Complete Genomic Sequence of Bacteriophage Felix O1 †


Abstract
:1. Introduction
2. Results and Discussion
2.1. Genome Assembly and DNA Sequence
2.2. Identification and Analysis of Open Reading Frames (ORFs)

2.3. Nucleotide Metabolism and DNA Replication
2.4. Codon Usage and tRNAs

{kind=link}
{kind=link}
{kind=link}
| tRNA # | tRNA beginning | tRNA end | Amino acid | Anticodon | Codon | Cove Score |
|---|---|---|---|---|---|---|
| 1 | 23699 | 23775 | Pro | TGG | CCA | 68.25 |
| 2 | 23783 | 23860 | Glu | TTC | GAA | 62.48 |
| 3 | 23952 | 24028 | Met | CAT | ATG | 28.90 |
| 4 | 24112 | 24188 | Asn | GTT | AAC | 38.74 |
| 5 | 24258 | 24345 | Pseudo (Tyr) | GTA | TAC | 39.92 |
| 6 | 24351 | 24427 | Asp | GTC | GAC | 61.37 |
| 7 | 24856 | 24931 | Lys | TTT | AAA | 60.20 |
| 8 | 25509 | 25584 | Ile | GAT | ATC | 49.05 |
| 9 | 26975 | 27052 | Leu | TAG | CTA | 52.24 |
| 10 | 27060 | 27135 | Lys(2) | CTT | AAG | 59.65 |
| 11 | 27142 | 27217 | Ala | TGC | GCA | 38.73 |
| 12 | 27224 | 27298 | Gly | TCC | GGA | 56.50 |
| 13 | 27728 | 27804 | Thr | TGT | ACA | 61.15 |
| 14 | 27900 | 27974 | Val | TAC | GTA | 51.84 |
| 15 | 27976 | 28053 | Leu(2) | CAA | TTG | 46.15 |
| 16 | 28168 | 28243 | Arg | ACG | CGT | 63.45 |
| 17 | 28828 | 28903 | Gln | TTG | CAA | 47.31 |
| 18 | 28906 | 28984 | Leu(3) | TAA | TTA | 57.88 |
| 19 | 28990 | 29065 | Gln(2) | CTG | CAG | 50.21 |
| 20 | 29097 | 29172 | Pseudo (His) | GTG | CAC | 42.31 |
| 21 | 29179 | 29254 | Phe | GAA | TTC | 43.77 |
| 22 | 30105 | 30180 | Cys | GCA | TGC | 47.75 |
2.5. Transcription
| Name | Position | Sequence | dG (- kcal/mol) |
|---|---|---|---|
| t2 | 3499..3531 | ggctgcttcggcggccttttttatttgtatttt | 14.00 |
| t5 | 5291..5316 | ggctccttcgggagcctttttcattt | 14.90 |
| t13 | 7947..7994 | gcctttcttatctggtaaatttttcaggtaaggagggctttttcattt | 21.80 |
| t23 | 12345..12368 | gcccctatttaaaggggctttttt | 11.70 |
| t29 | complement(16583..16616) | gggagctatcgagaggtagttccctttttagttt | 18.20 |
| t35 | complement(18652..18680) | ggctccttcgggagccttttttattttct | 14.90 |
| t38 | complement(20419..20445) | ggggctgatgcccctttaactatttat | 10.90 |
| t43 | complement(23542..23572) | gggtattaaacacattgtcaatacccttttt | 9.20 |
| t42 | 23547..23575 | gggtattgacaatgtgtttaatacccttt | 10.20 |
| t64 | 41180..41205 | gggggagactttaaaaggtcttccccttttttgtttctttt | 18.00 |
| t76 | 51080..51100 | gggggctgtacagccctcttt | 12.50 |
| t79 | 54120..54149 | ggctcctttttacgggagcctttttgtttt | 12.50 |
| t80 | complement(54105..54140) | ggctcccgtaaaaaggagccttaaaattttatttt | 11.50 |
| t104 | 69298..69323 | gccctgtacttagtatggggcttttt | 12.20 |
| t111 | 73004..73025 | gagcctcttcggaggctctttt | 16.10 |
| t116 | 77111..77131 | ggtctcttcggagaccttttt | 12.80 |
| t124 | 81488..81516 | gggaactgtaaaggttccctttttatttt | 10.60 |
| TIME (pi) | ||||
|---|---|---|---|---|
| ORFs maximally expressed | 5 min | 10 min | 25 min | 60 min |
| 13, 16, 20, 21, 22, 31, 45, 46, 48, 49, 54, 58, 62, 69, 70, 71, 74, 75, 79, 95, 105 | + | |||
| 2, 5,9,12, 81 | + | |||
| 1, 3, 4, 6,10,14,17,19, 23, 24, 25, 26, 28, 29, 32, 33, 34, 35, 38, 41, 42, 44, 52, 55, 57, 59, 61, 66, 67, 68, 78, 80, 83, 84, 87, 88, 89, 90, 93, 96, 97, 98, 99, b101, 104 , 108, 111, 112, 114, 115, 120, 122, 124, 125, 126, 127, 128, 129, 130 | + | |||
| 30, 36, 37, 51, 53, 63, 64, 72, 73, 76, 77, 106, 107, 109, 117, 121 | + | |||
2.6. Packaging and Morphogenesis
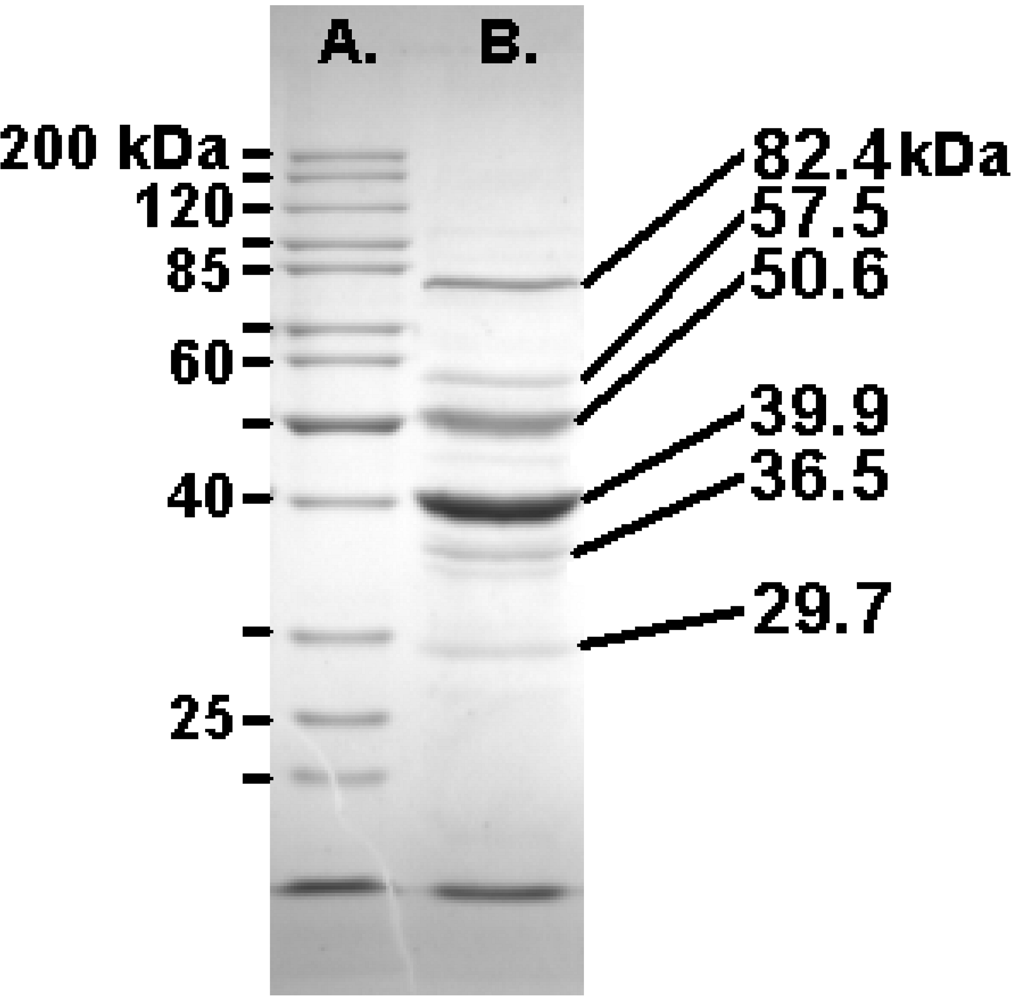
2.7. The Felix O1 Proteome
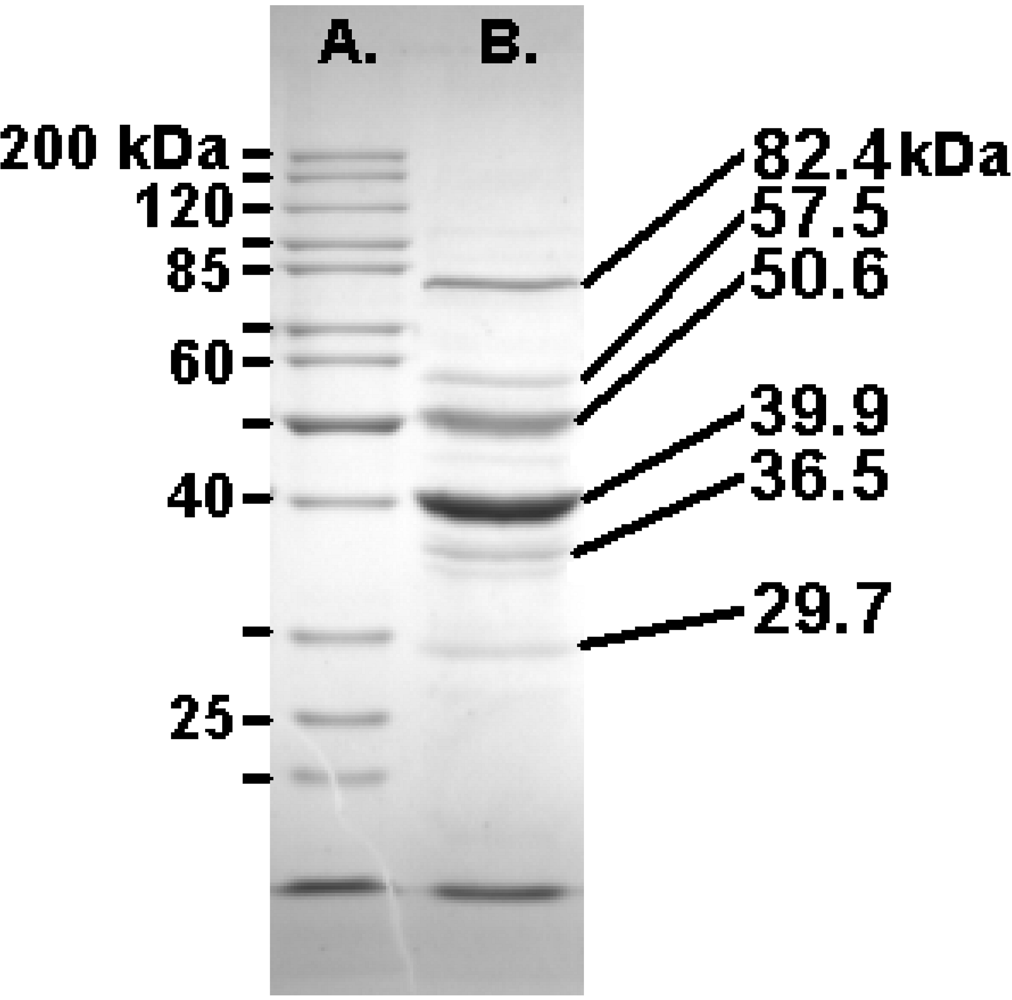
2.8. Lysis
| A | |||||
|---|---|---|---|---|---|
| Annotation | Mass | Score | Peptides | % Coverage | |
| gp77 | tail fiber protein | 84006 | 1310 | 19 | 31 |
| gp73 | conserved protein | 53071 | 1244 | 22 | 58 |
| gp67 | conserved protein | 80321 | 1218 | 24 | 40 |
| gp63 | conserved structural protein | 48918 | 1072 | 22 | 41 |
| gp58 | capsid protein | 41580 | 958 | 20 | 48 |
| gp36 | tail protein | 31610 | 813 | 14 | 44 |
| gp68 | conserved protein | 28700 | 612 | 10 | 29 |
| gp54 | hypothetical protein | 18526 | 498 | 10 | 52 |
| gp74 | hypothetical protein | 31597 | 444 | 10 | 42 |
| gp53 | conserved protein | 55525 | 399 | 12 | 25 |
| gp57 | hypothetical protein | 13728 | 372 | 8 | 44 |
| gp72 | conserved protein | 15648 | 309 | 5 | 38 |
| gp64 | conserved protein | 16268 | 262 | 5 | 40 |
| gp69 | conserved structural protein | 13289 | 211 | 4 | 41 |
| gp55 | hypothetical protein | 11715 | 165 | 3 | 30 |
| gp23 | hypothetical protein | 9679 | 134 | 4 | 53 |
| gp59 | hypothetical protein | 17233 | 119 | 5 | 28 |
| B | |||||
 | |||||
2.9. Introns and Homing Endonucleases
3. Experimental Section
3.1. Phage and Bacterial Strains
3.2. Isolation of Felix O1 DNA
3.3. Sequencing Strategy
3.4. RT-PCR Methods
3.5. DNA Sequence Analysis
3.6. Proteomics of Felix O1
3.7. Genome Sequence Accession Number
4. Conclusions
Acknowledgments
References
- Ackermann, H.W. Tailed bacteriophages: the order Caudovirales. Adv. Virus Res. 1998, 51, 135–201. [Google Scholar] [PubMed]
- Ackermann, H.W. Salmonella Phages Observed in the Electron Microscope. In: Salmonella Methods and Protocols. 2007, 213–234. [Google Scholar]
- Kropinski, A.M.; Sulakvelidze, A.; Konczy, P.; Poppe, C. Salmonella Phages and Prophages - Genomics and Practical Aspects. In Salmonella Methods and Protocols; Schatten, H., Eisenstark, A., Eds.; Humana Press: Totowa, NJ, USA, 2007; pp. 133–176. [Google Scholar]
- Villafane, R.; Zayas, M.; Gilcrease, E.B.; Kropinski, A.M.; Casjens, S.R. Genomic analysis of bacteriophage ε34 of Salmonella enterica serovar Anatum (15+). BMC Microbiol. 2008, 8, 227. [Google Scholar] [CrossRef]
- Kwon, H.J.; Cho, S.H.; Kim, T.E.; Won, Y.J.; Jeong, J.; Park, S.C.; Kim, J.H.; Yoo, H.S.; Park, Y.H.; Kim, S.J. Characterization of a T7-like lytic bacteriophage (phiSG-JL2) of Salmonella enterica serovar gallinarum biovar gallinarum. Appl. Environ. Microbiol. 2008, 74, 6970–6979. [Google Scholar] [CrossRef] [PubMed]
- Pickard, D.; Thomson, N.R.; Baker, S.; Wain, J.; Pardo, M.; Goulding, D.; Hamlin, N.; Choudhary, J.; Threfall, J.; Dougan, G. Molecular characterization of the Salmonella enterica serovar Typhi Vi-typing bacteriophage E1. J. Bacteriol. 2008, 190, 2580–2587. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W.; DuBow, M.S. Viruses of Prokaryotes; CRC Press: Boca Raton, FL, USA, 1987. [Google Scholar]
- Felix, A.; Callow, B.R. Typing of paratyphoid B bacilli by means of Vi bacteriophage. Brit. Med. J. 1943, 2, 4308–4310. [Google Scholar] [CrossRef]
- Welkos, S.; Schreiber, M.; Baer, H. Identification of Salmonella with the O-1 bacteriophage. Appl. Microbiol. 1974, 28, 618–622. [Google Scholar] [PubMed]
- Kallings, L.O.; Lindberg, A.A. Resistance to Felix 0-1 phage in salmonella bacteria. Acta Pathol. Microbiol. Scand. 1967, 70, 455–460. [Google Scholar] [PubMed]
- Kallings, L.O. Sensitivity of various salmonella strains to felix 0-1 phage . Acta Pathol. Microbiol. Scand. 1967, 70, 446–454. [Google Scholar] [PubMed]
- Hudson, H.P.; Lindberg, A.A.; Stocker, B.A. Lipopolysaccharide core defects in Salmonella typhimurium mutants which are resistant to Felix O phage but retain smooth character. J. Gen. Microbiol. 1978, 109, 97–112. [Google Scholar] [PubMed]
- MacPhee, D.G.; Krishnapillai, V.; Roantree, R.J.; Stocker, B.A. Mutations in Salmonella typhimurium conferring resistance to Felix O phage without loss of smooth character. J. Gen. Microbiol. 1975, 87, 1–10. [Google Scholar] [PubMed]
- Whichard, J.M.; Sriranganathan, N.; Pierson, F.W. Suppression of Salmonella growth by wild-type and large-plaque variants of bacteriophage Felix O1 in liquid culture and on chicken frankfurters. J. Food Prot. 2003, 66, 220–225. [Google Scholar] [PubMed]
- Hirsh, D.C.; Martin, L.D. Rapid detection of Salmonella spp. by using Felix-O1 bacteriophage and high-performance liquid chromatography. Appl. Environ. Microbiol. 1983, 45, 260–264. [Google Scholar] [PubMed]
- Kuhn, J.C. Detection of Salmonella by Bacteriophage Felix 01. In Salmonella Methods and Protocols; Schatten, H., Eisenstark, A., Eds.; Humana Press: Totowa, NJ, USA, 2007; pp. 21–37. [Google Scholar]
- Kuhn, J.; Suissa, M.; Wyse, J.; Cohen, I.; Weiser, I.; Reznick, S.; Lubinsky-Mink, S.; Stewart, G.; Ulitzur, S. Detection of bacteria using foreign DNA: the development of a bacteriophage reagent for Salmonella. Int. J. Food Microbiol. 2002, 74, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.; Suissa, M.; Wyse, J.; Cohen, I.; Weiser, I.; Reznick, S.; Lubinsky-Mink, S.; Stewart, G.; Ulitzur, S.; Kuhn, J.; Suissa, M.; Wyse, J.; Cohen, I.; Weiser, I.; Reznick, S.; Lubinsky-Mink, S.; Stewart, G.; Ulitzur, S. Detection of bacteria using foreign DNA: The development of a bacteriophage reagent for Salmonella. Int. J. Food Microbiol. 2002, 74, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE: Restriction enzymes and methyltransferases. Nucl. Acids Res. 2003, 31, 418–420. [Google Scholar] [CrossRef]
- Carver, T.; Thomson, N.; Bleasby, A.; Berriman, M.; Parkhill, J. DNAPlotter: Circular and linear interactive genome visualization. Bioinformatics 2009, 25, 119–120. [Google Scholar] [CrossRef] [PubMed]
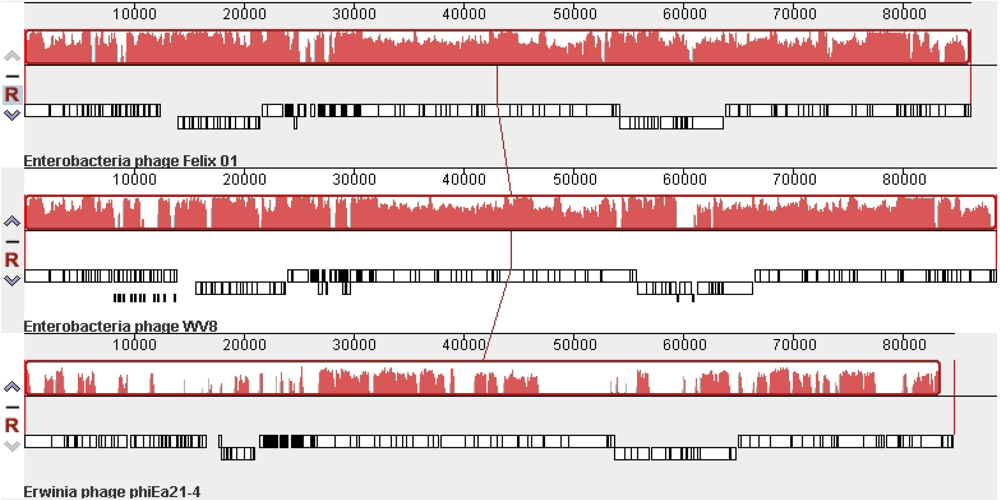
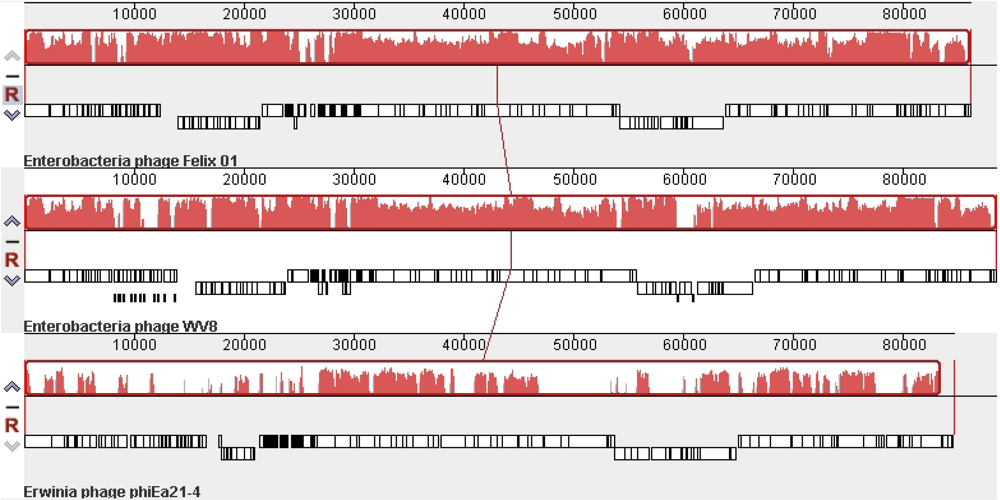
- Lehman, S.M.; Kropinski, A.M.; Castle, A.J.; Svircev, A.M. Complete genome of the broad-host-range Erwinia amylovora phage φEa21-4 and its relationship to Salmonella phage felix O1. Appl. Environ. Microbiol. 2009, 75, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Villegas, A.; She, Y.M.; Kropinski, A.M.; Lingohr, E.J.; Mazzocco, A.; Ojha, S.; Waddell, T.E.; Ackermann, H.W.; Moyles, D.M.; Ahmed, R.; Johnson, R.P. The genome and proteome of a virulent Escherichia coli O157:H7 bacteriophage closely resembling Salmonella phage Felix O1. Virology J. 2009, 6, 41. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Borodovsky, M.; Carver, T.J.; Cerdeno-Tarraga, A.M.; Darling, A.; Lomsadze, A.; Mahadevan, P.; Stothard, P.; Seto, D.; Van, D.G.; Wishart, D.S. In silico identification of genes in bacteriophage DNA. Bacteriophages Methods and Protocols 2009, 2, 57–89. [Google Scholar]
- Zafar, N.; Mazumder, R.; Seto, D. CoreGenes: A computational tool for identifying and cataloging "core" genes in a set of small genomes. BMC Bioinformatics 2002, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Darius, P.; Summer, E.J.; Seto, D.; Mahadevan, P.; Nilsson, A.S.; Ackermann, H.W.; Kropinski, A.M. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009, 9, 224. [Google Scholar] [CrossRef]
- Hove-Jensen, B.; Harlow, K.W.; King, C.J.; Switzer, R.L. Phosphoribosylpyrophosphate synthetase of Escherichia coli. Properties of the purified enzyme and primary structure of the prs gene. J.Biol.Chem. 1986, 261, 6765–6771. [Google Scholar]
- Lundin, D.; Torrents, E.; Poole, A.M.; Sjoberg, B.M. RNRdb, a curated database of the universal enzyme family ribonucleotide reductase, reveals a high level of misannotation in sequences deposited to Genbank. BMC Genomics. 2009, 10, 589. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, A.T.; George, M.; Basham, D.A.; Ford, M.E.; Houtz, J.M.; Pedulla, M.L.; Lawrence, J.G.; Hatfull, G.F.; Hendrix, R.W. Complete genomic sequence of the virulent Salmonella bacteriophage SP6 . J. Bacteriol. 2004, 186, 1933–1944. [Google Scholar] [CrossRef] [PubMed]
- Scholl, D.; Kieleczawa, J.; Kemp, P.; Rush, J.; Richardson, C.C.; Merril, C.; Adhya, S.; Molineux, I.J. Genomic analysis of bacteriophages SP6 and K1-5, an estranged subgroup of the T7 supergroup. J. Mol. Biol. 2004, 335, 1151–1171. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.J.; Studier, F.W. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 1983, 166, 477–535. [Google Scholar] [CrossRef] [PubMed]
- Villegas, A.; Kropinski, A.M. An analysis of initiation codon utilization in the Domain Bacteria - concerns about the quality of bacterial genome annotation. Microbiology 2008, 154, 2559–2661. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucl. Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Chibani-Chennoufi, S.; Dillmann, M.L.; Marvin-Guy, L.; Rami-Shojaei, S.; Brüssow, H. Lactobacillus plantarum bacteriophage LP65: A new member of the SPO1-like genus of the family Myoviridae. J. Bacteriol. 2004, 186, 7069–7083. [Google Scholar] [CrossRef] [PubMed]
- Carlton, R.M.; Noordman, W.H.; Biswas, B.; de Meester, E.D.; Loessner, M.J. Bacteriophage P100 for control of Listeria monocytogenes in foods: Genome sequence, bioinformatic analyses, oral toxicity study, and application. Regul. Toxicol. Pharmacol. 2005, 43, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Lopez, R.; Garcia, E.; Romero, P.; Lopez, R.; Garcia, E. Genomic organization and molecular analysis of the inducible prophage EJ-1, a mosaic myovirus from an atypical pneumococcus. Virology 2004, 322, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.R.; Gilcrease, E.B.; Huang, W.M.; Bunny, K.L.; Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Hatfull, G.F.; Hendrix, R.W.; Casjens, S.R.; Gilcrease, E.B.; Huang, W.M.; Bunny, K.L.; Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Hatfull, G.F.; Hendrix, R.W. The pKO2 linear plasmid prophage of Klebsiella oxytoca. J. Bacteriol. 2004, 186, 1818–1832. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Elkan, C. The value of prior knowledge in discovering motifs with MEME. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995, 3, 21–29. [Google Scholar] [PubMed]
- Lavigne, R.; Noben, J.P.; Hertveldt, K.; Ceyssens, P.J.; Briers, Y.; Dumont, D.; Roucourt, B.; Krylov, V.N.; Mesyanzhinov, V.V.; Robben, J.; Volckaert, G. The structural proteome of Pseudomonas aeruginosa bacteriophage φKMV. Microbiology 2006, 152, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Ceyssens, P.J.; Robben, J. Phage proteomics: Applications of mass spectrometry. In Bacteriophages: Methods and Protocols; Clokie, M.R.J., Kropinski, A., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 2, pp. 239–251. [Google Scholar]
- Miller, E.C.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, V.; Soldo, B.; Dusterhoft, A.; Hilbert, H.; Mauel, C.; Karamata, D. Introns and intein coding sequence in the ribonucleotide reductase genes of Bacillus subtilis temperate bacteriophage SPβ. Proc. Natl. Acad. Sci. USA 1998, 95, 1692–1697. [Google Scholar] [CrossRef]
- Mann, N.H.; Clokie, M.R.; Millard, A.; Cook, A.; Wilson, W.H.; Wheatley, P.J.; Letarov, A.; Krisch, H.M. The genome of S-PM2, a "photosynthetic" T4-type bacteriophage that infects marine Synechococcus strains. J. Bacteriol. 2005, 187, 3188–3200. [Google Scholar] [CrossRef] [PubMed]
- O'Flaherty, S.; Coffey, A.; Edwards, R.; Meaney, W.; Fitzgerald, G.F.; Ross, R.P. Genome of staphylococcal phage K: A new lineage of Myoviridae infecting gram-positive bacteria with a low G+C content. J. Bacteriol. 2004, 186, 2862–2871. [Google Scholar] [CrossRef] [PubMed]
- Yuzenkova, J.; Nechaev, S.; Berlin, J.; Rogulja, D.; Kuznedelov, K.; Inman, R.; Mushegian, A.; Severinov, K.; Yuzenkova, J.; Nechaev, S.; Berlin, J.; Rogulja, D.; Kuznedelov, K.; Inman, R.; Mushegian, A.; Severinov, K. Genome of Xanthomonas oryzae bacteriophage Xp10: An odd T-odd phage. J. Mol. Biol. 2003, 330, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D. Bacteriophages . Interscience Publishers, Inc.: New York, NY, USA, 1959. [Google Scholar]
- Borris, D.J. Temporal analysis of bacteriophage Felix O1 gene expression. M.Sc.Thesis. Virginia Polytechnic Institute and State University, Blacksburg, VA, USA, 2002; pp. 1–58. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucl. Acids Res. 1997, 25, 3389–4022. [Google Scholar] [CrossRef]
- Nakamura, Y.; Gojobori, T.; Ikemura, T. Codon usage tabulated from the international DNA sequence databases: Status for the year 2000. Nucl. Acids Res. 2000, 28, 292. [Google Scholar] [CrossRef]
- Ermolaeva, M.D.; Khalak, H.G.; White, O.; Smith, H.O.; Salzberg, S.L. Prediction of transcription terminators in bacterial genomes. J. Mol. Biol. 2000, 301, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Abreu-Goodger, C.; Merino, E. RibEx: A web server for locating riboswitches and other conserved bacterial regulatory elements . Nucl. Acids Res. 2005, 33, W690–W692. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 2001; pp. 1.1–7.94. [Google Scholar]
- Clauser, K.R.; Baker, P.; Burlingame, A.L. Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal. Chem. 1999, 71, 2871–2882. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.L.; Segall, A.M.; Steward, G.; Seguritan, V.; Breitbart, M.; Wolven, F.; Azam, F. The complete genomic sequence of the marine phage Roseophage SIO1shares homology with nonmarine phages. Limnol. Oceanogr. 2000, 45, 408–418. [Google Scholar] [CrossRef]
- Wietzorrek, A.; Schwarz, H.; Herrmann, C.; Braun, V. The genome of the novel phage Rtp, with a rosette-like tail tip, is homologous to the genome of phage T1. J. Bacteriol. 2006, 188, 1419–1436. [Google Scholar] [CrossRef] [PubMed]
- German, G.J.; Misra, R.; Kropinski, A.M. The T1-like bacteriophages. In The Bacteriophages, 2nd; Calendar, R.L., Ed.; Oxford University Press: New York, NY,USA, 2006; pp. 211–224. [Google Scholar]
- Ceyssens, P.J.; Lavigne, R.; Mattheus, W.; Chibeu, A.; Hertveldt, K.; Mast, J.; Robben, J.; Volckaert, G. Genomic analysis of Pseudomonas aeruginosa phages LKD16 and LKA1: Establishment of the φKMV subgroup within the T7 supergroup. J. Bacteriol. 2006, 188, 6924–6931. [Google Scholar] [CrossRef] [PubMed]
- Perry, L.L.; SanMiguel, P.; Minocha, U.; Terekhov, A.I.; Shroyer, M.L.; Farris, L.A.; Bright, N.; Reuhs, B.L.; Applegate, B.M. Sequence analysis of Escherichia coli O157:H7 bacteriophage φV10 and identification of a phage-encoded immunity protein that modifies the O157 antigen. FEMS Microbiol. Lett. 2009, 292, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.-W.; Kropinski, A.M. Unifying classical and molecular taxonomy-based classification: A rational classification system for the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 59, 406–414. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Whichard, J.M.; Weigt, L.A.; Borris, D.J.; Li, L.L.; Zhang, Q.; Kapur, V.; Pierson, F.W.; Lingohr, E.J.; She, Y.-M.; Kropinski, A.M.; et al. Complete Genomic Sequence of Bacteriophage Felix O1. Viruses 2010, 2, 710-730. https://doi.org/10.3390/v2030710
Whichard JM, Weigt LA, Borris DJ, Li LL, Zhang Q, Kapur V, Pierson FW, Lingohr EJ, She Y-M, Kropinski AM, et al. Complete Genomic Sequence of Bacteriophage Felix O1. Viruses. 2010; 2(3):710-730. https://doi.org/10.3390/v2030710
Chicago/Turabian StyleWhichard, Jean M., Lee A. Weigt, Douglas J. Borris, Ling Ling Li, Qing Zhang, Vivek Kapur, F. William Pierson, Erika J. Lingohr, Yi-Min She, Andrew M. Kropinski, and et al. 2010. "Complete Genomic Sequence of Bacteriophage Felix O1" Viruses 2, no. 3: 710-730. https://doi.org/10.3390/v2030710