Recent Advances in Hepatitis C Virus Cell Entry


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

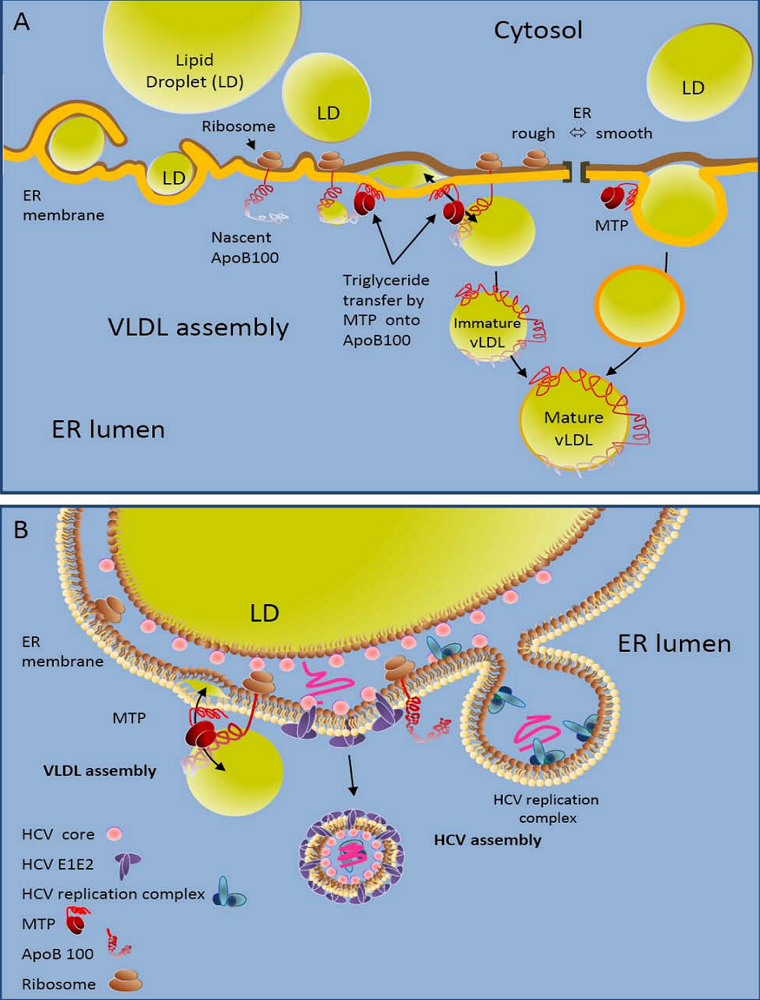
2. HCV Exists in Lipoprotein-Associated Forms
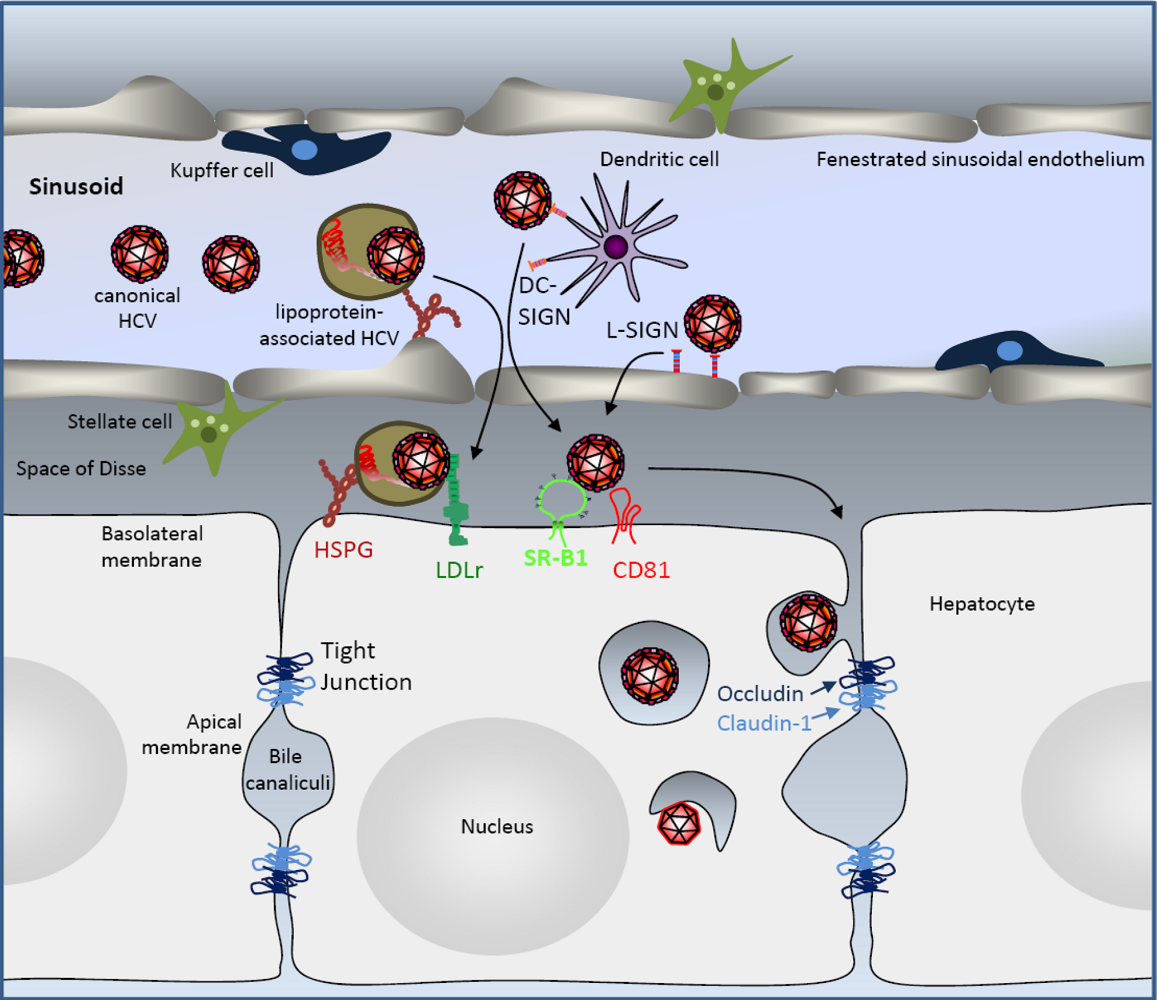
3 .HCV Liver Uptake
4. Hepatocyte Uptake of HCV Particles
4.1. The Tetraspanin CD81
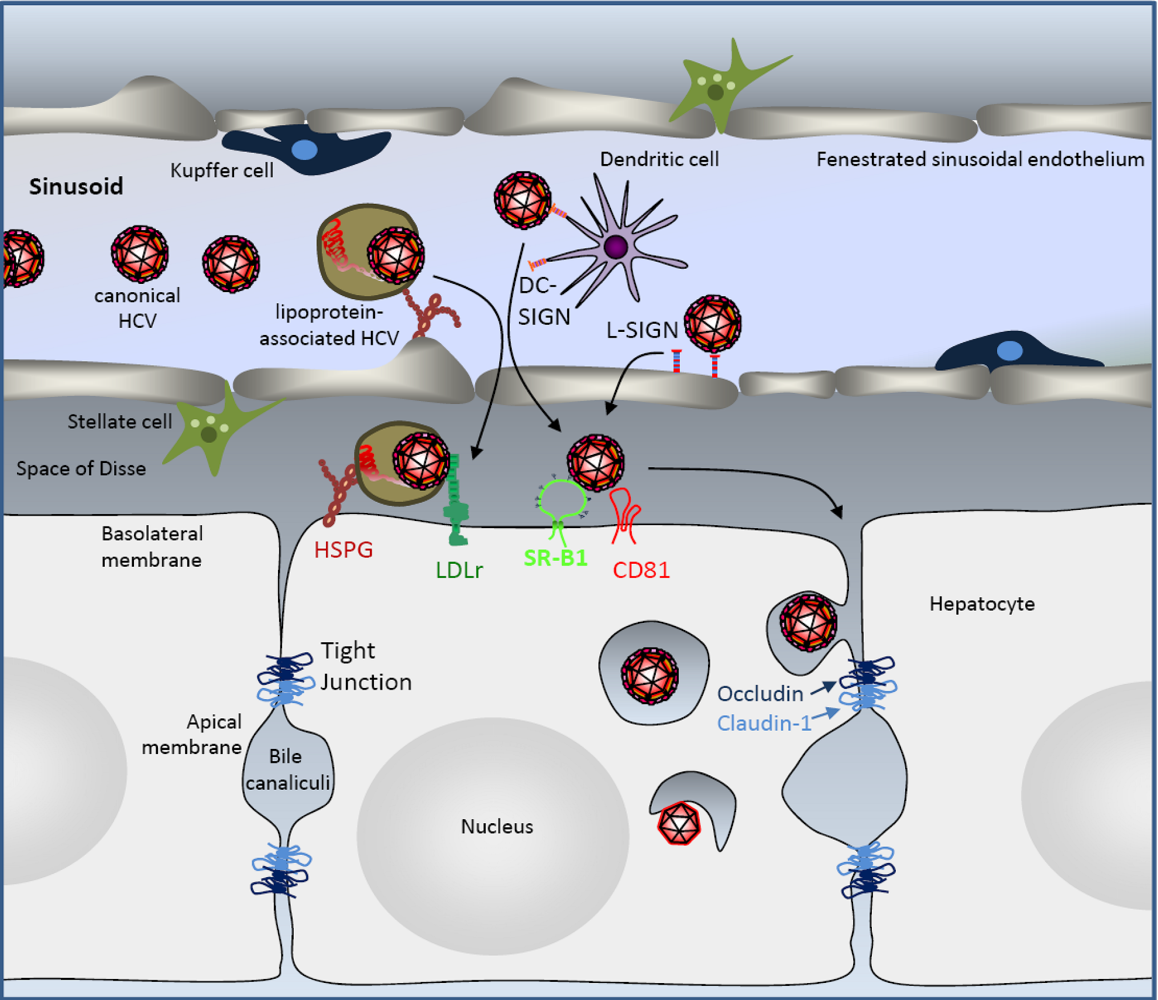
4.2. The Scavenger Receptor BI
4.3. The Tight Junction Factors Claudin-1 and Occludin
4.4. The Route of HCV Cell Entry
5. Contribution of VLDL to Hepatocyte Uptake of HCV
6. Conclusions
Acknowledgments
References
- Alter, M.J. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 2007, 13, 2436–2441. [Google Scholar] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Thiel, H.J.; Rice, C.M. Flaviviridae: The Viruses and Their Replication. in Fields Virology Chapter 1 , 5thKnipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: PA, USA, 2007. [Google Scholar] [PubMed]
- Negro, F. Mechanisms and significance of liver steatosis in hepatitis C virus infection. World J. Gastroenterol. 2006, 12, 6756–6765. [Google Scholar] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Thomssen, R.; Bonk, S.; Propfe, C.; Heermann, K.H.; Kochel, H.G.; Uy, A. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. 1992, 181, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.; McCaustland, K.; Krawczynski, K.; Spelbring, J.; Humphrey, C.; Cook, E.H. Hepatitis C virus: Buoyant density of the factor VIII-derived isolate in sucrose. J. Med. Virol. 1991, 34, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S ; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Meuleman, P.; Ploss, A.; Vanwolleghem, T.; ASyder, A.J.; McKeating, J.A.; Lanford, R.E.; Feinstone, S.M.; Major, M.E.; Leroux-Roels, G.; Rice, C. M. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. USA 2006, 103, 3805–3809. [Google Scholar] [CrossRef]
- Maillard, P.; Andreo, U.; Moreau, M.; Chapman, J.; Budkowska, A. The interaction of natural hepatitis C virus with human scavenger receptor SR-BI/Cla1 is mediated by ApoB-containing lipoproteins . FASEB J. 2006, 20, 735–737. [Google Scholar] [PubMed]
- Hussain, M.M.; Shi, J.; Dreizen, P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J. Lipid Res. 2003, 44, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Diaz, O.; Cubero, M.; Trabaud, M.A.; Quer, J.; Icard, V.;JI Esteban,; Lotteau, V.; Andre, P. Transmission of low-density hepatitis C viral particles during sexually transmitted acute resolving infection . J. Med. Virol. 2008, 80, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H. G.; Mizokami, M.; Bartenschlager, R.; Liang, T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.U.; Bassendine, M.F.; Burt, A.D.; Martin, C.; Pumeechockchai, W.; Toms, G.L. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 2006, 80, 2418–2428. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.U.; Bassendine, M.F.; Martin, C.; Lowther, D.; Purcell, P.J.; King, B.J.; Neely, D.; Toms, G. L. Characterization of hepatitis C RNA-containing particles from human liver by density and size. J. Gen. Virol. 2008, 89, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.M.; Huang, H.; Ye, J.; Gale Jr., M. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 2009, 394, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Perlemuter, G.; Sabile, A.; Letteron, P.; Vona, G.; Topilco, A.; Chretien, Y.; Koike, K.; Pessayre, D.; Chapman, J.; Barba, G.; Brechot, C. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: A model of viral-related steatosis. FASEB J. 2002, 16, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Alaei, M.; Negro, F. Hepatitis C virus and glucose and lipid metabolism. Diabetes Metab. 2008, 34, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Thomssen, R.; Bonk, S.; Thiele, A. Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med. Microbiol. Immunol. 1993, 182, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.C.; Russell, R.S.; Engle, R.E.; Faulk, K.N.; Purcell, R.H.; Emerson, S.U. Apolipoprotein c1 association with hepatitis C virus. J. Virol. 2008, 82, 9647–9656. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale, M.; Ye, J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins . Proc. Natl. Acad. Sci. USA 2007, 104, 5848–5853. [Google Scholar] [CrossRef]
- Diaz, O.; Delers, F.; Maynard, M.; Demignot, S.; Zoulim, F.; Chambaz, J.; Trepo, C.; Lotteau, V.; Andre, P. Preferential association of Hepatitis C virus with apolipoprotein B48-containing lipoproteins. J. Gen. Virol. 2006, 87, 2983–2991. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Nielsen, S.U.; Ibrahim, S.; Bassendine, M.F.; Toms, G.L. Binding of liver derived, low density hepatitis C virus to human hepatoma cells. J. Med. Virol. 2008, 80, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Daneker, G.W.; Lund, S.A.; Caughman, S.W.; Swerlick, R.A.; Fischer, A.H.; Staley, C.A.; Ades, E. W. Culture and characterization of sinusoidal endothelial cells isolated from human liver . In Vitro Cell Dev. Biol. Anim. 1998, 34, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Braet, F.; Riches, J.; Geerts, W.; Jahn, K.A.; Wisse, E.; Frederik, P. Three-dimensional organization of fenestrae labyrinths in liver sinusoidal endothelial cells. Liver Int. 2009, 29, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Yokomori, H. New insights into the dynamics of sinusoidal endothelial fenestrae in liver sinusoidal endothelial cells. Med. Mol. Morphol. 2008, 41, 1–4. [Google Scholar] [CrossRef]
- Breiner, K.M.; Schaller, H.; Knolle, P.A. Endothelial cell-mediated uptake of a hepatitis B virus: A new concept of liver targeting of hepatotropic microorganisms. Hepatology 2001, 34, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Bobardt, M.D.; Chatterji, U.; Selvarajah, S.; Van der Schueren, B.; David, G.; Kahn, B.; Gallay, P.A. Cell-free human immunodeficiency virus type 1 transcytosis through primary genital epithelial cells. J. Virol. 2007, 81, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.B.; Lu, B.; Wang, M.; Rivera, A.A.; Preuss, M.; Zhou, F.; Siegal, G.P.; Alvarez, R.D.; Curiel, D.T. Transport across a polarized monolayer of Caco-2 cells by transferrin receptor-mediated adenovirus transcytosis . Virology 2004, 325, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.S.; Gregorio, G.; Bitton, N.; Hendrickson, W.A.; Littman, D.R. DC-SIGN-mediated internalization of HIV is required for trans-enhancement of T cell infection. Immunity 2002, 16, 135–144. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.C.; Dhadly, P.; Cockerill, G.W.; Cuzzocrea, S.; Mota-Filipe, H.; Hinds, C.J.; Miller, N.E.; Thiemermann, C. Reconstituted high-density lipoprotein attenuates organ injury and adhesion molecule expression in a rodent model of endotoxic shock. Shock 2003, 20, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Simmons, G.; Pohlmann, S.; Baribaud, F.; Ni, H.; Leslie, G.J.; Haggarty, B.S.; Bates, P.; Weissman, D.; Hoxie, J.A.; Doms, R.W. Differential N-linked glycosylation of human immunodeficiency virus and Ebola virus envelope glycoproteins modulates interactions with DC-SIGN and DC-SIGNR. J. Virol. 2003, 77, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, H.; Mitchell, D.A.; Drickamer, K.; Weis, W.I. Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science 2001, 294, 2163–2166. [Google Scholar] [CrossRef] [PubMed]
- Appelmelk, B.J.; van Die, I.; van Vliet, S.J.; Vandenbroucke-Grauls, C.M.; Geijtenbeek, T.B.; van Kooyk, Y. Cutting edge: Carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J. Immunol. 2003, 170, 1635–1639. [Google Scholar] [PubMed]
- Guo, Y.; Feinberg, H.; Conroy, E.; Mitchell, D.A.; Alvarez, R.; Blixt, O.; Taylor, M.E.; Weis, W.I.; Drickamer, K. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR . Nat. Struct. Mol. Biol. 2004, 11, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Van Liempt, E.; Imberty, A.; Bank, C.M.; Van Vliet, S.J.; Van Kooyk, Y.; Geijtenbeek, T.B.; Van Die, I. Molecular basis of the differences in binding properties of the highly related C-type lectins DC-SIGN and L-SIGN to Lewis X trisaccharide and Schistosoma mansoni egg antigens. J. Biol. Chem. 2004, 279, 33161–33167. [Google Scholar] [CrossRef] [PubMed]
- Cormier, E.G.; Durso, R.J.; Tsamis, F.; Boussemart, L.; Manix, C.; Olson, W.C.; Gardner, J.P.; Dragic, T. L-SIGN (CD209L) and DC-SIGN (CD209) mediate transinfection of liver cells by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2004, 101, 14067–14072. [Google Scholar] [CrossRef]
- Gardner, J.P.; Durso, R.J.; Arrigale, R.R.; Donovan, G.P.; Maddon, P.J.; Dragic, T.; Olson, W.C. L-SIGN (CD 209L) is a liver-specific capture receptor for hepatitis C virus. Proc. Natl. Acad. Sci. USA 2003, 100, 4498–4503. [Google Scholar] [CrossRef]
- Lozach, P.Y.; Amara, A. C-type lectins L-SIGN and DC-SIGN capture and transmit infectious hepatitis C virus pseudotype particles. J. Biol. Chem. 2004, 279, 32035–32045. [Google Scholar] [CrossRef] [PubMed]
- Falkowska, E.; Gardner, J.P.; Cormier, E.G.; Arrigale, R.A.; Ogawa, R.N.; Donovan, G.P.; Maddon, P.J.; Olson, W.C.; Dragic, T. L-SIGN (CD209L) isoforms differently mediate trans-infection of hepatoma cells by hepatitis C virus pseudoparticles . J. Ge. Virol. 2006, 87, 2571–2576. [Google Scholar] [CrossRef]
- Williams, K.J.; Fless, G.M.; Petrie, K.A.; Snyder, M.L.; Brocia, R.W.; Swenson, T.L. Mechanisms by which lipoprotein lipase alters cellular metabolism of lipoprotein(a), low density lipoprotein, and nascent lipoproteins. Roles for low density lipoprotein receptors and heparan sulfate proteoglycans. J. Biol. Chem. 1992, 267, 13284–13292. [Google Scholar] [PubMed]
- Saxena, U.; Klein, M.G.; Goldberg, I.G. Identification and characterization of the endothelial cell surface lipoprotein lipase receptor. J. Biol. Chem. 1991, 266, 17516–17521. [Google Scholar] [PubMed]
- Mahley, R.W.; Ji, Z.S. Remnant lipoprotein metabolism: Key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J. Lipid Res. 1999, 40, 1–16. [Google Scholar] [PubMed]
- Sanan, D.A.; Fan, J.; Bensadoun, A.; Taylor, J.M. Hepatic lipase is abundant on both hepatocyte and endothelial cell surfaces in the liver. J. Lipid Res. 1997, 38, 1002–1013. [Google Scholar] [PubMed]
- Andreo, U.; Maillard, P.; Kalinina, O.; Walic, M.; Meurs, E.; Martinot, M.; Marcellin, P.; Budkowska, A. Lipoprotein lipase mediates hepatitis C virus (HCV) cell entry and inhibits HCV infection. Cell Microbiol. 2007, 9, 2445–56. [Google Scholar] [CrossRef]
- Cooper, A.D. Hepatic uptake of chylomicron remnants. J. Lipid Res. 1997, 38, 2173–2192. [Google Scholar] [PubMed]
- Stamataki, Z.; Shannon-Lowe, C.; Shaw, J.; Mutimer, D.; Rickinson, AB.; Gordon, J.; Adams, D.H.; Balfe, P.; McKeating, J.A. Hepatitis C virus association with peripheral blood B lymphocytes potentiates viral infection of liver-derived hepatoma cells. Blood 2009, 113, 585–93. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 2003, 100, 7271–7276. [Google Scholar] [CrossRef]
- Drummer, H.E.; Maerz, A.; Poumbourios, P. Cell surface expression of functional hepatitis C virus E1 and E2 glycoproteins. FEBS Lett. 2003, 546, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J. A.; Rice, C. M. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D. R.; Wieland, S. F.; Uprichard, S. L.; Wakita, T.; Chisari, F. V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef]
- Bartosch, B.; Cosset, F.L. Strategies for retargeted gene delivery using vectors derived from lentiviruses. Curr. Gene. Ther. 2004, 4, 427–443. [Google Scholar] [PubMed]
- Barth, H.; Schafer, C.; Adah, M.I.; Zhang, F.; Linhardt, R.J.; Toyoda, H.; Van Kuppevelt, T.H.; Depla, E.; Von Weizsacker, F.; Blum, H.E.; Baumert, T.F. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003, 278, 41003–41012. [Google Scholar] [CrossRef] [PubMed]
- Germi, R.; Crance, J.M.; Garin, D.; Guimet, J.; Lortat-Jacob, H.; Ruigrok, R.W.; Zarski, J.P.; Drouet, E. Cellular glycosaminoglycans and low density lipoprotein receptor are involved in hepatitis C virus adsorption. J. Med. Virol. 2002, 68, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; Abrignani, S. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Perugini, V.; Lavie, M.; Delgrange, D.; Canton, J.; Pillez, A.; Potel, J.; Lecoeur, C.; Rubinstein, E.; Dubuisson, J.; Wychowski, C.; Cocquerel, L. The association of CD81 with tetraspanin-enriched microdomains is not essential for Hepatitis C virus entry. BMC Microbiol. 2009, 9, 111. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F. L. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 2003, 278, 41624–41630. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.; Castet, V.; Fournier-Wirth, C.; Pichard-Garcia, L.; Avner, R.; Harats, D.; Roitelman, J.; Barbaras, R.; Graber, P.; Ghersa, P.; Smolarsky, M.; Funaro, A.; Malavasi, F.; Larrey, D.; Coste, J.; Fabre, J.M.; Sa-Cunha, A.; Maurel, P. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J. Hepatol. 2007, 46, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Randall, G.; Higginbottom, A.; Monk, P.; Rice, C.M.; McKeating, J.A. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 2004, 78, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Lavillette, D.; Tarr, A.W.; Voisset, C.; Donot, P.; Bartosch, B.; Bain, C.; Patel, A.H.; Dubuisson, J.; Ball, J. K.; Cosset, F.L. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 2005, 41, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Koutsoudakis, G.; Schnober, E.K.; Haberstroh, A.; Blum, H.E.; Cosset, F.L.; Wakita, T.; Jaeck, D.; Doffoel, M.; Royer, C.; Soulier, E.; Schvoerer, E.; Schuster, C.; Stoll-Keller, F.; Bartenschlager, R.; Pietschmann, T.; Barth, H.; Baumert, T.F. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 2007, 46, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Perugini, V.; Montpellier, C.; Delgrange, D.; Wychowski, C.; Helle, F.; Pillez, A.; Drobecq, H.; Le Naour, F.; Charrin, S.; Levy, S.; Rubinstein, E.; Dubuisson, J.; Cocquerel, L. The CD81 partner EWI-2wint inhibits hepatitis C virus entry . PLoS One 2008, 3, e1866. [Google Scholar] [CrossRef] [PubMed]
- Timpe, J.M.; Stamataki, Z.; Jennings, A.; Hu, K.; Farquhar, M.J.; Harris, H.J.; Schwarz, A.; Desombere, I.; Roels, G.L.; Balfe, P.; McKeating, J.A. Hepatit,is C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies . Hepatology 2008, 47, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Witteveldt, J.; Evans, M.J.; Bitzegeio, J.; Koutsoudakis, G.; Owsianka, A.M.; Angus, A.G.; Keck, Z.Y.; Foung, S.K.; Pietschmann, T.; Rice, C.M.; Patel, A.H. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J. Gen. Virol. 2009, 90, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Connelly, M.A. Comparison of class B scavenger receptors, CD36 and scavenger receptor BI (SR-BI), shows that both receptors mediate high density lipoprotein-cholesteryl ester selective uptake but SR-BI exhibits a unique enhancement of cholesteryl ester uptake. J. Biol. Chem. 1999, 274, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C. M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B; Verney, G.; Dreux, M.; Donot, P.; Morice, Y.; Penin, F.; Pawlotsky, J.M.; Lavillette, D.; Cosset, F.L. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies . J. Virol. 2005, 79, 8217–8229. [Google Scholar] [CrossRef] [PubMed]
- Voisset, C.; Callens, N.; Blanchard, E.; Op De Beeck, A.; Dubuisson, J.; Vu-Dac, N. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 2005, 280, 7793–7799. [Google Scholar] [CrossRef] [PubMed]
- von Hahn, T.; Lindenbach, B.D.; Boullier, A.; Quehenberger, O.; Paulson, M.; Rice, C.M.; McKeating, J.A. Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology 2006, 43, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Dao Thi, V. L.; Fresquet, J.; Guerin, M.; Julia, Z.; Verney, G.; Durantel, D.; Zoulim, F.; Lavillette, D.; Cosset, F.L.; Bartosch, B. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains . PLoS Pathog 2009, 5, e1000310. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Kocher, O.; and Krieger, M. Role of the adaptor protein PDZK1 in controlling the HDL receptor SR-BI. Curr. Opin. Lipidol. 2009, 20, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; TB, Xu S.; Acton, S.; Babitt, J.; Krieger, M. The efficient cellular uptake of high density lipoprotein lipids via scavenger receptor class B type I requires not only receptor-mediated surface binding but also receptor-specific lipid transfer mediated by its extracellular domain . J. Biol. Chem. 1998, 273, 26338–26348. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Bertaux, C.; Cukierman, L.; Cormier, E.; Lavillette, D.; Cosset, F.L.; Dragic, T. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J. Virol. 2008, 82, 3555–3560. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, W.; Shen, L.; Turner, J.R.; Coyne, C.B.; Wang, T. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J. Virol. 2009, 83, 2011–2014. [Google Scholar] [CrossRef] [PubMed]
- Mee, C.J.; Grove, J.; Harris, H.J.; Hu, K.; Balfe, P.; McKeating, J.A. Effect of cell polarization on hepatitis C virus entry. J. Virol. 2008, 82, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Mee, C.J.; Farquhar, M.J.; Harris, H.J.; Hu, K.; Ramma, W.; Ahmed, A.; Maurel, P.; Bicknell, R.; Balfe, P.; McKeating, J.A. Hepatitis C virus infection reduces hepatocellular polarity in a vascular endothelial growth factor dependent manner. Gastroenterology 2009, 138, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Shen, L.; Turner, J.R.; Bergelson, J.M. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2007, 2, 181–192. [Google Scholar] [CrossRef]
- Brazzoli, M.; Bianchi, A.; Filippini, S.; Weiner, A.; Zhu, Q.; Pizza, M.; Crotta, S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J. Virol. 2008, 82, 8316–8329. [Google Scholar] [CrossRef] [PubMed]
- Benedicto, I.; Molina-Jimenez, F.; Barreiro, O.; Maldonado-Rodriguez, A.; Prieto, J.; Moreno-Otero, R.; Aldabe, R.; Lopez-Cabrera, M.; Majano, P.L. Hepatitis C virus envelope components alter localization of hepatocyte tight junction-associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology 2008, 48, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Benedicto, I.;; Bartosch, B.; Cosset, F.L.; Lavillette, D.; Prieto, J.; Moreno-Otero, R.; Valenzuela-Fernandez, A.; Aldabe, R.; Lopez-Cabrera, M.; Majano, P. L. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection . J. Virol. 2009, 83, 8012–8020. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, L.; Meertens, L.; Bertaux, C.; Kajumo, F.; Dragic, T. Residues in a highly conserved claudin-1 motif are required for hepatitis C virus entry and mediate the formation of cell-cell contacts. J. Virol. 2009, 83, 5477–5484. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, E.; Belouzard, S.; Goueslain, L.; Wakita, T.; Dubuisson, J.; Wychowski, C.; Rouille, Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 2006, 80, 6964–6972. [Google Scholar] [CrossRef] [PubMed]
- Tscherne, D.M.; Jones, C.T.; Evans, M.J.; Lindenbach, B.D.; McKeating, J.A.; Rice, C.M. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 2006, 80, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Lavillette, D. Hepatitis C virus glycoproteins mediate low pH-dependent membrane fusion with liposomes. J. Biol. Chem. 2006, 281, 3909–3917. [Google Scholar] [CrossRef] [PubMed]
- Roohvand, F.; Maillard, P.; Lavergne, JP.; Boulant, S.; Walic, M.; Andreo, U.; Goueslain, L.; Helle, F.; Mallet, A.; McLauchlan, J.; Budkowska, A. Initiation of hepatitis C virus infection requires the dynamic microtubule network: role of the viral nucleocapsid protein. J. Biol. Chem. 2009, 284, 13778–13791. [Google Scholar] [CrossRef] [PubMed]
- Agnello, V.; Abel, G. Localization of hepatitis C virus in cutaneous vasculitic lesions in patients with type II cryoglobulinemia. Arthritis Rheum. 1997, 40, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Icard, V.; Diaz, O.; Scholtes, C.; Perrin-Cocon, L.; Ramiere, C.; Bartenschlager, R.; Penin, F.; Lotteau, V.; Andre, P. Secretion of hepatitis C virus envelope glycoproteins depends on assembly of apolipoprotein B positive lipoproteins . PLoS One 2009, 4, e4233. [Google Scholar] [CrossRef] [PubMed]
- Beglova, N.; Blacklow, S.C. The LDL receptor: How acid pulls the trigger. Trends Biochem. Sci. 2005, 30, 309–317. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Bartosch, B.; Dubuisson, J. Recent Advances in Hepatitis C Virus Cell Entry. Viruses 2010, 2, 692-709. https://doi.org/10.3390/v2030692
Bartosch B, Dubuisson J. Recent Advances in Hepatitis C Virus Cell Entry. Viruses. 2010; 2(3):692-709. https://doi.org/10.3390/v2030692
Chicago/Turabian StyleBartosch, Birke, and Jean Dubuisson. 2010. "Recent Advances in Hepatitis C Virus Cell Entry" Viruses 2, no. 3: 692-709. https://doi.org/10.3390/v2030692