Possibilities for RNA Interference in Developing Hepatitis C Virus Therapeutics

{kind=link}
Abstract
:1. A brief history of RNA therapeutics
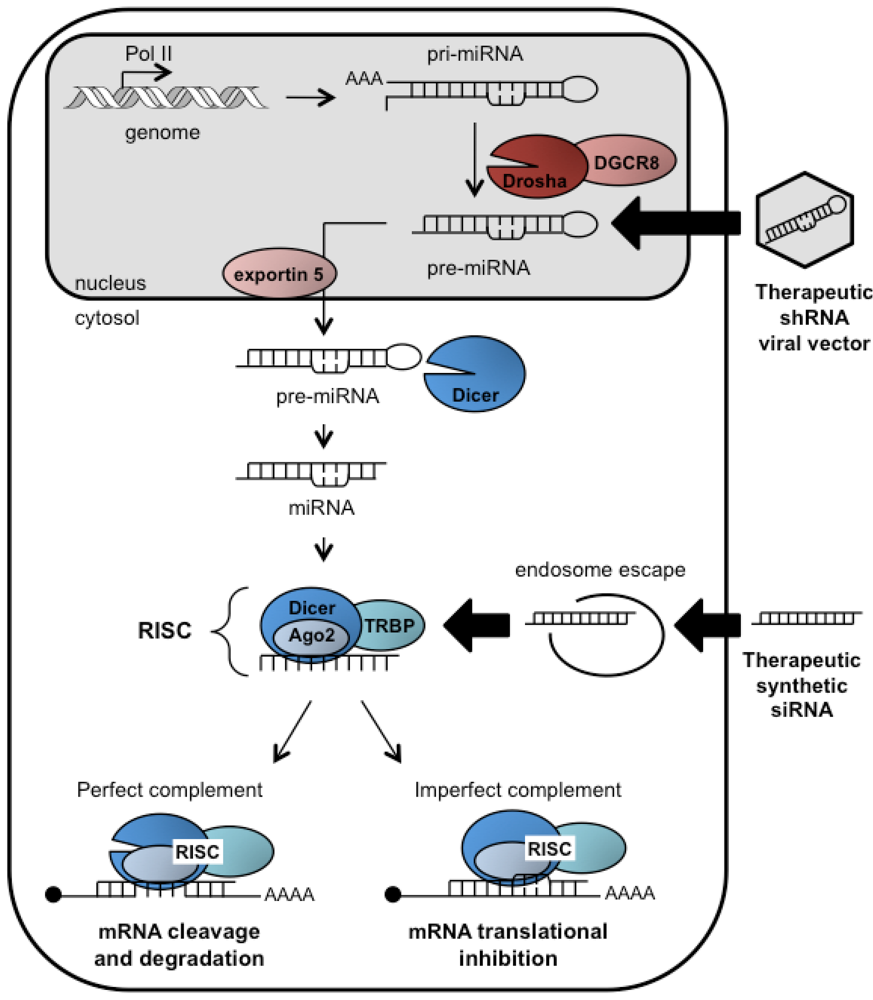
2. The RNAi pathway

3. RNAi and HCV
4. Advantages & pitfalls of RNAi therapeutics
5. siRNA drugs in clinical trials
6. The final frontier for RNAi therapeutics for HCV: effective siRNA delivery to the liver
7. Conclusions and outlook
Acknowledgments
References
- Dias, N.; Stein, C.A. Antisense oligonucleotides: basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar] [PubMed]
- Orr, R.M. Technology evaluation: fomivirsen, Isis Pharmaceuticals Inc/CIBA vision. Curr. Opin. Mol. Ther. 2001, 3, 288–294. [Google Scholar] [PubMed]
- Uhlenbeck, O.C. A small catalytic oligoribonucleotide. Nature 1987, 328, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, S.T.; Eckstein, F. Structure-function relationships of hammerhead ribozymes: from understanding to applications. Trends Biotechnol. 1995, 13, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Sullenger, B.A.; Gallardo, H.F.; Ungers, G.E.; Gilboa, E. Overexpression of TAR sequences renders cells resistant to human immunodeficiency virus replication. Cell 1990, 63, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Paddison, P.J.; Caudy, A.A.; Bernstein, E.; Hannon, G.J.; Conklin, D.S. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002, 16, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef] [PubMed]
- MacRae, I.J.; Zhou, K.; Doudna, J.A. Structural determinants of RNA recognition and cleavage by Dicer. Nat. Struct. Mol. Biol. 2007, 14, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kolb, F.A.; Jaskiewicz, L.; Westhof, E.; Filipowicz, W. Single processing center models for human Dicer and bacterial RNase III. Cell 2004, 118, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Chendrimada, T.P.; Cooch, N.; Shiekhattar, R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 2005, 123, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D.P.; Zamore, P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005, 123, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Hutvagner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. Embo J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, D.; Xue, H.; Taylor, D.W.; Patnode, H.; Mishima, Y.; Cheloufi, S.; Ma, E.; Mane, S.; Hannon, G.J.; Lawson, N.D.; et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 2010, 328, 1694–1698. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C. Micro RNAs are complementary to 3' UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002, 30, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; Nam, J.-W.; Farh, K.K.-H.; Chiang, H.R.; Shkumatava, A.; Bartel, D.P. Expanding the microRNA targeting code: functional sites with centered pairing. Mol. Cell. 2010, 38, 789–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, E.; Lee, S.K.; Wang, J.; Ince, N.; Ouyang, N.; Min, J.; Chen, J.; Shankar, P.; Lieberman, J. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat. Med. 2003, 9, 347–351. [Google Scholar] [CrossRef] [PubMed]
- de Fougerolles, A.; Vornlocher, H.P.; Maraganore, J.; Lieberman, J. Interfering with disease: a progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007, 6, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Aigner, A. Applications of RNA interference: current state and prospects for siRNA-based strategies in vivo. Appl. Microbiol. Biotechnol. 2007, 76, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; McManus, M.T.; Nguyen, T.; Shen, C.H.; Sharp, P.A.; Eisen, H.N.; Chen, J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 2718–2723. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, S.M.; Lo, C.Y.; Tumpey, T.M.; Epstein, S.L. Protection against lethal influenza virus challenge by RNA interference in vivo. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 8682–8686. [Google Scholar] [CrossRef] [PubMed]
- Bitko, V.; Musiyenko, A.; Shulyayeva, O.; Barik, S. Inhibition of respiratory viruses by nasally administered siRNA. Nat. Med. 2005, 11, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, H.; Kong, X.; Mohapatra, S.; San Juan-Vergara, H.; Hellermann, G.; Behera, S.; Singam, R.; Lockey, R.F.; Mohapatra, S.S. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat. Med. 2005, 11, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Merl, S.; Michaelis, C.; Jaschke, B.; Vorpahl, M.; Seidl, S.; Wessely, R. Targeting 2A protease by RNA interference attenuates coxsackieviral cytopathogenicity and promotes survival in highly susceptible mice. Circulation 2005, 111, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.J.; Guan, Y.; Tang, Q.; Du, C.; Xie, F.Y.; He, M.L.; Chan, K.W.; Wong, K.L.; Lader, E.; Woodle, M.C.; et al. Prophylactic and therapeutic effects of small interfering RNA targeting SARS-coronavirus. Antivir. Ther. 2004, 9, 365–374. [Google Scholar] [PubMed]
- Bai, F.; Wang, T.; Pal, U.; Bao, F.; Gould, L.H.; Fikrig, E. Use of RNA interference to prevent lethal murine west nile virus infection. J. Infect. Dis. 2005, 191, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Palliser, D.; Chowdhury, D.; Wang, Q.Y.; Lee, S.J.; Bronson, R.T.; Knipe, D.M.; Lieberman, J. An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature 2006, 439, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.W.; Li, H.; Lu, R.; Li, F.; Li, W.X. RNA silencing: a conserved antiviral immunity of plants and animals. Virus Res. 2004, 102, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, P.; Sklan, E.; Wilkins, C.; Burgon, T.; Samuel, M.A.; Lu, R.; Ansel, K.M.; Heissmeyer, V.; Einav, S.; Jackson, W.; et al. Six RNA viruses and forty-one hosts: viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems . PLoS Pathog. 2010, 6, e1000764. [Google Scholar] [CrossRef] [PubMed]
- Randall, G.; Panis, M.; Cooper, J.D.; Tellinghuisen, T.L.; Sukhodolets, K.E.; Pfeffer, S.; Landthaler, M.; Landgraf, P.; Kan, S.; Lindenbach, B.D.; et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12884–12889. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Schutz, S.; Sarnow, P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 2008, 4, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schuttler, C.G.; Fehr, C.; Junemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. Embo J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Jangra, R.K.; Yi, M.; Lemon, S.M. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J. Virol. 2010, 84, 6615–6625. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L. Targeting microRNA-122 to treat hepatitis C virus infection. Viruses 2010, 2, 1382–1393. [Google Scholar] [CrossRef]
- Cullen, B.R. Viruses and microRNAs . Nat. Genet. 2006, 38 (Suppl.), S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, S.B.; Brideau-Andersen, A.; Chisari, F.V. Interference of hepatitis C virus RNA replication by short interfering RNAs. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 2014–2018. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.A.; Jayasena, S.; Khvorova, A.; Sabatinos, S.; Rodrigue-Gervais, I.G.; Arya, S.; Sarangi, F.; Harris-Brandts, M.; Beaulieu, S.; Richardson, C.D. RNA interference blocks gene expression and RNA synthesis from hepatitis C replicons propagated in human liver cells. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 2783–2788. [Google Scholar] [CrossRef] [PubMed]
- Randall, G.; Grakoui, A.; Rice, C.M. Clearance of replicating hepatitis C virus replicon RNAs in cell culture by small interfering RNAs. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Sakamoto, N.; Enomoto, N.; Tanabe, Y.; Miyagishi, M.; Maekawa, S.; Yi, L.; Kurosaki, M.; Taira, K.; Watanabe, M.; et al. Inhibition of intracellular hepatitis C virus replication by synthetic and vector-derived small interfering RNAs. EMBO Rep. 2003, 4, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.Y.; Abrignani, S.; Houghton, M.; Han, J.H. Small interfering RNA-mediated inhibition of hepatitis C virus replication in the human hepatoma cell line Huh-7. J. Virol. 2003, 77, 810–812. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Kittler, R.; Buchholz, F.; Windisch, M.P.; Pietschmann, T.; Bartenschlager, R.; Frese, M. Alternative approaches for efficient inhibition of hepatitis C virus RNA replication by small interfering RNAs. J. Virol. 2004, 78, 3436–3446. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.A.; Richardson, C.D. Hepatitis C virus replicons escape RNA interference induced by a short interfering RNA directed against the NS5b coding region. J. Virol. 2005, 79, 7050–7058. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Contag, C.H.; Ilves, H.; Johnston, B.H.; Kaspar, R.L. Small hairpin RNAs efficiently inhibit hepatitis C IRES-mediated gene expression in human tissue culture cells and a mouse model. Mol. Ther. 2005, 12, 562–568. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, A.P.; Meuse, L.; Pham, T.T.; Conklin, D.S.; Hannon, G.J.; Kay, M.A. RNA interference in adult mice. Nature 2002, 418, 38–39. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Henry, S.D.; Metselaar, H.J.; Scholte, B.; Kwekkeboom, J.; Tilanus, H.W.; Janssen, H.L.; van der Laan, L.J. Combined antiviral activity of interferon-alpha and RNA interference directed against hepatitis C without affecting vector delivery and gene silencing. J. Mol. Med. 2009, 87, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Randall, G.; Higginbottom, A.; Monk, P.; Rice, C.M.; McKeating, J.A. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 2004, 78, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yamada, O.; Sakamoto, T.; Yoshida, H.; Iwai, T.; Matsushita, Y.; Shimamura, H.; Araki, H.; Shimotohno, K. Down-regulation of viral replication by adenoviral-mediated expression of siRNA against cellular cofactors for hepatitis C virus. Virology 2004, 320, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.I.; Mo, H.; Pilot-Matias, T.; He, Y.; Koev, G.; Krishnan, P.; Mondal, R.; Pithawalla, R.; He, W.; Dekhtyar, T.; et al. Identification of host genes involved in hepatitis C virus replication by small interfering RNA technology. Hepatology 2007, 45, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Supekova, L.; Supek, F.; Lee, J.; Chen, S.; Gray, N.; Pezacki, J.P.; Schlapbach, A.; Schultz, P.G. Identification of human kinases involved in hepatitis C virus replication by small interference RNA library screening. J. Biol. Chem. 2008, 283, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.L.; Cooper, J.D.; Heaton, N.S.; Yoon, R.; Oakland, T.E.; Jordan, T.X.; Mateu, G.; Grakoui, A.; Randall, G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7577–7582. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.W.; Benita, Y.; Peng, L.F.; Kim, S.S.; Sakamoto, N.; Xavier, R.J.; Chung, R.T. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009, 5, 298–307. [Google Scholar] [CrossRef]
- Vaillancourt, F.H.; Pilote, L.; Cartier, M.; Lippens, J.; Liuzzi, M.; Bethell, R.C.; Cordingley, M.G.; Kukolj, G. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 2009, 387, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Brass, A.L.; Ng, A.; Hu, Z.; Xavier, R.J.; Liang, T.J.; Elledge, S.J. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 16410–16415. [Google Scholar] [CrossRef] [PubMed]
- Randall, G.; Chen, L.; Panis, M.; Fischer, A.K.; Lindenbach, B.D.; Sun, J.; Heathcote, J.; Rice, C.M.; Edwards, A.M.; McGilvray, I.D. Silencing of USP18 potentiates the antiviral activity of interferon against hepatitis C virus infection. Gastroenterology 2006, 131, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- Trotard, M.; Lepere-Douard, C.; Regeard, M.; Piquet-Pellorce, C.; Lavillette, D.; Cosset, F.L.; Gripon, P.; Le Seyec, J. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. Faseb J. 2009, 23, 3780–3789. [Google Scholar] [CrossRef] [PubMed]
- Borawski, J.; Troke, P.; Puyang, X.; Gibaja, V.; Zhao, S.; Mickanin, C.; Leighton-Davies, J.; Wilson, C.J.; Myer, V.; Cornellataracido, I.; et al. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 2009, 83, 10058–10074. [Google Scholar] [CrossRef] [PubMed]
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Goncales Jr., F.L.; Haussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection . N. Engl. J. Med. 2002, 347, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Tuschl, T. On the art of identifying effective and specific siRNAs. Nat. Methods 2006, 3, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Konishi, M.; Wu, C.H.; Kaito, M.; Hayashi, K.; Watanabe, S.; Adachi, Y.; Wu, G.Y. siRNA-resistance in treated HCV replicon cells is correlated with the development of specific HCV mutations. J. Viral. Hepat. 2006, 13, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Lee, H.; Kim, S.I.; Yoon, Y.; Kim, M. Optimization of linear double-stranded RNA for the production of multiple siRNAs targeting hepatitis C virus. RNA 2009, 15, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Layzer, J.M.; McCaffrey, A.P.; Tanner, A.K.; Huang, Z.; Kay, M.A.; Sullenger, B.A. In vivo activity of nuclease-resistant siRNAs. RNA 2004, 10, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Dorsett, Y.; Tuschl, T. siRNAs: applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov. 2004, 3, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Choung, S.; Kim, Y.J.; Kim, S.; Park, H.O.; Choi, Y.C. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem. Biophys. Res. Commun. 2006, 342, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Allerson, C.R.; Sioufi, N.; Jarres, R.; Prakash, T.P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R.H.; Swayze, E.E.; Bhat, B. Fully 2'-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005, 48, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Dowler, T.; Bergeron, D.; Tedeschi, A.L.; Paquet, L.; Ferrari, N.; Damha, M.J. Improvements in siRNA properties mediated by 2'-deoxy-2'-fluoro-beta-D-arabinonucleic acid (FANA). Nucleic Acids Res. 2006, 34, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Abramov, M.; Van Aerschot, A.; Xu, D.; Juliano, R.L.; Herdewijn, P. Inhibition of MDR1 expression with altritol-modified siRNAs. Nucleic Acids Res. 2007, 35, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Burchard, J.; Schelter, J.; Chau, B.N.; Cleary, M.; Lim, L.; Linsley, P.S. Widespread siRNA "off-target" transcript silencing mediated by seed region sequence complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Burchard, J.; Leake, D.; Reynolds, A.; Schelter, J.; Guo, J.; Johnson, J.M.; Lim, L.; Karpilow, J.; Nichols, K.; et al. Position-specific chemical modification of siRNAs reduces "off-target" transcript silencing. RNA 2006, 12, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Guenthner-Biller, M.; Bourquin, C.; Ablasser, A.; Schlee, M.; Uematsu, S.; Noronha, A.; Manoharan, M.; Akira, S.; de Fougerolles, A.; et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005, 11, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.D.; Sood, V.; Shaw, J.R.; Fang, D.; McClintock, K.; MacLachlan, I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 2005, 23, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M. On the delivery of small interfering RNAs into mammalian cells. Expert. Opin. Drug Deliv. 2005, 2, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Minks, M.A.; West, D.K.; Benvin, S.; Baglioni, C. Structural requirements of double-stranded RNA for the activation of 2',5'-oligo(A) polymerase and protein kinase of interferon-treated HeLa cells. J. Biol. Chem. 1979, 254, 10180–10183. [Google Scholar] [PubMed]
- Kariko, K.; Bhuyan, P.; Capodici, J.; Ni, H.; Lubinski, J.; Friedman, H.; Weissman, D. Exogenous siRNA mediates sequence-independent gene suppression by signaling through toll-like receptor 3. Cells Tissues Organs 2004, 177, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, M.E.; Yamada, K.; Takeda, A.; Chandrasekaran, V.; Nozaki, M.; Baffi, J.Z.; Albuquerque, R.J.; Yamasaki, S.; Itaya, M.; Pan, Y.; et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 2008, 452, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Robbins, M.; Judge, A.; Liang, L.; McClintock, K.; Yaworski, E.; MacLachlan, I. 2'-O-methyl-modified RNAs act as TLR7 antagonists. Mol. Ther. 2007, 15, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Doehle, B.P.; Qin, Y.; Macara, I.G.; Cullen, B.R. Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA 2005, 11, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 11854–11859. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Fichou, Y.; Ferec, C. The potential of oligonucleotides for therapeutic applications. Trends Biotechnol. 2006, 24, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.N.; Schaffer, D.V. Antiviral RNAi therapy: emerging approaches for hitting a moving target. Gene Ther. 2006, 13, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Ashihara, E.; Kawata, E.; Maekawa, T. Future prospect of RNA interference for cancer therapies. Curr. Drug Targets 2010, 11, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Doug, M. As it matures, RNAi field sees failures of key clinical candidates . Gene Silencing News. 2009. Available online: http://www.genomeweb.com/rnai/it-matures-rnai-field-sees-failures-key-clinical-candidates (accessed 26 June 2010).
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Aleku, M.; Schulz, P.; Keil, O.; Santel, A.; Schaeper, U.; Dieckhoff, B.; Janke, O.; Endruschat, J.; Durieux, B.; Roder, N.; et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008, 68, 9788–9798. [Google Scholar] [CrossRef] [PubMed]
- United States National Institutes of Health. Clinical trials . Available online: http://www.clinicaltrials.gov (accessed 8 August 2010).
- DeVincenzo, J.; Lambkin-Williams, R.; Wilkinson, T.; Cehelsky, J.; Nochur, S.; Walsh, E.; Meyers, R.; Gollob, J.; Vaishnaw, A. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8800–8805. [Google Scholar] [CrossRef] [PubMed]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34+ cells in patients undergoing transplantation for AIDS-related lymphoma . 2010, 2, 36–43. [Google Scholar]
- Li, M.J.; Kim, J.; Li, S.; Zaia, J.; Yee, J.K.; Anderson, J.; Akkina, R.; Rossi, J.J. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol. Ther. 2005, 12, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjarn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Regulus Therapeutics. Regulus therapeutics and GlaxoSmithKline establish new collaboration to develop and commercialize microRNA therapeutics targeting miR-122 . Available online: http://www.regulusrx.com/news-events/press-release-details.php?id=34 (accessed 2 August 2010).
- Lewis, D.L.; Hagstrom, J.E.; Loomis, A.G.; Wolff, J.A.; Herweijer, H. Efficient delivery of siRNA for inhibition of gene expression in postnatal mice. Nat. Genet. 2002, 32, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Rigotti, A. Absorption, transport, and tissue delivery of vitamin E. Mol. Aspects Med. 2007, 28, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Nishina, K.; Unno, T.; Uno, Y.; Kubodera, T.; Kanouchi, T.; Mizusawa, H.; Yokota, T. Efficient in vivo delivery of siRNA to the liver by conjugation of alpha-tocopherol. Mol. Ther. 2008, 16, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, M.; Affolter, C.; Meyer, F.; Nguyen, I.; Richard, D.G.; Schuster, C.; Bartenschlager, R.; Voegel, J.C.; Ogier, J.; Baumert, T.F. Sustained delivery of siRNAs targeting viral infection by cell-degradable multilayered polyelectrolyte films. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 16320–16325. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.S.; Lee, A.C.; Akinc, A.; Bramlage, B.; Bumcrot, D.; Fedoruk, M.N.; Harborth, J.; Heyes, J.A.; Jeffs, L.B.; John, M.; et al. RNAi-mediated gene silencing in non-human primates. Nature 2006, 441, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Goldberg, M.; Qin, J.; Dorkin, J.R.; Gamba-Vitalo, C.; Maier, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Manoharan, M.; et al. Development of lipidoid-siRNA formulations for systemic delivery to the liver. Mol. Ther. 2009, 17, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Dominska, M.; Dykxhoorn, D.M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010, 123, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12982–12987. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Dahari, H.; Ribeiro, R.M.; Perelson, A.S. Rapid emergence of protease inhibitor resistance in hepatitis C virus . Sci. Transl. Med. 2010, 2, 30–32. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Berger, K.L.; Randall, G. Possibilities for RNA Interference in Developing Hepatitis C Virus Therapeutics. Viruses 2010, 2, 1647-1665. https://doi.org/10.3390/v2081647
Berger KL, Randall G. Possibilities for RNA Interference in Developing Hepatitis C Virus Therapeutics. Viruses. 2010; 2(8):1647-1665. https://doi.org/10.3390/v2081647
Chicago/Turabian StyleBerger, Kristi L., and Glenn Randall. 2010. "Possibilities for RNA Interference in Developing Hepatitis C Virus Therapeutics" Viruses 2, no. 8: 1647-1665. https://doi.org/10.3390/v2081647