The Natural Killer Cell Cytotoxic Function Is Modulated by HIV-1 Accessory Proteins

Department of Immunology and Microbiology, Rush University Medical Center, 1735 West Harrison St, Chicago, IL 60612, USA
*
Author to whom correspondence should be addressed.
Viruses 2011, 3(7), 1091-1111; https://doi.org/10.3390/v3071091
Submission received: 14 June 2011
/
Revised: 23 June 2011
/
Accepted: 24 June 2011
/
Published: 8 July 2011
(This article belongs to the Special Issue Antiviral Innate Immunity)
Abstract
:Natural killer (NK) cells’ major role in the control of viruses is to eliminate established infected cells. The capacity of NK cells to kill virus-infected cells is dependent on the interactions between ligands on the infected cell and receptors on the NK cell surface. Because of the importance of ligand-receptor interactions in modulating the NK cell cytotoxic response, HIV has developed strategies to regulate various NK cell ligands making the infected cell surprisingly refractory to NK cell lysis. This is perplexing because the HIV-1 accessory protein Vpr induces expression of ligands for the NK cell activating receptor, NKG2D. In addition, the accessory protein Nef removes the inhibitory ligands HLA-A and -B. The reason for the ineffective killing by NK cells despite the strong potential to eliminate infected cells is due to HIV-1 Vpu’s ability to down modulate the co-activation ligand, NTB-A, from the cell surface. Down modulation of NTB-A prevents efficient NK cell degranulation. This review will focus on the mechanisms through which the HIV-1 accessory proteins modulate their respective ligands, and its implication for NK cell killing of HIV-infected cells.
1. Natural Killer Cells Control Virus Production
An important first line of defense against viruses involves natural killer (NK) cells. Unlike cytotoxic T-lymphocytes, which also destroy virus-infected cells, NK cells respond to infected cells without prior exposure to infected cells or their components. Hence, the action of NK cells to immediately eliminate virus-infected cells and prevent the spread of the virus is essential during the early stages of viral infections since adaptive anti-viral immune responses are not fully developed. The importance of NK cells in controlling virus infections, in vivo, was initially shown in mice depleted of NK cells. Without NK cells, infected mice are unable to control a plethora of viruses [1,2,3]. Humans lacking or having dysfunctional NK cells develop fatal disseminated herpes infections [4,5,6,7,8]. Since NK cells are effective at eliminating virus-infected cells, many viruses that chronically infect humans utilize strategies to avoid detection and elimination by NK cells [9,10,11,12].
NK cells control viruses in one of three ways: (1) by directed release of the contents of lytic granules onto infected cells, (2) by expressing ligands (i.e., Fas ligand and TRAIL) that engage death receptors on infected cells, and (3) by secreting cytokines (e.g., IFN-γ, MIP-1β) that impede or prevent virus replication [13,14,15,16,17]. In fact, secretion of IFN-γ by NK cells has been shown to enhance MHC class I expression on antigen presenting cells to enhance CD8+ T-cell recognition of infected cells [18]. NK cell cytotoxic granules contain perforin and granzymes. Perforin forms pores on the target cell, and granzymes enter through the pores and initiate apoptosis within the target cell [19,20]. The directed release of the contents of cytolytic granules (degranulation) is triggered by two distinct mechanisms. Antibody-dependent cellular cytotoxicity (ADCC) results in the release of perforin and granzymes when antibody-coated cells engage the low affinity receptor for IgG, CD16 (or FcγRIIIa), on NK cells [21]. Secondly, engagement of germline-encoded receptors on NK cells by their invariant ligands on the target cells regulates degranulation [22,23,24,25]. This review will focus on the latter NK cell response. In particular, we will discuss how HIV-1-infected cells manipulate surface ligands that trigger NK receptors essential in regulating degranulation.
2. Polarization of Lytic Granules in NK Cells Requires Stable Interactions with Target Cells
Directed degranulation by NK cells ensures that a specific, infected cell is killed leaving healthy neighboring cells unharmed [25,26]. Polarization of lytic granules towards the target cells and subsequent degranulation requires the participation of three major classes of receptors: (1) adhesion receptors, (2) activating receptors, and (3) co-activating receptors [26]. The initial step of cytolysis involves a loose association (Kd = ~ 100 μM) between target cells and NK cells that require carbohydrate Ag LewisX (e.g., CD15, Gal-β1-4 GlcNAc α1-3Fuc) on target cells to interact with CD2 or selectins on NK cells [27]. Loose interactions between Ag LewisX and lectin-like receptors such as CD94 and NKG2D may allow for the NK cell to slow down and initiate longer contact with its target [28]. This loose adhesion between NK and target cells is soon followed by a stable interaction involving integrins (e.g., lymphocyte function-associated antigen (LFA)-1) on NK cells and the intercellular adhesion molecules (ICAM) family on target cells [29,30]. Once a stable interaction occurs, “firm” adhesion follows [31,32]. “Firm” adhesion involves a conformation change from a “closed” low affinity (Kd = ~ 100 μM) state to an “open” configuration in which the integrins bind to ICAMs with higher affinity (Kd = ~ 0.4 μM) [33,34]. The engagement of activating receptors on the NK cell with their ligands on the target cell enhances the “open” configuration state [34].
Following the binding of integrins on NK cells to ICAMs on target cells, several events occur within the NK cells which brings the cytolytic granules towards the target cell [35]. Initially, the cytotoxic granules migrate in the direction of the microtubule-organizing center (MTOC) towards the minus ends of the microtubules found at the MTOC [36]. This is followed by actin cytoskeleton rearrangement that recruits activating receptors into lipid rafts. The clustering of activation receptors between the points of contact of the NK cell and its target forms the immunological synapse [37,38,39]. Following recruitment of activation receptors to lipid rafts, the MTOC containing lytic granules polarizes toward the immunological synapse due to the reorganization of the actin network [35,40]. The consequence of the MTOC containing granules polarizing to the synapse will allow for the eventual release of cytotoxic granules in an area adjacent to the activating receptors (Figure 1).
3. Degranulation Follows Engagement of NK Cell Activating Receptors
While integrin engagement leads to polarization of lytic granules towards the target cell, triggering integrins alone is insufficient for NK cells to degranulate [41]. “Activating” receptors (aNKRs) on NK cells being bound by their respective ligands on the target cell are required to elicit an NK cell to degranulate [42]. aNKRs lack signaling motifs in their intracellular domains. Instead, these receptors associate non-covalently with adaptor proteins through a charged residue in the activating receptor transmembrane region [24,43]. aNKRs (e.g., NKp30, NKp44, NKp46) and the killer immunoglobulin-like receptors with short cytoplasmic tails (e.g., KIR2DS1, KIR3DS1) interact with adaptor proteins containing immunoreceptor tyrosine-based activation motifs (ITAMs), defined as YXX(L/I)X6–8YXX(L/I) (where X6–8 are 6 to 8 residues between the two tyrosines) [22,44,45,46,47,48]. ITAM containing adaptor molecules include: DAP12, FcεRIγ, and CD3ζ [25,49]. These adaptor proteins become phosphorylated by tyrosine kinases such as Syk and ZAP-70 allowing for NK cell cytotoxicity (See Table 1 for list of aNKRs) [50].
A second group of aNKRs does not associate with adaptor molecules containing ITAMs [24]. For example, NKG2D associates with the adaptor protein DAP10, which contains an YXXM motif. After phosphorylation, DAP10 recruits phosphatidylinositol-3-kinase (PI3K) and growth factor receptor-bound protein (Grb)-2 [51,52].
4. Phospholipase C-γ is the Key Mediator for NK Cell Degranulation
Engagement of both adhesion and activating receptors lead to recruitment and phosphorylation of Vav1, a guanine exchange factor [53]. Vav1 ultimately regulates actin cytoskeleton rearrangement for polarization of granules and activation receptor clustering [39]. Phosphorylation of Vav1 ultimately leads to phosphorylation of phospholipase C-γ (PLCγ) following LFA-1 and/or NKG2D engagement (Figure 2). Phosphorylated PLCγ in turn hydrolyzes phosphatidylinositol 4, 5 bisphosphate (PIP2) to inositol 1, 4, 5 trisphosphate (IP3) and diacylglycerol (DAG) [54,55]. DAG recruits mammalian uncoordinated (Munc) 13-4, an essential component of the vesicle fusion complex crucial for regulated degranulation, to the plasma membrane [56]. Vesicle fusion to the plasma membrane is further facilitated by Rab27a, a Rab GTPase. Rab27a is recruited to the cell surface after LFA-1 engagement allowing for the granules to dock with Munc 13-4 at the plasma membrane [57]. IP3 binds to inositol trisphosphate receptor on the endoplasmic reticulum membrane and opens a calcium channel resulting in the release of calcium into the cytoplasm [58]. Calcium is necessary for the final fusion between the cytolytic granule and plasma membrane [59].
The amount of Vav1 phosphorylation induced by integrins and activating receptor(s) is insufficient to lead to degranulation. However, there may be a sufficient amount of phosphorylated Vav1 to lead to granule polarization and immunological synapse formation. This is because Vav1 is negatively regulated by the ubiquitin ligase c-Cbl. c-Cbl is induced upon engagement of an activating receptor (e.g., NKG2D), to dampen NK cell cytotoxicity, which in turn, leads to Vav1’s ubiquitination and proteosomal degradation. The loss of Vav1 decreases the levels of phosphorylated PLCγ and prevents NK cell degranulation [55]. To overcome the effects of c-Cbl’s inhibition of Vav1, NK cells require concomitant engagement of activating and co-activating receptors [55].
Co-activation receptors include the CD2 family of activating receptors (e.g., 2B4, CRACC, NTB-A) (See Table 2 for list of caNKR) [60,61,62]. The CD2 family of receptors associates with signaling lymphocyte activation molecule (SLAM)-associated protein (SAP) to induce cellular cytotoxicity [63,64,65]. When these receptors bind their respective ligands, tyrosine residues in the cytoplasmic tail become phosphorylated allowing for the recruitment of SAP. SAP binds to the tyrosine kinase, Fyn, through an arginine at position 78 in its SH2 domain [66,67]. Once bound to SAP, Fyn phosphorylates both the SLAM-family receptor as well as molecules such as PLCγ to induce the NK cell to degranulate [68]. More importantly, degranulation of resting NK cells requires at least a specific pair of activating and/or co-activating receptors to be engaged. Such pairs include NKG2D and NKp46, NKp46 and DNAM-1, and 2B4 and DNAM-1. However, combinations such as NKG2D and DNAM-1 do not result in NK cell degranulation [42].
5. Inhibitory Receptors Regulate Cytoskeleton Rearrangement and Synapse Formation
In addition to activation receptors, NK cell responses are regulated by inhibitory receptors (iNKRs) present on NK cells. iNKRs contain immunoreceptor tyrosine-based inhibitory motifs (ITIMs) in the cytoplasmic domain defined by the sequence (I/L/V/S)XYXX(L/V) [25]. Once the ITIMs become phosphorylated they recruit phosphatases such as Src homology region 2 domain containing phosphatase (SHP)-1 and SHP-2 [69,70]. These phosphatases lead to the dephosphorylation of signaling molecules involved in calcium influx, actin rearrangement, and granule polarization initiated by adhesion receptors [71,72,73]. Inhibitory receptors also activate kinases such as c-Abl which play a role in inhibiting actin rearrangement [74].
One major set of ligands to iNKRs is the major histocompatibility complex (MHC) class I molecules [45,75,76,77]. The MHC-I recognizing families of iNKRs are the killer-immunoglobulin (Ig)-like receptors (KIRs) with ITIM-containing cytoplasmic tails, which recognizes HLA-A, -B, and -C; the receptor pair of NKG2A and CD94, which recognizes HLA-E; and the interleukin-like transcript type 2 (ILT-2), which recognizes multiple HLA molecules. As MHC class I molecules are constitutively expressed on all healthy nucleated cells, iNKRs recognition of their MHC-I ligand inhibits the NK cell’s ability to kill healthy “self” cells. Many viruses down modulate MHC class I molecules on infected cells to avoid CD8+ T-cell responses [78]. However, MHC-I down modulation leaves the target cell susceptible to destruction by NK cells by removing inhibitory ligands [10,11]. Simply lacking or having impaired expression of MHC class I molecules is insufficient for NK cell lysis. NK cells will only degranulate and kill the infected cell after engagement of activating receptors. Moreover, NK cells are capable of degranulating target cells that have normal expression of MHC-I on its surface if activating receptors are engaged to overcome the inhibition [79].
6. HIV-1 Alters NK Cell Degranulation by Modulating Ligands to NK Cell Receptors
Nearly 25 years ago NK cells were found to be inefficient at killing autologous HIV-1 infected primary T-cells [80,81,82]. However, the mechanism to explain the inability of NK cells to lyse HIV-infected cells has only recently been discovered. Below we will discuss how HIV-1 gene products impact NK cell cytolytic function and ultimately how HIV-1 avoids destruction by NK cells.
6.1. HIV-1 Vpr Induces Expression of Ligands for the NK Cell Activating Receptor NKG2D
NKG2D is a type II transmembrane C-type lectin that associates with the adaptor protein DAP10 [83,84]. It is expressed on all NK cells in the blood as well as subsets of CD8+ T-cells, γδ T-cells, and NKT cells [21]. NKG2D ligands include the MHC class I polypeptide-related sequence (MIC)-A and -B, as well as the cytomegalovirus unique long 16-binding protein (ULBP)-1 thru -6 [85,86]. These ligands typically are not expressed on healthy cells; however, NKG2D ligands are expressed at relatively high levels on cells undergoing genotoxic stress [87]. DNA damage leads to NKG2D ligand expression through induction of the DNA damage sensor ataxia telangiectasia-mutated (ATM) and ataxia telangiectasia-mutated and Rad 3-related (ATR) [87]. ATM detects double stranded DNA breaks, whereas ATR detects single stranded DNA breaks, typically at replication forks [88,89]. Activation of either of these kinases will result in cell cycle arrest and subsequent induction of NKG2D ligands [90].
ATR and not ATM is phosphorylated and activated during HIV-1 infection by the viral protein Vpr [88,91]. One of the benefits of ATR’s induction for HIV-1 is to induce G2 cell cycle arrest. The long terminal repeat (LTR) of HIV-1 is highly active during the G2 phase of the cell cycle resulting in higher viral production [92,93,94]. It has been hypothesized that G2 cell cycle arrest is associated with Vpr binding to DCAF1, a substrate recognition subunit for an E3 ubiquitin ligase complex, through Vpr’s leucine rich motif from residue 60–68 (Figure 3). Vpr binds not only to DCAF1 but also an unknown cell cycle protein through an arginine at residue 80 of Vpr [95,96,97]. The Vpr complex in turn associates with the E3 ubiqutin ligase complex, Cullin 4a/damaged DNA binding protein I (Cul4aDDB1/DCAF1) [98]. When the unknown cell cycle protein is ubiquitinated and destroyed by the E3 ubiquitin ligase complex, G2 arrest is induced and ATR becomes phosphorylated [98]. ATR’s phosphorylation through Vpr not only results in G2 arrest, but also leads to the expression NKG2D ligands. Vpr leads to ULBP-1 and -2 expression but not ULBP-3 through -6, or MIC-A and -B [99,100]. The specificity of NKG2D ligand expression may be due to ATR’s influence on the transcription factors Specificity Protein (SP)1 and SP3 resulting in ULBP-1 transcription [101]. Induction of ULBP-1 and -2 by Vpr provides an activating signal for NK cells to degranulate [99,100].
Nef has also been implicated in down modulating ligands for the activating receptor NKG2D [102]. Namely, it was shown that Nef down modulates MIC-A, ULBP-1, and ULBP-2. However, in more recent study, deleting Nef from HIV-1 resulted in surface expression of NKG2D ligands to the same level as wild-type infected cells [99]. Furthermore, we were never able to detect MIC-A cell surface expression on HIV-1 infected primary CD4+ T-cells. The discrepancies between the two studies have yet to be determined.
6.2. Nef down Modulates Ligands for Inhibitory Receptors on NK Cells
It was reported over 20 years ago that HIV-1 down modulates MHC class I molecules [103]. It was later discovered that MHC class I down modulation was due to the HIV-1 protein negative regulatory factor (Nef) [104]. In subsequent studies, Nef was found to selectively down modulate the MHC class I molecules HLA-A and -B, but not HLA-C and -E [105]. Nef is a 27 kD protein expressed early in HIV-1 infection. Nef selectively down modulates HLA-A and -B by binding to two key residues (a tyrosine at position 321 and an aspartate at position 328) in the cytoplasmic tails of HLA-A and -B. These residues are not present in the cytoplasmic tail of HLA-C preventing Nef from down modulating HLA-C [105]. While HLA-E cytoplasmic tail has the two residues found in HLA-A and -B, Nef is incapable of down modulating HLA-E. Exchanging the cytoplasmic tail of HLA-E for the cytoplasmic tail of HLA-A resulted in HLA-E’s down modulation, indicating that the cytoplasmic tail of HLA-E differs in numerous other residues to HLA-A and -B preventing Nef from down modulating HLA-E [105,106].
Upon binding to HLA-A and -B, Nef re-routes both MHC class I molecules from the trans-Golgi network (TGN) to endosomes by recruiting adaptor protein (AP)-1, a clathrin-mediated protein that facilitates trafficking between the TGN and endosomes [107] (Figure 4). The Nef/MHC-I complex binds to the μ subunit of AP-1 [108,109,110]. Nef then recruits coatomer protein-1β (β-COP), a component of COP-1 coats for transport through the early secretory pathways [111]. β-COP binds to an arginine motif (RXR) on the N-terminal of Nef [112]. Nef’s ability to bind to β-COP causes HLA-A and -B to be directed to lysosomes for their degradation. Additionally, Nef increases the internalization of MHC-I molecules from the plasma membrane through Nef’s ability to utilize PACS-1, a phosphofurin acidic cluster sorting protein [113]. PACS-1 controls endosome to TGN trafficking by binding to acidic motifs of a protein and forms a bridge to AP-1. Nef contains a cluster of glutamic acids from residue 62-65 that binds with PACS-1 connecting Nef with AP-1 [114]. In this instance, Nef forces the endocytosis of HLA-A and -B into the TGN. However, recent studies have shown that Nef primarily acts to re-route HLA-A and -B from the TGN to lysosomes, instead of promoting the internalization of MHC-I molecules via the PACS-1 mechanism [107,115,116].
It was thought that the selectivity of Nef’s down modulation of only HLA-A and -B, and not HLA-C and -E, was necessary for HIV-1 to evade responses from NK cells [105]. However, as NK cell expression of iNKRs is variegated, there are a significant number of NK cells not expressing iNKRs to HLA-C and HLA-E [117]. Because of the difference in NK cell expression of iNKRs, it stands to reason that NK cells lacking iNKRs for HLA-C and HLA-E should efficiently lyse HIV-1 infected T-cells due to Vpr’s induction of NKG2D ligands and Nef’s down modulation of HLA-A and -B. In fact, NK cells lacking iNKRs for HLA-C and -E are able to lyse HIV-1-infected cells when HLA-A and -B are down modulated [117]. These studies indicate that NK cells have the ability to kill HIV-1-infected cells. However, it should be noted that NK cell killing of HIV-1-infected cells is still two-thirds less than that observed with NK cell killing of the NK-sensitive cell line, K562 [117]. Furthermore, HIV-1 infected CD4+ T-cells have the capability to activate NK cells to the same extent as K562 cells, yet the infected T-cells induce NK cells to degranulate to a third of the level of NK cells exposed to K562 cells [118].
6.3. NK Cell Degranulation is Hindered by Down Modulation of NTB-A on HIV-1 Infected Cells by Vpu
Why NK cells are activated but unable to degranulate in the presence of HIV-1 infected cells was a mystery until recent work illustrating the need for not only an activating receptor but also a co-activating receptor to be engaged for NK cell degranulation [42]. NK, T-cell, B-cell antigen (NTB-A) is one such co-activating receptor that is expressed on all blood NK cells as well as CD4+ T-cells [119]. In addition, NTB-A is down modulated on infected T-cells by HIV-1 [120,121].
NTB-A is a type I transmembrane protein and a member of the signaling lymphocytic activation molecule (SLAM) receptor family [119]. It is a part of the Ig superfamily containing a distal V-type and a proximal C2-type domain in the extracellular portion of the receptor. NTB-A forms a homotypic ligand-receptor pair. When bound by its ligand, two of NTB-A’s three immunoreceptor tyrosine-based switch motifs (TXYXXV/I) are phosphorylated by tyrosine kinases such as Lck [43,122]. Phosphorylation of tyrosine 284 results in binding of Ewing’s sarcoma-activated transcript (EAT)-2, a SAP family member. EAT-2 binding to NTB-A has been implicated in cytokine production by primary NK cells [123]. Phosphorylation of tyrosine 319 results in SAP binding to NTB-A [63,124]. SAP then binds the protein kinase Fyn, which in turn, triggers NK cells degranulation [67]. While triggering NTB-A alone is insufficient to induce NK cells to release their granules, when triggered together with NKG2D, NK cells are able to degranulate [118].
Since ligand binding of NTB-A on NK cells has been shown to induce NK cell cytotoxicity, it seems likely that HIV-1-infected cells down modulate NTB-A to evade NK cells. More specifically, recent work has shown the HIV accessory protein Vpu to be both necessary and sufficient for NTB-A’s down modulation [118]. Vpu’s transmembrane region binds to NTB-A and sequesters NTB-A within the TGN [125]. Furthermore, Vpu does not increase the internalization of NTB-A and does not alter the steady-state levels of NTB-A expressed in the cell [118].
Vpu’s down modulation of NTB-A is distinct from the down modulation of CD4 and tetherin/BST-2. For both CD4 and tetherin/BST-2, Vpu serves as an adaptor to β-TrCP, a human F box protein that serves as a substrate recognition receptor for the E3 ubiquitin ligase complex. Phosphoserines at positions 52 and 56 of Vpu are necessary for the interaction between β-TrCP and Vpu. Ubiquitinized CD4 is degraded by proteasomes, whereas ubiquitinized tetherin/BST-2 is transported to acidified endosomes though this may not be the only mechanism of Vpu’s down modulation of tetherin/BST-2 [126,127,128,129,130,131,132]. However, this is not the case with Vpu’s down modulation of NTB-A. Mutations of the serine residues at position 52 and 56 result in down modulation of NTB-A to similar levels as wild-type Vpu. Additionally, NTB-A’s association with Vpu does not lead to its degradation by proteasomes nor is NTB-A trafficked to acidified endosomes for its destruction [118].
Thus, it is likely that NTB-A is unable to recycle back to the cell surface, removing the second activating signal needed to trigger NK cell degranulation. NTB-A’s down-modulation by Vpu prevents NK cell degranulation and lysis of HIV-1-infected T-cells by three-fold [118]. Interestingly, even in the presence of Vpu, NK cells expressing iNKRs for HLA-C and HLA-E were able to degranulate albeit two-fold less than NK cells not expressing iNKRs for HLA-C and HLA-E [118].
7. Conclusion
Our recent studies indicate that HIV-1 evades NK cells. It would seem likely that NK cells would be skewed toward killing infected cells since HIV-1 infection leads to increased expression of ICAMs on the infected cell surface [133], induces expression of ULBP-1 and ULBP-2, and down modulates HLA-A and HLA-B. However, Vpu’s down modulation of NTB-A and the retention of HLA-C and -E prevent efficient degranulation of NK cells (Figure 5). Future work needs to be conducted to better understand the mechanism through which Vpu down modulates NTB-A, and furthermore, why NTB-A, but not other co-activating ligands, is crucial for NK cell lysis of HIV-1-infected cells.
Acknowledgments
We wish to thank Zachary B. Davis for his assistance in reviewing and editing this manuscript. We would also like to acknowledge support from NIH grants AI081681 and AI165361.
Conflict of Interest
The authors declare no conflict of interest.
References and Notes
- Bukowski, J.F.; Woda, B.A.; Habu, S.; Okumura, K.; Welsh, R.M. Natural killer cell depletion enhances virus synthesis and virus-induced hepatitis in vivo. J. Immunol. 1983, 131, 1531–1538. [Google Scholar] [CrossRef]
- Habu, S.; Akamatsu, K.; Tamaoki, N.; Okumura, K. In vivo significance of NK cell on resistance against virus (HSV-1) infections in mice. J. Immunol. 1984, 133, 2743–2747. [Google Scholar] [CrossRef] [PubMed]
- Stein-Streilein, J.; Guffee, J.; Fan, W. Locally and systemically derived natural killer cells participate in defense against intranasally inoculated influenza virus. Reg. Immunol. 1988, 1, 100–105. [Google Scholar] [PubMed]
- Biron, C.A.; Byron, K.S.; Sullivan, J.L. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989, 320, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Joncas, J.; Monczak, Y.; Ghibu, F.; Alfieri, C.; Bonin, A.; Ahronheim, G.; Rivard, G. Brief report: killer cell defect and persistent immunological abnormalities in two patients with chronic active Epstein-Barr virus infection. J. Med. Virol. 1989, 28, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Dalloul, A.; Oksenhendler, E.; Chosidow, O.; Ribaud, P.; Carcelain, G.; Louvet, S.; Massip, P.; Lebon, P.; Autran, B. Severe herpes virus (HSV-2) infection in two patients with myelodysplasia and undetectable NK cells and plasmacytoid dendritic cells in the blood. J. Clin. Virol. 2004, 30, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Aoukaty, A.; Lee, I.F.; Wu, J.; Tan, R. Chronic active Epstein-Barr virus infection associated with low expression of leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1) on natural killer cells. J. Clin. Immunol. 2003, 23, 141–145. [Google Scholar] [CrossRef]
- Etzioni, A.; Eidenschenk, C.; Katz, R.; Beck, R.; Casanova, J.L.; Pollack, S. Fatal varicella associated with selective natural killer cell deficiency. J. Pediatr. 2005, 146, 423–425. [Google Scholar] [CrossRef]
- Crotta, S.; Stilla, A.; Wack, A.; D’Andrea, A.; Nuti, S.; D’Oro, U.; Mosca, M.; Filliponi, F.; Brunetto, R.M.; Bonino, F.; et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J. Exp. Med. 2002, 195, 35–41. [Google Scholar] [CrossRef]
- Wills, M.R.; Ashiru, O.; Reeves, M.B.; Okecha, G.; Trowsdale, J.; Tomasec, P.; Wilkinson, G.W.; Sinclair, J.; Sissons, J.G. Human cytomegalovirus encodes an MHC class I-like molecule (UL142) that functions to inhibit NK cell lysis. J. Immunol. 2005, 175, 7457–7465. [Google Scholar] [CrossRef]
- Zou, Y.; Bresnahan, W.; Taylor, R.T.; Stastny, P. Effect of human cytomegalovirus on expression of MHC class I-related chains A. J. Immunol. 2005, 174, 3098–3104. [Google Scholar] [CrossRef] [PubMed]
- Reinis, M.; Simova, J.; Indrova, M.; Bieblova, J.; Pribylova, H.; Moravcova, S.; Jandlova, T.; Bubenik, J. Immunization with MHC class I-negative but not -positive HPV16-associated tumour cells inhibits growth of MHC class I-negative tumours. Int. J. Oncol. 2007, 30, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, T.; Djeu, J.Y.; Dougherty, S.F.; Oppenheim, J.J. Capacity of human large granular lymphocytes (LGL) to produce multiple lymphokines: interleukin 2, interferon, and colony stimulating factor. J. Immunol. 1983, 131, 2379–2385. [Google Scholar] [CrossRef] [PubMed]
- Arase, H.; Arase, N.; Saito, T. Fas-mediated cytotoxicity by freshly isolated natural killer cells. J. Exp. Med. 1995, 181, 1235–1238. [Google Scholar] [CrossRef]
- Bluman, E.M.; Bartynski, K.J.; Avalos, B.R.; Caligiuri, M.A. Human natural killer cells produce abundant macrophage inflammatory protein-1 alpha in response to monocyte-derived cytokines. J. Clin. Invest. 1996, 97, 2722–2727. [Google Scholar] [CrossRef]
- Bratke, K.; Kuepper, M.; Bade, B.; Virchow, J.C., Jr.; Luttmann, W. Differential expression of human granzymes A, B, and K in natural killer cells and during CD8+ T cell differentiation in peripheral blood. Eur. J. Immunol. 2005, 35, 2608–2616. [Google Scholar] [CrossRef]
- Fellows, E.; Gil-Parrado, S.; Jenne, D.E.; Kurschus, F.C. Natural killer cell-derived human granzyme H induces an alternative, caspase-independent cell-death program. Blood 2007, 110, 544–552. [Google Scholar] [CrossRef]
- Mocikat, R.; Braumuller, H.; Gumy, A.; Egeter, O.; Ziegler, H.; Reusch, U.; Bubeck, A.; Louis, J.; Mailhammer, R.; Riethmuller, G.; et al. Natural killer cells activated by MHC class I(low) targets prime dendritic cells to induce protective CD8 T cell responses. Immunity 2003, 19, 561–569. [Google Scholar] [CrossRef]
- Criado, M.; Lindstrom, J.M.; Anderson, C.G.; Dennert, G. Cytotoxic granules from killer cells: specificity of granules and insertion of channels of defined size into target membranes. J. Immunol. 1985, 135, 4245–4251. [Google Scholar] [CrossRef]
- Keefe, D.; Shi, L.; Feske, S.; Massol, R.; Navarro, F.; Kirchhausen, T.; Lieberman, J. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity 2005, 23, 249–262. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Moretta, L.; Moretta, A. Unravelling natural killer cell function: triggering and inhibitory human NK receptors. EMBO J. 2004, 23, 255–259. [Google Scholar] [CrossRef]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol. Rev. 2006, 214, 73–91. [Google Scholar] [CrossRef]
- Warren, H.S.; Altin, J.G.; Waldron, J.C.; Kinnear, B.F.; Parish, C.R. A carbohydrate structure associated with CD15 (Lewis x) on myeloid cells is a novel ligand for human CD2. J. Immunol. 1996, 156, 2866–2873. [Google Scholar] [CrossRef]
- Higai, K.; Ichikawa, A.; Matsumoto, K. Binding of sialyl Lewis X antigen to lectin-like receptors on NK cells induces cytotoxicity and tyrosine phosphorylation of a 17-kDa protein. Biochim. Biophys. Acta 2006, 1760, 1355–1363. [Google Scholar] [CrossRef]
- Sugie, K.; Nakamura, K.; Teshigawara, K.; Diamond, M.S.; Springer, T.A.; Nakamura, Y.; Leonard, W.J.; Uchida, A.; Yodoi, J. Activation of natural killer cells by the mAb YTA-1 that recognizes leukocyte function-associated antigen-1. Int. Immunol. 1995, 7, 763–769. [Google Scholar]
- Orange, J.S. Formation and function of the lytic NK-cell immunological synapse. Nat. Rev. Immunol. 2008, 8, 713–725. [Google Scholar] [CrossRef]
- Davignon, D.; Martz, E.; Reynolds, T.; Kurzinger, K.; Springer, T.A. Lymphocyte function-associated antigen 1 (LFA-1): A surface antigen distinct from Lyt-2,3 that participates in T lymphocyte-mediated killing. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 4535–4539. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Kawashima, H.; Lowe, J.B.; Lanier, L.L.; Fukuda, M. Suppression of tumor formation in lymph nodes by L-selectin-mediated natural killer cell recruitment. J. Exp. Med. 2005, 202, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Lollo, B.A.; Chan, K.W.; Hanson, E.M.; Moy, V.T.; Brian, A.A. Direct evidence for two affinity states for lymphocyte function-associated antigen 1 on activated T cells. J. Biol. Chem. 1993, 268, 21693–21700. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; Ljunggren, H.G.; Long, E.O. Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood 2009, 114, 2657–2666. [Google Scholar] [CrossRef]
- Barber, D.F.; Faure, M.; Long, E.O. LFA-1 contributes an early signal for NK cell cytotoxicity. J. Immunol. 2004, 173, 3653–3659. [Google Scholar] [CrossRef] [PubMed]
- Mentlik, A.N.; Sanborn, K.B.; Holzbaur, E.L.; Orange, J.S. Rapid lytic granule convergence to the MTOC in natural killer cells is dependent on dynein but not cytolytic commitment. Mol. Biol. Cell 2010, 21, 2241–2256. [Google Scholar] [CrossRef]
- Davis, D.M.; Chiu, I.; Fassett, M.; Cohen, G.B.; Mandelboim, O.; Strominger, J.L. The human natural killer cell immune synapse. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 15062–15067. [Google Scholar] [CrossRef]
- Riteau, B.; Barber, D.F.; Long, E.O. Vav1 phosphorylation is induced by beta2 integrin engagement on natural killer cells upstream of actin cytoskeleton and lipid raft reorganization. J. Exp. Med. 2003, 198, 469–474. [Google Scholar] [CrossRef]
- Graham, D.B.; Cella, M.; Giurisato, E.; Fujikawa, K.; Miletic, A.V.; Kloeppel, T.; Brim, K.; Takai, T.; Shaw, A.S.; Colonna, M.; et al. Vav1 controls DAP10-mediated natural cytotoxicity by regulating actin and microtubule dynamics. J. Immunol. 2006, 177, 2349–2355. [Google Scholar] [CrossRef]
- Wulfing, C.; Purtic, B.; Klem, J.; Schatzle, J.D. Stepwise cytoskeletal polarization as a series of checkpoints in innate but not adaptive cytolytic killing. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7767–7772. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; March, M.E.; Barber, D.F.; Ljunggren, H.G.; Long, E.O. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J. Exp. Med. 2005, 202, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Bottino, C.; Castriconi, R.; Moretta, L.; Moretta, A. Cellular ligands of activating NK receptors. Trends Immunol. 2005, 26, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Sivori, S.; Vitale, M.; Pende, D.; Morelli, L.; Augugliaro, R.; Bottino, C.; Moretta, L. Existence of both inhibitory (p58) and activatory (p50) receptors for HLA-C molecules in human natural killer cells. J. Exp. Med. 1995, 182, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Dohring, C.; Scheidegger, D.; Samaridis, J.; Cella, M.; Colonna, M. A human killer inhibitory receptor specific for HLA-A1,2. J. Immunol. 1996, 156, 3098–3101. [Google Scholar] [CrossRef]
- Pessino, A.; Sivori, S.; Bottino, C.; Malaspina, A.; Morelli, L.; Moretta, L.; Biassoni, R.; Moretta, A. Molecular cloning of NKp46: a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 1998, 188, 953–960. [Google Scholar] [CrossRef]
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999, 190, 1505–1516. [Google Scholar] [CrossRef]
- Chiesa, S.; Mingueneau, M.; Fuseri, N.; Malissen, B.; Raulet, D.H.; Malissen, M.; Vivier, E.; Tomasello, E. Multiplicity and plasticity of natural killer cell signaling pathways. Blood 2006, 107, 2364–2372. [Google Scholar] [CrossRef]
- MacFarlane, A.W., 4th; Campbell, K.S. Signal transduction in natural killer cells. Curr. Top. Microbiol. Immunol. 2006, 298, 23–57. [Google Scholar]
- Colucci, F.; Schweighoffer, E.; Tomasello, E.; Turner, M.; Ortaldo, J.R.; Vivier, E.; Tybulewicz, V.L.; Di Santo, J.P. Natural cytotoxicity uncoupled from the Syk and ZAP-70 intracellular kinases. Nat. Immunol. 2002, 3, 288–294. [Google Scholar] [CrossRef]
- Chang, C.; Dietrich, J.; Harpur, A.G.; Lindquist, J.A.; Haude, A.; Loke, Y.W.; King, A.; Colonna, M.; Trowsdale, J.; Wilson, M.J. Cutting edge: KAP10, a novel transmembrane adapter protein genetically linked to DAP12 but with unique signaling properties. J. Immunol. 1999, 163, 4651–4654. [Google Scholar] [CrossRef] [PubMed]
- Upshaw, J.L.; Arneson, L.N.; Schoon, R.A.; Dick, C.J.; Billadeau, D.D.; Leibson, P.J. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat. Immunol. 2006, 7, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Tybulewicz, V.L. Vav-family proteins in T-cell signalling. Curr. Opin. Immunol. 2005, 17, 267–274. [Google Scholar] [CrossRef]
- Carpenter, G.; Ji, Q. Phospholipase C-gamma as a signal-transducing element. Exp. Cell Res. 1999, 253, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Das, A.; Gross, C.C.; Bryceson, Y.T.; Long, E.O. Synergistic signals for natural cytotoxicity are required to overcome inhibition by c-Cbl ubiquitin ligase. Immunity 2010, 32, 175–186. [Google Scholar] [CrossRef]
- Menager, M.M.; Menasche, G.; Romao, M.; Knapnougel, P.; Ho, C.H.; Garfa, M.; Raposo, G.; Feldmann, J.; Fischer, A.; de Saint Basile, G. Secretory cytotoxic granule maturation and exocytosis require the effector protein hMunc13-4. Nat. Immunol. 2007, 8, 257–267. [Google Scholar] [CrossRef]
- Wood, S.M.; Meeths, M.; Chiang, S.C.; Bechensteen, A.G.; Boelens, J.J.; Heilmann, C.; Horiuchi, H.; Rosthoj, S.; Rutynowska, O.; Winiarski, J.; et al. Different NK cell-activating receptors preferentially recruit Rab27a or Munc13-4 to perforin-containing granules for cytotoxicity. Blood 2009, 114, 4117–4127. [Google Scholar] [CrossRef]
- Yang, J.; McBride, S.; Mak, D.O.; Vardi, N.; Palczewski, K.; Haeseleer, F.; Foskett, J.K. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca(2+) release channels. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7711–7716. [Google Scholar] [CrossRef]
- Maul-Pavicic, A.; Chiang, S.C.; Rensing-Ehl, A.; Jessen, B.; Fauriat, C.; Wood, S.M.; Sjoqvist, S.; Hufnagel, M.; Schulze, I.; Bass, T.; et al. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc. Natl. Acad. Sci. U. S. A. 108 108, 3324–3329. [CrossRef]
- Brown, M.H.; Boles, K.; van der Merwe, P.A.; Kumar, V.; Mathew, P.A.; Barclay, A.N. 2B4, the natural killer and T cell immunoglobulin superfamily surface protein, is a ligand for CD48. J. Exp. Med. 1998, 188, 2083–2090. [Google Scholar] [CrossRef]
- Kumaresan, P.R.; Lai, W.C.; Chuang, S.S.; Bennett, M.; Mathew, P.A. CS1, a novel member of the CD2 family, is homophilic and regulates NK cell function. Mol. Immunol. 2002, 39, 1–8. [Google Scholar] [CrossRef]
- Falco, M.; Marcenaro, E.; Romeo, E.; Bellora, F.; Marras, D.; Vely, F.; Ferracci, G.; Moretta, L.; Moretta, A.; Bottino, C. Homophilic interaction of NTBA, a member of the CD2 molecular family: induction of cytotoxicity and cytokine release in human NK cells. Eur. J. Immunol. 2004, 34, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Sayos, J.; Wu, C.; Morra, M.; Wang, N.; Zhang, X.; Allen, D.; van Schaik, S.; Notarangelo, L.; Geha, R.; Roncarolo, M.G.; et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature 1998, 395, 462–469. [Google Scholar] [CrossRef]
- Chen, R.; Relouzat, F.; Roncagalli, R.; Aoukaty, A.; Tan, R.; Latour, S.; Veillette, A. Molecular dissection of 2B4 signaling: implications for signal transduction by SLAM-related receptors. Mol. Cell. Biol. 2004, 24, 5144–5156. [Google Scholar] [CrossRef] [PubMed]
- Eissmann, P.; Beauchamp, L.; Wooters, J.; Tilton, J.C.; Long, E.O.; Watzl, C. Molecular basis for positive and negative signaling by the natural killer cell receptor 2B4 (CD244). Blood 2005, 105, 4722–4729. [Google Scholar] [CrossRef]
- Archuleta, M.M.; Schieven, G.L.; Ledbetter, J.A.; Deanin, G.G.; Burchiel, S.W. 7,12-Dimethylbenz[a]anthracene activates protein-tyrosine kinases Fyn and Lck in the HPB-ALL human T-cell line and increases tyrosine phosphorylation of phospholipase C-gamma 1, formation of inositol 1,4,5-trisphosphate, and mobilization of intracellular calcium. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 6105–6109. [Google Scholar] [PubMed]
- Chan, B.; Lanyi, A.; Song, H.K.; Griesbach, J.; Simarro-Grande, M.; Poy, F.; Howie, D.; Sumegi, J.; Terhorst, C.; Eck, M.J. SAP couples Fyn to SLAM immune receptors. Nat. Cell Biol. 2003, 5, 155–160. [Google Scholar] [CrossRef]
- Ozdener, F.; Dangelmaier, C.; Ashby, B.; Kunapuli, S.P.; Daniel, J.L. Activation of phospholipase Cgamma2 by tyrosine phosphorylation. Mol. Pharmacol. 2002, 62, 672–679. [Google Scholar] [CrossRef]
- Binstadt, B.A.; Brumbaugh, K.M.; Dick, C.J.; Scharenberg, A.M.; Williams, B.L.; Colonna, M.; Lanier, L.L.; Kinet, J.P.; Abraham, R.T.; Leibson, P.J. Sequential involvement of Lck and SHP-1 with MHC-recognizing receptors on NK cells inhibits FcR-initiated tyrosine kinase activation. Immunity 1996, 5, 629–638. [Google Scholar] [CrossRef]
- Burshtyn, D.N.; Scharenberg, A.M.; Wagtmann, N.; Rajagopalan, S.; Berrada, K.; Yi, T.; Kinet, J.P.; Long, E.O. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity 1996, 4, 77–85. [Google Scholar] [CrossRef]
- Kaufman, D.S.; Schoon, R.A.; Robertson, M.J.; Leibson, P.J. Inhibition of selective signaling events in natural killer cells recognizing major histocompatibility complex class I. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 6484–6488. [Google Scholar] [CrossRef] [PubMed]
- Valiante, N.M.; Phillips, J.H.; Lanier, L.L.; Parham, P. Killer cell inhibitory receptor recognition of human leukocyte antigen (HLA) class I blocks formation of a pp36/PLC-gamma signaling complex in human natural killer (NK) cells. J. Exp. Med. 1996, 184, 2243–2250. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, C.C.; Watzl, C.; Billadeau, D.D.; Leibson, P.J.; Burshtyn, D.N.; Long, E.O. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol. Cell. Biol. 2003, 23, 6291–6299. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.E.; Long, E.O. Inhibitory receptor signaling via tyrosine phosphorylation of the adaptor Crk. Immunity 2008, 29, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, E.; Pende, D.; Viale, O.; Than, A.; Di Donato, C.; Orengo, A.M.; Biassoni, R.; Verdiani, S.; Amoroso, A.; Moretta, A.; et al. Involvement of HLA class I alleles in natural killer (NK) cell-specific functions: expression of HLA-Cw3 confers selective protection from lysis by alloreactive NK clones displaying a defined specificity (specificity 2). J. Exp. Med. 1992, 176, 963–971. [Google Scholar] [CrossRef]
- Moretta, A.; Biassoni, R.; Bottino, C.; Pende, D.; Vitale, M.; Poggi, A.; Mingari, M.C.; Moretta, L. Major histocompatibility complex class I-specific receptors on human natural killer and T lymphocytes. Immunol. Rev. 1997, 155, 105–117. [Google Scholar] [CrossRef]
- Natarajan, K.; Dimasi, N.; Wang, J.; Mariuzza, R.A.; Margulies, D.H. Structure and function of natural killer cell receptors: Multiple molecular solutions to self, nonself discrimination. Annu. Rev. Immunol. 2002, 20, 853–885. [Google Scholar] [CrossRef]
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [CrossRef]
- Oberg, L.; Johansson, S.; Michaelsson, J.; Tomasello, E.; Vivier, E.; Karre, K.; Hoglund, P. Loss or mismatch of MHC class I is sufficient to trigger NK cell-mediated rejection of resting lymphocytes in vivo—Role of KARAP/DAP12-dependent and -independent pathways. Eur. J. Immunol. 2004, 34, 1646–1653. [Google Scholar] [CrossRef]
- Ruscetti, F.W.; Mikovits, J.A.; Kalyanaraman, V.S.; Overton, R.; Stevenson, H.; Stromberg, K.; Herberman, R.B.; Farrar, W.L.; Ortaldo, J.R. Analysis of effector mechanisms against HTLV-I- and HTLV-III/LAV-infected lymphoid cells. J. Immunol. 1986, 136, 3619–3624. [Google Scholar] [CrossRef]
- Zheng, Z.Y.; Zucker-Franklin, D. Apparent ineffectiveness of natural killer cells vis-a-vis retrovirus-infected targets. J. Immunol. 1992, 148, 3679–3685. [Google Scholar] [CrossRef] [PubMed]
- Bonaparte, M.I.; Barker, E. Inability of natural killer cells to destroy autologous HIV-infected T lymphocytes. AIDS 2003, 17, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Houchins, J.P.; Yabe, T.; McSherry, C.; Bach, F.H. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J. Exp. Med. 1991, 173, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Song, Y.; Bakker, A.B.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999, 285, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Cosman, D.; Mullberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef]
- Harrison, J.C.; Haber, J.E. Surviving the breakup: The DNA damage checkpoint. Annu. Rev. Genet. 2006, 40, 209–235. [Google Scholar] [CrossRef]
- Andersen, J.L.; Le Rouzic, E.; Planelles, V. HIV-1 Vpr: Mechanisms of G2 arrest and apoptosis. Exp. Mol. Pathol. 2008, 85, 2–10. [Google Scholar] [CrossRef]
- Roshal, M.; Kim, B.; Zhu, Y.; Nghiem, P.; Planelles, V. Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J. Biol. Chem. 2003, 278, 25879–25886. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.C.; Rogel, M.E.; Kinsey, C.M.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.H.; Emerman, M. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of Vpr in vivo. Nat. Med. 1998, 4, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Hrimech, M.; Yao, X.J.; Bachand, F.; Rougeau, N.; Cohen, E.A. Human immunodeficiency virus type 1 (HIV-1) Vpr functions as an immediate-early protein during HIV-1 infection. J. Virol. 1999, 73, 4101–4109. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.S.; Chen, J.; Andersen, J.L.; Ardon, O.; Dehart, J.L.; Blackett, J.; Choudhary, S.K.; Camerini, D.; Nghiem, P.; Planelles, V. Human immunodeficiency virus type 1 Vpr-mediated G2 arrest requires Rad17 and Hus1 and induces nuclear BRCA1 and gamma-H2AX focus formation. Mol. Cell Biol. 2004, 24, 9286–9294. [Google Scholar] [CrossRef]
- Zhao, L.J.; Wang, L.; Mukherjee, S.; Narayan, O. Biochemical mechanism of HIV-1 Vpr function. Oligomerization mediated by the N-terminal domain. J. Biol. Chem. 1994, 269, 32131–32137. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Belaidouni, N.; Estrabaud, E.; Morel, M.; Rain, J.C.; Transy, C.; Margottin-Goguet, F. HIV1 Vpr Arrests the Cell Cycle by Recruiting DCAF1/VprBP, a Receptor of the Cul4-DDB1 Ubiquitin Ligase. Cell Cycle 2007, 6, 182–188. [Google Scholar] [CrossRef]
- DeHart, J.L.; Zimmerman, E.S.; Ardon, O.; Monteiro-Filho, C.M.; Arganaraz, E.R.; Planelles, V. HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol. J. 2007, 4, 57. [Google Scholar] [CrossRef]
- DeHart, J.L.; Planelles, V. Human immunodeficiency virus type 1 vpr links proteasomal degradation and checkpoint activation. J. Virol. 2008, 82, 1066–1072. [Google Scholar] [CrossRef]
- Ward, J.; Davis, Z.; DeHart, J.; Zimmerman, E.; Bosque, A.; Brunetta, E.; Mavilio, D.; Planelles, V.; Barker, E. HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog. 2009, 5, e1000613. [Google Scholar] [CrossRef]
- Richard, J.; Sindhu, S.; Pham, T.N.; Belzile, J.P.; Cohen, E.A. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood 115, 1354–1363. [CrossRef]
- Lopez-Soto, A.; Quinones-Lombrana, A.; Lopez-Arbesu, R.; Lopez-Larrea, C.; Gonzalez, S. Transcriptional regulation of ULBP1, a human ligand of the NKG2D receptor. J. Biol. Chem. 2006, 281, 30419–30430. [Google Scholar] [CrossRef]
- Cerboni, C.; Neri, F.; Casartelli, N.; Zingoni, A.; Cosman, D.; Rossi, P.; Santoni, A.; Doria, M. Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J. Gen. Virol. 2007, 88, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Kerkau, T.; Schmitt-Landgraf, R.; Schimpl, A.; Wecker, E. Downregulation of HLA class I antigens in HIV-1-infected cells. AIDS Res. Hum. Retroviruses 1989, 5, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, O.; Marechal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.B.; Gandhi, R.T.; Davis, D.M.; Mandelboim, O.; Chen, B.K.; Strominger, J.L.; Baltimore, D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 1999, 10, 661–671. [Google Scholar] [CrossRef]
- Williams, M.; Roeth, J.F.; Kasper, M.R.; Fleis, R.I.; Przybycin, C.G.; Collins, K.L. Direct binding of human immunodeficiency virus type 1 Nef to the major histocompatibility complex class I (MHC-I) cytoplasmic tail disrupts MHC-I trafficking. J. Virol. 2002, 76, 12173–12184. [Google Scholar] [CrossRef]
- Le Gall, S.; Buseyne, F.; Trocha, A.; Walker, B.D.; Heard, J.M.; Schwartz, O. Distinct trafficking pathways mediate Nef-induced and clathrin-dependent major histocompatibility complex class I down-regulation. J. Virol. 2000, 74, 9256–9266. [Google Scholar] [CrossRef]
- Le Gall, S.; Erdtmann, L.; Benichou, S.; Berlioz-Torrent, C.; Liu, L.; Benarous, R.; Heard, J.M.; Schwartz, O. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 1998, 8, 483–495. [Google Scholar] [CrossRef]
- Noviello, C.M.; Benichou, S.; Guatelli, J.C. Cooperative binding of the class I major histocompatibility complex cytoplasmic domain and human immunodeficiency virus type 1 Nef to the endosomal AP-1 complex via its mu subunit. J. Virol. 2008, 82, 1249–1258. [Google Scholar] [CrossRef]
- Wonderlich, E.R.; Williams, M.; Collins, K.L. The tyrosine binding pocket in the adaptor protein 1 (AP-1) mu1 subunit is necessary for Nef to recruit AP-1 to the major histocompatibility complex class I cytoplasmic tail. J. Biol. Chem. 2008, 283, 3011–3022. [Google Scholar] [CrossRef]
- Duden, R.; Griffiths, G.; Frank, R.; Argos, P.; Kreis, T.E. Beta-COP, a 110 kd protein associated with non-clathrin-coated vesicles and the Golgi complex, shows homology to beta-adaptin. Cell 1991, 64, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.R.; Wonderlich, E.R.; Roeth, J.F.; Leonard, J.A.; Collins, K.L. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog. 2008, 4, e1000131. [Google Scholar] [CrossRef] [PubMed]
- Piguet, V.; Wan, L.; Borel, C.; Mangasarian, A.; Demaurex, N.; Thomas, G.; Trono, D. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell Biol. 2000, 2, 163–167. [Google Scholar] [CrossRef]
- Greenberg, M.E.; Iafrate, A.J.; Skowronski, J. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. EMBO J. 1998, 17, 2777–2789. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.R.; Collins, K.L. Nef-mediated disruption of HLA-A2 transport to the cell surface in T cells. J. Virol. 2003, 77, 3041–3049. [Google Scholar] [CrossRef]
- Lubben, N.B.; Sahlender, D.A.; Motley, A.M.; Lehner, P.J.; Benaroch, P.; Robinson, M.S. HIV-1 Nef-induced down-regulation of MHC class I requires AP-1 and clathrin but not PACS-1 and is impeded by AP-2. Mol. Biol. Cell 2007, 18, 3351–3365. [Google Scholar] [CrossRef] [PubMed]
- Bonaparte, M.I.; Barker, E. Killing of human immunodeficiency virus-infected primary T-cell blasts by autologous natural killer cells is dependent on the ability of the virus to alter the expression of major histocompatibility complex class I molecules. Blood 2004, 104, 2087–2094. [Google Scholar] [CrossRef]
- Shah, A.H.; Sowrirajan, B.; Davis, Z.B.; Ward, J.P.; Campbell, E.M.; Planelles, V.; Barker, E. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 2010, 8, 397–409. [Google Scholar] [CrossRef]
- Bottino, C.; Falco, M.; Parolini, S.; Marcenaro, E.; Augugliaro, R.; Sivori, S.; Landi, E.; Biassoni, R.; Notarangelo, L.D.; Moretta, L.; et al. NTB-A [correction of GNTB-A], a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J. Exp. Med. 2001, 194, 235–246. [Google Scholar] [CrossRef]
- Ward, J.; Bonaparte, M.; Sacks, J.; Guterman, J.; Fogli, M.; Mavilio, D.; Barker, E. HIV modulates the expression of ligands important in triggering natural killer cell cytotoxic responses on infected primary T-cell blasts. Blood 2007, 110, 1207–1214. [Google Scholar] [CrossRef]
- Fogli, M.; Mavilio, D.; Brunetta, E.; Varchetta, S.; Ata, K.; Roby, G.; Kovacs, C.; Follmann, D.; Pende, D.; Ward, J.; et al. Lysis of endogenously infected CD4+ T cell blasts by rIL-2 activated autologous natural killer cells from HIV-infected viremic individuals. PLoS Pathog. 2008, 4, e1000101. [Google Scholar] [CrossRef] [PubMed]
- Flaig, R.M.; Stark, S.; Watzl, C. Cutting edge: NTB-A activates NK cells via homophilic interaction. J. Immunol. 2004, 172, 6524–6527. [Google Scholar] [CrossRef] [PubMed]
- Meinke, S.; Watzl, C. Functions of NTB-A’s adaptor proteins. University Heidelberg: Heidelberg, Germany, 2010; unpublished work. [Google Scholar]
- Claus, M.; Meinke, S.; Bhat, R.; Watzl, C. Regulation of NK cell activity by 2B4, NTB-A and CRACC. Front. Biosci. 2008, 13, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Sowrirajan, B.; Cambell, E.; Barker, E. HIV-1 Vpu Sequesters NTB-A in the trans-Golgi Network. Rush University Medical Center, Stritch School of Medicine, Loyola University Chicago: Maywood, IL, USA, 2011; unpublished work. [Google Scholar]
- Schubert, U.; Schneider, T.; Henklein, P.; Hoffmann, K.; Berthold, E.; Hauser, H.; Pauli, G.; Porstmann, T. Human-immunodeficiency-virus-type-1-encoded Vpu protein is phosphorylated by casein kinase II. Eur. J. Biochem. 1992, 204, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [CrossRef]
- Schubert, U.; Anton, L.C.; Bacik, I.; Cox, J.H.; Bour, S.; Bennink, J.R.; Orlowski, M.; Strebel, K.; Yewdell, J.W. CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 1998, 72, 2280–2288. [Google Scholar] [CrossRef] [PubMed]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Fruh, K.; Moses, A.V. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. J. Virol. 2009, 83, 7931–7947. [Google Scholar] [CrossRef]
- Goffinet, C.; Homann, S.; Ambiel, I.; Tibroni, N.; Rupp, D.; Keppler, O.T.; Fackler, O.T. Antagonism of CD317 restriction of human immunodeficiency virus type 1 (HIV-1) particle release and depletion of CD317 are separable activities of HIV-1 Vpu. J. Virol. 2010, 84, 4089–4094. [Google Scholar] [CrossRef]
- Ward, J.; Barker, E. HIV-1 increases cell surface levels of ICAMs. Rush University Medical Center, Chicago, IL, USA. 2008, unpublished work.
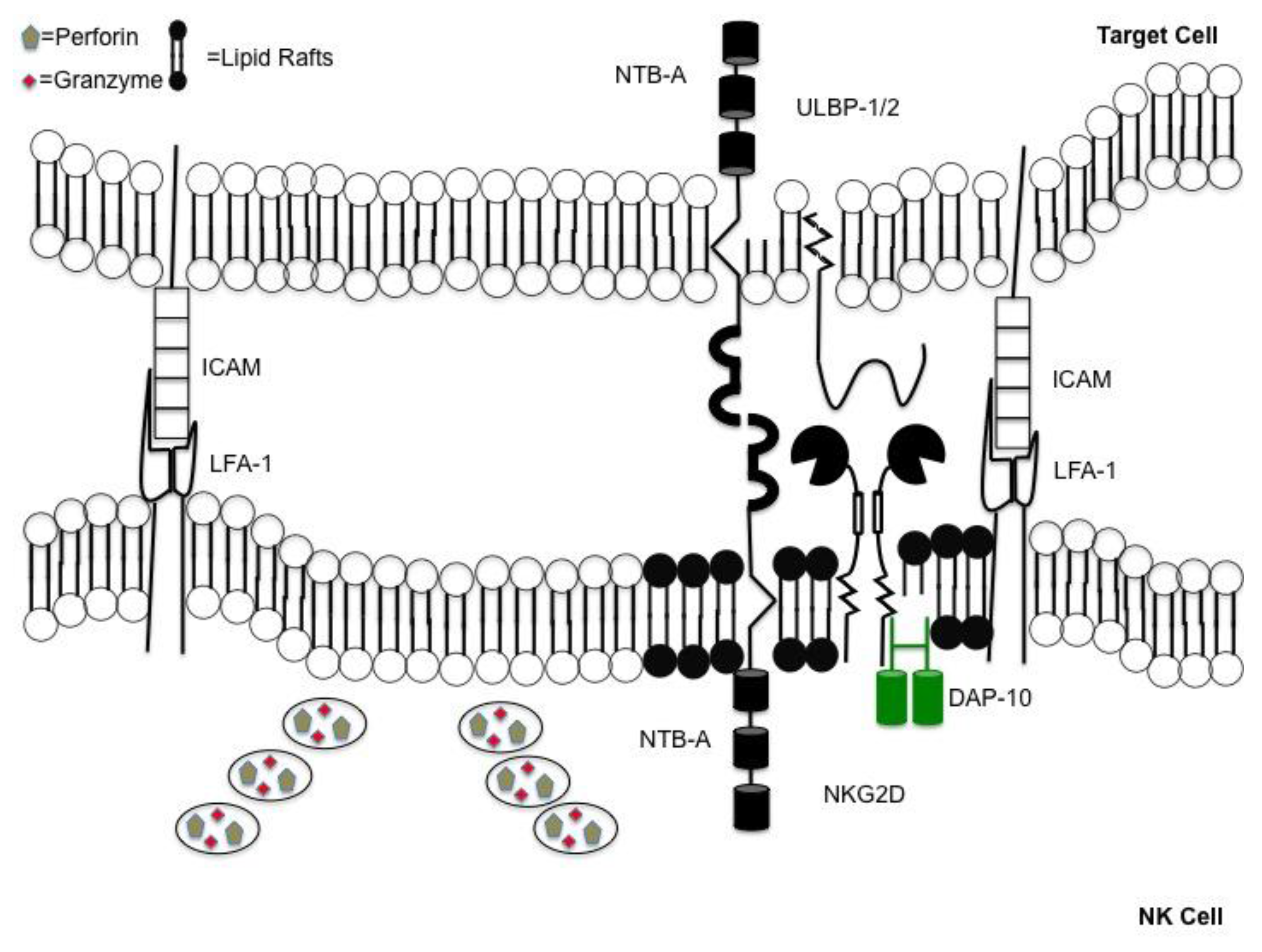
Figure 1.
Coordinated events occur when forming an immunological synapse. Directed release of cytotoxic granules requires coordinated series of events. Synapse formation is accomplished following engagement of lymphocyte function-associated antigen (LFA)-1 on natural killer (NK) cells with intercellular adhesion molecules (ICAMs) on the target. Immunological synapse contains a “lipid raft” domain (shown in black) containing activating receptors. This is adjacent to a section of the plasma membrane allowing for the unobstructed release of perforin and granzymes, into the space between the NK cell and target.
Figure 1.
Coordinated events occur when forming an immunological synapse. Directed release of cytotoxic granules requires coordinated series of events. Synapse formation is accomplished following engagement of lymphocyte function-associated antigen (LFA)-1 on natural killer (NK) cells with intercellular adhesion molecules (ICAMs) on the target. Immunological synapse contains a “lipid raft” domain (shown in black) containing activating receptors. This is adjacent to a section of the plasma membrane allowing for the unobstructed release of perforin and granzymes, into the space between the NK cell and target.

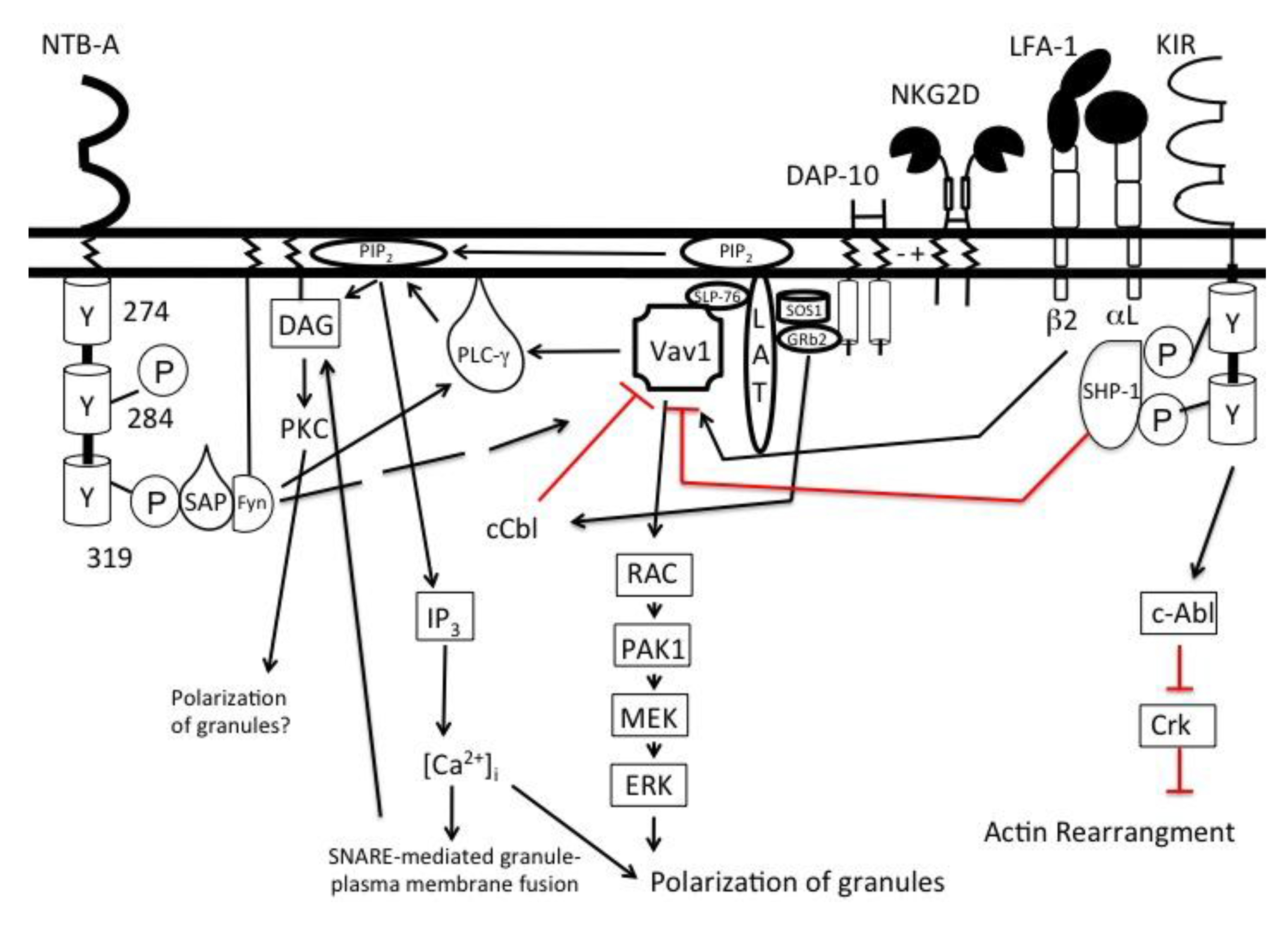
Figure 2.
Two activating signals are required to initiate NK cell degranulation. Signals from activating (NKG2D) and co-activating receptor (NTB-A) converge to activate phospholipase-Cγ (PLCγ). Vav1, a guanine exchange factor, is able to phosphorylate PLCγ when triggered by activating receptors. However, engagement of activating receptors also phosphorylates c-Cbl, an ubiquitin ligase that negatively regulates Vav1. To overcome this inhibition, a co-activating receptor must be engaged to phosphorylate sufficient PLCγ to trigger degranulation. Degranulation is mediated by the cleavage of phosphatidylinositol into diacylglycerol (DAG) and inositol 1,4,5 triphosphate (IP3). DAG recruits key proteins to the plasma membrane to initiate fusion of the granule and plasma membrane. IP3, once cleaved, binds its receptor, IP3 receptor, on the membrane of the endoplasmic reticulum to open calcium channels. Ca2+ influx allows for the exocytosis of the cytolytic granules. Inhibitory receptors such as the KIRs can recruit phosphatases that lead to the dephosphorylation of Vav1. However, the KIRs also have the ability to activate tyrosine kinases like c-Abl leading to the inhibition of actin rearrangement.
Figure 2.
Two activating signals are required to initiate NK cell degranulation. Signals from activating (NKG2D) and co-activating receptor (NTB-A) converge to activate phospholipase-Cγ (PLCγ). Vav1, a guanine exchange factor, is able to phosphorylate PLCγ when triggered by activating receptors. However, engagement of activating receptors also phosphorylates c-Cbl, an ubiquitin ligase that negatively regulates Vav1. To overcome this inhibition, a co-activating receptor must be engaged to phosphorylate sufficient PLCγ to trigger degranulation. Degranulation is mediated by the cleavage of phosphatidylinositol into diacylglycerol (DAG) and inositol 1,4,5 triphosphate (IP3). DAG recruits key proteins to the plasma membrane to initiate fusion of the granule and plasma membrane. IP3, once cleaved, binds its receptor, IP3 receptor, on the membrane of the endoplasmic reticulum to open calcium channels. Ca2+ influx allows for the exocytosis of the cytolytic granules. Inhibitory receptors such as the KIRs can recruit phosphatases that lead to the dephosphorylation of Vav1. However, the KIRs also have the ability to activate tyrosine kinases like c-Abl leading to the inhibition of actin rearrangement.

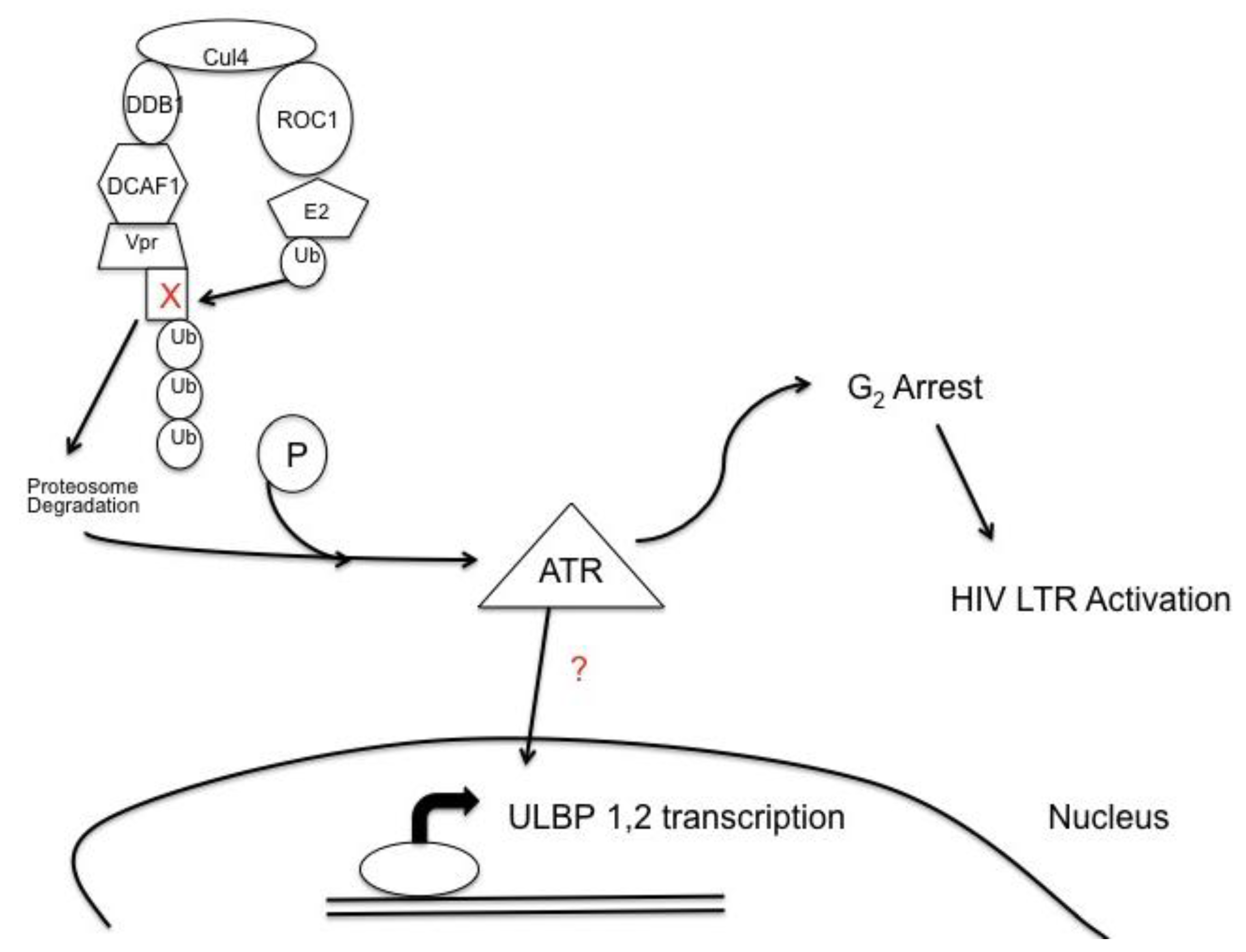
Figure 3.
HIV-1 Vpr leads to expression of NKG2D ligands, unique long 16-binding protein (ULBP)-1 and -2. Viral protein R, Vpr, bridges DCAF1, a substrate recognition factor for an E3 ubiquitin ligase system to an unknown cell cycle protein causing for the cell cycle protein to become ubiquitinated and destroyed. The degradation of the cell cycle protein induces phosphorylation of the DNA damage sensor ataxia telangiectasia-mutated and Rad 3-related (ATR), which in turn induces expression of the NKG2D ligands, ULBP-1 and -2. Phosphorylation of ATR also leads to G2 cell cycle arrest creating a favorable environment for transcription of the HIV long terminal repeat (LTR).
Figure 3.
HIV-1 Vpr leads to expression of NKG2D ligands, unique long 16-binding protein (ULBP)-1 and -2. Viral protein R, Vpr, bridges DCAF1, a substrate recognition factor for an E3 ubiquitin ligase system to an unknown cell cycle protein causing for the cell cycle protein to become ubiquitinated and destroyed. The degradation of the cell cycle protein induces phosphorylation of the DNA damage sensor ataxia telangiectasia-mutated and Rad 3-related (ATR), which in turn induces expression of the NKG2D ligands, ULBP-1 and -2. Phosphorylation of ATR also leads to G2 cell cycle arrest creating a favorable environment for transcription of the HIV long terminal repeat (LTR).

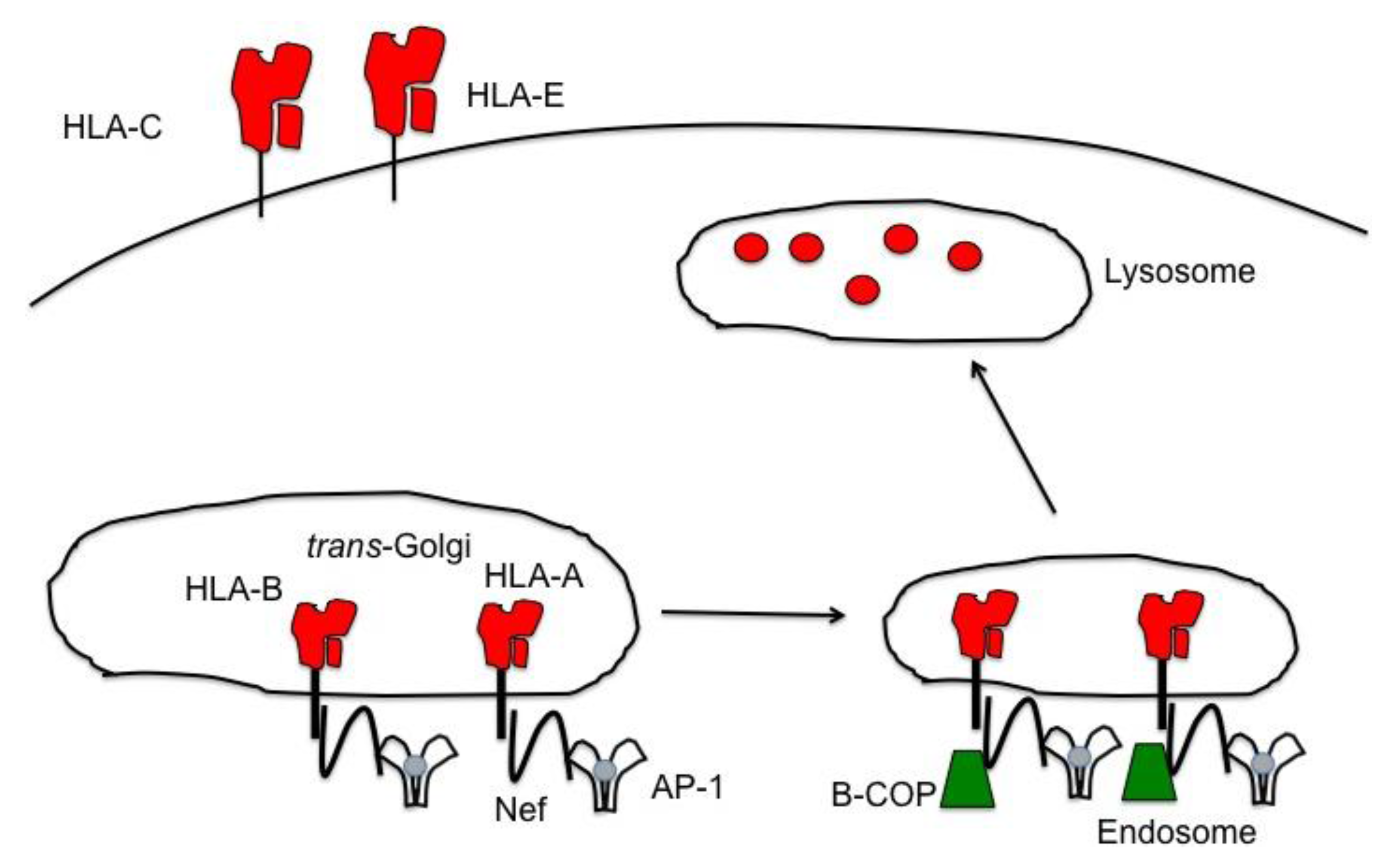
Figure 4.
HLA-A and -B are down modulated by HIV Nef, removing an inhibitory signal for NK cells. HIV-1 Nef selectively down modulates HLA-A and -B, but leaves HLA-C and -E on the infected cell surface. Nef re-routes both HLA-A and -B from the TGN to endosomes and eventually to lysosomes for the degradation of the MHC-I molecules. Nef is able to bind to both the tail of the MHC-I molecules as well as the trafficking molecule, AP-1, to facilitate the transport of HLA-A and -B to the endosomes. Once in the endosome, Nef recruits β-COP for further transport into lysosomes. The down modulation of HLA-A and -B removes the inhibitory ligands for the NK cell inhibitory receptor KIR3DL2 and KIR3DL1, respectively. Yet, because Nef is unable to down modulate HLA-C and -E, NK cells expressing inhibitory receptors for these two MHC-I molecules will inhibit NK cell degranulation of HIV infected cells.
Figure 4.
HLA-A and -B are down modulated by HIV Nef, removing an inhibitory signal for NK cells. HIV-1 Nef selectively down modulates HLA-A and -B, but leaves HLA-C and -E on the infected cell surface. Nef re-routes both HLA-A and -B from the TGN to endosomes and eventually to lysosomes for the degradation of the MHC-I molecules. Nef is able to bind to both the tail of the MHC-I molecules as well as the trafficking molecule, AP-1, to facilitate the transport of HLA-A and -B to the endosomes. Once in the endosome, Nef recruits β-COP for further transport into lysosomes. The down modulation of HLA-A and -B removes the inhibitory ligands for the NK cell inhibitory receptor KIR3DL2 and KIR3DL1, respectively. Yet, because Nef is unable to down modulate HLA-C and -E, NK cells expressing inhibitory receptors for these two MHC-I molecules will inhibit NK cell degranulation of HIV infected cells.

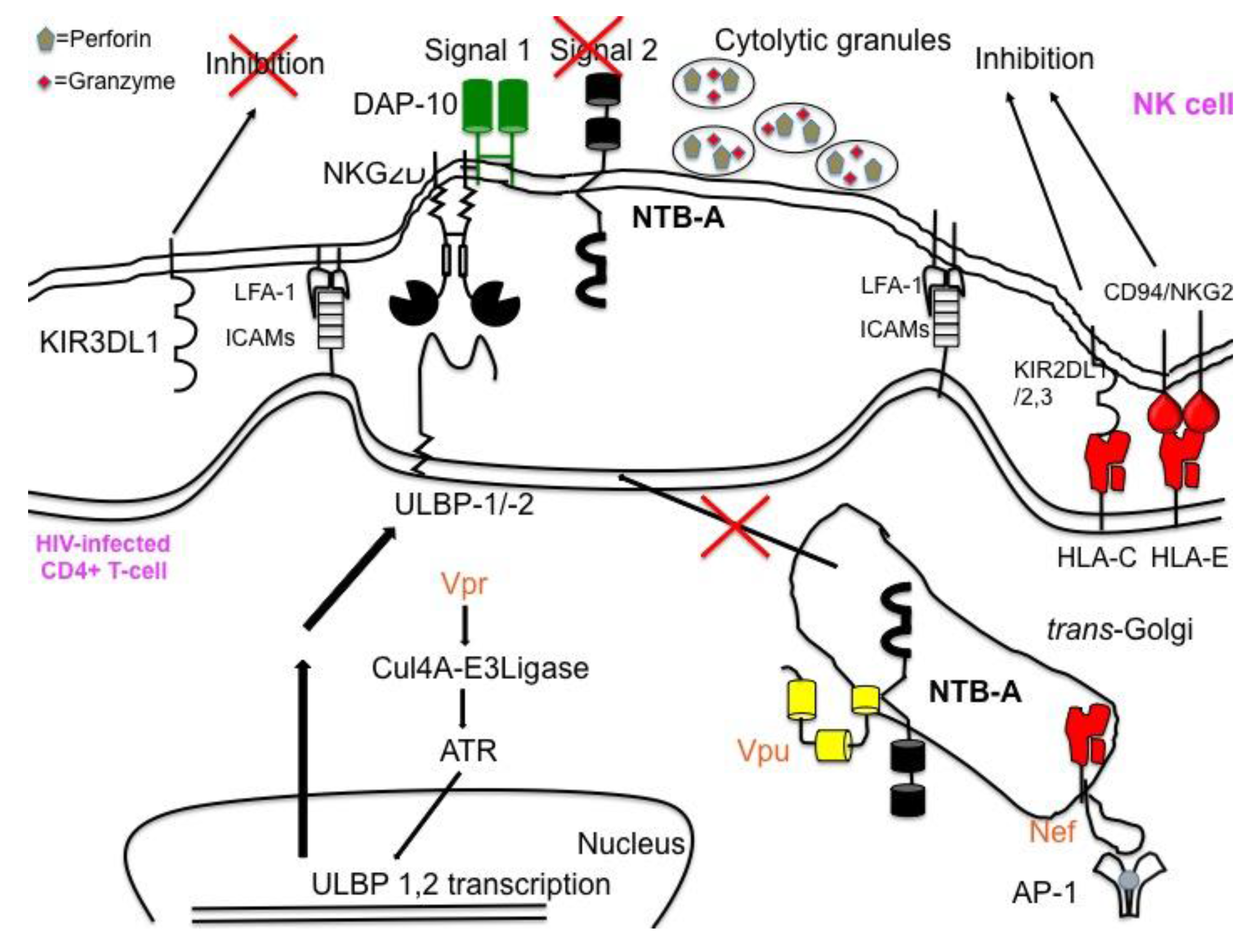
Figure 5.
HIV-1 Vpu’s down modulation of NTB-A prevents NK cells from efficiently killing infected cells. Though Vpr induces expression of ligands for the activating receptor NKG2D and Nef down modulates ligands for the NK cell inhibitory receptor, NK cells are still unable to efficiently lyse HIV infected CD4+ T-cells. Vpu’s sequestration of NTB-A in the trans-Golgi network in the infected cell prevents NK cells from obtaining a second activating signal to initiate NK cell degranulation.
Figure 5.
HIV-1 Vpu’s down modulation of NTB-A prevents NK cells from efficiently killing infected cells. Though Vpr induces expression of ligands for the activating receptor NKG2D and Nef down modulates ligands for the NK cell inhibitory receptor, NK cells are still unable to efficiently lyse HIV infected CD4+ T-cells. Vpu’s sequestration of NTB-A in the trans-Golgi network in the infected cell prevents NK cells from obtaining a second activating signal to initiate NK cell degranulation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Natural Killer Cell Activation Receptors.
| Name | Ligand | Adaptor Protein |
|---|---|---|
| NKp30 | Unknown | FcεRIγCD3ζ |
| NKp44 | Unknown | DAP12 |
| NKp46 | Unknown | FcεRIγCD3ζ |
| CD158h/KIR2DS1 | HLA-C | DAP12 |
| CD158i/KIR2DS4 | HLA-Cw4 | DAP12 |
| CD158e2/KIR3DS1 | Unknown | DAP12 |
| CD158j/KIR2DS2 | Unknown | DAP12 |
| NKG2D | ULBP-1-6, MIC-A,-B | DAP10 |
| CD16 | IgG | FcεRIγCD3ζ |
Modified from [25].
Table 2.
Natural Killer Cell co-Activation Receptors.
| Name | Ligand | Adaptor Protein |
|---|---|---|
| NTB-A | NTB-A | SAP/EAT-2 |
| DNAM-1 | CD155/CD112 | Protein Kinase C |
| CRACC | CRACC | EAT-2 |
| 2B4 | CD48 | SAP |
Modified from [25].
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2011 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sowrirajan, B.; Barker, E. The Natural Killer Cell Cytotoxic Function Is Modulated by HIV-1 Accessory Proteins. Viruses 2011, 3, 1091-1111. https://doi.org/10.3390/v3071091
AMA Style
Sowrirajan B, Barker E. The Natural Killer Cell Cytotoxic Function Is Modulated by HIV-1 Accessory Proteins. Viruses. 2011; 3(7):1091-1111. https://doi.org/10.3390/v3071091
Chicago/Turabian StyleSowrirajan, Bharatwaj, and Edward Barker. 2011. "The Natural Killer Cell Cytotoxic Function Is Modulated by HIV-1 Accessory Proteins" Viruses 3, no. 7: 1091-1111. https://doi.org/10.3390/v3071091