Arenaviruses and Lethal Mutagenesis. Prospects for New Ribavirin-based Interventions


Abstract
:1. Introduction: complexity of LCMV populations
2. Contributions of LCMV to lethal mutagenesis

{kind=link}
| Observation and implications | References |
|---|---|
| • J.J. Holland and colleagues explore for the first time quasispecies error catastrophe with real viruses and show that poliovirus and vesicular stomatitis virus have very limited tolerance to increased mutagenesis. | [69] |
| This study was the birth of experimental studies on the application of the concept of error catastrophe developed by M. Eigen, P. Schuster and colleagues. | |
| • L.A. Loeb, J. Mullins and colleagues show that a mutagenic pyrimidine analogue impairs HIV-1 replication in cell culture. They coin the term “lethal mutagenesis”. | [70] |
| This study suggested the use of mutagenic agents as antiretroviral drugs. | |
| • E. Domingo, P. Lowenstein and colleagues show that lymphocytic choriomeningitis virus and foot-and-mouth disease virus can be extinguished by mutagenic agents, and that low viral load and low viral fitness favor extinction. | [35,71] |
| These experiments suggested that modification of virus population parameters, specifically a decrease in fitness and low viral load, may help in producing virus extinction. | |
| • S. Crotty, R. Andino, C.E. Cameron and colleagues demonstrate that the antiviral ribonucleoside analogue ribavirin is mutagenic for poliovirus. | [72] |
| This important discovery implied that ribavirin might be exerting some of its antiviral clinical activity as a mutagen. This is still a debated issue, but there is evidence that ribavirin is mutagenic for a number of RNA viruses including LCMV. | |
| • 5-Fluorouracil impeded the establishment of a persistent LCMV infection in mice. | [37] |
| This experiment constitutes a proof of principle of the feasibility of a lethal mutagenesis-based antiviral approach in vivo. | |
| • Experimental and theoretical evidence for the lethal defection model of virus extinction. | [67] |
| These results introduced the concept that a mutagenic agent may not only “kill” virus but that, more subtly, the agent may be generating interfering genomes that participate in the impairment of viral replication and eventual extinction. The results suggested also the possibility of guiding internal interactions within mutant spectra to achieve extinction through modest mutagenic intensities. | |
| • When a mutagen participates in therapy, a sequential inhibitor-mutagen administration might have an advantage over the corresponding combination treatment. | [73,74,75] |
| These studies illustrate that the interactions among drugs must be considered with regard to efficacy, in particular in the case of antiviral inhibitors used with virus-specific mutagenic agents. The general advantage of a combination therapy need not apply when a mutagen is involved in therapy. | |
| • 5-Azacytidine can induce lethal mutagenesis of HIV-1 | [76] |
| This result suggests that some nucleotide analogues can be incorporated both into RNA and DNA during the retroviral life cycle. It shows also that there is room for classic antiviral and anti-cancer agents to find an application in lethal mutagenesis. | |
| • First clinical trial involving administration of a pyrimidine analogue to AIDS patients. The resident HIV-1 was mutagenized although no virus extinction was achieved. | [51] |
| In addition to representing the first clinical trial based on lethal mutagenesis, this result opens the possibility of improved efficacy in vivo using either combination or sequential administration of inhibitors and mutagens, in conjunction with provirus mobilization from carrier cells, a point under active investigation. |
4. Sequential versus combination antiviral treatments
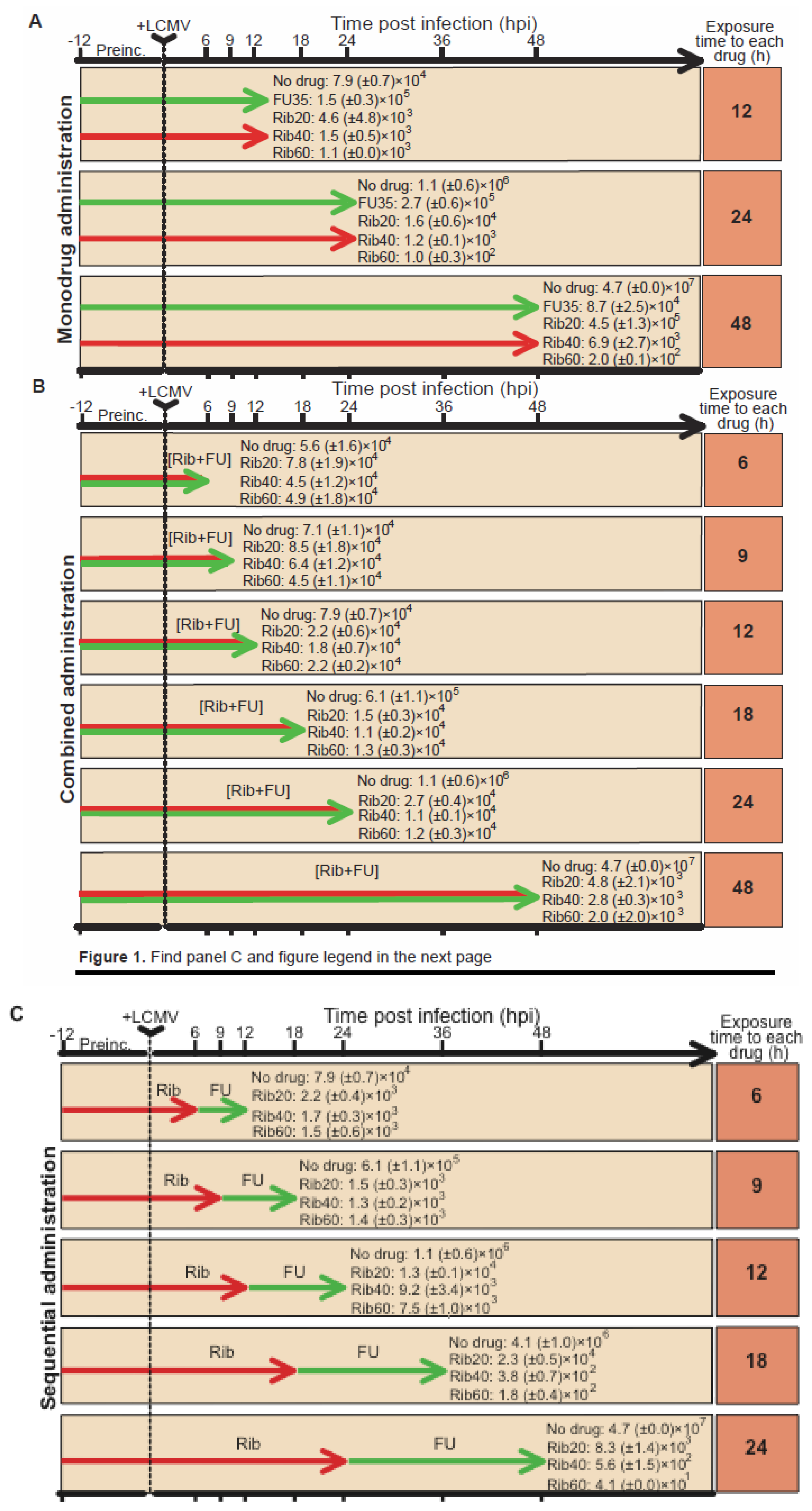
4.2. Ribavirin in sequential versus combination administration for lethal mutagenesis of LCMV
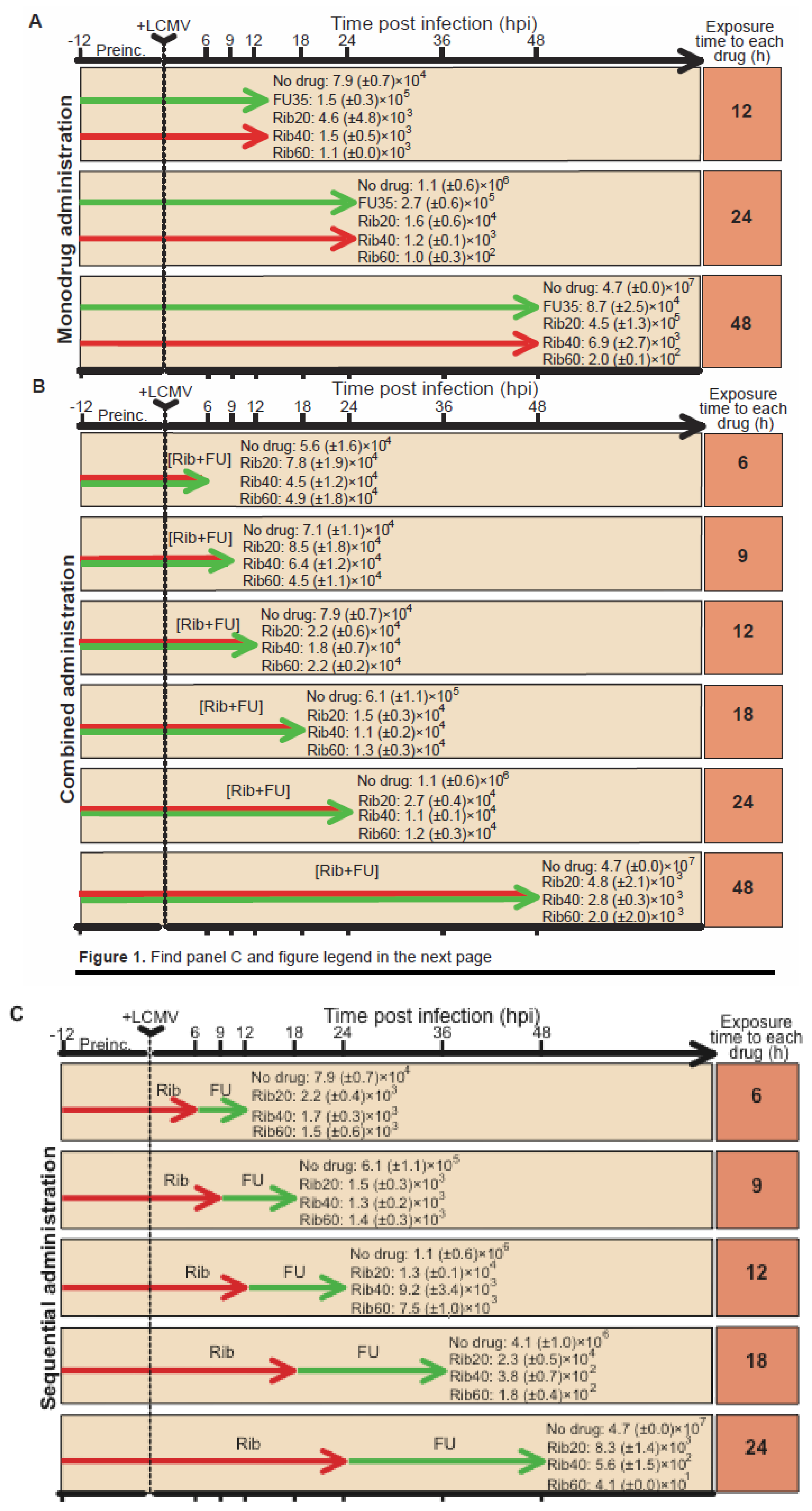
4.3. Implications and prospects for anti-arenavirus interventions
 |
5. Concluding remarks
Conflict of Interest
Acknowledgements
References
- Oldstone, M.B.A. Arenaviruses I and III. Current Topics in Microbiol. and Immunol: Berlin, Heidelberg, 2002; Vol. 262 and 263, Springer-Verlag. [Google Scholar]
- Buchmeier, M.J.; de la Torre, J.C.; Peters, C.J. Arenaviridae: the viruses and their replication. D.M. Knipe P.M., Howley, et al., Eds.; Lappincott Williams & Wilkins: Phyladelphia, 2007; pp. 1791–1827. [Google Scholar]
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; Nichol, S.T.; Lipkin, W.I. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 2009, 5, e1000455. [Google Scholar]
- Goeijenbier, M.; Wagenaar, J.; Goris, M.; Martina, B.; Henttonen, H.; Vaheri, A.; Reusken, C.; Hartskeerl, R.; Osterhaus, A.; Van Gorp, E. Rodent-borne hemorrhagic fevers: under-recognized, widely spread and preventable - epidemiology, diagnostics and treatment. Critical reviews in microbiology 2012, in press. [Google Scholar]
- Barton, L.L. Lymphocytic choriomeningitis virus: a neglected central nervous system pathogen. Clin. Infect. Dis. 1996, 22, 197. [Google Scholar] [CrossRef]
- Fischer, S.A.; Graham, M.B.; Kuehnert, M.J.; Kotton, C.N.; Srinivasan, A.; Marty, F.M.; Comer, J.A.; Guarner, J.; Paddock, C.D.; DeMeo, D.L.; Shieh, W.J.; Erickson, B.R.; Bandy, U.; DeMaria, A., Jr.; Davis, J.P.; Delmonico, F.L.; Pavlin, B.; Likos, A.; Vincent, M.J.; Sealy, T.K.; Goldsmith, C.S.; Jernigan, D.B.; Rollin, P.E.; Packard, M.M.; Patel, M.; Rowland, C.; Helfand, R.F.; Nichol, S.T.; Fishman, J.A.; Ksiazek, T.; Zaki, S.R. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 2006, 354, 2235–2249. [Google Scholar]
- Jamieson, D.J.; Kourtis, A.P.; Bell, M.; Rasmussen, S.A. Lymphocytic choriomeningitis virus: an emerging obstetric pathogen? Am. J. Obstet. Gynecol. 2006, 194(6), 1532–1536. [Google Scholar]
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Conlan, S.; Quan, P.L.; Hui, J.; Marshall, J.; Simons, J.F.; Egholm, M.; Paddock, C.D.; Shieh, W.J.; Goldsmith, C.S.; Zaki, S.R.; Catton, M.; Lipkin, W.I. A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med. 2008, 358, 991–998. [Google Scholar] [CrossRef]
- Ambrosio, A.; Saavedra, M.; Mariani, M.; Gamboa, G.; Maiza, A. Argentine hemorrhagic fever vaccines. Human vaccines 2011, 7, 694–700. [Google Scholar] [CrossRef]
- Goicochea, M.A.; Zapata, J.C.; Bryant, J.; Davis, H.; Salvato, M.S.; Lukashevich, I.S. Evaluation of Lassa virus vaccine immunogenicity in a CBA/J-ML29 mouse model. Vaccine 2012, 30, 1445–1452. [Google Scholar] [CrossRef]
- Sepulveda, C.S.; Garcia, C.C.; Fascio, M.L.; D'Accorso, N.B.; Docampo Palacios, M.L.; Pellon, R.F.; Damonte, E.B. Inhibition of Junin virus RNA synthesis by an antiviral acridone derivative. Antiviral. Res. 2012, 93, 16–22. [Google Scholar] [CrossRef]
- Mendenhall, M.; Russell, A.; Smee, D.F.; Hall, J.O.; Skirpstunas, R.; Furuta, Y.; Gowen, B.B. Effective oral favipiravir (T-705) therapy initiated after the onset of clinical disease in a model of arenavirus hemorrhagic Fever. PLoS neglected tropical diseases 2011, 5, e1342. [Google Scholar] [CrossRef]
- Gowen, B.B.; Smee, D.F.; Wong, M.H.; Hall, J.O.; Jung, K.H.; Bailey, K.W.; Stevens, J.R.; Furuta, Y.; Morrey, J.D. Treatment of late stage disease in a model of arenaviral hemorrhagic fever: T-705 efficacy and reduced toxicity suggests an alternative to ribavirin. PLoS One 2008, 3, e3725. [Google Scholar]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Scribner, C.L.; Craven, R.B.; Johnson, K.M.; Elliott, L.H.; Belmont-Williams, R. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986, 314, 20–26. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiology and Molecular Biology Reviews 2012, 76, 159–216. [Google Scholar] [CrossRef]
- Jabara, C.B.; Jones, C.D.; Roach, J.; Anderson, J.A.; Swanstrom, R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl. Acad. Sci. USA 2011, 108, 20166–20171. [Google Scholar]
- Fernandez, G.; Clotet, B.; Martinez, M.A. Fitness landscape of human immunodeficiency virus type 1 protease quasispecies. J. Virol. 2007, 81, 2485–2496. [Google Scholar] [CrossRef]
- Ciurea, A.; Klenerman, P.; Hunziker, L.; Horvath, E.; Senn, B.M.; Ochsenbein, A.F.; Hengartner, H.; Zinkernagel, R.M. Viral persistence in vivo through selection of neutralizing antibody- escape variants. Proc. Natl. Acad. Sci. USA 2000, 97, 2749–2754. [Google Scholar]
- Aebischer, T.; Moskophidis, D.; Rohrer, U.H.; Zinkernagel, R.M.; Hengartner, H. In vitro selection of lymphocytic choriomeningitis virus escape mutants by cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 11047–11051. [Google Scholar] [CrossRef]
- Ahmed, R.; Hahn, C.S.; Somasundaram, T.; Villarete, L.; Matloubian, M.; Strauss, J.H. Molecular basis of organ-specific selection of viral variants during chronic infection. J. Virol. 1991, 65, 4242–4247. [Google Scholar]
- Ahmed, R.; Simon, R.S.; Matloubian, M.; Kolhekar, S.R.; Southern, P.J.; Freedman, D.M. Genetic analysis of in vivo-selected viral variants causing chronic infection: importance of mutation in the L RNA segment of lymphocytic choriomeningitis virus. J. Virol. 1988, 62, 3301–3308. [Google Scholar]
- Buesa-Gomez, J.; Teng, M.N.; Oldstone, C.E.; Oldstone, M.B.; de la Torre, J.C. Variants able to cause growth hormone deficiency syndrome are present within the disease-nil WE strain of lymphocytic choriomeningitis virus. J. Virol. 1996, 70, 8988–8992. [Google Scholar]
- Ciurea, A.; Hunziker, L.; Martinic, M.M.; Oxenius, A.; Hengartner, H.; Zinkernagel, R.M. CD4+ T-cell-epitope escape mutant virus selected in vivo. Nat. Med. 2001, 7, 795–800. [Google Scholar] [CrossRef]
- Dockter, J.; Evans, C.F.; Tishon, A.; Oldstone, M.B. Competitive selection in vivo by a cell for one variant over another: implications for RNA virus quasispecies in vivo. J. Virol. 1996, 70, 1799–1803. [Google Scholar]
- Evans, C.F.; Borrow, P.; de la Torre, J.C.; Oldstone, M.B. Virus-induced immunosuppression: kinetic analysis of the selection of a mutation associated with viral persistence. J. Virol. 1994, 68, 7367–7373. [Google Scholar]
- Matloubian, M.; Kolhekar, S.R.; Somasundaram, T.; Ahmed, R. Molecular determinants of macrophage tropism and viral persistence: importance of single amino acid changes in the polymerase and glycoprotein of lymphocytic choriomeningitis virus. J. Virol. 1993, 67, 7340–7349. [Google Scholar]
- Matloubian, M.; Somasundaram, T.; Kolhekar, S.R.; Selvakumar, R.; Ahmed, R. Genetic basis of viral persistence: single amino acid change in the viral glycoprotein affects ability of lymphocytic choriomeningitis virus to persist in adult mice. J. Exp. Med. 1990, 172, 1043–1048. [Google Scholar] [CrossRef]
- Pircher, H.; Moskophidis, D.; Rohrer, U.; Burki, K.; Hengartner, H.; Zinkernagel, R.M. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature 1990, 346, 629–633. [Google Scholar] [CrossRef]
- Salvato, M.; Borrow, P.; Shimomaye, E.; Oldstone, M.B. Molecular basis of viral persistence: a single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with suppression of the antiviral cytotoxic T-lymphocyte response and establishment of persistence. J. Virol. 1991, 65, 1863–1869. [Google Scholar]
- Teng, M.N.; Borrow, P.; Oldstone, M.B.; de la Torre, J.C. A single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with the ability to cause growth hormone deficiency syndrome. J. Virol. 1996, 70, 8438–8443. [Google Scholar]
- Hunziker, L.; Ciurea, A.; Recher, M.; Hengartner, H.; Zinkernagel, R.M. Public versus personal serotypes of a viral quasispecies. Proc. Natl. Acad. Sci. USA 2003, 100, 6015–6020. [Google Scholar] [CrossRef]
- Teng, M.N.; Oldstone, M.B.; de la Torre, J.C. Suppression of lymphocytic choriomeningitis virus-induced growth hormone deficiency syndrome by disease-negative virus variants. Virology 1996, 223, 113–119. [Google Scholar] [CrossRef]
- Sevilla, N.; Domingo, E.; de la Torre, J.C. Contribution of LCMV towards deciphering biology of quasispecies in vivo. Curr. Top. Microbiol. Immunol. 2002, 263, 197–220. [Google Scholar] [CrossRef]
- Sevilla, N.; de la Torre, J.C. Arenavirus Diversity And Evolution: Quasispecies In Vivo. Cur. Top. Microbiol. Immunol. 2006, Vol. 299, 315–335. [Google Scholar]
- Grande-Pérez, A.; Sierra, S.; Castro, M.G.; Domingo, E.; Lowenstein, P.R. Molecular indetermination in the transition to error catastrophe: systematic elimination of lymphocytic choriomeningitis virus through mutagenesis does not correlate linearly with large increases in mutant spectrum complexity. Proc. Natl. Acad. Sci. USA 2002, 99, 12938–12943. [Google Scholar] [CrossRef]
- Grande-Pérez, A.; Gómez-Mariano, G.; Lowenstein, P.R.; Domingo, E. Mutagenesis-induced, large fitness variations with an invariant arenavirus consensus genomic nucleotide sequence. J. Virol. 2005, 79, 10451–10459. [Google Scholar] [CrossRef]
- Ruiz-Jarabo, C.M.; Ly, C.; Domingo, E.; de la Torre, J.C. Lethal mutagenesis of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV). Virology 2003, 308, 37–47. [Google Scholar] [CrossRef]
- Martin, V.; Abia, D.; Domingo, E.; Grande-Perez, A. An interfering activity against lymphocytic choriomeningitis virus replication associated with enhanced mutagenesis. J. Gen. Virol. 2010, 91, 990–1003. [Google Scholar] [CrossRef]
- Moreno, H.; Gallego, I.; Sevilla, N.; de la Torre, J.C.; Domingo, E.; Martin, V. Ribavirin can be mutagenic for arenaviruses. J. Virol. 2011, 85, 7246–7255. [Google Scholar] [CrossRef]
- Moreno, H.; Tejero, H.; de la Torre, J.C.; Domingo, E.; Martin, V. Mutagenesis-mediated virus extinction: virus-dependent effect of viral load on sensitivity to lethal defection. PLoS One 2012, 7, e32550. [Google Scholar]
- Graci, J.D.; Cameron, C.E. Therapeutically targeting RNA viruses via lethal mutagenesis. Future Virol. 2008, 3, 553–566. [Google Scholar] [CrossRef]
- Eigen, M.; Schuster, P. The hypercycle. A principle of natural self-organization. Springer: Berlin, 1979. [Google Scholar]
- Biebricher, C.K.; Eigen, M. The error threshold. Virus Res. 2005, 107, 117–127. [Google Scholar] [CrossRef]
- Clay, P.G.; McRae, M.; Laurent, J.P. Safety, Tolerability, and Pharmacokinetics of KP-1461 in Phase I Clinical Studies: A Single Oral Dose Study in Non-HIV-Infected Adults, and a 14-Day Dose-Escalating Study in Highly Antiretroviral-Experienced HIV-Infected Adults. J. Int. Assoc. Physicians AIDS Care (Chic) 2011, 10, 232–238. [Google Scholar] [CrossRef]
- Harris, K.S.; Brabant, W.; Styrchak, S.; Gall, A.; Daifuku, R. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antiviral Res. 2005, 67, 1–9. [Google Scholar] [CrossRef]
- Hicks, C.; Clay, P.; Redfield, R.; Lalezari, J.; Liporace, R.; Schneider, S.; Sension, M.; McRae, M.; Laurent, J.P. Safety, Tolerability, and Efficacy of KP-1461 as Monotherapy for 124 Days in Antiretroviral-Experienced, HIV Type 1-Infected Subjects. AIDS research and human retroviruses 2012, in press. [Google Scholar]
- Harki, D.A.; Graci, J.D.; Edathil, J.P.; Castro, C.; Cameron, C.E.; Peterson, B.R. Synthesis of a universal 5-nitroindole ribonucleotide and incorporation into RNA by a viral RNA-dependent RNA polymerase. Biochemistry 2007, 8, 1359–1362. [Google Scholar]
- Harki, D.A.; Graci, J.D.; Galarraga, J.E.; Chain, W.J.; Cameron, C.E.; Peterson, B.R. Synthesis and antiviral activity of 5-substituted cytidine analogues: identification of a potent inhibitor of viral RNA-dependent RNA polymerases. J. Med. Chem. 2006, 49, 6166–6169. [Google Scholar] [CrossRef]
- Harki, D.A.; Graci, J.D.; Korneeva, V.S.; Ghosh, S.K.; Hong, Z.; Cameron, C.E.; Peterson, B.R. Synthesis and antiviral evaluation of a mutagenic and non-hydrogen bonding ribonucleoside analogue: 1-beta-D-Ribofuranosyl-3-nitropyrrole. Biochemistry 2002, 41, 9026–9033. [Google Scholar]
- Chu, C.K. Recent advances in nucleosides: Chemistry. and chemotherapy; Elsevier: Amsterdam, 2002. [Google Scholar]
- Mullins, J.I.; Heath, L.; Hughes, J.P.; Kicha, J.; Styrchak, S.; Wong, K.G.; Rao, U.; Hansen, A.; Harris, K.S.; Laurent, J.P.; Li, D.; Simpson, J.H.; Essigmann, J.M.; Loeb, L.A.; Parkins, J. Mutation of HIV-1 genomes in a clinical population treated with the mutagenic nucleoside KP1461. PLoS One 2011, 6, e15135. [Google Scholar]
- Scheidel, L.M.; Durbin, R.K.; Stollar, V. Sindbis virus mutants resistant to mycophenolic acid and ribavirin. Virology 1987, 158, 1–7. [Google Scholar] [CrossRef]
- Scheidel, L.M.; Stollar, V. Mutations that confer resistance to mycophenolic acid and ribavirin on Sindbis virus map to the nonstructural protein nsP1. Virology 1991, 181, 490–499. [Google Scholar] [CrossRef]
- Rosenblum, C.I.; Stollar, V. SVMPA, a mutant of sindbis virus resistant to mycophenolic acid and ribavirin, shows an increased sensitivity to chick interferon. Virology 1999, 259, 228–233. [Google Scholar] [CrossRef]
- Agudo, R.; Ferrer-Orta, C.; Arias, A.; de la Higuera, I.; Perales, C.; Perez-Luque, R.; Verdaguer, N.; Domingo, E. A multi-step process of viral adaptation to a mutagenic nucleoside analogue by modulation of transition types leads to extinction-escape. PLoS Pathog. 2010, 6, e1001072. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Kirkegaard, K. Increased fidelity reduces poliovirus fitness under selective pressure in mice. PLoS Pathog. 2005, 1, 102–110. [Google Scholar]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef]
- Arias, A.; Arnold, J.J.; Sierra, M.; Smidansky, E.D.; Domingo, E.; Cameron, C.E. Determinants of RNA-dependent RNA polymerase (in)fidelity revealed by kinetic analysis of the polymerase encoded by a foot-and-mouth disease virus mutant with reduced sensitivity to ribavirin. J. Virol. 2008, 82, 12346–12355. [Google Scholar]
- Ferrer-Orta, C.; Sierra, M.; de la Higuera, I.; Agudo, R.; Arias, A.; Perez-Luque, R.; Escarmis, C.; Domingo, E.; Verdaguer, N. Structure of foot-and-mouth disease virus mutant polymerases with reduced sensitivity to ribavirin. J. Virol. 2010, 84, 6188–6199. [Google Scholar]
- Sierra, M.; Airaksinen, A.; González-López, C.; Agudo, R.; Arias, A.; Domingo, E. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: implications for error catastrophe. J. Virol. 2007, 81, 2012–2024. [Google Scholar] [CrossRef]
- Feigelstock, D.A.; Mihalik, K.B.; Feinstone, S.M. Selection of hepatitis C virus resistant to ribavirin. Virology journal 2011, 8, 402. [Google Scholar] [CrossRef]
- Coffey, L.L.; Beeharry, Y.; Borderia, A.V.; Blanc, H.; Vignuzzi, M. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16038–16043. [Google Scholar]
- Pfeiffer, J.K.; Kirkegaard, K. Ribavirin resistance in hepatitis C virus replicon-containing cell lines conferred by changes in the cell line or mutations in the replicon RNA. J. Virol. 2005, 79, 2346–2355. [Google Scholar] [CrossRef]
- Levi, L.I.; Gnadig, N.F.; Beaucourt, S.; McPherson, M.J.; Baron, B.; Arnold, J.J.; Vignuzzi, M. Fidelity variants of RNA dependent RNA polymerases uncover an indirect, mutagenic activity of amiloride compounds. PLoS Pathog. 2010, 6, e1001163. [Google Scholar] [CrossRef]
- Young, K.C.; Lindsay, K.L.; Lee, K.J.; Liu, W.C.; He, J.W.; Milstein, S.L.; Lai, M.M. Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology 2003, 38, 869–878. [Google Scholar]
- Grande-Pérez, A.; Lazaro, E.; Lowenstein, P.; Domingo, E.; Manrubia, S.C. Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. USA 2005, 102, 4448–4452. [Google Scholar]
- Eigen, M. Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. USA 2002, 99, 13374–13376. [Google Scholar] [CrossRef]
- Holland, J.J.; Domingo, E.; de la Torre, J.C.; Steinhauer, D.A. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J. Virol. 1990, 64, 3960–3962. [Google Scholar]
- Loeb, L.A.; Essigmann, J.M.; Kazazi, F.; Zhang, J.; Rose, K.D.; Mullins, J.I. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. USA 1999, 96, 1492–1497. [Google Scholar]
- Sierra, S.; Dávila, M.; Lowenstein, P.R.; Domingo, E. Response of foot-and-mouth disease virus to increased mutagenesis. Influence of viral load and fitness in loss of infectivity. J. Virol. 2000, 74, 8316–8323. [Google Scholar]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.N.; Hong, Z.; Andino, R.; Cameron, C.E. The broad-spectrum antiviral ribonucleotide, ribavirin, is an RNA virus mutagen. Nature Medicine 2000, 6, 1375–1379. [Google Scholar] [CrossRef]
- Perales, C.; Agudo, R.; Tejero, H.; Manrubia, S.C.; Domingo, E. Potential benefits of sequential inhibitor-mutagen treatments of RNA virus infections. PLoS Pathog. 2009, 5, e1000658. [Google Scholar] [CrossRef]
- Perales, C.; Iranzo, J.; Manrubia, S.C.; Domingo, E. The impact of quasispecies dynamics on the use of therapeutics. Trends in microbiology 2012, in press. [Google Scholar]
- Iranzo, J.; Perales, C.; Domingo, E.; Manrubia, S.C. Tempo and mode of inhibitor-mutagen antiviral therapies: A multidisciplinary approach. Proc. Natl. Acad. Sci. USA 2011, 108, 16008–16013. [Google Scholar]
- Dapp, M.J.; Clouser, C.L.; Patterson, S.; Mansky, L.M. 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J. Virol. 2009, 83, 11950–11958. [Google Scholar] [CrossRef]
- Iranzo, J.; Manrubia, S.C. Stochastic extinction of viral infectivity through the action of defectors. Europhys. Lett. 2009, 85, 18001. [Google Scholar] [CrossRef]
- Turner, P.E.; Chao, L. Prisoner's dilemma in an RNA virus. Nature 1999, 398, 441–443. [Google Scholar] [CrossRef]
- Wilke, C.O.; Reissig, D.D.; Novella, I.S. Replication at periodically changing multiplicity of infection promotes stable coexistence of competing viral populations. Evolution Int. J. Org. Evolution 2004, 58, 900–905. [Google Scholar]
- Perales, C.; Mateo, R.; Mateu, M.G.; Domingo, E. Insights into RNA virus mutant spectrum and lethal mutagenesis events: replicative interference and complementation by multiple point mutants. J. Mol. Biol. 2007, 369, 985–1000. [Google Scholar] [CrossRef]
- Youngner, J.S.; Whitaker-Dowling, P. Interference. In Encyclopedia of Virology; Granoff, A., Webster, R.G., Eds.; Academic Press: San Diego, California, 1999; Vol. 2, pp. 850–854. [Google Scholar]
- de la Torre, J.C.; Holland, J.J. RNA virus quasispecies populations can suppress vastly superior mutant progeny. J. Virol. 1990, 64, 6278–6281. [Google Scholar]
- Chumakov, K.M.; Powers, L.B.; Noonan, K.E.; Roninson, I.B.; Levenbook, I.S. Correlation between amount of virus with altered nucleotide sequence and the monkey test for acceptability of oral poliovirus vaccine. Proc. Natl. Acad. Sci. USA 1991, 88, 199–203. [Google Scholar]
- Crowder, S.; Kirkegaard, K. Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nature Genetics 2005, 37, 701–709. [Google Scholar] [CrossRef]
- González-López, C.; Arias, A.; Pariente, N.; Gómez-Mariano, G.; Domingo, E. Preextinction viral RNA can interfere with infectivity. J. Virol. 2004, 78, 3319–3324. [Google Scholar] [CrossRef]
- Perales, C.; Lorenzo-Redondo, R.; López-Galíndez, C.; Martínez, M.A.; Domingo, E. Mutant spectra in virus behavior. Future Virology 2010, 5, 679–698. [Google Scholar] [CrossRef]
- Ojosnegros, S.; Perales, C.; Mas, A.; Domingo, E. Quasispecies as a matter of fact: viruses and beyond. Virus. Res. 2011, 162, 203–215. [Google Scholar] [CrossRef]
- Xiang, D.; Sharma, V.R.; Freter, C.E.; Yan, J. Anti-tumor Monoclonal Antibodies in Conjunction with beta-Glucans: a Novel Anti-cancer Immunotherapy. Current medicinal chemistry 2012, in press. [Google Scholar]
- Murphy, C.G.; Morris, P.G. Recent advances in novel targeted therapies for HER2-positive breast cancer. Anti-cancer drugs 2012, 23, 765–776. [Google Scholar] [CrossRef]
- Pariente, N.; Sierra, S.; Lowenstein, P.R.; Domingo, E. Efficient virus extinction by combinations of a mutagen and antiviral inhibitors. J. Virol. 2001, 75, 9723–9730. [Google Scholar] [CrossRef]
- Tapia, N.; Fernandez, G.; Parera, M.; Gomez-Mariano, G.; Clotet, B.; Quinones-Mateu, M.; Domingo, E.; Martinez, M.A. Combination of a mutagenic agent with a reverse transcriptase inhibitor results in systematic inhibition of HIV-1 infection. Virology 2005, 338, 1–8. [Google Scholar] [CrossRef]
- Agudo, R.; Arias, A.; Pariente, N.; Perales, C.; Escarmis, C.; Jorge, A.; Marina, A.; Domingo, E. Molecular characterization of a dual inhibitory and mutagenic activity of 5-fluorouridine triphosphate on viral RNA synthesis. Implications for lethal mutagenesis. J. Mol. Biol. 2008, 382, 652–666. [Google Scholar] [CrossRef]
- Perales, C.; Agudo, R.; Domingo, E. Counteracting quasispecies adaptability: extinction of a ribavirin-resistant virus mutant by an alternative mutagenic treatment. PLoS ONE 2009, 4, e5554. [Google Scholar]
- Dapp, M.J.; Holtz, C.M.; Mansky, L.M. Concomitant lethal mutagenesis of human immunodeficiency virus type 1. J. Mol. Biol. 2012, 419, 158–170. [Google Scholar] [CrossRef]
- Torella, J.P.; Chait, R.; Kishony, R. Optimal drug synergy in antimicrobial treatments. PLoS computational biology 2010, 6, e1000796. [Google Scholar] [CrossRef]
- Pasquato, A.; Rochat, C.; Burri, D.J.; Pasqual, G.; de la Torre, J.C.; Kunz, S. Evaluation of the anti-arenaviral activity of the subtilisinkexin isozyme-1/site-1 protease inhibitor PF-429242. Virology 2012, 423, 14–22. [Google Scholar] [CrossRef]
- Kolokoltsov, A.A.; Adhikary, S.; Garver, J.; Johnson, L.; Davey, R.A.; Vela, E.M. Inhibition of Lassa virus and Ebola virus infection in host cells treated with the kinase inhibitors genistein and tyrphostin. Arch. Virol. 2012, 157, 121–127. [Google Scholar] [CrossRef]
- Lee, A.M.; Pasquato, A.; Kunz, S. Novel approaches in anti-arenaviral drug development. Virology 2011, 411, 163–169. [Google Scholar] [CrossRef]
- Narayanan, A.; Bailey, C.; Kashanchi, F.; Kehn-Hall, K. Developments in antivirals against influenza, smallpox and hemorrhagic fever viruses. Expert. Opin. Investig. Drugs 2011, 20, 239–254. [Google Scholar] [CrossRef]
- Neuman, B.W.; Bederka, L.H.; Stein, D.A.; Ting, J.P.; Moulton, H.M.; Buchmeier, M.J. Development of peptide-conjugated morpholinooligomers as pan-arenavirus inhibitors. Antimicrob. Agents Chemother. 2011, 55, 4631–4638. [Google Scholar] [CrossRef]
- Urata, S.; Yun, N.; Pasquato, A.; Paessler, S.; Kunz, S.; de la Torre, J.C. Antiviral activity of a small-molecule inhibitor of arenavirus glycoprotein processing by the cellular site 1 protease. J. Virol. 2011, 85, 795–803. [Google Scholar]
- Eigen, M. Self-organization of matter and the evolution of biological macromolecules. Naturwissenschaften. 1971, 58, 465–523. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moreno, H.; Grande-Pérez, A.; Domingo, E.; Martín, V. Arenaviruses and Lethal Mutagenesis. Prospects for New Ribavirin-based Interventions. Viruses 2012, 4, 2786-2805. https://doi.org/10.3390/v4112786
Moreno H, Grande-Pérez A, Domingo E, Martín V. Arenaviruses and Lethal Mutagenesis. Prospects for New Ribavirin-based Interventions. Viruses. 2012; 4(11):2786-2805. https://doi.org/10.3390/v4112786
Chicago/Turabian StyleMoreno, Héctor, Ana Grande-Pérez, Esteban Domingo, and Verónica Martín. 2012. "Arenaviruses and Lethal Mutagenesis. Prospects for New Ribavirin-based Interventions" Viruses 4, no. 11: 2786-2805. https://doi.org/10.3390/v4112786