivirus Kunjin NS5 protein but not of the NS1 Protein requires its coexpression with other components of the viral replicase. J. Virol. 1999, 73, 10272–10280. [Google Scholar] [PubMed]
- Meylan, E.; Tschopp, J. Toll-like receptors and RNA helicases: Two parallel ways to trigger antiviral responses. Mol. Cell 2006, 22, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Yang, L.; Nakhaei, P.; Sun, Q.; Sharif-Askari, E.; Julkunen, I.; Hiscott, J. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J. Biol. Chem. 2006, 281, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Chen, H.B.; Wang, X.J.; Huang, H.; Khromykh, A.A. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A In inhibition of beta interferon promoter-driven transcription. J. Virol. 2004, 78, 12225–12235. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Muñoz-Jordán, J.L.; Fredericksen, B.L. How Flaviviruses Activate and Suppress the Interferon Response. Viruses 2010, 2, 676-691. https://doi.org/10.3390/v2020676
Muñoz-Jordán JL, Fredericksen BL. How Flaviviruses Activate and Suppress the Interferon Response. Viruses. 2010; 2(2):676-691. https://doi.org/10.3390/v2020676
Chicago/Turabian StyleMuñoz-Jordán, Jorge L., and Brenda L. Fredericksen. 2010. "How Flaviviruses Activate and Suppress the Interferon Response" Viruses 2, no. 2: 676-691. https://doi.org/10.3390/v2020676